

Abstract
Polypeptide GalNAc-transferase T3 (GalNAc-T3) regulates fibroblast growth factor 23 (FGF23) by O-glycosylating Thr178 in a furin proprotein processing motif RHT178R↓S. FGF23 regulates phosphate homeostasis and deficiency in GALNT3 or FGF23 results in hyperphosphatemia and familial tumoral calcinosis. We explored the molecular mechanism for GalNAc-T3 glycosylation of FGF23 using engineered cell models and biophysical studies including kinetics, molecular dynamics and X-ray crystallography of GalNAc-T3 complexed to glycopeptide substrates. GalNAc-T3 uses a lectin domain mediated mechanism to glycosylate Thr178 requiring previous glycosylation at Thr171. Notably, Thr178 is a poor substrate site with limiting glycosylation due to substrate clashes leading to destabilization of the catalytic domain flexible loop. We suggest GalNAc-T3 specificity for FGF23 and its ability to control circulating levels of intact FGF23 is achieved by FGF23 being a poor substrate. GalNAc-T3’s structure further reveals the molecular bases for reported disease-causing mutations. Our findings provide an insight into how GalNAc-T isoenzymes achieve isoenzyme-specific nonredundant functions.
GalNAc-T3 belongs to the large family of GalNAc-Ts (CAZy27) that initiate mucin-type (GalNAc-type) O-glycosylation in most eukaryotes1–3. These isoenzymes transfer a GalNAc moiety from uridine diphosphate-GalNAc (UDP-GalNAc) onto Ser/Thr residues of proteins. GalNAc-Ts are differentially expressed and have distinct albeit partly overlapping acceptor substrate specificities3–5. GalNAc-Ts are type II transmembrane proteins with a Golgi lumen located catalytic, GT-A fold domain connected by a flexible linker to a ricin-type lectin domain. This latter domain not only modulates substrate specificity, but is also responsible for the enzyme’s long-range or remote prior glycosylation activity, recognizing GalNAcα1-O-Thr/Ser residues on glycopeptide substrates ~6–17 residues distant to the acceptor site6–10.
Deficiency of the GALNT3 gene results in the congenital diseases familial tumoral calcinosis (FTC) and hyperostosis-hyperphosphatemia syndrome, characterized by hyperphosphatemia and ectopic calcifications11. We previously demonstrated that GalNAc-T3 specifically glycosylates FGF23 at Thr178 in its subtilisin-like proprotein convertase (PC) site (RHT178R↓S), and that site-specific O-glycosylation at Thr178 inhibits furin PC processing and enables secretion of intact active FGF23 in Chinese hamster ovary (CHO) cells12. The importance of Thr178 site-specific O-glycosylation is demonstrated by mutations in the RHTR179 sequon, which prevent PC cleavage of FGF23, and result in hypophosphatemic Rickets disease13 due to high levels of intact FGF23. In contrast, mutations in FGF23 that impair its folding, intracellular transport or stability result in FTC clinically indistinguishable from the loss of GALNT3 (ref.14). Regulation of FGF23 and its signaling through FGFR1c, is further complicated by phosphorylation of Ser180 adjacent to the PC processing site, by the Golgi-located FAM20C kinase15,16 and the need for the coreceptor alpha-Klotho17 (Fig. 1a).
Fig. 1 |. In vitro evidence that GalNAc-T3 glycosylates FGF23 Thr178 using its lectin domain.
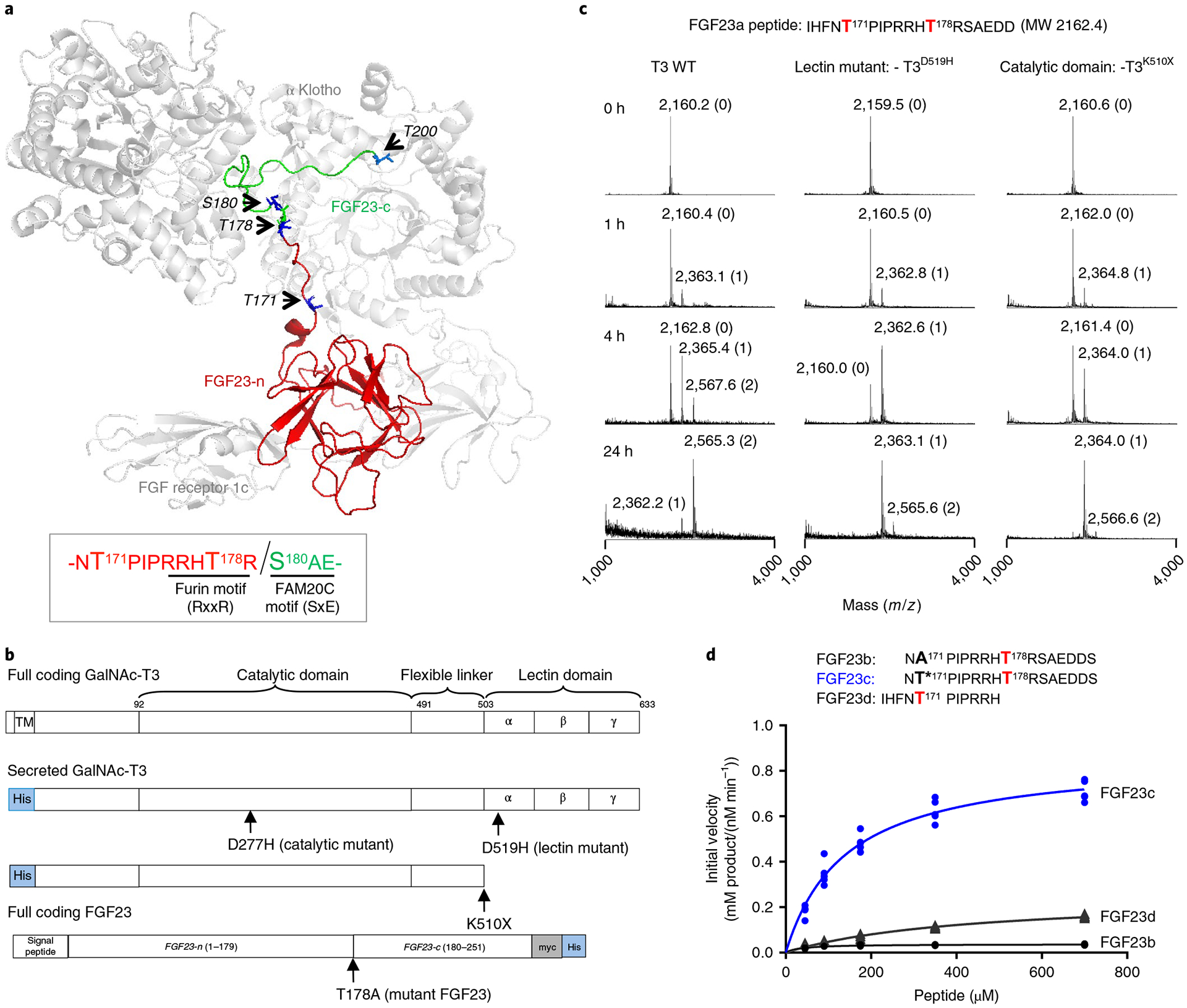
a, FGF23 bound to FGFR1c and α-Klotho showing the extended structure of the C-terminal portion of FGF23 (PDB 5W21, ref.25). Arrows point to the side chains (in blue) of known phosphorylated (Ser180) and O-glycosylated sites (Thr171, Thr178 and Thr200) on FGF23. The globular FGF23 N-terminal fragment (n-FGF23) and extended unstructured C-terminal fragment (c-FGF23) are colored red and green, respectively. Sequence motifs for FGF23’s furin cleavage site and FAM20C phosphorylation site are illustrated in the box. Note that the noncleavable FGF23 R176Q-R178Q was used for crystallization25. b, GalNAc-T3 and FGF23 expression constructs, where the GalNAc-T3 catalytic and lectin domain inactivating/truncation mutants are shown. c, Glycosylation of the FGF23a substrate as a function of time for WT and mutant GalNAc-T3 monitored by MALDI–TOF–MS showing the role of GalNAc-T3 lectin domain. The number of GalNAc residues incorporated at each peak are given in parenthesis next to the mass values. These experiments were performed in quintuplicate each giving similar results. d, Peptide glycosylation kinetics of the human GalNAc-T3 against a series of FGF23 (glyco)peptides; FGF23b (A171–T178), FGF23c (T*171–T178) and FGF23d (T171). Plotted lines represent the Michaelis–Menten nonlinear fit (using GraphPad Prism 7.03) of the initial velocity data using the kinetic values given in Supplementary Table 2. Initial velocities (shown as individual points) were obtained in triplicate or higher for each peptide concentration, giving a total of 15, 24 and 20 independent determinations for FGF23b, FGF23c and FGF23d, respectively.
GalNAc-T3 has rather broad acceptor substrate specificity5,18,19 and contributes substantially to the overall O-glycoproteome of cells19,20, but the specific O-glycosylation of FGF23 at Thr178 may represent its most important biological role. Other GalNAc-T isoenzymes exhibit similar highly specific roles that drive subtle biological processes despite their seemingly broad substrate specificities21,22. It has been a conundrum as to how GalNAc-T isoenzymes with relatively broad glycosylation specificities can serve in fine-tuned regulation of protein functions through glycosylation of highly specific glycosites in proteins.
Here, we applied a multidisciplinary approach to elucidate that GalNAc-T3 uses its lectin-mediated long-range properties to glycosylate Thr178 in FGF23 both in vitro and in vivo. We demonstrate that GalNAc-T3 selectively rescues a minor fraction of intact FGF23 regardless of furin processing, which we attribute to FGF23 being a relatively poor and slow substrate. Thus, GalNAc-T3 is proposed to be the primary regulator of a stabilized intact FGF23. Finally, we explore the molecular basis of many known disease-causing mutations in GALNT3.
Results
FGF23 Thr178 in vitro glycosylation by GalNAc-T3.
Since GalNAc-T3 shows long-range prior glycosylation preferences6,9, we tested whether glycosylation of Thr178 in a FGF23 model peptide, FGF23a12, was lectin dependent, requiring prior Thr171 glycosylation (Fig. 1b–d). Time-course assays with wild type (WT) GalNAc-T3 and lectin domain inactivating mutants (GalNAc-T3D519H and GalNAc-T3D510X)23 against FGF23a demonstrated that GalNAc-T3WT initiated incorporation of GalNAc at Thr171 followed by glycosylation at Thr178 (Fig. 1c). In contrast, both GalNAc-T3 lectin domain inactivating mutants failed to substantially glycosylate the second Thr178 site (Fig. 1c).
Kinetic studies performed with shorter FGF23 model (glyco) peptide substrates to further access the role of Thr171 (FGF23b (A171–T178), FGF23c (T*171–T178, where T* = Thr-O-GalNAc) and FGF23d (T171); Fig. 1d and Supplementary Table 1) showed that glycosylation of Thr178 was several-fold faster in the presence of glycosylated Thr171 by both their Kcat values (turnover of subtrate molecules) and catalytic efficiencies (Fig. 1d and Supplementary Table 2). This confirms that glycosylation of FGF23 is initiated at Thr171 by any of several GalNAc-Ts12 and only subsequently at Thr178 by GalNAc-T3 through a lectin-mediated mechanism.
FGF23 Thr178 glycosylation by GalNAc-T3 in cells.
We proceeded to confirm these in vitro findings in CHO cells by coexpressing C-terminal myc/His-tagged versions of WT FGF23T178, and the FGF23T178A mutant, with or without coexpression of GalNAc-T3WT (Figs. 1b and 2a and Supplementary Fig. 1). We and others12,16 typically observe that the secreted intact FGF23 migrates as two main bands (Fig. 2a, labeled nos. 1 and 2, where the lower band no. 2 may further appear as two additional bands), which we attribute to differences in O-glycosylation state at Thr171, Thr178 and Thr200 (Fig. 1a and Supplementary Fig. 2)12,16,24,25. Without coexpression of GalNAc-T3, FGF23T178 migrated as two weak bands, while FGF23T178A migrated as only the lower band (no. 2). Coexpression of GalNAc-T3 substantially increases the secretion and/or stability of WT FGF23T178 (no. 1, lane 5) compared to FGF23A178 (no. 2, lane 4). This confirms the identification of the upper band (no. 1) as intact FGF23 with O-glycosylation at Thr178, and it also indicates that CHO cells have endogenous capacity for a very low level of glycosylation at Thr178, presumably by low expression of GalNAc-T3. To probe the role of the catalytic and lectin domains of GalNAc-T3, we then coexpressed inactive lectin (D519H)26 and catalytic domain (D277H)23 GalNAc-T3 mutants with WT FGF23T178 (Fig. 2a). That both mutants failed to induce the higher FGF23 band (no. 1, lane 5 versus lanes 6 and 7) confirms that both activities of GalNAc-T3 are required. As a control we included coexpression of GalNAc-T2, which does not glycosylate Thr178 in vitro12. As predicted, this did not induce accumulation of the upper band (no. 1) giving a banding pattern identical to WT cells (compare lanes 8 and 3, Fig. 2a). This was confirmed by the 35S-labeling experiment in Fig. 2b. Note that the expression of the GalNAc-T3 mutants (Fig. 2a, lanes 6 and 7) showed an increase in the intensity of the lower band (no. 2) compared to WT cells, indicating the coexpression of these GalNAc-T3 mutants somehow increases the glycosylation of glycosites other than Thr178.
Fig. 2 |. Glycosylation of Thr178 in FGF23 by GalNAc-T3 in cells requires its lectin domain activity.
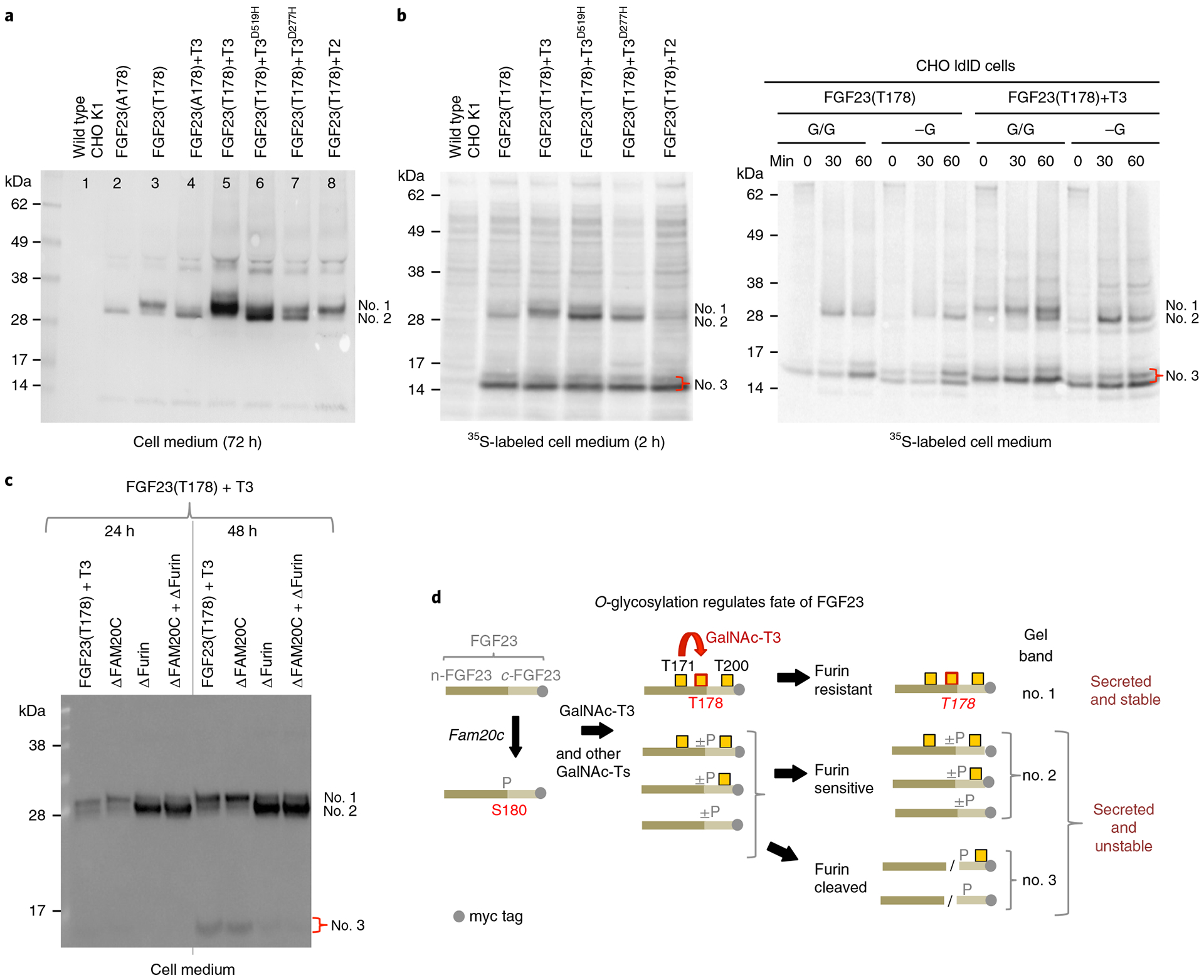
a, Anti-myc NuPAGE western blot analysis of culture medium of CHO K1 cells stably transfected with C-terminal myc labeled WT (FGF23T178) and mutant (FGF23A178) FGF23 with or without coexpression of WT and mutant GalNAc-T3 and WT GalNAc-T2. Medium was collected and analyzed after 72 h growth. The different glycosylation states of full length FGF23 are labeled: upper band (no. 1) FGF23 fully glycosylated at Thr178, lower band (no. 2) FGF23 lacking glycosylation at Thr178 but glycosylated at Thr171 and/or Thr200. b, Left panel shows NuPAGE analysis of 35S-Met labeled (2 h without chase) FGF23 (C-terminal myc tagged) expressed in CHO K1 cells with or without coexpression of WT and mutant GalNAc-T3 and with GalNAc-T2. Right panel shows NuPAGE analysis of 35S-Met labeled FGF23 expressed in CHO ldlD cells with or without coexpression of GalNAc-T3 under (G/G) or lacking (-G) conditions for O-glycosylation. The bands at (no. 3) represent the furin cleaved FGF23 C-terminal fragment. c, Western blot analysis of culture medium of CHO, CHO-ΔFAM20c, CHO-ΔFURIN and CHO-ΔFAM20c-ΔFURIN cells stably transfected with WT FGF23 and GalNAc-T3 demonstrating GalNAc-T3 only partially glycosylates FGF23 Thr178 under these conditions. d, Schematic representation of the possible fates of FGF23. Numbers to the right of the species correspond to the gel bands labeled in a–c. All experiments were performed in sextuplicate.
Since these cell experiments were performed after 72 h culture, we explored the same experimental design using 35S-Met/35S-Cys metabolic labeling at 2 h to better evaluate the secretion of FGF23 (Fig. 2b, left panel). As expected, only coexpression of GalNAc-T3WT with WT FGF23T178 resulted in the appearance of the Thr178 glycosylated band (no. 1) (Fig. 2b). The apparent secretion levels of FGF23 without and with coexpression of GalNAc-T3 at this timepoint were not markedly different, and furthermore the C-terminal tagged fragment of FGF23 resulting from furin cleavage (band no. 3, ~15 kDa) was detectable regardless of coexpression of GalNAc-T3 (Fig. 2b). We reproduced these findings in the CHO ldlD cell system12,27, where supplementation with GalNAc is required for initiation of O-glycosylation and supplementation with Gal for elongation of O-glycans (and N-glycans) are required (Fig. 2b, right panel). CHO cells with (G/G) and without (-G) capacity for O-glycosylation, secreted partially glycosylated FGF23 (no. 2), whereas only with the coexpression of GalNAc-T3 under G/G conditions was Thr178 glycosylated FGF23 secreted (no. 1; Fig. 2b). With the coexpression of GalNAc-T3, no markedly enhanced secretion of intact FGF23 (bands nos. 1 and 2) was observed with O-glycosylation (G/G) compared to the absence of O-glycosylation (-G) (Fig. 2b). Note also that the C-terminal fragment (no. 3) is not affected by the general glycosylation capacity of CHO cells (compare G/G and -G lanes), with (or without) coexpression of GalNAc-T3 (Fig. 2b). These findings indicate that a substantial fraction of FGF23 undergoes furin cleavage regardless of glycosylation by GalNAc-T3, and that the main reason for the higher amounts of intact fully glycosylated FGF23 observed after 72 h (Fig. 2a) is due to its stability after secretion rather than an increase in the rate of secretion.
GalNAc-T3 glycosylates and rescues only a fraction of FGF23.
Since a substantial amount of FGF23 undergoes furin PC processing regardless of coexpression of GalNAc-T3 (Fig. 2b), we explored the role of furin as well as FAM20C on this process. The sequence surrounding Thr178 (~RRHTR↓SAEDD~) contains a selective FAM20C phosphorylation site (underlined)16 and a furin cleavage site (italics). FAM20C phosphorylation of Ser180 was reported to inhibit GalNAc-T3 O-glycosylation at Thr178 (ref.16) (see also below), indicating that these processes may compete to regulate FGF23 processing16 (see Fig. 2d). We therefore explored the effects of eliminating furin cleavage and phosphorylation by targeted knockout (KO) of the Furin and Fam20c genes in CHO K1 cells stably expressing GalNAc-T3 and FGF23 (Fig. 2c). Analysis of FGF23 after 24 or 48 h revealed that the upper band (no. 1) representing intact FGF23 with Thr178 O-glycosylation was largely unaffected by KO of Fam20c and/or Furin, while KO of Furin resulted in a marked increase of the lower FGF23 band (no. 2) without Thr178 glycosylation (Fig. 2c). These results are in agreement with the above findings that GalNAc-T3 may only glycosylate a fraction of FGF23 in CHO cells regardless of the availability of excess intact FGF23 substrate. In addition, this also implies that GalNAc-T3 inefficiently glycosylates FGF23 in CHO K1 cells.
GalNAc-T3 glycosylation of FGF23 is a slow process.
GalNAc-T3 glycosylates FGF23c with a Kcat of 17 min−1, which is on the order of some of the slowest enzymes in nature (for example lysozyme, Kcat of 5 min−1, Supplementary Fig. 3)28. To understand GalNAc-T3’s range in activity, GalNAc-T3 was characterized against a series of remote glycosylation probing (glyco)peptides (P1, -TPGP-; P2, -TPGP–T*- and P3, -T*–TPGP-) that we previously used to characterize kinetic properties of GalNAc-T2 and T4 (refs.9,29) (Fig. 3a and Supplementary Table 2). These substrates contain the ~TPGP~ sequence motif common to most GalNAc-Ts including GalNAc-T3 (refs.5,6). GalNAc-T3 against P3 (-T*–TPGP-) was ~5- and ~17-fold more active compared to P1 or P2 (-TPGP-, -TPGP–T*-) and FGF23c, respectively (Fig. 3b and Supplementary Table 2). The enhanced activity with P3 over P1 and P2 is due to the long-range glycosylation preference of GalNAc-T3 (ref.6), while the low activity with the FGF23c must be due to its poor sequence motif. Thus, the N-terminal glycosylated residue of FGF23c is not sufficient to bring its activity up to that of P3.
Fig. 3 |. Glycosylation kinetics of human GalNAc-T3 against a series of (glyco)peptides.
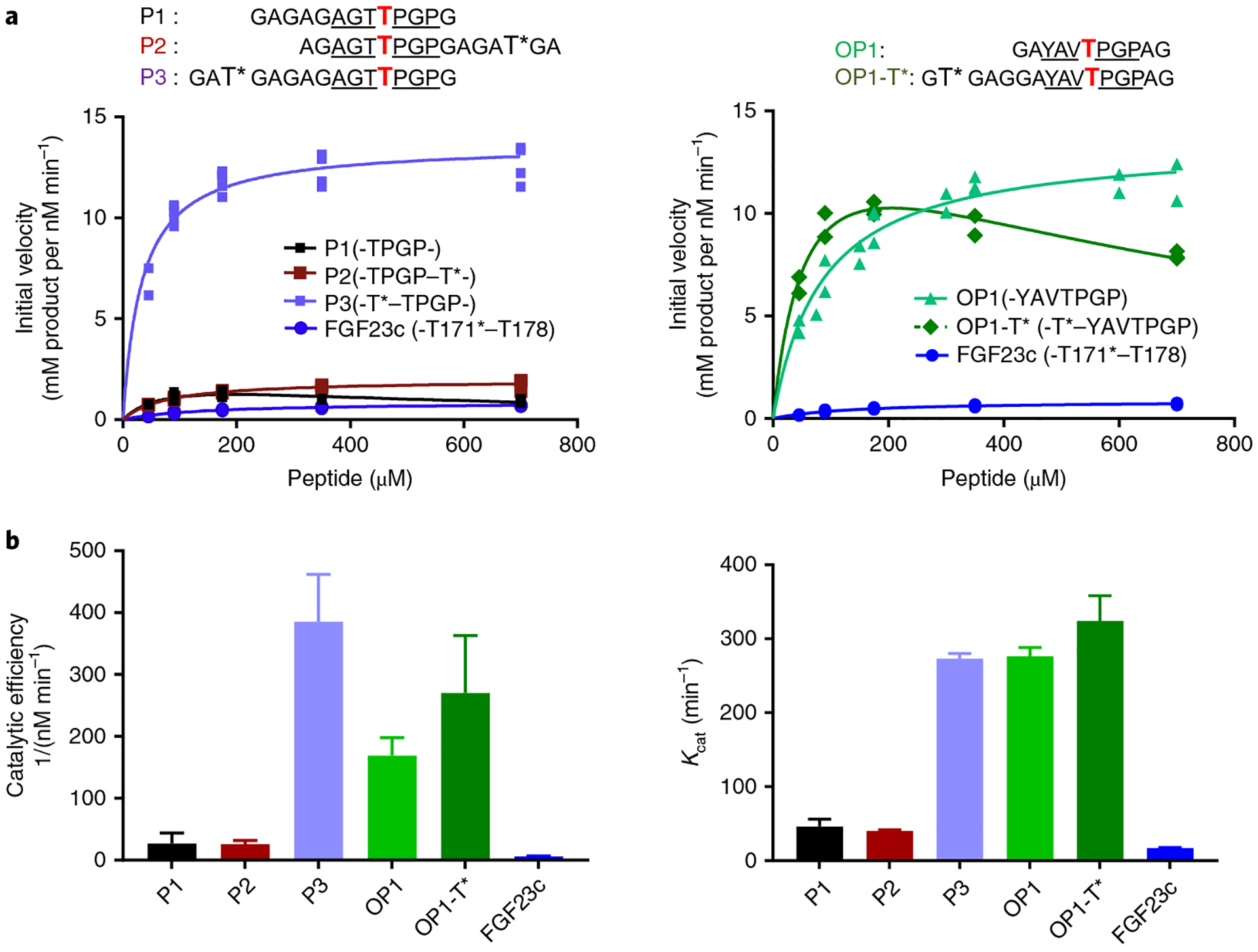
a, Glycosylation kinetics of GalNAc-T3 against the N-terminal prior glycosylated FGF23c (T*171–T178) glycopeptide in comparison to P1, P2, P3 (left) and OP1 and OP1-T* (right). Plotted lines represent the Michaelis–Menten nonlinear fit (using GraphPad Prism 7.03) of the initial velocity data using the kinetic values given in Supplementary Table 2. Initial velocities (individual points) were obtained in triplicate or higher for each peptide concentration (except for OP1-T*), giving a total of 19, 16, 16, 21, 21, 13 and 24 independent determinations for P1, P2, P3, OP1, OP1-T* and FGF23c, respectively. b, Summary of Kcat and catalytic efficiency values derived from the GraphPad Prism fit of the data in a (see Supplementary Table 2). Error bars represent the standard deviation calculated by the GraphPad Prism fit of the data sets as described in the Methods. Note that the fitting of OP1-T* included substrate inhibition.
We next sought to determine an optimally active ‘naked’ peptide motif for GalNAc-T3. For this, we surveyed the activity of GalNAc-T3 against a series of peptides (designated OP1 to OP10) designed from our random peptide substrate preferences obtained for a panel of GalNAc-T isoenzymes (Supplementary Fig. 4)19. The proposed GalNAc-T3 optimal substrate sequence (GAYAVTPGPAG, called OP1) was indeed the most active of the series (Fig. 3a and Supplementary Table 2). Interestingly, the naked OP1 peptide was nearly as active as P3 (Fig. 3). We next asked whether a remote N-terminal previously glycosylated site would further amplify the activity of OP1 using the OP1-T* glycopeptide (-T*–YAVTPGP-) containing an N-terminal GalNAc moiety. Very similar Kcat and catalytic efficiency values were found for OP1-T*, OP1 and P3, indicating that the maximal turnover and catalytic efficiency for GalNAc-T3 was achieved (Fig. 3 and Supplementary Table 2). OP1 is therefore a nearly optimal substrate for GalNAc-T3, which cannot be significantly improved by the addition of previous N-terminal glycosylation. The OP1 and P3 substrates reflect the two different enzymatic processes for GalNAc-T isoenzymes in which OP1 probes optimal binding to the catalytic domain of GalNAc-T3, and P3 probes the lectin-assisted binding with glycopeptide substrates. Since all of our other transferase optimal peptides (OP) show lower but still significant activities with GalNAc-T3, we conclude that a unique sequence specific for glycosylation by GalNAc-T3 would have to be a suboptimal sequence, lacking any of the common GalNAc-T isoenzyme substrate motifs.
Architecture of the ternary complexes of GalNAc-T3.
To provide insight into GalNAc-T3’s glycosylation of FGF23, several failed attempts to crystallize the human GalNAc-T3 and other GalNAc-T3 orthologs were made. We succeeded with the crystallization of the Taeniopygia guttata (Tg) GalNAc-T3 (80% sequence identity to the human (Hs) GalNAc-T3; Supplementary Fig. 5), which gave kinetic properties comparable to the HsGalNAc-T3 (Supplementary Fig. 6 and Supplementary Table 2). Crystals of the TgGalNAc-T3 were obtained in the space group P212121, subsequently soaked with UDP-MnCl2 and either P3 or FGF23c. Structures were obtained at 2.12 Å (with P3) and 1.96 Å (with FGF23c) (Methods, Fig. 4 and Supplementary Table 3), showing compact structures with a N-terminal catalytic domain GT-A fold and a C-terminal lectin domain (Fig. 4a). Except for the four C-terminal residues of FGF23c and the terminal residues of P3, the density for both glycopeptides and UDP/Mn+2 were well defined in both crystal structures (Fig. 4b). TgGalNAc-T3, HsGalNAc-T2 and T4 displays high similarities at the secondary structure level (Supplementary Fig. 7), however, at the tertiary structural level, the GalNAc-binding site of their lectin domains (that is, T2 versus T3/T4) are located on opposite sides relative to their catalytic domains (Fig. 4c). We previously reported that the differences in the orientations of the lectin domains of GalNAc-T2 and T4 are due to the nature of their flexible linkers9. This readily explains the basis of GalNAc-T3’s long-range glycosylation preferences with FGF23c, P3 and other glycopeptide substrates, similar to GalNAc-T4 (refs.9,29). By atomic force microscopy (AFM) and solution small-angle X-ray scattering (SAXS) an ensemble of compact structures were observed for the HsGalNAc-T3, in contrast to the compact, extended and dimeric forms reported for the HsGalNAc-T2 (Supplementary Figs. 8–10a,b and Supplementary Table 4)8. GalNAc-T3’s compact structures are likely due to domain–domain interactions lacking in GalNAc-T2 (Supplementary Figs. 9 and 10c).
Fig. 4 |. Crystal structures of TgGalNAc-T3 in ternary complexes with UDP-Mn+2-P3 and UDP-Mn+2-FGF23c.
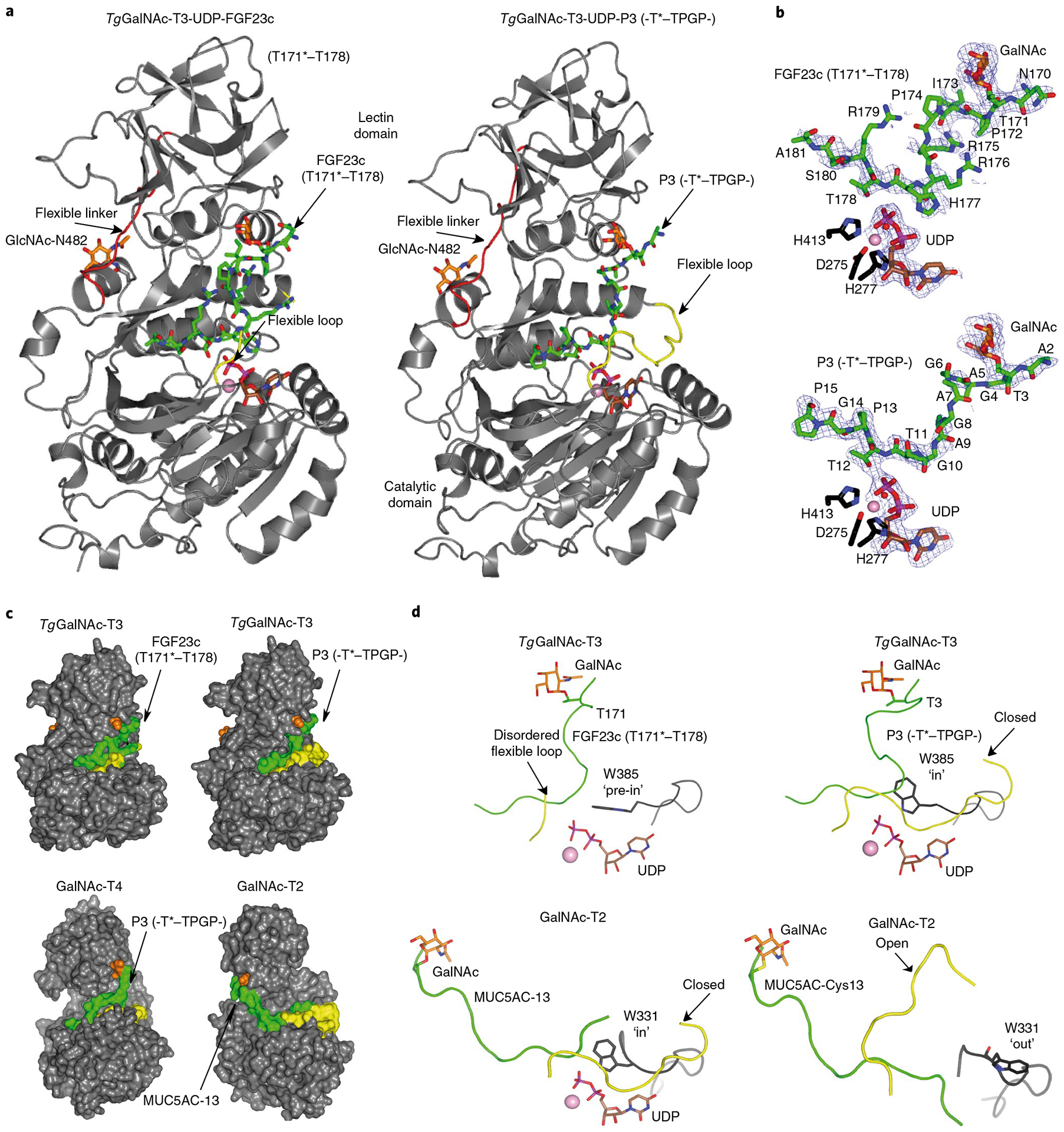
a, Ribbon structures of TgGalNAc-T3 ternary structures with glycopeptides FGF23c (left) and P3 (right). Note that the FGF23c glycopeptide is unstructured, resembling the unstructured region of the FGF23 C terminus within the alpha-Klotho–FGF23–FGFR1c complex25. b, Zoomed in structures showing the bound Mn+2, UDP and peptides (FGF23c, upper, and P3, lower). Electron density maps are Fo–Fc (blue) contoured at 2.2 σ for P3/FGF23c and UDP. Note that no electron density was visible for any of the Arg side chains and the EDDS sequence. In a and b the catalytic and lectin domains are colored in gray, the flexible linker and catalytic domain active site loops are depicted in red and yellow, respectively. The GalNAc moieties of P3 and FGF23c are shown as orange carbon atoms, while the remaining residues are shown as green carbon atoms. The UDP nucleotide is depicted with brown carbon atoms whereas the manganese atom is shown as a pink sphere. The remaining N-linked GlcNAc moiety bound to Asn482 is shown with orange carbon atoms. c, Surface representations of the complexes of GalNAc-T3–FGF23c–P3, GalNAc-T4–P3 (PDB 5NQA) and GalNAc-T2–MUC5AC-13 (PDB 5AJP) showing the different orientations of the lectin domain (compare GalNAc-T3/T4 with GalNAc-T2). The proteins, flexible loops, nucleotides, GalNAc moieties and peptide substrates are colored in gray, yellow, brown, orange and green, respectively. d, Expanded views of the TgGalNAc-T3–UDP–P3, TgGalNAc-T3–UDP–FGF23c, GalNAc-T2–UDP–MUC5AC-13 and GalNAc-T2–MUC5AC–Cys13 complexes showing their bound glycopeptide and the catalytic domain active site flexible loops. Note that Trp331 in GalNAc-T2–UDP–MUC5AC-13 and GalNAc-T2–MUC5AC–Cys13 complexes adopt ‘in’ and ‘out’ conformations, respectively, while Trp385 in the TgGalNAc-T3–UDP–P3 and TgGalNAc-T3–UDP–FGF23c complexes adopt in and ‘pre-in’ conformations, respectively.
Our structures clearly show the catalytic domain flexible loop (residues 417–428) found in all other GalNAc-Ts, whose movement is necessary for the binding of substrate and transfer of GalNAc. This loop is disordered in the TgGalNAc-T3–UDP–FGF23c complex and ordered/closed in the TgGalNAc-T3–UDP–P3 complex (Fig. 4a,c,d). Oscillations of this loop between open and closed conformations define the inactive and active states of these isoenzymes3,8–10,30,31. Another feature of the structures is the conformation adopted by the catalytic Trp385 residue that interacts with the pyrophosphate group of UDP-GalNAc that moves in concert with the flexible loop (Fig. 4d). This residue with bound P3 adopts an ‘in’ conformation and with FGF23c a ‘pre-in’ conformation, where an ‘in’ conformation represents a closed flexible loop and an active enzymatic state and the ‘pre-in’ conformation a step immediately preceding the ‘in’ conformation8. An ‘out’ conformation for Trp385 would be associated with an open flexible loop and an inactive enzymatic state (Fig. 4a,c,d).
Lectin domain substrate binding of GalNAc-T3.
Both structures reveal the GalNAc residue of each monoglycopeptide substrate bound to GalNAc-T3’s lectin α-subdomain GalNAc-binding site (Fig. 5a,b). GalNAc interacts with conserved residues Asp517, Asn539, Tyr531 and His534 (in GalNAc-T2, ref.8 and -T4, ref.9), and nonconserved residues Glu520 and Leu536 (unique to GalNAc-T3) (Fig. 5a,b and Supplementary Figs. 5 and 7). Most interactions are through hydrogen bonds in GalNAc-T2/T3/T4 (ref.9), although CH/π interactions are also observed between the GalNAc and Tyr471/Tyr531/Phe475 (ref.8), respectively. An extra CH3–CH3 interaction is found between Leu536 and the acetamido group (Fig. 5a,b). As found in GalNAc-T4 (ref.29), there is a solvent exposed gap in glycopeptide substrate binding between the lectin-domain GalNAc-binding site and the catalytic domain peptide-binding site (residues Gly4-Ala9 of P3 and Pro172-Arg176 of FGF23c, Figs. 4c and 5a,b) indicating similarities between GalNAc-T3 and T4 in their binding of glycopeptide substrates. Such a gap in substrate binding is not observed for GalNAc-T2 (ref.8). Overall, FGF23c and P3 are recognized very similarly by the TgGalNAc-T3 lectin domain. Such binding is further supported by saturation transfer difference–nuclear magnetic resonance (STD–NMR) experiments that show high STD enhancements for the GalNAc moiety and low enhancements for the peptide residues (Supplementary Figs. 11 and 12 and Supplementary Tables 5 and 6).
Fig. 5 |. Structural features of the peptide, UDP and lectin-domain GalNAc-binding sites of TgGalNAc-T3.
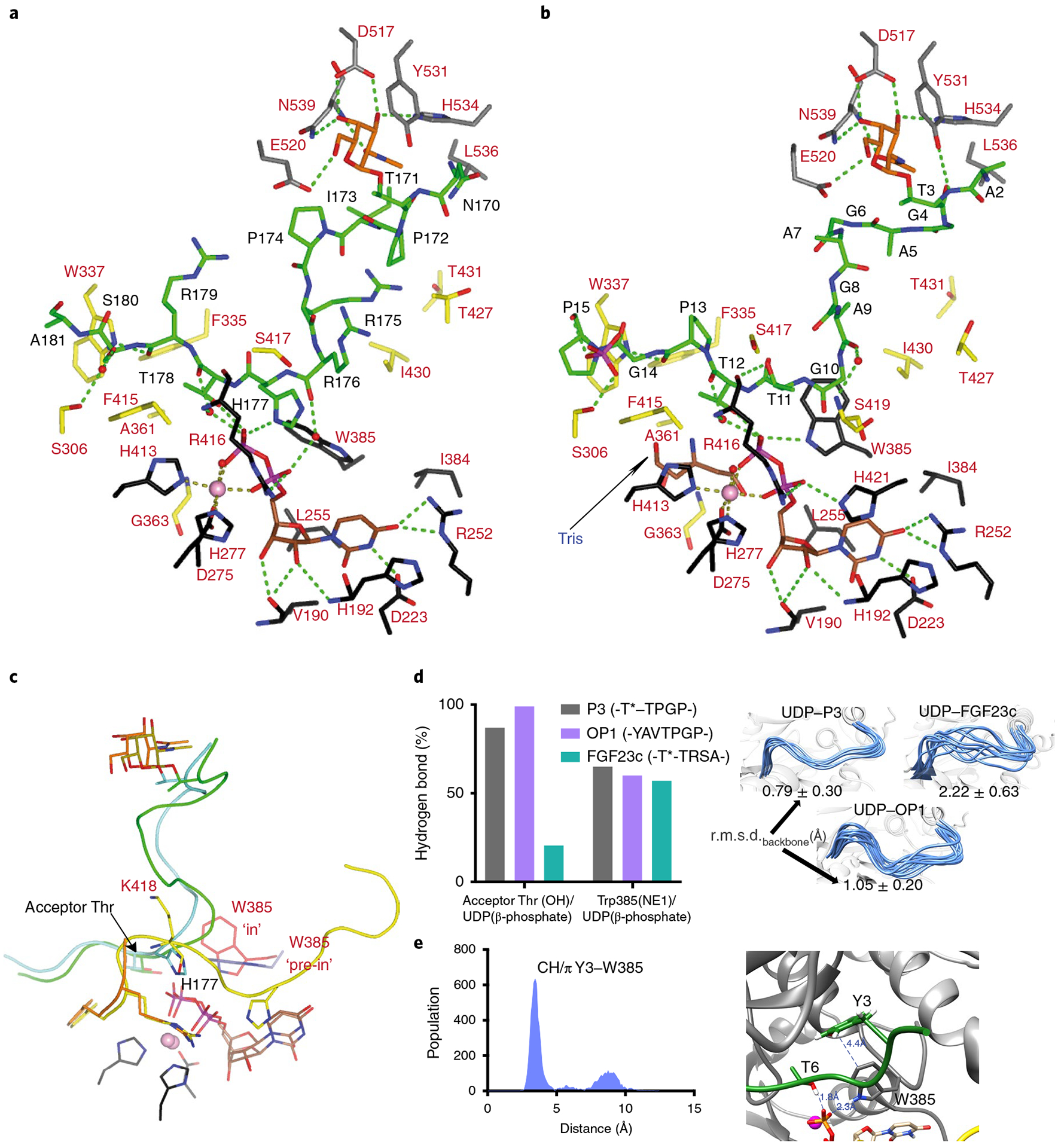
a,b, View of the complete sugar-nucleotide, peptide and lectin-domain binding sites of TgGalNAc-T3–UDP–FGF23c (a) and TgGalNAc-T3–UDP–P3 (b) complexes. Residues forming the sugar-nucleotide, peptide and lectin-domain binding sites are depicted as black, yellow and gray carbon atoms, respectively. UDP and the glycopeptide are shown as brown and green carbon atoms, respectively. Mn2+ and GalNAc moiety are depicted as a pink sphere and orange carbon atoms, respectively. Hydrogen bond interactions are shown as dotted green lines. Water molecules are depicted as red spheres. Note that only water-mediated interactions in which only one water molecule acts as a bridge between the residues are shown. In the TgGalNAc-T3–UDP–FGF23c complex, a phosphate and a Tris molecule are identified in the catalytic domain active site. c, Close-up view of the superimposed TgGalNAc-T3–UDP–P3 (flexible loop in yellow) and TgGalNAc-T3–UDP–FGF23c (flexible loop in orange) complexes. Amino acid residues of P3 and FGF23c are colored green and aquamarine, respectively, while their GalNAc moieties are colored olive and orange, respectively. Trp385 in either the in or pre-in conformation is colored as red and black carbon atoms, respectively. d, Extent of hydrogen bond formation between the acceptor Thr OH and UDP phosphate and Trp385 and UDP phosphate obtained from 0.5 μs MD simulations of TgGalNAc-T3–UDP–P3, TgGalNAc-T3–UDP–FGF23c and TgGalNAc-T3–UDP–OP1. Note that for the MD simulations of TgGalNAc-T3–UDP–FGF23c we used the FGF23c without the EDDS sequence as no electron density for the EDDS sequence was visualized in the crystal structure (left). Ensemble of structures obtained from the MD simulations showing differences in the stability of the flexible loop for the closed conformation (right). e, Analysis of the CH–π interaction between the optimal OP1 Tyr3 and GalNAc-T3 Trp385 obtained by MD simulations (left). Close-up view of the CH–π interaction between Tyr3 and Trp385 (right).
Catalytic domain peptide-binding site of GalNAc-T3.
The peptide-binding site of TgGalNAc-T3 reveals that both substrates are bound by a limited number of direct and water-mediated hydrogen bonds. Direct hydrogen bonds are observed with Thr11P3 side chain and Arg416 backbone, and Pro13P3/Arg179FGF23c backbones and Trp337 side chain, while CH–π interactions occur between Pro15P3/Ala181FGF23c side chains and Trp337/Phe415 (Ala181FGF23c only interacts with Trp337; Fig. 5a,b). Water-mediated hydrogen bonds are observed through the Ala9/Thr12P3 backbones and Ser419/Arg416 backbones, Thr178FGF23c backbone and Arg416 backbone, and the Ser180FGF23c side chain and Ser306 side chain (Fig. 5a,b). Note that water-mediated hydrogen bonds form between the Thr12P3/Thr178FGF23c backbones with UDP β-phosphate, and the Arg176FGF23c backbone with UDP α-phosphate (Fig. 5a,b). Assuming that the interactions between the GalNAc moiety of the two monoglycopeptides with the lectin domain are conserved, the observed differences in Km for these peptides (Supplementary Table 2) must be due to dissimilarities of their interactions at the peptide-binding site (that is, P3 establishes more direct interactions than FGF23c). Furthermore, the differences in position and mobility of the catalytic domain flexible loop are also likely to contribute to their different kinetic properties (Figs. 4a,c,d and 5c). For bound FGF23c, His177FGF23c sterically clashes with Lys418 of the flexible loop (Fig. 5c) leading to increased flexibility and instability, which is also observed from our molecular dynamics (MD) simulations (see below). Such a steric clash would impede (but not exclude) the adoption of a stable closed conformation (Fig. 5d). Thus, the higher number of interactions with P3 together with a more stable and closed flexible loop likely explains the better Km of P3 over FGF23c.
Thus, three structural features of the catalytic domain binding site likely dictate enzyme turnover, potentially accounting for the differences in Kcat between the two monoglycopeptides (Supplementary Table 2): (1) the stability of the hydrogen bond between the acceptor Thr (Thr12P3/Thr178FGF23c) and the UDP β-phosphate (simplified as O-HThr⋯OUDP); (2) the stability of the flexible loop closed conformation and (3) the stability of the Trp385’s ‘in’ conformation that relies on the hydrogen bond between Trp358 and the UDP β-phosphate. This in conformation also forms a stacking interaction between the GalNAc moiety of UDP-GalNAc and the Trp385 side chain in GalNAc-T2 (ref.4). Our MD simulations further show that these features are more stable in the TgGalNAc-T3–UDP–P3 complex than the TgGalNAc-T3–UDP–FGF23c complex (Fig. 5d), providing an explanation for their different Kcat values.
In the GalNAc-T3 optimal OP1 peptide substrate the Tyr at the position 3 appears to be key for optimal catalysis (Fig. 3, Supplementary Fig. 4 and Supplementary Table 2). To address the role of this residue molecular docking and MD simulations were conducted that demonstrated that the Tyr forms a CH–π interaction with Trp385 (Fig. 5e). This interaction is key for enhancing binding of the peptide and guiding catalysis toward the acceptor Thr. This indicates that the binding of the -TyrAlaVal- motif at the catalytic domain may be equivalent energetically to the binding of a remote glycopeptide to the lectin domain; that is, OP1 versus P3 (Fig. 5d), thus providing the molecular basis for how these two peptides can produce the maximum turnover rate for GalNAc-T3 catalysis.
Role of positively charged residues surrounding Thr178.
To access the importance of the Arg and His residues around the FGF23 Thr178 acceptor site (RHTR179), we characterized three additional glycopeptides, FGF23e-g against GalNAc-T3 (Supplementary Table 1). FGF23e mimics a rare mutation (H177N) found in the African population (gnomAD browser) that exhibits no known deleterious effects, while FGF23f (R176A-H177A) and FGF23g (R179A) address the effects of Arg176/His177 and Arg179, respectively. Note that the Arg176/Arg179 residues are essential for furin cleavage16. Kinetic studies of these substrates with HsGalNAc-T3 revealed decreased Kcat values (~50–75%) but similar or reduced (~50%) catalytic efficiencies compared to FGF23c (Supplementary Fig. 13 and Supplementary Table 2). These results indicate that the Arg and His residues flanking Thr178 of FGF23c indeed contribute to GalNAc-T3’s substrate binding and catalysis5.
Impact of FGF23 Ser180 phosphorylation on glycosylation.
It has been reported that Ser180 phosphorylation negatively affects GalNAc-T3 O-glycosylation of a short FGF23 peptide without Thr171 in vitro16. We tested a longer FGF23 peptide containing both Thr171 and Thr178 acceptor sites with and without Ser180 phosphorylation (FGF23i and FGF23h, respectively) (Supplementary Fig. 14a and Supplementary Table 1). Time-course analysis of GalNAc-T3 glycosylation monitored by matrix-assisted laser desorption/ionization–time of flight–mass spectrometry (MALDI–TOF–MS) revealed glycosylation of Thr171 with both substrates, while the lectin-mediated glycosylation of Thr178 was severely affected, but not completely blocked by Ser180 phosphorylation in FGF23i (Supplementary Fig. 14a). MD simulations of TgGalNAc-T3 complexed with UDP and FGF23i, shows that the peptide, including Thr178, is released from the peptide-binding site (Supplementary Fig. 14b). Eventually, in the MD trajectory a distance of 11 Å is reached between Thr178-OH and the UDP β-phosphate. In addition, an interaction forms between pSer180 and Lys526, a residue conserved in the HsGalNAc-T3 (Supplementary Fig. 5) that is located in the lectin domain far from the peptide-binding site (Supplementary Fig. 14b). Overall, our data confirms that FAM20C phosphorylation of FGF23 slows Thr178 glycosylation notably. Tagliabracci and coworkers suggested that phosphorylation of FGF23 slightly accelerate cleavage of FGF23 by furin16. This is in keeping with the finding that no substantial difference in FGF23 cleavage is observed with the overexpression of FAM20C in osteocyte IDG-SW3 cells32 and consistent with our findings (Fig. 2c).
The nucleotide-sugar binding site of GalNAc-T3.
The structures of the UDP binding site among the GalNAc-T isoenzymes are the most conserved feature of GalNAc-Ts8,29. This is true for GalNAc-T3 where the pyrimidine moiety of UDP is sandwiched between His192 and Ile384, and Mn2+ is hexagonally coordinated by the UDP pyrophosphate group, His413, the D275XH277 motif, and a water molecule (Fig. 5a,b). UDP is tethered by hydrogen bonds with Val190, His192, Asp223 and Arg252 while its pyrophosphate interacts with His421, Trp385 and Arg416. However, in the TgGalNAc-T3–FGF23c complex, His421 and Trp385 do not interact with the pyrophosphate, as His421 is disordered and Trp385 adopts a ‘pre-in’ conformation. This is consistent with the low activity of GalNAc-T3 against FGF23c.
GalNAc-T6 is highly active against FGF23c.
Next we performed kinetic studies with GalNAc-T6, the closest paralog of GalNAc-T3 (Supplementary Fig. 15), which has been shown to have nearly identical substrate specificity as GalNAc-T3 (ref.33). We also included two other isoenzymes, GalNAc-T4 and T12, which display the same long-range glycosylation preference as GalNAc-T3 (ref.6) (Supplementary Fig. 16). The activity of GalNAc-T4 and T12 against nonglycosylated FGF23b was very low (Supplementary Fig. 16a), while both GalNAc-T3 and T6 showed the highest Kcat values as would be predicted for GalNAc-Ts within the same subfamily3,33. The FGF23c was also only used by GalNAc-T3 and T6, and GalNAc-T6 gave a ~3.5-fold better Kcat than that of GalNAc-T3 (Supplementary Fig. 16b). GalNAc-T6 also had a slightly better Km toward FGF23c, indicating that GalNAc-T6 is a better enzyme against FGF23c than GalNAc-T3. These kinetic differences were further supported by MD simulations of the TgGalNAc-T3 and HsGalNAc-T4, T6 and T12 complexed with UDP and FGF23c (Supplementary Videos 1–4; numbered in order according to the isoenzyme number). According to the simulations these complexes are stable throughout the entire MD trajectory, except for GalNAc-T12, where the peptide departs from the lectin-domain GalNAc-binding site. The calculations also show that the FGF23c Thr178 hydroxyl approaches UDP in the correct orientation for transfer only with GalNAc-T3 and T6. In contrast, with GalNAc-T4 and T12, the hydroxyl group of Thr178 is greater than 8 Å from the phosphate group of UDP. The latter would likely prevent glycosylation of the peptide.
HsGalNAc-T6 has a limited expression pattern in normal tissues compared to HsGalNAc-T3 (refs.2,34), and is not expressed in bone and osteocyte/blasts18,33 where FGF23 is produced. Note that both GalNAc-T3 and T6 are overexpressed or de novo expressed in many types of carcinoma, which may contribute to the tumor-induced osteomalacia associated with overexpression of FGF23 in benign tumors35.
Evaluation of disease-causing mutations in GALNT3.
The structure of TgGalNAc-T3 enables us to examine the structural roles of reported FTC and hyperostosis-hyperphosphatemia syndrome disease-causing mutations36,37 (http://www.hgmd.cf.ac.uk/ac/index.php) (Fig. 6a,b and Supplementary Figs. 5 and 17). Fourteen frameshift and point-nonsense mutations were identified that would lead to truncated inactive proteins (Fig. 6b). Another fourteen single point missense mutations have been reported at residues conserved in both HsGalNAc-T3 and TgGalNAc-T3, ten of these are located in the catalytic domain and four in the lectin domain (Fig. 6a). Four mutations (G256V, C280Y, L366R and H415Q) are located in or very close to the UDP–GalNAc-binding site, and likely disrupt the binding of UDP–GalNAc (Supplementary Fig. 17a). At least three mutations (T359K, A440E and I220N) potentially destabilize the enzyme by placing a charged or polar residue into a hydrophobic environment (Supplementary Fig. 17c,f,g). Two other mutations (E281G and R162Q) likely destabilize the human enzyme by the loss of ionic interactions between neighboring amino acid residues (Supplementary Fig. 17d,e). One missense mutation, C574G, disrupts a disulfide bridge in the lectin domain that may disrupt function and/or structure (Supplementary Fig. 17b), but regardless of whether the protein product is misfolded and degraded or has only lost its lectin function, the present results predict that the essential glycosylation function of FGF23 is lost. The last four FTC mutations (R180H, T272K and R438H/C) will require additional experimental study because they are partly or completely exposed to solvent and are not obviously involved in other interactions (Fig. 6a).
Fig. 6 |. Mapping HsGalNAc-T3 FTC mutations to the structure of TgGalNAc-T3.
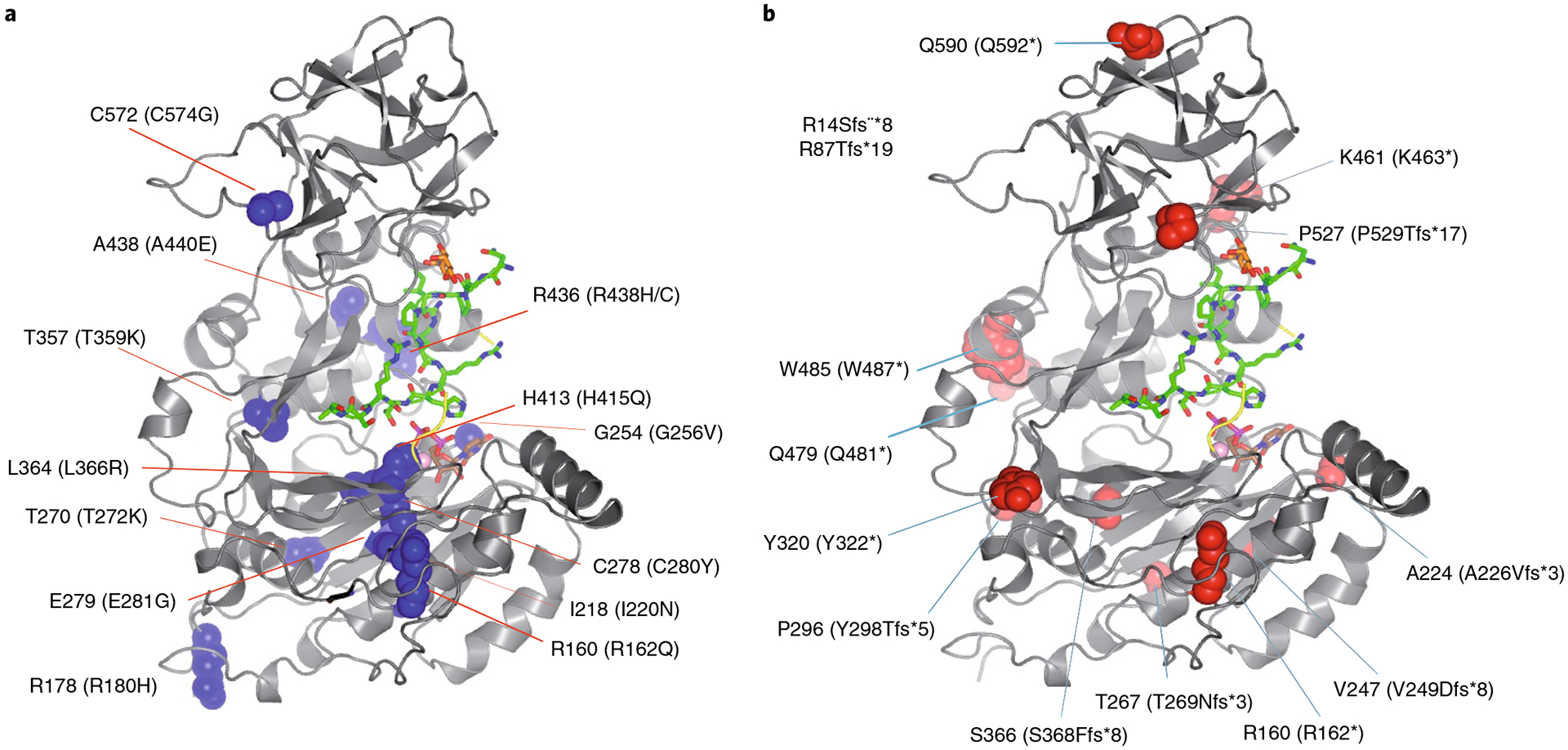
a, Structure of TgGalNAc-T3 where human FTC missense mutations are mapped as blue spheres (human residue numbers in parentheses). b, TgGalNAc-T3 structure showing the FTC frameshift and point-nonsense mutations mapped as red spheres (human residue numbers in parentheses). Note the R14Sfs*8 and the R87Tfg*19 mutants in the inset are located in the GalNAc-T3 stem. See Supplementary Figs. 5 and 17 for more information on the structural consequences of these mutations.
Discussion
The key findings of this study are that the unique selectivity of GalNAc-T3 for the critical Thr178 glycosite in FGF23 is obtained via lectin-mediated substrate binding and that poor catalysis leads to glycosylation of only a fraction of FGF23. We demonstrated that glycosylation of Thr178 both in vitro and in cells require previous GalNAc-glycosylation at Thr171 and a functioning GalNAc-T3 lectin domain, representing a confirmation of the lectin-mediated function of a GalNAc-T being used in a cellular context. Furthermore, our findings provide insights into how GalNAc-T isoenzymes with apparent extensive overlapping specificities achieve nonredundant isoenzyme-specific functions. Such specificity is achieved by using catalytically poor substrates, at the expense of optimal kinetic properties, and in the case of GalNAc-T3 and FGF23 through the assistance of its lectin domain. This may provide for optimal fine-tuning of O-glycosylation of highly specific glycosites to regulate important biological functions. Indeed, the expression of GalNAc-T3 in bone appears to be regulated by the level of circulating phosphate as a feedback loop for the secretion of stabilized FGF23 (ref.38). Similarly, we have recently demonstrated for GalNAc-T11 that its specific nonredundant substrate sites are glycosylated in a tight enzyme dose-dependent fashion that also appears subsaturated in cell lines39.
In summary, we propose that the site-specific and inefficient glycosylation of FGF23 is the key limiting step in regulating intact biological active FGF23 and phosphate homeostasis. We predict that these findings will apply to other isoenzymes of the GalNAc-T family to control distinct biological processes.
Methods
Stable expression of FGF23 mutants and GalNAc-transferases in CHO ldlD, CHO K1 and CHO GS−/− cells.
Expression constructs of human FGF23 (FGF23(RHTR)) with C-terminal myc and His tags and full-coding human GalNAc-T3 in the pcDNA3.1 vector (Invitrogen) were prepared as previously reported12 (Fig. 1b). Mutant FGF23(A178) constructs were obtained by PCR using FGF23(T178) as a template with the following primer pairs: RHAR(A178)-5′, 5′-CCACGGCGGCACGCCCGGAGCGCC-3′ and RHAR(A178)-3′, 5′-GGCGCTCCGGGCGTGCCGCCGTGG-3′. Full length GalNAc-T2 was amplified from human salivary gland RNA (Clontech) using primer pairs EBHC69 (5′-GCGAATTCGCCGAGTTGGGAGAATGCGGC-3′)/68 (5′-GCGAATTCCTACTGCTGCAGGTTGAGC-3′). EBHC68 and M-MLV RT (Promega) were used to generate template complementary DNA using High-Fidelity polymerase (Roche). EcoRI digested GalNAc-T2 product was inserted into the EcoRI site of pcDNA3-zeo (Invitrogen). Based on the previously reported secreted GalNAc-T3 construct a histidine tagged expression construct was generated by PCR using primer pairs T3HIS (5′-GCGGGATCCCCACCATCACCATCACCATCCAAGGATGGAAAGGAACATG-3′)/EBHC215, pAcGP67-GalNAc-T3sol DNA as a template and High-Fidelity polymerase (Roche). BamHI digested product was inserted into the BamHI site of pAcGP67 (Pharmingen) generating histidine tagged secreted GalNAc-T3his-sol. Quick Change kit (Stratagene) was used as previously described40 to generate mutant constructs of GalNAc-T3: full-coding GalNAc-T3D519H using primers T3D519HF (5’-AGCCTCTATGTCTGCATGTTGGAGAAAAC-3′)/T3D519HR (5′-GTTTTCTCCAACATGCAGACATAGAGGCT-3′) using full-coding pcDNA3-GalNAc-T3 as a template; secreted His-tagged GalNAc-T3D519H using T3D519HF/T3D519HR and secreted His-tagged GalNAc-T3D277H using T3D273HF (5′-CGCTCACATTTTTACATGCTCACTGTGAG-3′)/T3H273HR (5′-CTCACAGTGAGCATGTAAAAATGTGAGCG-3′ using pAcGP67-GalNAc-T3his-sol as a template. CHO ldlD27 cells and CHO K1 cells were grown in Ham’s F12/DMEM (1:1) supplemented with 3% FBS and 1 mM glutamine. CHO GS−/− cells were grown as suspension cultures in serum-free media (EX-CELL CHO CD Fusion, supplemented with 4 mM l-glutamine. Cells were transfected with pcDNA3.1 constructs using Metafectine reagent (Biontex Laboratories) and stable transfectants were screened by selecting with 0.4 mg ml−1 G418 and/or 0.4 mg ml−1 Zeocin (Invitrogen), and the expression of FGF23, GalNAc-T3 and GalNAc-T2 (ref.39) were confirmed by cell staining with anti-myc antibody (Invitrogen), anti-6xHis antibody (Thermo) and monoclonal antibodies raised against human GalNAc-T3 (2D10) and human GalNAc-T2 (4C4)41. Monoclonal mouse anti-human GalNAc-T4 antibody (4G2) was used for negative control. To evaluate the effects of O-glycosylation, CHO ldlD transfectant cells were grown in medium supplemented with 0.1 mM Gal, 1 mM GalNAc or a combination. Culture media was collected and used for western blot analysis.
Precise gene targeting using CRISPR/Cas9 in CHO GS−/− cells.
Gene targeting was performed as described before42 in stably FGF23 expressed CHO GS−/− cell lines with targeted KI of human GalNAc-T3. Briefly, 2 × 106 of CHO cells were transfected with Amaxa Nucleofector 2B (Amaxa kit V and program U24) with 1 μg each of endotoxin free plasmid DNA of Cas9-GFP and guide RNA-targeted FURIN or FAM20C gene. Then, 48 h after transfection, green fluorescent protein (GFP-)positive cells were enriched by fluorescence activated cell sorting, and following 1 week of culture, cells were single-sorted in 96-well plates. KO clones with frameshift mutations were identified by indel detection by amplicon analysis as described43 and selected clones were further verified by Sanger sequencing.
SDS–PAGE western blotting.
CHO GS−/− stably FGF23 expressed cells were seeded at 0.5 × 10 cells per ml in 50 ml TPP TubeSpin Bioreactors with 180 r.p.m. shaking speed at 37 °C and 5% CO2. After 48 h of cultivation the medium was centrifuged at 500g for 5 min and collected as supernatant used for western blot analysis. The supernatants were separated on 16/10/6% Tris-Tricine gels at 125 V for 150 min and transferred onto 0.22 μm polyvinylidene difluoride membrane using the iBlot system (Invitrogen). Membranes were blocked in 5% skimmed milk in TBS-T, washed three times in TBS-T before overnight incubation at 4 °C with anti-6xHis mouse IgG (Thermo), followed by washing and incubation with HRP-conjugated secondary antibodies (Dako) for 1 h at room temperature.
Metabolic labeling.
CHO K1 or CHO ldlD cells were seeded in 12-well plates with serum-containing medium, and allowed to grow until 80% confluent. Depletion of intracellular methionine was achieved by culturing in l-methionine/l-cysteine-free Dulbecco’s modified Eagle medium containing 3% FBS (dialyzed molecular weight cutoff, 3,500) for 1 h. Cells were labeled with 100 μCi per ml of promix 35S-Label (which includes radioactive l-methionine and l-cysteine) for 1 h or 2 h in the same medium. Immunoprecipitation of FGF23 using anti-myc from cell lysates and culture medium was performed as follows: the supernatant culture medium was subject to immunoprecipitation together with the culture medium. Supernatant and medium were precleared with 1:10 vol./vol. protein-G Sepharose (Amersham Biosciences), incubated with anti-myc antibody overnight (5 μg ml−1), and the IgG1 antibody precipitated with protein-G Sepharose (1:20 vol./vol.). The precipitate was analyzed by 4–12% Bis-Tris gradient NuPAGE (Invitrogen) blotted on nitrocellulose membranes (Bio-Rad) and detected using a STORM 820 PhosphorImager.
Immunocytology.
Immunostaining of CHO K1 cells was performed with washed cells on multi-well slides using fixation with 3% paraformaldehyde and permeabilization with 0.5% Triton X-100. Slides were incubated overnight at 4 °C with anti-myc, GALNT2 (4C4), GALNT3 (2D10) and GALNT4 (4G2), followed by fluorescein isothiocyanate-conjugated rabbit anti-mouse immunoglobulins (F261, Dako) and detection on a Zeiss fluorescence microscope using epi-illumination.
In vitro glycosyltransferase assays.
Recombinant human WT and mutant GalNAc-T3 enzymes for portions of the study were produced as soluble truncated proteins in insect cells as previously described44. The recombinant GalNAc-transferases were purified on Ni-NTA (QIAGEN) and assessed the purity by Coomassie stained SDS–PAGE gels and western blot analysis with monoclonal anti-GalNAc-T3 antibody (2F6). In vitro glycosylation assays for GalNAc-T3 glycosylation of peptides and phosphopeptides were performed as described before and monitored with MALDI–TOF19. Reactions were performed as product development assays in 25 μl buffer (25 mM cacodylic acid sodium, pH 7.4, 10 mM MnCl2, 0.25% Triton X-100), 2 mM UDP-GalNAc (Sigma), 0.4 μg of purified GalNAc-T3 and 10 μg of acceptor peptides (FGF23a, FGF23h and FGF23i). Reactions were incubated at 37 °C and 1 μl of the reaction mixtures were taken at the indicated time points and analyzed by MALDI–TOF–MS. To determine sites of incorporation, the glycosylated peptides were digested with endo-Arg and products analyzed by MALDI–TOF.
Cloning and purification of human GalNAc-T3.
The DNA sequence encoding amino acid residues 92–633 of the human GalNAc-T3 (HsGalNAc-T3) was codon optimized and synthesized by GenScript for expression in Pichia pastoris. The DNA, containing at the 5′ end a recognition sequence for XhoI and a KEX2 cleavage signal, and at the 3′ end a sequence encoding a histidine tag, a stop codon and a recognition sequence for SacII, was cloned into the pUC57 vector (GenScript). Following digestion with XhoI and SacII, the construct was subcloned into the protein expression vector pPICZαA, resulting in the plasmid pPICZαA-HsGalNAc-T3. The linearized plasmid after digestion with SacI-HF was transformed into the SMD1168 P. pastoris strain. Transformants were selected, grown and harvested as previously described30.
For the HsGalNAc-T3 purification, supernatant was dialyzed against buffer A (25 mM Tris-HCl pH 7.5, 10 mM β-mercaptoethanol) and loaded into a His-Trap Column (GE Healthcare). Each protein was eluted with an imidazole gradient in buffer A from 10 mM up to 400 mM. Buffer exchanged to buffer B (20 mM MES pH 6.7, 10 mM β-mercaptoethanol) was carried out using a HiPrep 26/10 Desalting Column (GE Healthcare) to trim the two potential N-glycosylation sites of HsGalNAc-T3 with the maltose binding protein (MBP)-Endo N-acetyl-β-d-glucosaminidase H (EndoH) fusion protein. A ratio of 0.012 mg of MBP-EndoH per milligram of enzyme was used to N-deglycosylate HsGalNAc-T3. After 20 h of reaction at 18 °C, the difference in size between noncleaved HsGalNAc-T3 and the cleaved preparation was verified through SDS–PAGE.
Once HsGalNAc-T3 EndoH deglycosylation was completed, buffer B was exchanged to buffer C (25 mM Tris-HCl pH 7.5, 150 mM NaCl) using again a HiPrep 26/10 desalting Column (GE Healthcare). The MBP-EndoH fusion protein was removed from the solution by the affinity binding to a MBPTrap HP Column (GE Healthcare). HsGalNAc-T3 was finally purified by size-exclusion chromatography using a HiLoad 26/60 Superdex 75 Colum (GE Healthcare), previously equilibrated with buffer C. Fractions containing HsGalNAc-T3 were dialyzed against buffer D (25 mM Tris-HCl pH 7.5, 1 mM tris(2-carboxyethyl) phosphine (TCEP)), concentrated and used for biophysical experiments. The quantification of the enzyme was carried out by absorbance at 280 nm using its theoretical extinction coefficient (90,145 M−1 cm−1).
Cloning and purification of T. guttata GalNAc-T3.
Although multiple attempts were performed, we were unable to obtain crystals of HsGalNAc-T3. Due to this, we conducted a search for the HsGalNAc-T3 orthologs that were expressed and purified as shown above. Hereafter, we focused on the TgGalNAc-T3 as it was the only ortholog for which we could obtain crystals and because of its high sequence homology (~80%) with HsGalNAc-T3. The DNA sequence encoding amino acid residues 91–631 of the TgGalNAc-T3 was codon optimized and synthesized by GenScript for expression in P. pastoris. The TgGalNAc-T3-encoding DNA construct contained the same features as described for the HsGalNAc-T3-encoding DNA construct except for the absence of the sequence encoding a histidine tag. All the subsequent steps were conducted in the same manner as described above. For the TgGalNAc-T3 purification, the supernatant was first dialyzed against buffer E (50 mM sodium acetate pH 5.4, 10 mM β-mercaptoethanol) and loaded into a HiTrap SP Sepharose FF Colum (GE Healthcare). The enzyme was eluted by a NaCl gradient up to 1 M in buffer A. Subsequent steps of the purification (N-glycan trimming and size-exclusion chromatography) were the same as the described for the HsGalNAc-T3. The quantification of the enzyme was carried out by absorbance at 280 nm using its theoretical extinction coefficient (86,290 M−1 cm−1).
Expression and purification of human GalNAc-T6 and T12.
The pGEn2-HsGalNAc-T6 and pGEn2-HsGalNAc-T12 plasmids encoding HsGalNAc-T6 and HsGalNAc-T12 were courteously provided by K. Moremen from the University of Georgia, Complex Carbohydrate Research Center. Both proteins were cloned as a GFP fusion construct that contained a N-terminal six-histidine-tag together with a tobacco etch virus (TEV) cleavage site between the GFP and the GalNAc-T6/T12.
Both plasmids were transfected into the human embyronic kidney 293F (HEK293F) cell line as described below. Cells in 2× 500 ml cell culture flasks were first grown to 1–1.5 × 106 cells per ml in F17 serum-free media with 2% Glutamax and 0.1% P188. For each 500 ml of culture, 500 μg of HsGalNAc-T6 or HsGalNAc-T12 plasmid (1 μg μl−1) was diluted to 8.4 ml with sterile 300 mM NaCl. Meanwhile, 500 μg of PEI-MAX (1 mg ml−1) was diluted to 7.85 ml with sterile 300 mM NaCl and added to the plasmid DNA solution. This mixture was incubated for 10 min and carefully added to each 500 ml cell culture flask. Cells were harvested 4 d posttransfection by spinning down at 300g for 5 min, after which the supernatants were collected and centrifuged at 40,000g for 10 min.
Supernatants were dialyzed against buffer A (25 mM Tris-HCl pH 7.5, 10 mM β-mercaptoethanol) and loaded into a His-Trap Column (GE Healthcare). Each protein was eluted with an imidazol gradient in buffer A from 10 mM up to 400 mM. Buffer exchange to buffer B (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM β-mercaptoethanol) was carried out using a HiPrep 26/10 Desalting Column (GE Healthcare). TEV protease (Sigma Aldrich) was then added in a ratio of 1/50 (TEV/protein) to both fusion constructs to cleavage the bound His-GFP. After 20 h of reaction at 18 °C, the cleavage was verified through SDS–PAGE.
TEV protease and GFP were later removed using a His-Trap Column (GE Healthcare), to which they bound. Isolated HsGalNAc-T6 and HsGalNAc-T12 were finally dialyzed against buffer C (25 mM Tris-HCl pH 7.5, 1 mM TCEP), concentrated and used for biophysical experiments. Quantification of both enzymes was carried out by absorbance at 280 nm using their theoretical extinction coefficients (ε280nmHsGalNAc-T6 = 93,850 M−1 cm−1 and ε280nmHsGalNAc-T12 = 88,810 M−1 cm−1).
Crystallization.
For the TgGalNAc-T3–UDP–P3 complex, crystals of the TgGalNAc-T3 were grown by sitting drop experiments at 18 °C by mixing 0.5 μl of protein solution (3.6 mg ml−1 TgGalNAc-T3, 5 mM UDP, 2 mM MnCl2 and 5 mM of MUC5AC-3 peptide in the buffer D) with an equal volume of a reservoir solution (1.3 M Na/K hydrogen phosphate pH 6.5). Note that MUC5AC-3 is an additive that we have previously found to assist GalNAc-T crystallization9. Under these conditions, crystals appeared within 15–30 d. The obtained crystals were then soaked for 20 min with a solution of 70 mM P3, 20 mM UDP and 5 mM MnCl2. Crystals were finally cryoprotected in a saturated solution of lithium sulfate and immediately frozen in a nitrogen gas stream cooled to 100 K.
For the TgGalNAc-T3–UDP–FGF23c complex, crystals of the TgGalNAc-T3 were grown by sitting drop experiments at 18 °C by mixing 0.5 μl of protein solution (3.6 mg ml−1 TgGalNAc-T3, 5 mM UDP, 2 mM MnCl2 and 5 mM of MUC5AC-3 peptide in the buffer D) with an equal volume of a reservoir solution (Molecular Dimensions, Morpheus precipitant mix 4, 0.1 M nitrate phosphate buffer and 0.2 M Morpheus buffer System 1). Under these conditions, crystals appeared within 15–30 d. The obtained crystals where then soaked for 40 min with a solution of 30 mM FGF23c, 15 mM UDP and 4 mM MnCl2. Crystals were finally cryoprotected in the precipitant solution containing an additional 20% ethylene glycol and immediately frozen in a nitrogen gas stream cooled to 100 K. Note that the soaking experiments were performed using a large excess of either FGF23c or P3 regardless of the different incubation times and concentration reported for each glycopeptide substrate.
Structure determination and refinement.
Diffraction data were collected on synchrotron beamline I24 of the Diamond Light Source (Harwell Science and Innovation Campus) at a wavelength of 0.97 Å and a temperature of 100 K. Data were processed and scaled using XDS45 and CCP4 (refs.46,47) software packages. Relevant statistics are given in Supplementary Table 3. The crystal structure was solved by molecular replacement with Phaser46,47 using the Protein Data Bank (PDB) entry 5NQA, for the human GalNAc-T4, as the template. Initial phases were further improved by cycles of manual model building in Coot48 and refinement with REFMAC5 (ref.49). ARP/wARP50 was used to build 503 out of 541 residues. Further rounds of Coot and refinement with REFMAC5 were performed to obtain the final structure. Note that the crystal structure of TgGalNAc-T3 cocrystallized with MUC5AC-3 and UDP alone displayed no electron density for either the peptide or UDP. The final models of crystals soaked with P3/FGF23c and UDP were validated with PROCHECK, model statistics are given in Supplementary Table 3. The asymmetric unit of the P212121 crystal contained one molecule of TgGalNAc-T3. The Ramachandran plots for the TgGalNAc-T3-UDP-P3 and TgGalNAc-T3–UDP–FGF23c complexes show that 95.1 and 96.4%, 4.4 and 3.4%, and 0.5 and 0.2% of the amino acids are in most favored, allowed and disallowed regions, respectively.
MD simulations in explicit water.
The crystal structures of the TgGalNAc-T3 in complex with P3 and FGF23c (without the unobserved C-terminal ethylenediamine-N,N’-disuccinic acid (EDDS) residues) were used as starting coordinates for most of the simulations. For comparing the conformational state of the FGF23c bound to GalNAc-T3, -T4, -T6 and –T12 during the simulations, we generated homology models of the human GalNAc-T6 and T12 by using the TgGalNAc-T3 and the human GalNAc-T4 (PDB 6H0B, ref.29) crystal structures as templates, respectively. The homology models were generated using the swiss-model server51. Whenever it was necessary, the glycopeptides found in these crystal structures were replaced by the peptides studied in this work by editing the original residues with Pymol 2.0 (https://pymol.org/2/). MD simulations were carried out with AMBER 18, implemented with ff14SB and Gycam06 (ref.52) force fields. Each conjugate was immersed in a 10 Å water box with TIP3P water molecules and charge neutralized by adding explicit Na+ counter ions. A two-stage geometry optimization approach was performed. The first stage minimized only the solvent molecules and ions, while the second stage was a unrestrained minimization of all the atoms in the simulation cell. The system was then heated by incrementing the temperature from 0 to 300 K under a constant pressure of 1 atmosphere under periodic boundary conditions. Harmonic restraints of 10 kcal mol−1 were applied to the solute, and the Andersen temperature coupling scheme was used to control and equalize the temperature. The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Hydrogen atoms were kept fixed through the simulations using the SHAKE algorithm. Long-range electrostatic effects were modeled using the particle-mesh-Ewald method. An 8 Å cutoff was applied to Lennard–Jones interactions. Each system was equilibrated for 2 ns with a 2-fs time step at a constant volume and temperature of 300 K. Production trajectories were then run for additional 0.5 μs under the same simulation conditions.
Enzyme kinetics.
In vitro glycosylation reactions were performed against a series of peptide/glycopeptide substrates (listed in Supplementary Table 1) with human GalNAc-T6, T4, T12, T2, -T3 isoenzymes and TgGalNAc-T3. Reactions (20 μl) were carried out with 0.02–0.03 μM enzyme, 0.02 mM to 0.7 mM substrate, 100 mM sodium cacodylate (pH 6.5), 1 mM β-mercaptoethanol, 10 mM MnCl2 and 0.2 mM UDP-[3H]-GalNAc (~5.5 × 107 DPM per μmole, American Radiolabeled Chemicals Inc.). Reactions were incubated at 37 °C and quenched with 40 μl of 250 mM EDTA at the appropriate reaction time. Reaction time points were chosen based on preliminary experiments to optimize each peptide’s initial rate determination. To account for the loss of transferase activity over long storage time, rates were normalized to the activity of glycopeptide P3 obtained on fresh transferase. After quenching, nonhydrolyzed UDP-[3H]-GalNAc was removed on a small Dowex 1 × 8 anion exchange column. Overall GalNAc use was determined by scintillation counting (Beckman LS6500 scintillation counter), by comparing [3H]-GalNAc in the Dowex pass through to the starting UDP-[3H]-GalNAc. GalNAc transfer to peptide substrate was determined by Sephadex G10 gel filtration chromatography, which separates [3H]-GalNAc transferred to substrate from [3H]-GalNAc transferred to water (the latter due to UDP-[3H]-GalNAc hydrolysis that is nonproductive GalNAc transfer).
Kinetics values were calculated from initial rates, where <30% of the UDP-GalNAc donor was consumed and less than 15% product formed. Experiments at each substrate concentration (5–7 different concentrations) were repeated 2–5 times, and values fit to nonlinear Michaelis–Menten or Michaelis–Menten with substrate inhibition programs in GraphPad Prism 7.03 from which the kinetic constants of Km, Vmax, Ki and Kcat along with their standard deviations were obtained. Kcat or turnover number (min−1) was obtained in terms of enzyme concentration in mM, while the catalytic efficiency (nmol−1 min−1) was obtained from Vmax per Km in terms of total enzyme amount in nanomoles. Reported standard deviations for the catalytic efficiency were calculated from the extreme maximum and minimum values of the Vmax and Km values based on their respective GraphPad Prism reported standard deviations.
Glycosylation site determination by Edman amino acid sequencing.
The site of glycosylation of FGF23b (Thr178) was determined by Edman amino acid sequencing using a Shimadzu PPS-Q53A Edman protein sequencer as previously described. G10 purified products were spotted onto polybrene precycled glass fiber disk (GFD) and sequenced using a modified GFD program. The glycosylated-PTH-Thr derivatives, were collected directly into scintillation vials on a Shimadzu fraction collector and scintillation counted for [3H]-GalNAc content (Beckman LS6500 scintillation counter). The presence of [3H]-GalNAc lag after a site of glycosylation is commonly observed in these determinations due to the poor extraction of the hydrophilic glycosylated-PTH residues from the GFDs.
Solid-phase peptide synthesis.
(Glyco)peptides were synthesized by stepwise microwave assisted solid-phase synthesis on a Liberty Blue synthesizer using the Fmoc strategy on Rink Amide MBHA resin (0.1 mmol). Fmoc-Thr[GalNAc(Ac)3-α-D]-OH (2.0 equiv.) was synthesized as described in the literature53 and manually coupled using HBTU [(2(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate], while all other Fmoc amino acids (5.0 equiv.) were automatically coupled using oxyma pure/DIC (N,N’-diisopropylcarbodiimide). The O-acetyl groups of GalNAc moiety were removed in a mixture of NH2NH2/MeOH (7:3). (Glyco)peptides were then released from the resin, and all acid sensitive side-chain protecting groups were simultaneously removed using TFA 95%, TIS (triisopropylsilane) 2.5% and H2O 2.5%, followed by precipitation with cold diethyl ether. The crude products were purified by high-performance liquid chromatography on a Phenomenex Luna C18(2) column (10 μm, 250 × 21.2 mm2) and a dual absorbance detector, with a flow rate of 10 ml min−1.
Peptide preparation.
All the peptides used in this work were dissolved at 100 mM in buffer 25 mM Tris-HCl pH 7.5. The pH of each solution was measured with pH strips and when needed adjusted to pH 7–8 through the addition of 0.1–5 μl of 2 M NaOH.
STD–NMR studies of FGF23c and P3 glycopeptide substrates.
All NMR experiments were recorded on a Bruker Avance 600 MHz spectrometer equipped with a triple channel cryoprobe head. The 1H NMR resonances of the peptides were completely assigned through standard two-dimensional–total correlation spectroscopy (2D–TOCSY) (30 and 80 ms mixing time) and 2D–nuclear Overhauser enhancement spectroscopy (2D–NOESY) experiments (400 ms mixing time). Solution conditions used for the NMR characterization studies were 1–3 mM glycopeptide, 25 mM perdeuterated Tris-d11 in 90:10 H2O/D2O pH 7.4, 7.5 mM NaCl and 1 mM DTT. The assignments were accomplished at 278 K (Supplementary Tables 5 and 6). The resonance of 2,2,3,3-tetradeutero-3-trimethylsilylpropionic acid (TSP) was used as a chemical shift reference in the 1H NMR experiments (δ TSP = 0 ppm). Peak lists for the 2D–TOCSY and 2D–NOESY spectra were generated by interactive peak picking using the Computer Aided Resonance Assignment software.
Samples for STD experiments were prepared in perdeuterated 25 mM Tris-d11 in deuterated water pH 7.4, 7.5 mM NaCl and 1 mM DTT. STD–NMR experiments of glycopeptides with GalNAc-Ts (human GalNAc-T3 and TgGalNAc-T3) were performed in the presence of UDP, MnCl2 at 298 K and with a molar ratio adjusted to 65:1 peptide/GalNAc-T.
The STD–NMR spectra were acquired with 1,920 transients in a matrix with 64 k data points in T2 in a spectral window of 12,335.53 Hz centered at 2,819.65 Hz, using the protocol previously described8,32. Control experiments were performed with the ligands in the presence and absence of the protein to optimize the frequency for protein saturation (−1 ppm) and to ensure that the ligand signals were not affected. However, all glycopeptides, when irradiated at −1 ppm in the absence of protein, showed residual saturation on the aliphatic methyl groups in the STD–NMR spectra. This nonspecific saturation was corrected for by subtraction, when quantifying the STD–NMR data in the presence of transferase. A blank STD experiment with only the protein was also recorded, which on subtraction eliminated the background signal of the protein. The STD–NMR total intensities were normalized with respect to the highest STD–NMR response. The signal of the anomeric proton of GalNAc, as well as, the Hα protons of the Ala residues could not be analyzed in the STD–NMR spectra due to their close proximity to the HDO resonance. The H6s protons of P3 could not be analyzed due to the close proximity to the Tris buffer signal.
AFM imaging.
HsGalNAc-T3 in buffer A (100 nM transferase, 25 mM MES pH 6.0, 1 mM TCEP and 400 nM MnCl2) was incubated on freshly cleaved muscovite mica pieces (Electron Microscopy Sciences) for 10 min at room temperature. The enzyme was further incubated with 1 μM UDP, 1–4 μM FGF23c and 1–4 μM MUC5AC-3 for 15 min under mild stirring at room temperature. Note that MUC5AC-3 was previously reported to be a monoglycopeptide substrate for GalNAc-T3 (ref.23). After sample incubation, the substrate was washed extensively with the buffer to remove unbound molecules. AFM measurements were performed in buffer A at 20 °C using the Tapping Mode on a MultiMode 8 AFM system (Bruker) at 18 kHz, using a 2 nm ultrasharp V-shaped silicon nitride cantilevers (SNL-D; Bruker Probes) with a spring constant of 0.06 N m−1 and nominal resonant frequency of 18 kHz.
AFM images were further analyzed with the WSxM software54. At least, three samples per condition were analyzed using data obtained from ten images of ten different 500 nm2 area regions55. Each feature was further analyzed using the zoom function of the WSxM program, performed without losing image information and discarding nonclear artifacts56.
SAXS.
SAXS experiments were conducted on the SWING beamline at the SOLEIL synchrotron (Gif-sur-Yvette, France) at a wavelength of λ = 1.033 Å. The Aviex charge-coupled device detector was positioned to collect data in the q-range 0.006–0.60 Å−1 (q = 4πsinθ λ−1, where 2θ is the scattering angle). All solutions were mixed in a fixed-temperature (15 °C) quartz capillary with a diameter of 1.5 mm and a wall thickness of 10 μm, positioned within a vacuum chamber. Then, 50 μl of monodisperse HsGalNAc-T3 at a concentration of 7 mg ml−1 was injected onto a size-exclusion column (SEC-3, 300 Å Agilent) using an Agilent HPLC system, and eluted directly into the SAXS flow-through capillary cell at a flow rate of 0.3 ml min−1. The elution buffer consisted of 25 mM Tris-HCl pH 7.5 and 1 mM TCEP. Identical experiments were performed in the presence of 1 mM of UDP/MnCl2, and with 1 mM of UDP/MnCl2 and 1 mM MUC5AC-3. Note that the ligands were also in the elution buffer at the indicated concentrations. SAXS data were collected continuously, with data collecting frames of 1 s and with a dead time between frames of 0.25 s. Selected frames corresponding to the main elution peak were averaged using FOXTROT. A large number of frames were collected during the first minutes of the elution and averaged to account for buffer/substrate scattering for subsequent subtraction from the signal obtained during protein elution. Data reduction to absolute units, frame averaging and subtraction were done using FOXTROT (synchrotron SOLEIL). Data were analyzed using the ATSAS v.2.7.3 software suite57.
Ranch2.0 (ref.58) was used to generate 10,000 structures using the homology model of the HsGalNAc-T3, which was obtained by using the swiss-model server51 and the TgGalNAc-T3 crystal structure as the template. Three structural pools were generated that differed in the presence or absence of ligand in the PDB files (either UDP–Mn+2 or UDP–Mn+2-MUC5AC-3 (where MUC5AC-3 was added using the modeler software59) and the TgGalNAc-T3–UDP–P3 complex. Only the flexible linker (residues 492–504) was considered as a flexible region. Two-hundred independent cycles of the EOM2.0 Genetic Algorithm (Gajoe)60 were run on each structural pool to estimate the variability in the distribution of the Rg values. The relatively high values of χ2 observed for the ensemble optimization method (Supplementary Fig. 10) may be explained by the fact that flexibility was not allowed in the catalytic domain flexible loop. Atomic models were refined using DADIMODO (software for rigid-body refinement of atomic models of multi-domain proteins against SAXS data; https://dadimodo.synchrotron-soleil.fr) enabling flexibility in the loop (residues 417–428) and the flexible linker (residues 492–504). Ten models for each sample were generated and ranked according to their χ2.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank the Diamond Light Source (Oxford) synchrotron beamline I24 (experiment nos. MX14739-6 and MX14739-11) and the SOLEIL synchrotron (Gif-sur-Yvette) SWING beamline (experiment nos. 99170088). We thank ARAID, MEC (grant no. CTQ2013-44367-C2-2-P, BFU2016-75633-P and RTI2018-099592-B-C21), the National Institutes of Health (grant no. GM113534 and instrument grant no. GM113534-01S), the Danish National Research Foundation (grant no. DNRF107), the FCT-Portugal (grant no. UID/Multi/04378/2013) and Gobierno de Aragón (grant nos. E34_R17, E35_17R and LMP58_18) with FEDER (grant no. 2014-2020) funds for ‘Building Europe from Aragón’ for financial support. I.C. thanks the Universidad de La Rioja for the FPI grant. F.M. and H.C. thank FCT-Portugal for IF Investigator (IF/00780/2015), PTDC/BIA-MIB/31028/2017 and UID/Multi/04378/2019 projects, and PTNMR (grant no. ROTEIRO/0031/2013 and PINFRA/22161/2016). P.B. acknowledges support from the Labex EpiGenMed, an ‘Investissements d’avenir’ program (grant no. ANR-10-LABX-12-01). The CBS (Montpellier) is a member of France-BioImaging (FBI, ANR-10-INBS-04-01) and the French Infrastructure for Integrated Structural Biology (FRISBI, ANR-10-INBS-05). The research leading to these results has also received funding from the FP7 (2007-2013) under BioStruct-X (grant agreement nos. 283570 and BIOSTRUCTX_5186). We also thank I. Echániz for technical support and K. Moremen from the University of Georgia, Complex Carbohydrate Research Center, for supplying the pGEn2-HsGalNAc-T6 and pGEn2-HsGalNAc-T12 plasmids.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-019-0444-x.
Data availability
The crystal structures of TgGalNAc-T3-UDP-P3 and TgGalNAc-T3-UDP–FGF23c complexes were deposited at the RCSB PDB with accession codes 6S24 and 6S22, respectively.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-019-0444-x.
References
- 1.Hurtado-Guerrero R Recent structural and mechanistic insights into protein O-GalNAc glycosylation. Biochem Soc. Trans 44, 61–67 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Bennett EP et al. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Las Rivas M, Lira-Navarrete E, Gerken TA & Hurtado-Guerrero R Polypeptide GalNAc-Ts: from redundancy to specificity. Curr. Opin. Struct. Biol 56, 87–96 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lira-Navarrete E et al. Substrate-guided front-face reaction revealed by combined structural snapshots and metadynamics for the polypeptide N-acetylgalactosaminyltransferase 2. Angew. Chem. Int. Ed. Engl 53, 8206–8210 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Gerken TA et al. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J. Biol. Chem 286, 14493–14507 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerken TA et al. The lectin domain of the polypeptide GalNAc transferase family of glycosyltransferases (ppGalNAc Ts) acts as a switch directing glycopeptide substrate glycosylation in an N- or C-terminal direction, further controlling mucin type O-glycosylation. J. Biol. Chem 288, 19900–19914 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revoredo L et al. Mucin-type O-glycosylation is controlled by short- and long-range glycopeptide substrate recognition that varies among members of the polypeptide GalNAc transferase family. Glycobiology 26, 360–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lira-Navarrete E et al. Dynamic interplay between catalytic and lectin domains of GalNAc-transferases modulates protein O-glycosylation. Nat. Commun 6, 6937 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivas ML et al. The interdomain flexible linker of the polypeptide GalNAc transferases dictates their long-range glycosylation preferences. Nat. Commun 8, 1959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Las Rivas M et al. Structural analysis of a GalNAc-T2 mutant reveals an induced-fit catalytic mechanism for GalNAc-Ts. Chemistry 24, 8382–8392 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Topaz O et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet 36, 579–581 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Kato K et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem 281, 18370–18377 (2006). [DOI] [PubMed] [Google Scholar]
- 13.White KE et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet 26, 345–348 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Benet-Pages A, Orlik P, Strom TM & Lorenz-Depiereux B An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet 14, 385–390 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Simpson MA et al. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin. Genet 75, 271–276 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Tagliabracci VS et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl Acad. Sci. USA 111, 5520–5525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urakawa I et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Bennett EP, Hassan H & Clausen H cDNA cloning and expression of a novel human UDP-N-acetyl-alpha-d-galactosamine. Polypeptide N-acetylgalactosaminyltransferase, GalNAc-t3. J. Biol. Chem 271, 17006–17012 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Kong Y et al. Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology 25, 55–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schjoldager KT et al. Deconstruction of O-glycosylation–GalNAc-T isoforms direct distinct subsets of the O-glycoproteome. EMBO Rep 16, 1713–1722 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khetarpal SA et al. Loss of function of GALNT2 lowers high-density lipoproteins in humans, nonhuman primates, and rodents. Cell Metab. 24, 234–245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J. Biol. Chem 293, 7408–7422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshimura Y et al. Elucidation of the sugar recognition ability of the lectin domain of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase 3 by using unnatural glycopeptide substrates. Glycobiology 22, 429–438 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Frishberg Y et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J. Bone Min. Res 22, 235–242 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Chen G et al. alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553, 461–466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagen FK, Hazes B, Raffo R, deSa D & Tabak LA Structure-function analysis of the UDP-N-acetyl-d-galactosamine:polypeptide N-acetylgalactosa minyltransferase. Essential residues lie in a predicted active site cleft resembling a lactose repressor fold. J. Biol. Chem 274, 6797–6803 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Kozarsky K, Kingsley D & Krieger M Use of a mutant cell line to study the kinetics and function of O-linked glycosylation of low density lipoprotein receptors. Proc. Natl Acad. Sci. USA 85, 4335–4339 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bar-Even A et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011). [DOI] [PubMed] [Google Scholar]
- 29.de Las Rivas M et al. Structural and mechanistic insights into the catalytic-domain-mediated short-range glycosylation preferences of GalNAc-T4. ACS Cent. Sci 4, 1274–1290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lira-Navarrete E et al. Structural insights into the mechanism of protein O-fucosylation. PLoS ONE 6, e25365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghirardello M et al. Glycomimetics targeting glycosyltransferases: synthetic, computational and structural studies of less-polar conjugates. Chemistry 22, 7215–7224 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto H et al. Posttranslational processing of FGF23 in osteocytes during the osteoblast to osteocyte transition. Bone 84, 120–130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett EP et al. Cloning and characterization of a close homologue of human UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase-T3, designated GalNAc-T6. Evidence for genetic but not functional redundancy. J. Biol. Chem 274, 25362–25370 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Joshi HJ et al. Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology 28, 284–294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minisola S et al. Tumour-induced osteomalacia. Nat. Rev. Dis. Prim 3, 17044 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Rafaelsen S, Johansson S, Raeder H & Bjerknes R Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet. 15, 98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramnitz MS, Gafni RI & Collins MT Hyperphosphatemic familial tumoral calcinosis. in GeneReviews (R) (eds Adams MP et al. ) https://www.ncbi.nlm.nih.gov/books/NBK476672/ (Univ. Wash., Seattle, 2018). [PubMed] [Google Scholar]
- 38.Takashi Y et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl Acad. Sci. USA 116, 11418–11427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hintze J et al. Probing the contribution of individual polypeptide GalNAc-transferase isoforms to the O-glycoproteome by inducible expression in isogenic cell lines. J. Biol. Chem 293, 19064–19077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wandall HH et al. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology 17, 374–387 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Steentoft C et al. A validated collection of mouse monoclonal antibodies to human glycosyltransferases functioning in mucin-type O-glycosylation. Glycobiology 29, 645–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol 33, 842–844 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Lonowski LA et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protoc 12, 581–603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wandall HH et al. Substrate specificities of three members of the human UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase family, GalNAc-T1, -T2, and -T3. J. Biol. Chem 272, 23503–23514 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W Xds. Acta Crystallogr. D 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 48.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langer G, Cohen SX, Lamzin VS & Perrakis A Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waterhouse A et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maier JA et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plattner C, Hofener M & Sewald N One-pot azidochlorination of glycals. Org. Lett 13, 545–547 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Horcas I et al. WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum 78, 013705 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Lostao A, Peleato ML, Gomez-Moreno C & Fillat MF Oligomerization properties of FurA from the cyanobacterium Anabaena sp. PCC 7120: direct visualization by in situ atomic force microscopy under different redox conditions. Biochim. Biophys. Acta 1804, 1723–1729 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Sun T, Lin FH, Campbell RL, Allingham JS & Davies PL An antifreeze protein folds with an interior network of more than 400 semi-clathrate waters. Science 343, 795–798 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Franke D et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr 50, 1212–1225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tria G, Mertens HD, Kachala M & Svergun DI Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCr. J 2, 207–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sali A & Blundell TL Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 60.Bernado P, Mylonas E, Petoukhov MV, Blackledge M & Svergun DI Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc 129, 5656–5664 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.