

Abstract
The conventional use of E. coli system for protein expression is limited to non-glycosylated proteins. While yeast, insect and mammalian systems are available to produce heterologous glycoproteins, developing an engineered E. coli-based glycosylation platform will provide a faster, more economical, and more convenient alternative. In this work, we present a two-step approach for production of a homogeneously glycosylated eukaryotic protein using the E. coli expression system. Human interferon α-2b (IFNα) is used as a model protein to illustrate this glycosylation scheme. In the first step, the N-glycosyltransferase from Actinobacillus pleuropneumoniae (ApNGT) is co-expressed for in vivo transfer of a glucose residue to IFNα at an NX(S/T) N-glycosylation sequon. Several E. coli systems were examined to evaluate the efficiency of IFNα N-glucosylation, In the second step, the N-glucosylated protein is efficiently elaborated with biantennary sialylated complex-type N-glycan using an in vitro chemoenzymatic method. The N-glycosylated IFNα product was found to be biologically active and displayed significantly improved proteolytic stability. This work presents a feasible E. coli-based glycosylation machinery for producing therapeutic eukaryotic glycoproteins.
Keywords: E. coli glycosylation, N-glycosyltransferase, endoglycosidase, transglycosylation, sugar oxazoline, eukaryotic protein
1. Introduction
N-glycosylation is a key post-translational modification that can significantly influence the structural and functional properties of proteins (1). In the case of therapeutic proteins, glycosylation can govern their biological activity and pharmacokinetic attributes (2). Efficient post-translational modifications and protein folding make eukaryotic systems attractive hosts for the expression of recombinant therapeutic proteins (3). However, the high cost of production, longer culture duration and meticulous handling of eukaryotic cells make the bacterial system an appealing alternative, with E. coli acting as a model organism (4,5). Indeed, most therapeutic non-glycosylated proteins are expressed using E. coli and yeast expression systems. However, E. coli expression of heterologous glycoproteins is problematic, as E. coli lacks a protein glycosylation machinery.
Toward this end, several research groups have attempted to design a glycosylation system in E. coli. In collaboration with Aebi group, we have demonstrated a combined method that involves the functional transfer of a glycoengineered Campylobacter jejuni glycosylation machinery into E. coli to produce N-glycosylated eukaryotic proteins (6). While this design allows N-glycosylation of the protein, the resulting glycoprotein contains an Asn-linked (GalNAc)5GlcNAc-glycan, which is not a eukaryotic N-glycan. Further in vitro trimming of the outer GalNAc moieties by a bacterial α-N-acetylgalactosaminidase is required to generate the GlcNAc-protein, which then serves as the key intermediate for an endo-²-N-acetylglucosaminidase (ENGase)-catalyzed glycosylation to afford a eukaryotic glycoprotein. Moreover, it has been observed that expression of a heterologous eukaryotic glycoprotein such as the CH2 domain of immunoglobulin G (IgG) and a single-chain antibody F8 results in a low glycosylation efficiency (5-10% yield), which represents a bottleneck of the method (6). In another study, DeLisa and co-workers have reported a bottom-up glycoengineering method for producing eukaryotic N-glycoproteins in E. coli (7). In this approach, the genes responsible for constructing the N-glycan core (Man3GlcNAc2) are engineered into E. coli to build up the N-glycan glycolipid precursor. Then, PglB, the key bacterial oligosaccharyltransferase from C. jejuni is co-expressed to produce a recombinant glycosylated protein carrying a Man3GlcNAc2 N-glycan. However, the glycosylation yield is very low (ca. 1%), probably due to the low efficiency of the PglB-catalyzed transfer of a eukaryotic N-glycan from the corresponding glycolipid precursor (7). Further study to improve the glycosylation efficiency of this method is required.
The discovery of a cytoplasmic N-glycosyltransferase in Actinobacillus pleuropneumoniae (ApNGT) that can transfer glucose (Glc) and galactose from a sugar donor to peptides and proteins at NX(S/T) N-glycosylation site represents an alternate avenue to protein glycosylation (8,9). Several groups have demonstrated that ApNGT can transfer glucose from UDP-Glc to polypeptides and proteins carrying the consensus NX(S/T) N-glycosylation sequence (where X is any natural amino acid except proline). We have previously reported that in vitro transfer of glucose to peptides by ApNGT, coupled with subsequent sugar chain elongation provides an efficient way to synthesize glycopeptides carrying a full-length N-glycan (10). Peng George Wang and co-workers have designed an engineered ApNGT (Q469A) that displayed a relaxed substrate specificity and higher efficiency of in vitro glucosylation with peptides and a bacterial protein containing multiple N-glycosylation sites (11). In a later study, they have employed ApNGT Q469A, which can accept UDP-glucosamine (GlcN) as a donor substrate, to generate GlcN-peptides. Acetylation of the GlcN-peptides by an acetyltransferase resulted in GlcNAc-peptides, which were then extended with complex-type N-glycans by EndoM-N175Q to produce glycopeptides (12). More recently, Jewett, MrKsich, and co-workers have explored the peptide acceptor sequence preference of a panel of NGT variants, which provides an exciting opportunity for sequential glycosylation of a protein to make glycoproteins with multiple glycosylation sites (13,14).
For N-glucosylation in E. coli, Aebi and co-workers have reported that co-expression of ApNGT in E. coli is able to transfer glucose to bacterial adhesins (15). In addition, N-glucosylation of heterologous proteins such as human erythropoietin (EPO) was detected in E. coli when ApNGT is co-expressed, although the human EPO forms inclusion bodies in E. coli and the extent of glucosylation is not determined (15). More recently, Aebi and co-workers have shown that co-expression of ApNGT and three other glycosyltransferases in E. coli cytoplasm is able to generate a polysialylated protein (16).
While these studies have examined the application of ApNGT for in vivo glucosylation and explored further glycan modifications, attachment of natural eukaryotic N-glycans to these E. coli-expressed Glc-containing proteins has not been attempted so far. A closely related effort of transferring eukaryotic N-glycan to O-linked GlcNAc in proteins has been studied by Wang and co-workers (17). O-GlcNAc transferase (OGT) was used to generate O-GlcNAc containing bovine protein in E. coli and a glycosynthase EndoM-N175Q was tested for in vitro transfer of sialylated complex type glycan, resulting in a 30% yield.
In the present study, we report a case study of in vivo glucosylation of a eukaryotic protein by ApNGT in E. coli coupled with in vitro chemoenzymatic sugar chain elongation to produce a heterologous glycoprotein carrying a sialylated N-glycan. Human interferon α-2b (hIFNα-2b, referred to as IFNα hereafter) was used as a model eukaryotic protein and the overall experimental design is shown in Figure 1. Upon successful in vivo glucosylation with ApNGT in E. coli, EndoCC-N180H glycosynthase was found to efficiently transfer a sialylated N-glycan to IFNα-Glc. The purified sialylated IFNα displayed the expected anti-proliferative activity in a cell proliferation test and a significantly enhanced stability towards protease degradation.
Figure 1. Schematic representation of in vivo ApNGT-catalyzed glucosylation and in vitro chemoenzymatic transglycosylation of eukaryotic proteins in E. coli.
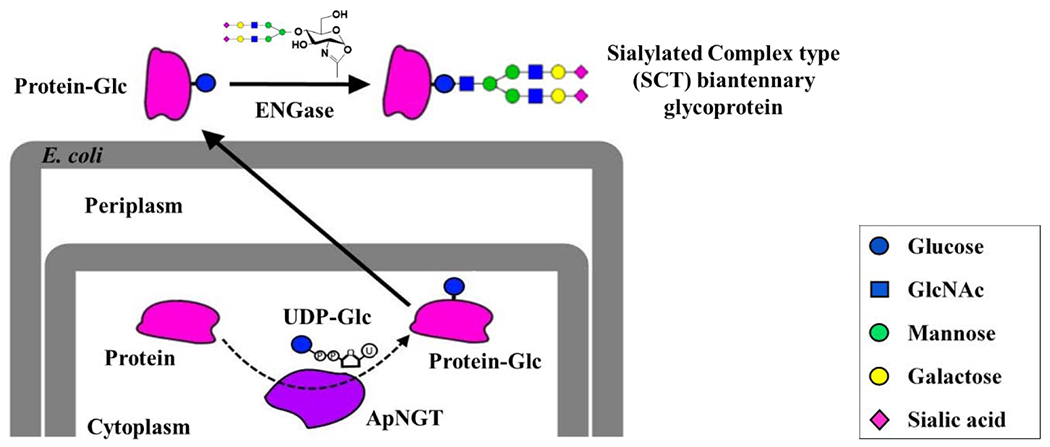
The N-glycosylated protein contains an innermost glucose instead of GlcNAc present in natural N-glycans.
2. Results and Discussion
2.1. Introduction of N-glycosylation sites into IFNα
IFNα is a 19.2 kDa α-helical protein consisting of 165 amino acids with a single O-glycosylation site at Thr-106. It is a cytokine that possesses anti-viral and anti-proliferative properties and is commonly used to treat Hepatitis C and several forms of cancers (18). N-glycosylation of a modified IFNα variant with four engineered N-glycan sites that was expressed in a mammalian system shows up to a 25-fold increase in its serum half-life (19). Based on this study, we chose the Pro-4 (P4) and/or Gln-158 (Q158) as the position to introduce an N-glycosylation site to test ApNGT-catalyzed in vivo glucosylation in E. coli. P4 lies in the flexible region close to the protein N-terminus and Q158 is at the end of an α-helix close to the C-terminus. Each of the chosen sites were modified to attain an ANAT sequence at the X−1NX+1X+2 positions to achieve high efficiency of glucosylation by ApNGT, as suggested by the previous report (15). The structure of IFNα and the glycoengineered sequences are shown in Figure 2.
Figure 2. The structure of IFNα and the introduced N-glycosylation site.
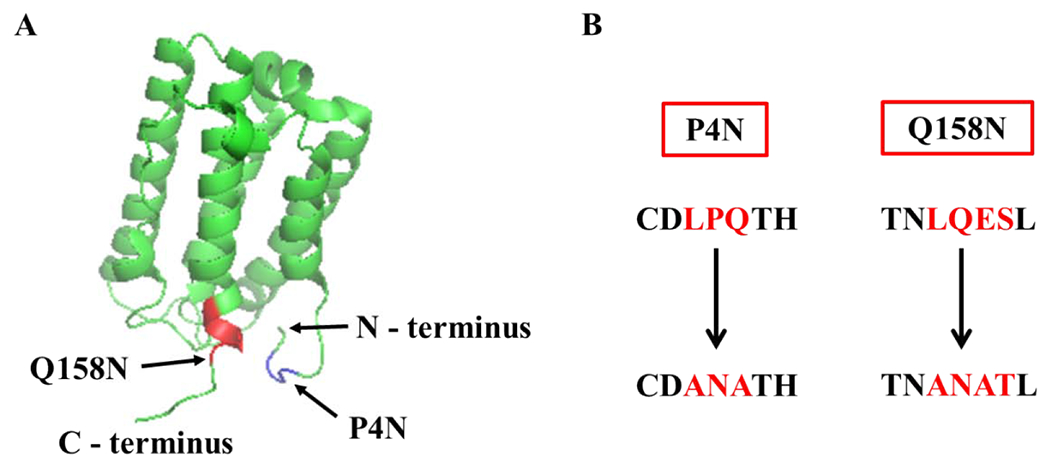
A) The structure of human IFNα (PDB code 2hym). The two positions chosen for site-directed mutagenesis are highlighted. Mutagenesis at P4 is shown in blue and at Q158 in red. B) The engineered N-glycosylation sites. The modified amino acids in both the mutants are highlighted in red.
2.2. In vivo glucosylation of GST-IFNα in E. coli
In the initial study, IFNα wild-type (WT) and mutant proteins were expressed as fusion proteins with a Glutathione-S-Transferase (GST) tag at the N-terminus to enable soluble expression in E. coli cytoplasm (20). The fusion protein design allows for release of IFNα by thrombin cleavage at an inserted thrombin protease site. The mutants, GST-IFNα-P4N and GST-IFNα-Q158N were each co-expressed with ApNGT for in vivo glucosylation in E. coli with a protein yield of 50-70 mg/L. Mass spectrometric analyses of the purified fusion proteins showed completely glucosylated GST-IFNα-P4N (ESI-MS: Calculated, M = 45638 Da; found, M = 45639 Da after deconvolution) (Figure 3A) and about 80% glucosylated GST-IFNα-Q158N (ESI-MS: Calculated, M = 45620 Da; found, M = 45621 after deconvolution) (Figure 3B). SDS-PAGE profiles of the purified fusion proteins are shown in Figure S1.
Figure 3. Co-expression of GST-IFNα mutants with ApNGT in E. coli.
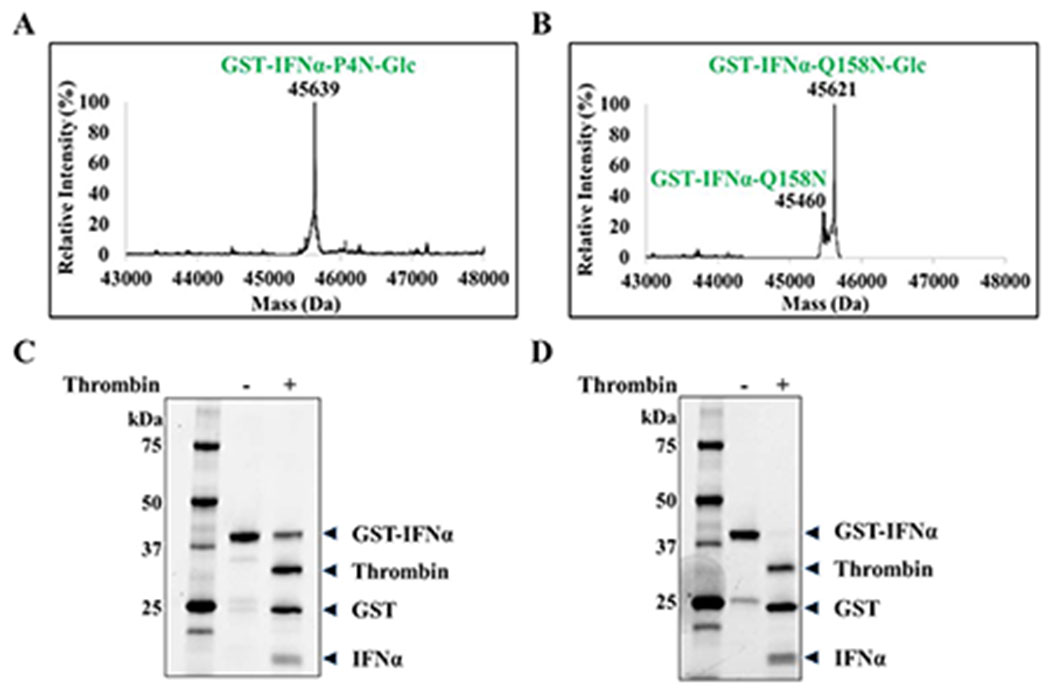
A) Deconvoluted LC-MS profiles of GST-IFNα-P4N. Calculated mass with two disulfide bonds = 45476 Da, calculated mass post glucosylation = 45638 Da. (M+H)+ peak observed at 45639 Da. B) Deconvoluted LC-MS profiles of GST-IFNα-Q158N. Calculated mass with two disulfide bonds = 45458 Da, calculated mass post glucosylation = 45620 Da. (M+H)+ peaks observed at 45460 and 45621 Da. C) SDS-PAGE showing thrombin cleavage of GST-IFNα-WT expressed in E. coli BL21(DE3) with overnight incubation. D) SDS-PAGE showing thrombin cleavage of GST-IFNα-WT expressed in E. coli Origami 2 (DE3) with overnight incubation.
Contrary to the efficient glucosylation of GST-IFNα fusion proteins, protease release of the GST tag posed some unexpected challenges. We found that the two mutants and the wild-type GST-IFNα, expressed in E. coli BL21(DE3) strain, could not be efficiently cleaved by the protease thrombin. The reaction, when carried out with an excess of enzyme showed a maximum of 50% cleavage with overnight incubation (Figure 3C) and increased only up to 70% when prolonged to incubation for two days. Intensive optimization of the reaction conditions did not show any further improvement in the GST tag removal. A possible explanation of this observation is improper folding of the fusion protein making the cleavage site hard to access by thrombin. In common E. coli strains, the cytoplasm hosts a reductive environment which may prevent the formation of disulfide bonds in proteins, thus causing misfolding of heterologous proteins. To test such a possibility, we expressed GST-IFNα in an Origami 2 (DE3) strain, which carries mutations in thioredoxin reductase (trxB) and glutathione reductase (gor) genes to provide an oxidative environment in the cytoplasm. Encouragingly, GST-IFNα expressed in Origami showed complete cleavage of GST tag by thrombin with overnight incubation under previously used reaction conditions (Figure 3D). This observation prompted us to perform refolding of the soluble GST-IFNα expressed in E. coli BL21(DE3) (21) and to examine thrombin cleavage of the refolded protein. Under the previously described reaction conditions, ca. 70% release of the GST tag was observed with overnight incubation indicating a moderate improvement in the protease cleavage.
To test in vivo glucosylation in Origami 2 (DE3) strain, GST-IFNα-P4N and GST-IFNα-Q158N were co-expressed with ApNGT. Surprisingly, neither of the proteins showed any glucose transfer (data not shown). To probe this observation further, the two fusion proteins were also co-expressed with ApNGT Q469A, as the mutant enzyme has shown a broader substrate specificity and better glucosylation efficiency in a previous report (11). Unfortunately, neither of the fusion proteins showed any glucosylation. While this result was unexpected, it provides an important implication in the mechanism of glucosylation by ApNGT in E. coli. It has been suggested that ApNGT requires substrate proteins in a not fully folded state for in vivo glucosylation to occur (15). The lack of in vivo glucosylation in Origami 2 (DE3) strain compared to the highly efficient glucose transfer in BL21(DE3) supports this hypothesis and suggests co-translational glucosylation by ApNGT of the nascent recombinant protein (IFNα).
In parallel to the examination of GST fusion protein, we also tested in vivo glucosylation of IFNα without any fusion tag. This work was focused on one of the mutants, Q158N. IFNα-Q158N was cloned into pET28a vector under T7 promoter for expression in E. coli BL21(DE3). We purified IFNα from inclusion bodies (IBs) using established refolding protocols (21–23). A final yield of ca. 120 mg/L was achieved. SDS-PAGE profile of the purified refolded protein is shown in Figure S2A. Interestingly, co-expression of ApNGT and IFNα-Q158N showed a relatively low efficiency of glucosylation in IBs with a maximum glucose transfer of 35% under optimized culture conditions (Figure S3). The low glucose transfer efficiency could be due to insufficient exposure of IFNα expressed in IBs to the cytoplasmic ApNGT.
Finally, we attempted to express soluble glucosylated 6xHis-IFNα-Q158N in E. coli. by optimizing the conditions. First, we tried to induce protein production at a relatively low temperature (16-25°C), which resulted in soluble expression of 6xHis-IFNα-Q158N with a yield of ca. 20 mg/L. SDS-PAGE profile of the purified protein is shown in Figure S2B. Second, to reach optimum glucose transfer, we executed stepwise induction of ApNGT and IFNα-Q158N. ApNGT was induced by arabinose 2 h prior to the induction of IFNα by IPTG. In this way, a maximum glucosylation of 55% was achieved at 16°C (Figure 4A). In comparison, co-induction of ApNGT and IFNα-Q158N at 16°C resulted in 40% glucosylation. Comparing these results to the 80% glucosylation of GST-IFNα-Q158N (containing the same acceptor sequence), we speculate that the local conformation of the glycosylation site and the protein folding kinetics likely affect the efficiency of glucose transfer by ApNGT.
Figure 4. LC-MS analysis of in vivo glucosylation of IFNα-Q158N, subsequent in vitro transglycosylation, and enrichment of the transglycosylation product.
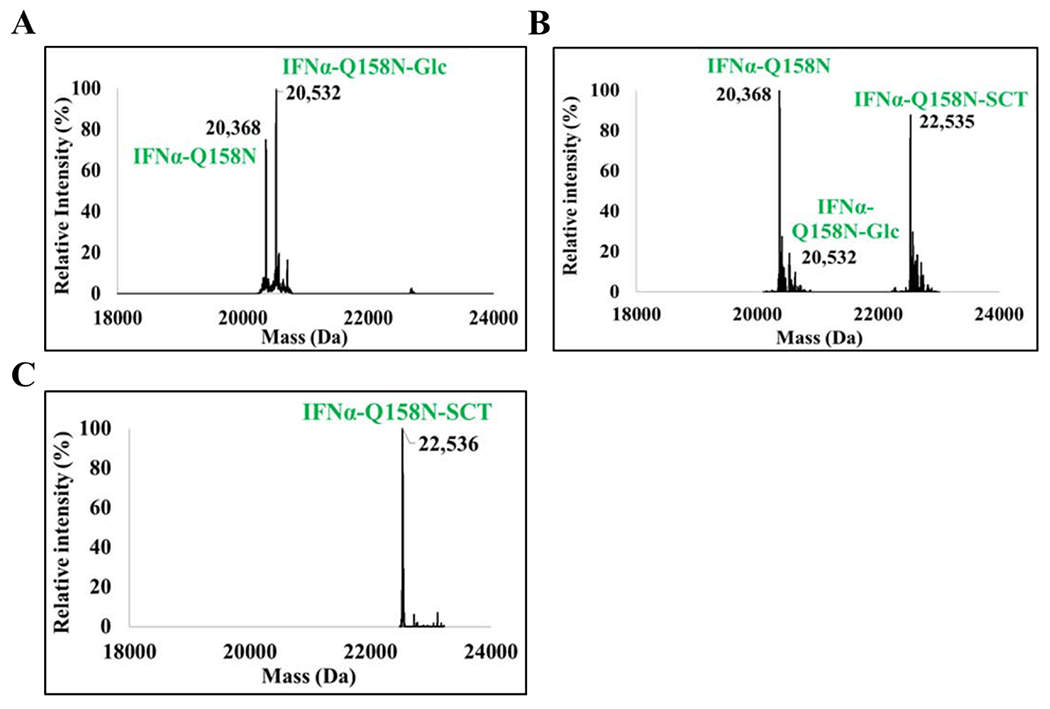
Deconvoluted LC-MS profile of A) IFNα-Q158N co-expressed with ApNGT in E. coli. Calculated mass of IFNα-Q158N with two disulfide bonds, M = 20370 Da. Calculated mass of the glucosylated protein, M = 20532 Da. Additional higher mass peak observed in the profile is an unidentified non-glucose adduct. B) In vitro chemoenzymatic transglycosylation of IFNα-Q158N by EndoCC-N180H. Calculated mass of IFNα-Q158N-SCT, M = 22534 Da. 90% transglycosylation was observed. It should be noted that the protein is only 55% glucosylated resulting in a total yield of 50% sialylated protein. C) The IFNα-Q158N-SCT enriched using Lectenz affinity chromatography. The IFNα-Q158N referred to in this figure is the His-tagged protein.
2.3. In vitro chemoenzymatic transglycosylation of glucosylated IFNα-Q158N
Several glycosynthases, which are endoglycosidase mutants that demonstrate reduced product hydrolysis activity but are able to use sugar oxazolines as an activated donor substrate for transglycosylation, can transfer N-glycan en bloc to GlcNAc- or glucose-linked peptides and proteins to form homogeneous glycoproteins (24). Such in vitro glycoengineering of glycoproteins relies strongly on the substrate specificities of the parent endoglycosidases. Glycosynthases that can transfer different glycoforms to peptide and protein substrates have been developed. Previously we have shown that EndoA or EndoM mutants could transfer N-glyeans to glucose-Asn-linked polypeptides that are generated by glucosylation with ApNGT (10). However, very few studies have reported the enzymatic N-glycan transfer to intact glucose-proteins. To examine the in vitro enzymatic N-glycan transfer, we chose the glucosylated 6xHis-IFNα-Q158N recombinant protein as the acceptor substrate, which contains ca. 55% glucosylation (Figure 4A). An initial attempt to use EndoM-N175A and EndoM-N175Q mutants to transfer a sialylated N-glycan to the glucosylated IFNα failed to provide glycosylation product (data not shown). This result is consistent with previous reports that EndoM mutants (N175Q or N175A) can efficiently transfer complex type N-glycans from the corresponding glycan oxazolines to GlcNAc- or Glc-polypeptides, but the EndoM mutants are much less efficient to act on folded and intact GlcNAc-proteins (25,26).
Finally, an efficient in vitro N-glycan transfer was achieved by using the N180H mutant of EndoCC, an endoglycosidase from Coprinopsis cinerea that has been previously reported to be able to efficiently transfer a complex type N-glycan to deglycosylated ribonuclease B (GlcNAc-RNase B) (27). Hence, we tested EndoCC-N180H for the transfer of SCT-glycan to the partially glucosylated 6xHis-IFNα-Q158N. Gratifyingly, the enzyme exhibited high efficiency in transferring SCT-glycan to the glucosylated IFNα substrate, with a 90% yield (Figure 4B). Thus, an E. coli co-expression of IFNα and ApNGT coupled with in vitro sugar chain extension with the EndoCC-N180H provides a feasible approach to produce glycosylated IFNα, which may be applicable to other therapeutic proteins.
After the successful transglycosylation, we attempted to purifty the transglycosylation product with a SiaRich™ Pan-Specific Sialic Acid Affinity Column (Lectenz® Bio). The lectin column is expected to bind to α-2,3/6/8 linked sialic acid conjugates which are then eluted out using high salt concentration. The 6xHis-IFNα-Q158N mixture was applied to the lectin column, then the column was eluted with a gradient of salt solution and two protein fractions were obtained. However, to our surprise, the 6xHis-IFNα-Q158N-SCT was eluted first at a lower salt concentration, followed by the non-glycosylated 6xHis-IFNα-Q158N protein. The results suggest that the sialylated IFNα has only weak binding affinity with the lectin. To verify that the lectin binds to sialylated glycoprotein efficiently, we tested the lectin column with fetuin as a positive control, which contains multiple sialylated N- and O-glycans. As predicted, fetuin bound tightly to the affinity column and was eluted out with only high concentration of salt. The unexpected behavior of the sialylated and non-glycosylated IFNα suggests that the non-glycosylated IFNα exerts a relatively strong non-specific protein-protein interaction with the lectin while the large sialylated glycan attached might cancel out the non-specific protein-protein interaction, although at the same time the lectin-glycan interaction also plays a role in the affinity. The isolated sialylated IFNα (6xHis-IFNα-Q158N-SCT) was confirmed by ESI-MS as a homogeneously glycosylated protein with an expected mass spectrometry profile (Calculated, M = 22534 Da; found, M = 22536 Da; deconvoluted data) (Figure 4C).
2.4. Biological activity and proteolytic stability of glycoengineered IFNα
Glycosylation of proteins is known to partially interfere with receptor binding and reduce the in vitro biological activity of proteins (28). IFNα is a therapeutic protein that exhibits anti-viral and anti-proliferative properties and is used in the treatment of cancers and hepatitis C (18). To test the effect of sialylated glycosylation on the biological activity of IFNα-Q158N, we conducted an in vitro anti-proliferative assay using Burkitt’s lymphoma Daudi cells (19,29). The growth of Daudi cells as a function of IFNα concentration was studied in microtiter plates using a colorimetric method. The anti-proliferative activities of the glucosylated and sialylated 6xHis-IFNα-Q158N were examined alongside a reference standard. Results from duplicate experiments showed the activity of 55% glucosylated 6xHis-IFNα-Q158N to be comparable with that of the reference standard. Attaching a sialylated glycan to the protein showed a 3-fold decrease in its biological activity (Figure 5A, Table S1). This result is consistent with a previous report of IFNα-2b displaying a 10-fold reduction in biological activity upon glycosylation at four engineered sites (19). The loss in activity was however, compensated by a 25-fold increase in serum half-life of the glycosylated IFNα-2b in mice (19). Another study reports a 4-fold reduction in in vitro activity of a recombinant human EPO when attached with two additional N-glycan chains (30). But the increase in sialic acid content shows improved serum half-life and an overall 4-fold increase in the in vivo potency of the modified glycoprotein (30).
Figure 5. Effects of sialylated glycosylation on the anti-proliferative activity and proteolytic stability.
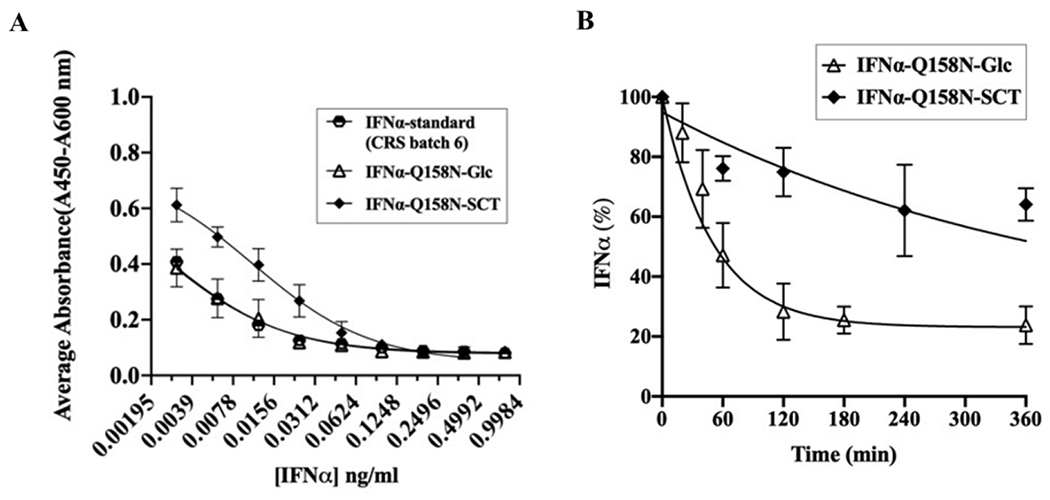
A) In vitro anti-proliferative activity of glucosylated and sialylated IFNα-Q158N in comparison with IFNα reference standard. Data from duplicate experiments were fitted to obtain the dose response curves and half maximal inhibitory concentrations (IC50) of the IFNα proteins. B) In vitro trypsin digestion of glucosylated and sialylated IFNα-Q158N. Protein decay data from duplicate experiments were used to obtain the half-life of the proteins towards trypsin digestion.
In addition to the effect of sialylation on serum half-life of proteins, glycosylation also confers enhanced resistance to protease cleavage, mainly through steric hindrance (31). Glycosylation of IFNγ at Asn-25 has been reported to drastically improve its proteolytic stability towards several proteases in in vitro assays (31). The glycosylated IFNγ retains significant anti-viral activity post protease treatment, unlike the non-glycosylated and N25Q variants of IFNγ. Similarly, O-glycosylation of an engineered insulin at Thr-27 of B-chain doubled its proteolytic stability towards in vitro chymotrypsin treatment (32). To examine the effect of glycosylation on the proteolytic stability of IFNα-Q158N, we studied the half-life of IFNα towards trypsin digestion which has the same cleavage specificity as plasmin found in blood. The decay of the proteins (33) was monitored using LC-MS with the reference standard used as an internal standard for quantitation (32). Indeed, the sialylated IFNα (6xHis-IFNα-Q158N-SCT) showed at least a 7-fold higher resistance to tryptic digestion in comparison to the glucosylated protein (Figure 5B, Table S2). This increased stability is advantageous to IFNα as cytokines are known to be susceptible to proteolytic degradation (34). In summary, these results are in good agreement with the previous reports and indicate that while sialylated glycosylation resulted in some decrease in in vitro anti-proliferative activity, it significantly enhanced the proteolytic stability which could lead to increased overall biological activity in vivo.
3. Conclusion
An E. coli-based method to generate a homogeneously glycosylated eukaryotic protein is described. We used ApNGT co-expression to produce glucosylated human IFNα in E. coli which was then efficiently glycosylated in vitro using EndoCC-N180H to generate homogeneously sialylated IFNα. Biological activity of the purified product was confirmed alongside an IFNα reference standard using an anti-proliferative assay. This study demonstrates the feasibility of utilizing the ApNGT-based glycosylation system in combination with enzymatic sugar chain elongation to generate fully glycosylated heterologous eukaryotic proteins.
4. Experimental procedures
4.1. Gene constructs and plasmids
The coding region of wild type IFNα (Uniprot: P01563, IFNA2_HUMAN, Δ1-23) was modified for a P4N mutation as shown in Figure 2B. Codon-optimized synthetic gene of IFNα-P4N with an N-terminal GST tag, cloned into pGEX-4T-1 between BamHI and NotI sites was procured from GenScript. pGEX-GST-IFNα-WT and pGEX-GST-IFNα-Q158N constructs were generated from pGEX-GST-IFNα-P4N using Q5® Site-Directed Mutagenesis Kit (New England BioLabs) and respective primers. IFNα-Q158N gene was amplified from pGEX-GST-IFNα-Q158N using PfuUltra II Fusion High-fidelity DNA Polymerase (Agilent) and cloned into pET28a(+) vector (Novagen) between NcoI and BamHI sites. N-terminal 6xHis-tag was introduced into pET28a-IFNα-Q158N using the Q5 mutagenesis kit. Wild type ApNGT sequence (Uniprot:A3N2T3, NGT_ACTP2) was cloned into pET45b vector using KpnI/XhoI sites, into pET28a vector using NdeI/XhoI sites and into pBAD33.1 vector using NdeI/SalI sites. pBAD33.1 was a gift from Christian Raetz (Addgene plasmid # 36267; http://n2t.net/addgene:36267;RRID:Addgene_36267 (35).
4.2. Expression and purification of GST-IFNα in E. coli
Overnight grown pre-cultures of E. coli BL21(DE3) or Origami 2 (DE3) cells co-transformed with plasmids pGEX-GST-IFNα (respective mutants or WT) and pET28a-ApNGT were diluted 50 times into Terrific Broth (TB) medium supplemented with 100 μg/ml carbenicillin and 50 μg/ml kanamycin and cultured at 37°C, 250 RPM. The broth was induced with 0.5 mM IPTG when OD600 reached 0.6 - 0.8, and continued to culture overnight at 22°C. Cells were harvested by centrifugation at 4000 RPM for 15 minutes at 4°C. The cell pellet was resuspended in PBS buffer, pH-7.4. Cells were lysed by sonication (12 cycles of 30s burst/30s cooling). The cell lysate was centrifuged at 20000 RPM for 12 minutes at 4°C. The supernatant containing soluble proteins was filtered and loaded onto a Reduced Glutathione resin column (Pierce™ Glutathione Agarose, Cat. # 16100) and allowed to bind overnight at 4°C. The flowthrough was collected and the column was washed with 10 column volumes of PBS buffer. Bound proteins were eluted using 33 mM glutathione in 50 mM Tris-HCl, 200 mM NaCl at pH – 8.0. The eluted proteins were dialyzed against PBS buffer, pH-7.4 at 4°C.
4.3. Removal of GST tag from GST-IFNα-WT by Thrombin
5 μg of GST-IFNα-WT was treated with 2.5 U of Thrombin (Human Plasma, High Activity from Millipore Inc., Cat. # 605195) in PBS buffer, pH – 7.4. The reaction mixture (20 μl) was incubated at 22°C overnight. Samples were analyzed by SDS-PAGE.
4.4. Expression and purification of His-tagged IFNα-Q158N in E. coli
Overnight grown pre-cultures of E. coli BL21(DE3) cells co-transformed with plasmids pET28a-IFNα-Q158N and pBAD33.1-ApNGT were diluted 50 times into TB medium supplemented with 50 μg/ml kanamycin and 25 μg/ml chloramphenicol and cultured at 37°C, 250 RPM. ApNGT expression was induced with 0.4% L-arabinose at OD600 ~ 0.2 two hours prior to IFNα induction with 0.5 mM IPTG at OD600 ~ 0.6 - 0.8 and cultured overnight at 16-25 °C after another addition of 0.4% L-arabinose. Protein purification was done as mentioned in Section 4.2. The supernatant containing His-tagged soluble proteins was filtered and purified using HisTrap™ columns (Cytiva, Cat. # 17524701), following manufacturer’s protocol. The eluted proteins were dialyzed against 50 mM Tris-HCl, 2.5% sucrose, pH-8.5 at 4°C.
4.5. Refolding of IFNα-Q158N expressed in E. coli inclusion bodies
Protein expression was done as outlined in Section 4.2 using constructs pET28a-IFNα-Q158N and pET45b-ApNGT. At the end of culture, cells were harvested by centrifugation at 4000 RPM for 15 minutes at 4°C. The cell pellet was resuspended in 50 mM Tris-HCl, pH-8.5 containing 2 mM Ethylenediaminetetraacetic acid (EDTA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed by sonication (15 cycles of 30s burst/30s cooling). The cell lysate was centrifuged at 10000 g for 20 minutes at 4°C. The cell pellet containing IBs was washed in 50 mM Tris-HCl, pH-8.0 containing 1% Triton X-100 by stirring for 1 h at room temperature. Washed IBs were centrifuged at 10000 g for 10 minutes at 4°C. The IBs were washed again in 50 mM Tris-HCl, pH-8.0 containing 5 M urea, 20 mM DTT by stirring at room temperature for 1 h. This was followed by centrifugation at 15000 g for 30 minutes at 4°C. The washed IBs were then solubilized in 6 M guanidine hydrochloride, 50 mM Tris-HCl, pH-8.0 containing 100 mM BME at room temperature for 1 h. The suspension was centrifuged at 10000 g for 30 minutes at 4°C. Refolding of solubilized IBs was done by dialysis of the supernatant against refolding buffer containing 50 mM Tris-HCl, pH-8.0, 4 M guanidine hydrochloride, 0.2 mM EDTA, 1 mM reduced and 0.1 mM oxidized glutathiones at 4°C. Refolding buffers with decreasing amount of guanidine hydrochloride in steps of 2 M were used for subsequent dialyses. The final buffer contained 50 mM Tris-HCl, 2.5% sucrose, 0.2 mM EDTA at pH-8.5.
4.6. SDS-PAGE sample preparation and analysis
Approximately 1 μg of purified protein diluted in water was mixed with 5 μl of 2X SDS loading dye containing β-mercaptoethanol to make a final volume of 10 μl. The samples were denatured at 95°C for 5-10 minutes and cooled to 4°C before loading. 6 μl of protein ladder (Precision Plus Protein Standards, Bio-Rad, Cat. # 1610363) was loaded for reference. Precast polyacrylamide stain-free gels were used for electrophoresis (Mini-PROTEAN™ TGX Stain-Free™ Protein Gels, Bio-Rad, Cat. # 4568086, 4568106). The gels were run in 1X Tris-Glycine-SDS buffer at 220V for 35 minutes. Gel imaging was done using Gel Doc™ EZ Imager (Bio-Rad).
4.7. Mass spectrometric analysis of IFNα proteins
IFNα samples were prepared in 0.1% formic acid (FA) and injected into a QExactivePlus OrbiTrap mass spectrometer in line with Ultimate 3000 HPLC system (ThermoFisher) for liquid chromatography-electrospray ionization mass spectrometry (LC-MS) analysis. Protein chromatography was performed on a C4 column (Xbridge™ BEH300, 3.5 μm, 2.1 x 50 mm, 300 Å, Waters) at a flow rate of 0.4 mL/min and a column temperature of 23°C. Water containing 0.1% FA and acetonitrile containing 0.1% FA were used as solvents A and B respectively. Column equilibration was done with 5% B for 2 minutes, followed by sample injection and a 6 minute linear gradient of 5 - 95% B for protein separation. Column washing was done with 95% B for 2 minutes, followed by column equilibration with 5% B for 2 minutes. MS scan range was set at 400 to 3000 m/z with a scan speed of 0.9 Hz and 140,000 resolution. MagTran software (Amgen) was used to deconvolute the m/z profiles.
4.8. Expression and purification of EndoCC-N180H in E. coli
Expression of EndoCC-N180H was done using a protocol adapted from previously reported literature (27). Briefly, E. coli BL21(DE3) cells transformed with plasmid pET41b-EndoCC-N180H were cultured at 37°C, 250 RPM overnight. The pre-culture was diluted 20 times into 1L Luria Bertani (LB) broth supplemented with 50 μg/ml kanamycin and cultured overnight at 30°C, 250 RPM. Cells were harvested by centrifugation at 4000 RPM for 15 minutes at 4°C. The cell pellet was resuspended in 20 ml bacterial cell lysis buffer (GoldBio®) supplemented with 45 μg of lysozyme and 40 U of DNase and incubated at 37°C for 1 h. The cell lysate was centrifuged at 20000 RPM for 30 minutes at 4°C. The supernatant containing soluble 6xHis-tagged enzyme was filtered and purified using a HisTrap™ column. The eluted protein was dialyzed against PBS, pH-7.4 at 4°C and stored at −80°C.
4.9. Transglycosylation of glucosylated 6xHis-IFNα-Q158N by EndoCC-N180H
A mixture of the glucosylated 6xHis-IFNα-Q158N (400 μg, 0.8 mg/ml, about 55% is glucosylated), SCT-oxazoline (36) (500 μg), and the N180H mutant of EndoCC (5 μg, 0.01 μg/μl) was incubated in a Tris-HCl buffer (20 mM, pH 7.5) at 30°C. The enzymatic transglycosylation was monitored by LC-MS analysis. After 2 h, an additional portion of SCT-oxazoline was added to push the reaction to completion. Then the sialylated IFNα-Q158N product was isolated by lectin affinity chromatography.
4.10. Enrichment of sialylated 6xHis-IFNα-Q158N
SiaRich™ Pan-Specific Sialic Acid Affinity Column (Lectenz® Bio) was used for enrichment of 6xHis-IFNα-Q158N-SCT using the recommended protocol, on an ÄKTA FPLC System (Cytiva). The column was equilibrated with at least 5 column volumes of binding buffer (10 mM EPPS, 10 mM NaCl, pH-7.5). The transglycosylation reaction mixture of 6xHis-IFNα-Q158N was dialyzed against the binding buffer to get rid of excessive SCT-oxazoline and salts. The dialyzed solution was diluted to 1 ml with binding buffer before loading on to the column at 0.2 – 0.5 ml/min. The column was then washed with 10 column volumes of binding buffer at 1 ml/min. Elution was performed with 5 ml of 10 mM EPPS, 500 mM NaCl buffer, pH 7.5. 0.5 ml fractions were collected, and samples were analyzed using LC-MS. Column regeneration (10 mM EPPS, 1 M NaCl buffer, pH-7.5) and storage conditions were followed as recommended in the product datasheet provided by the manufacturer.
4.11. In vitro anti-proliferative assay
Daudi cells (ATCC® CCL-213™) were maintained in suspension in RPMI-1640 medium (ATCC) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin in T-75 flasks (Sarstedt). Cells were sub-cultured every 2-3 days, when the cell density reached 1-2 x 106 cells/ml, with a seeding density of 2-4 x 105 cells/ml. The anti-proliferative assay was conducted following previously reported protocols (19,29). Interferon alfa-2b CRS batch 6 (EDQM Catalogue code: I0320301, Millipore Sigma) was used as a reference standard for comparison of the biological activity of engineered IFNα. Serial dilutions of IFNα (reference standard and test samples) were made in a 96-well plate to attain a final concentration range of 0.779 – 0.003 ng/ml (10 μl volume in each well). Each of the concentration was tested in duplicates. To each well, 200 μl of Daudi cells at a density of 5 x 104 cells/ml of culture medium was added. Wells with only 200 μl cells (no IFNα) and only 200 μl culture medium (no IFNα or cells) were run as controls. The well plate was incubated at 37°C, 5% CO2 for 96 h. 10 μl of Cell Counting Kit-8 (Sigma) was added to each well and the plate was incubated at 37°C, 5% CO2 for 3-4 h. The absorbance of formazan released by viable cells by reducing WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] dye was measured at 450 nm using a spectrophotometer. Dose-response curves of [IFNα] and absorbance (A450nm – A600nm) were plotted in each case using GraphPad Prism to obtain IC50 values.
4.12. In vitro trypsin digestion of 6xHis-IFNα-Q158N
10 μg of IFNα-Q158N-Glc or IFNα-Q158N-SCT was mixed with trypsin at a final enzyme concentration of 0.1 μg/ml in 50 mM Tris-HCl, pH-8.0 and incubated at 37°C. 1 μg sample was drawn from the reaction mixture at 0, 20, 40, 60, 120, 180, 360 min for IFNα-Q158N-Glc and 0, 60, 120, 240, 360, 840, 1680 min for IFNα-Q158N-SCT. Drawn samples were added to 0.1% formic acid containing 0.1 μg Interferon alfa-2b CRS batch 6 for LC-MS analysis. The decrease in relative intensity of IFNα-Q158N (100*It/I0) with respect to the internal standard was monitored over time, where It, is the relative intensity of IFNα-Q158N at time t and I0 is the relative intensity of IFNα-Q158N at t=0. Half-lives of the proteins towards trypsin digestion were obtained by fitting the data to first-order decay as reported in the past (32,33) using GraphPad Prism.
Supplementary Material
Acknowledgements
We thank Chun Mun Loke for technical assistance and Chao Li for stimulating discussions. This work was supported by the US National Institutes of Health (NIH grant R01 GM080374).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.O’Connor SE, Imperiali B. Modulation of protein structure and function by asparagine-linked glycosylation. Chemistry & Biology 1996;3:803–12 [DOI] [PubMed] [Google Scholar]
- 2.Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs 2010;24:9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 2006;24:1241–52 [DOI] [PubMed] [Google Scholar]
- 4.Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 2014;5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khow O, Suntrarachun S. Strategies for production of active eukaryotic proteins in bacterial expression system. Asian Pac J Trop Biomed 2012;2:159–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwarz F, Huang W, Li C, Schulz BL, Lizak C, Palumbo A, et al. A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat Chem Biol 2010;6:264–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valderrama-Rincon JD, Fisher AC, Merritt JH, Fan YY, Reading CA, Chhiba K, et al. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat Chem Biol 2012;8:434–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi KJ, Grass S, Paek S, St Geme JW, Yeo HJ. The Actinobacillus pleuropneumoniae HMW1C-like glycosyltransferase mediates N-linked glycosylation of the Haemophilus influenzae HMW1 adhesin. PLoS One 2010;5:e15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz F, Fan YY, Schubert M, Aebi M. Cytoplasmic N-glycosyltransferase of Actinobacillus pleuropneumoniae is an inverting enzyme and recognizes the NX(S/T) consensus sequence. J Biol Chem 2011;286:35267–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lomino JV, Naegeli A, Orwenyo J, Amin MN, Aebi M, Wang LX. A two-step enzymatic glycosylation of polypeptides with complex N-glycans. Bioorg Med Chem 2013;21:2262–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song Q, Wu Z, Fan Y, Song W, Zhang P, Wang L, et al. Production of homogeneous glycoprotein with multisite modifications by an engineered N-glycosyltransferase mutant. J Biol Chem 2017;292:8856–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y, Wu Z, Zhang P, Zhu H, Song Q, Wang L, et al. A novel enzymatic method for synthesis of glycopeptides carrying natural eukaryotic N-glycans. Chem Commun (Camb) 2017;53:9075–7 [DOI] [PubMed] [Google Scholar]
- 13.Kightlinger W, Lin L, Rosztoczy M, Li W, DeLisa MP, Mrksich M, et al. Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat Chem Biol 2018;14:627–35 [DOI] [PubMed] [Google Scholar]
- 14.Lin L, Kightlinger W, Prabhu SK, Hockenberry AJ, Li C, Wang LX, et al. Sequential Glycosylation of Proteins with Substrate-Specific N-glycosyltransferases. ACS Cent Sci 2020;6:144–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naegeli A, Neupert C, Fan YY, Lin CW, Poljak K, Papini AM, et al. Molecular analysis of an alternative N-glycosylation machinery by functional transfer from Actinobacillus pleuropneumoniae to Escherichia coli. J Biol Chem 2014;289:2170–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keys TG, Wetter M, Hang I, Rutschmann C, Russo S, Mally M, et al. A biosynthetic route for polysialylating proteins in Escherichia coli. Metab Eng 2017;44:293–301 [DOI] [PubMed] [Google Scholar]
- 17.Wu Z, Jiang K, Zhu H, Ma C, Yu Z, Li L, et al. Site-Directed Glycosylation of Peptide/Protein with Homogeneous O-Linked Eukaryotic N-Glycans. Bioconjug Chem 2016;27:1972–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang YS, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Adv Drug Deliv Rev 2002;54:547–70 [DOI] [PubMed] [Google Scholar]
- 19.Ceaglio N, Etcheverrigaray M, Kratje R, Oggero M. Novel long-lasting interferon alpha derivatives designed by glycoengineering. Biochimie 2008;90:437–49 [DOI] [PubMed] [Google Scholar]
- 20.Rabhi-Essafi I, Sadok A, Khalaf N, Fathallah DM. A strategy for high-level expression of soluble and functional human interferon alpha as a GST-fusion protein in E. coli. Protein Eng Des Sel 2007;20:201–9 [DOI] [PubMed] [Google Scholar]
- 21.Srivastava P, Bhattacharaya P, Pandey G, Mukherjee KJ. Overexpression and purification of recombinant human interferon alpha2b in Escherichia coli. Protein Expr Purif 2005;41:313–22 [DOI] [PubMed] [Google Scholar]
- 22.Ahmed N, Bashir H, Zafar AU, Khan MA, Tahir S, Khan F, et al. Optimization of conditions for high-level expression and purification of human recombinant consensus interferon (rh-cIFN) and its characterization. Biotechnol Appl Biochem 2015;62:699–708 [DOI] [PubMed] [Google Scholar]
- 23.Valente CA, Monteiro GA, Cabral JM, Fevereiro M, Prazeres DM. Optimization of the primary recovery of human interferon alpha2b from Escherichia coli inclusion bodies. Protein Expr Purif 2006;45:226–34 [DOI] [PubMed] [Google Scholar]
- 24.Li C, Wang LX. Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem Rev 2018;118:8359–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou G, Ochiai H, Huang W, Yang Q, Li C, Wang LX. Chemoenzymatic synthesis and Fcγ receptor binding of homogeneous glycoforms of antibody Fc domain. Presence of a bisecting sugar moiety enhances the affinity of Fc to FcγIIIa receptor. J Am Chem Soc 2011;133:18975–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q, An Y, Zhu S, Zhang R, Loke CM, Cipollo JF, et al. Glycan Remodeling of Human Erythropoietin (EPO) Through Combined Mammalian Cell Engineering and Chemoenzymatic Transglycosylation. ACS Chem Biol 2017;12:1665–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi Y, Eshima Y, Huang Y, Kinoshita T, Sumiyoshi W, Nakakita SI, et al. Highly efficient transglycosylation of sialo-complex-type oligosaccharide using Coprinopsis cinerea endoglycosidase and sugar oxazoline. Biotechnol Lett 2017;39:157–62 [DOI] [PubMed] [Google Scholar]
- 28.Elliott S, Egrie J, Browne J, Lorenzini T, Busse L, Rogers N, et al. Control of rHuEPO biological activity: the role of carbohydrate. Exp Hematol 2004;32:1146–55 [DOI] [PubMed] [Google Scholar]
- 29.Silva MM, Lamarre B, Cerasoli E, Rakowska P, Hills A, Bailey MJ, et al. Physicochemical and biological assays for quality control of biopharmaceuticals: interferon alpha-2 case study. Biologicals 2008;36:383–92 [DOI] [PubMed] [Google Scholar]
- 30.Egrie JC, Dwyer E, Browne JK, Hitz A, Lykos MA. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp Hematol 2003;31:290–9 [DOI] [PubMed] [Google Scholar]
- 31.Sareneva T, Pirhonen J, Cantell K, Julkunen I. N-glycosylation of human interferon-gamma: glycans at Asn-25 are critical for protease resistance. Biochem J 1995;308 ( Pt 1):9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan X, Chaffey PK, Wei X, Gulbranson DR, Ruan Y, Wang X, et al. Chemically Precise Glycoengineering Improves Human Insulin. ACS Chem Biol 2018;13:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramon J, Saez V, Baez R, Aldana R, Hardy E. PEGylated interferon-alpha2b: a branched 40K polyethylene glycol derivative. Pharm Res 2005;22:1374–86 [DOI] [PubMed] [Google Scholar]
- 34.Zhao W, Oskeritzian CA, Pozez AL, Schwartz LB. Cytokine production by skin-derived mast cells: endogenous proteases are responsible for degradation of cytokines. J Immunol 2005;175:2635–42 [DOI] [PubMed] [Google Scholar]
- 35.Chung HS, Raetz CR. Interchangeable domains in the Kdo transferases of Escherichia coli and Haemophilus influenzae. Biochemistry 2010;49:4126–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giddens JP, Wang LX. Chemoenzymatic Glyco-engineering of Monoclonal Antibodies. Methods Mol Biol 2015;1321:375–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.