

Significance
Cullin-RING E3 ubiquitin ligases (CRLs) direct protein degradation to impact a myriad of physiological and pathological processes including cancer. This work reports the small-molecule compounds 33-11 and KH-4-43 as inhibitors of E3 CRL4 with anticancer potential. These compounds have provided an opportunity for developing tool compounds to address mechanistic and phenotypic questions about CRL4 in biochemical, cell-based, and animal studies. The results of correlation studies between cullin4 protein level and the compounds’ cytotoxic response as well as cullin4 depletion experiments suggest a role for low E3 abundance in sensitizing tumor cells for apoptosis. Collectively, the 33-11/KH-4-43–based CRL4 inhibitors may provide new exploitable therapeutic opportunities to target a subset of tumor lines characterized by low CUL4 expression.
Keywords: protein degradation, E3 CRL4, cullin4, small-molecule inhibitors, tumor inhibition
Abstract
Cullin-RING (really intersting new gene) E3 ubiquitin ligases (CRLs) are the largest E3 family and direct numerous protein substrates for proteasomal degradation, thereby impacting a myriad of physiological and pathological processes including cancer. To date, there are no reported small-molecule inhibitors of the catalytic activity of CRLs. Here, we describe high-throughput screening and medicinal chemistry optimization efforts that led to the identification of two compounds, 33-11 and KH-4-43, which inhibit E3 CRL4 and exhibit antitumor potential. These compounds bind to CRL4’s core catalytic complex, inhibit CRL4-mediated ubiquitination, and cause stabilization of CRL4’s substrate CDT1 in cells. Treatment with 33-11 or KH-4-43 in a panel of 36 tumor cell lines revealed cytotoxicity. The antitumor activity was validated by the ability of the compounds to suppress the growth of human tumor xenografts in mice. Mechanistically, the compounds’ cytotoxicity was linked to aberrant accumulation of CDT1 that is known to trigger apoptosis. Moreover, a subset of tumor cells was found to express cullin4 proteins at levels as much as 70-fold lower than those in other tumor lines. The low-cullin4–expressing tumor cells appeared to exhibit increased sensitivity to 33-11/KH-4-43, raising a provocative hypothesis for the role of low E3 abundance as a cancer vulnerability.
Cullin-RING (really intersting new gene) E3 ubiquitin (Ub) ligases (CRLs) are the largest RING-type E3 family, consisting of ∼300 members, ∼50% of the E3s identified in humans (1, 2). CRLs target many critical regulators of cell division and signaling. Canonical CRLs are modular complexes, in which a cullin (CUL) subunit’s N-terminal domain assembles interchangeably with CUL-specific substrate receptors capable of binding a substrate. On the other hand, a CUL’s C-terminal domain (CTD) binds a RING finger protein, ROC1/RBX1 for CUL1 to 4 or ROC2 for CUL5, to form a core ligase complex. CRLs’ core ligase collaborates with E2 Ub-conjugating enzymes for transferring Ub(s) to the bound substrate or a Ub moiety on a growing Ub chain.
A selective small-molecule modulator of CRLs’ function allows us to address mechanistic and phenotypic questions about its targets in biochemical, cell-based, and animal studies. To date, there is only one Food and Drug Administration (FDA)-approved E3 drug class that targets the substrate receptor cereblon (thalidomide/lenalidomide) (3). Current drug/probe discovery efforts against the Ub-proteasome system depend heavily on traditional methods that exploit the ability of small-molecule agents to disable an enzyme’s catalytic pocket. However, RING-type E3s are atypical enzymes and contribute to ubiquitination by mediating protein–protein interactions with substrate, E2, and Ub (1). High-resolution structural studies have shown that interactions involving E3’s RING domain, E2, and Ub are characterized by large, relatively flat interfaces (4). Such perceived “undruggable” features impose a significant barrier to structure-based ligand search using either virtual or fragment-based physical screening.
To date, there are no reported small-molecule lead compounds targeting the catalytic activity of any CRL. To address this need, we have recently created a novel high-throughput screen (HTS) platform using the fluorescence (Förster) resonance energy transfer (FRET) K48 di-Ub assay (5). In this system, a FRET signal is generated as a result of covalent conjugation of two Ub molecules carrying a pair of matching fluorophores in a reaction that requires E1, E2 Cdc34, and an E3 CRL1 subcomplex (ROC1–CUL1 CTD). Fully functional Ub variants were created to allow only one nucleophilic attack that produces a single Ub–Ub isopeptide bond, thereby eliminating the complexity associated with polyubiquitin chain assembly to ensure a high degree of reproducibility for effective HTS. Each fluorophore is placed to either donor or receptor Ub at a specific site in a manner that satisfies optimal energy transfer. A pilot HTS identified a small-molecule compound, suramin (an antitrypanosomal drug), that can inhibit E3 CRL1 activity by disrupting its ability to recruit E2 Cdc34 (5). These observations have provided proof-of-principle evidence that an E2–E3 interface can be perturbed through small-molecule modulators. The current study describes a large-scale HTS and extensive follow-up hit-to-lead studies, which identified a class of small-molecule inhibitors against E3 CRLs.
Results
The initial efforts from the HTS of >105 compounds and subsequent follow-up characterization studies have identified hit compound 33 (SI Appendix, Fig. S1) as an inhibitor of E3 CRL1/SCF using multiple assays including reconstituted substrate ubiquitination (SI Appendix, Figs. S2 and S3) and K48 di-Ub synthesis (SI Appendix, Fig. S4). Collectively, the results revealed the inhibitory effects of 33 on the ubiquitination of E3 SCFβTrCP2 substrates IκBα (SI Appendix, Fig. S2) and β-catenin (SI Appendix, Fig. S3), and on di-Ub chain assembly catalyzed by the E3 subcomplex ROC1–CUL1 CTD (SI Appendix, Fig. S4), without significantly affecting Ub thiol ester formation with E1 and E2 Cdc34a (SI Appendix, Fig. S5). Compound 33 exhibited ability to 1) inhibit ubiquitination of IκBα by SCFβTrCP2 with E2 Cdc34a or Cdc34b equally (SI Appendix, Fig. S2D); 2) block the activity of SCFβTrCP2 in either a Nedd8-modified or unmodified form, with the unmodified E3 appearing slightly more sensitive to the compound (SI Appendix, Fig. S2 B and C); and 3) inhibit ubiquitination reactions by E2 Cdc34 more potently than those by E2 UbcH5c (compare SI Appendix, Fig. S3B and SI Appendix, Fig. S3C). In addition, the results of immobilization experiments showed that the inhibitory effects of 33 were diminished by washing the preassembled E3–inhibitor complex (SI Appendix, Fig. S6), suggesting that the compound acts in a reversible manner.
Subsequent structure–activity relationship (SAR) studies using both commercially available analogs and medicinal chemistry have led to the identification of lead E3 CRL4 inhibitors 33-11 and KH-4-43 (SI Appendix, Fig. S1), respectively.
KH-4-43/33-11 Bind to E3 ROC1–CUL4A CTD.
We analyzed ligand–target interactions using microscale thermophoresis (MST), which measures the motion of molecules along microscopic temperature gradients and detects changes in their hydration shell, charge, or size (6). MST allows analysis of binding interaction events directly in solution without the need of immobilization to a surface. KH-4-43, 33-11, and 33 directly bind to the purified E3 ROC1–CUL4A CTD complex with a Kd of 83, 223, or 688 nM, respectively (Fig. 1 B and C). By comparison, KH-4-43, 33-11, and 33 bind to the purified, highly related E3 ROC1–CUL1 CTD complex with a Kd of 9.4, 4.5, or 1.6 µM, respectively (Fig. 1C and SI Appendix, Fig. S7A). Thus, while 33 binds to ROC1–CUL4A CTD and ROC1–CUL1 CTD with comparable affinity (∼0.7 vs. 1.6 µM in Kd), KH-4-43 and 33-11 bind to ROC1–CUL4A CTD about 100- or 20-fold more effectively than ROC1–CUL1 CTD. In addition, 33 exhibits virtually no binding activity to ROC1 alone, with a Kd of >500 µM (Fig. 1). Thus, the KH-4-43/33-11–based scaffold binds selectively to ROC1–CUL4A CTD. The MST binding data also suggest that the pyrazole ring substitution of 33-11/KH-4-43 enhances the selective binding to ROC1–CUL4A CTD over the oxygen atom linker of 33 to the pendant phenyl group (compare the structures of these three compounds in Fig. 1A).
Fig. 1.

Class of E3 CRL inhibitors and their selective interactions with ROC1–CUL4A CTD. (A) Chemical structures. (B and C) Ligand–E3 binding measured by MST. Purified ROC1–CUL4A CTD was mixed with increasing amounts of KH-4-43 and the resulting mixtures were analyzed by MST as detailed in SI Appendix, Methods. The fitting binding curve was generated with a calculated Kd (B). The error bars represent the range of three different readings. Similar binding experiments were performed with various complexes or a single protein agent along with the indicated compounds. The binding Kd is indicated in C. With respect to ROC1–CUL1 CTD, the average Kd is shown for each compound based on results of multiple MST experiments as detailed in SI Appendix, Fig. S7A. CUL1 CTD, CUL1 amino acids 411 to 776; CUL4A CTD, CUL4A amino acids 400 to 759.
Additional points are worth noting. First, 33-11/KH-4-43 showed little effect on Ub thiol ester formation with E1/E2 Cdc34 (SI Appendix, Fig. S5). Second, in keeping with observations that 33 binds to ROC1–CUL1 CTD more effectively than 33-11/KH-4-43 (SI Appendix, Fig. S7A), 33 was found more potent than 33-11/KH-4-43 in inhibiting FRET K48 di-Ub synthesis catalyzed by the E3 subcomplex ROC1–CUL1 (SI Appendix, Fig. S7B). This effect was further confirmed by the results of gel-based di-Ub synthesis experiments (SI Appendix, Fig. S8). Third, 33 binds to ROC1–CUL1 more effectively than ROC1–CUL1–Nedd8 (SI Appendix, Fig. S9A), and showed the ability to inhibit ubiquitination by ROC1–CUL1 more potently than that by ROC1–CUL1–Nedd8 (SI Appendix, Fig. S9B). These results are consistent with SI Appendix, Fig. S2 B and C, supporting the hypothesis that modification of CUL1 by Nedd8 renders E3 slightly less sensitive to compound 33.
KH-4-43/33-11 Inhibit Ubiquitination by E3 CRL4 In Vitro.
Previous studies have established lenalidomide-dependent ubiquitination of CK1α by E3 CRL4CRBN in vivo and in vitro (7, 8). To evaluate and quantify inhibitory effects of KH-4-43 on CRL4, we developed a biochemical assay to monitor CK1α ubiquitination with UbcH5c as a priming E2 and Cdc34b as an elongating E2 (Fig. 2 A, Top scheme). This approach utilizes a two-step reaction employing two Ub molecules labeled with distinct fluorophores, fluorescein-Ub (F-Ub) and iFluor555-Ub-Q31C-K48R [I-Ub-K48R (5)], respectively. After a brief incubation of the immobilized Flag-CK1α with CRL4CRBN, lenalidomide, F-Ub, E1 and E2 UbcH5c, E2 Cdc34b and I-Ub-K48R were then added for further incubation followed by an extensive washing step to remove materials unbound to Flag-CK1α. Ub-K48R was used because it allows E2 Cdc34b to attach only one Ub moiety to a receptor Ub that is linked to CK1α, thereby facilitating quantification. Fluorescence imaging showed time-dependent accumulation of CK1α–I-Ub-K48R conjugates (Fig. 2 A, Upper, lanes 2 to 4, red) that in size corresponded to the substrate modified by two or three Ub moieties. These products were significantly diminished in the absence of lenalidomide (lane 5) or UbcH5c/F-Ub (lane 7), demonstrating the dependency on both lenalidomide, which is required for E3–substrate interactions (8), and UbcH5c, which is necessary for priming the ubiquitination (9). Omission of E2 Cdc34b/I-Ub-K48R abolished the formation of CK1α–I-Ub-K48R conjugates, but instead accumulated CK1α–F-Ub (lane 6, green). To unequivocally determine whether CK1α–F-Ub can be utilized by CRL4CRBN/Cdc34b to form CK1α–I-Ub-K48R, preformed CK1α–F-Ub was purified and subjected to ubiquitination reaction with freshly added E3/E2/Ub reagents. The results showed lenalidomide-dependent formation of CK1α–I-Ub-K48R (red), concomitant with the disappearance of CK1α–F-Ub (green) (Fig. 2A, lanes 8 and 9). Note that the reaction products revealed by fluorescence scanning are verified by immunoblot analysis (Fig. 2 A, Lower). Collectively, these data demonstrate that CK1α is ubiquitinated by sequential actions of UbcH5c and Cdc34b: The substrate is first monoubiquitinated by UbcH5c followed by Ub chain elongation by Cdc34b. This mechanism closely resembles the ubiquitination of IκBα by E3 SCFβTrCP (9).
Fig. 2.

Effects of compounds on the ubiquitination of CK1α by E3 CRL4CRBN in vitro. (A) Lenalidomide-dependent ubiquitination of CK1α by E3 CRL4CRBN driven by E2 UbcH5c and Cdc34b. (A, Top) The reaction scheme using a procedure as described in SI Appendix, Methods. F-Ub, green; I-Ub-K48R, red. The reaction products were analyzed by both merged fluorescence imaging and immunoblot using an anti-CK1α antibody. (B) KH-4-43 inhibits the ubiquitination of CK1α by CRL4CRBN with Ub-K48R. (C) KH-4-43 inhibits the ubiquitination of CK1α by CRL4CRBN with the wild-type Ub. (D and E) Effects of 33-11 and 33 on the ubiquitination of CK1α by CRL4CRBN. The effects of 33-11 and 33 shown in D and E were determined using assays similar to B and C, respectively.
Using the above assay, we determined the effects of KH-4-43, which was added in the elongation phase of the reaction along with E2 Cdc34b/I-Ub-K48R. The results of fluorescence imaging or immunoblot analysis revealed the ability of KH-4-43 to inhibit the formation of CK1α–I-Ub-K48R conjugates in a dose-dependent fashion (Fig. 2B). To confirm this effect, KH-4-43 was subjected to CK1α ubiquitination with nonfluorescent, wild-type Ub to allow Ub chain formation, and both UbcH5c and Cdc34b were added simultaneously. The reaction products were detected by immunoblot. As shown, this reaction scheme supported Ub chain formation on CK1α that required adenosine triphosphate (ATP) (Fig. 2C, lanes 2 and 3), UbcH5c, and Cdc34b (Fig. 2E, lanes 1 to 3), as well as lenalidomide (Fig. 2E, lanes 7 and 8). KH-4-43 was able to block the formation of Ub chains on CK1α in a concentration-dependent manner (Fig. 2C).
We next examined the effects of compounds 33-11 and 33 on the ubiquitination of CK1α by CRL4CRBN using both the fluorescence assay with I-Ub-K48R (Fig. 2D) and a straight immunoblot experiment with the wild-type Ub (Fig. 2E). Significant inhibitory effects by 33-11 and 33 were observed at 30 µM. Comparison of the effects by KH-4-43, 33–11, and 33 suggests that KH-4-43 was the most potent among the three compounds, exhibiting inhibition at lower-dose ranges of 7.5 and 15 µM (compare Fig. 2 B and C and Fig. 2 D and E). It should be cautioned that the immobilization procedure used in our experiments inevitably causes significant variations, which precludes precise determination of the quantitative difference between these compounds. Despite this limitation, the above results unequivocally establish that KH-4-43/33-11/33 inhibit the ubiquitination of CK1α by CRL4CRBN in vitro.
Recent studies have shown that UBE2G1 works as an elongating E2 in the ubiquitination of E3 CRL4CRBN substrates including IKZFs and GSPT1 (10, 11). When UBE2G1 was used in lieu of Cdc34b for the ubiquitination of CK1α by CRL4CRBN, UBE2G1 was found to support ubiquitination poorly (Fig. 2E, compare lanes 3, 8, and 13). Data quantification suggests that in this case, Cdc34b was ∼4-fold more effective than UBE2G1 in converting the UbcH5c-primed mono-Ub to longer Ub chains. Whether these differences reflect the different substrates utilized awaits future investigation.
To more precisely determine the compounds’ inhibitory effects on E3 CRL4, we employed a CUL4A–Ub elongation assay that measures ubiquitination catalyzed by the CRL4 core ligase complex ROC1–CUL4A (SI Appendix, Fig. S10A). Both KH-4-43 and 33-11 were able to inhibit this ubiquitination reaction, with KH-4-43 appearing more potent than 33-11 (SI Appendix, Fig. S10B). In addition, the results of immobilization experiments showed that the inhibitory effects of compound KH-4-43 were diminished by washing the preformed ROC1–CUL4A/inhibitor complex (SI Appendix, Fig. S11), suggesting that the compound acts reversibly. To aid quantitative analysis, we developed a [32P]Ub chain elongation by ROC1–CUL4A assay (SI Appendix, Fig. S12). The results revealed the order of the compound’s inhibitory strength against ROC1–CUL4A as KH-4-43 (IC50 of 10 µM; IC50 is the concentration of an inhibitor required to inhibit ubiquitination by 50%.), 33-11 (IC50 of 21 µM), and 33 (IC50 of 67 µM).
The results of additional experiments shed light onto the properties of KH-4-43/33-11/33. First, side-by-side comparison found 33-11/KH-4-43 less active than 33 in inhibiting the ubiquitination of E3 SCFβTrCP2 substrates IκBα (SI Appendix, Fig. S13) and β-catenin (SI Appendix, Fig. S14). Consistent with these observations, 33-11/KH-4-43 were less potent than 33 in inhibiting di-Ub chain assembly by ROC1–CUL1 (SI Appendix, Figs. S7B and S8) and showed no inhibition of reactions by ROC1–CUL2/ROC1–CUL3 (SI Appendix, Fig. S16). In addition, 33-11 inhibited ubiquitination mediated by Nedd8–ROC1–CUL4A more effectively than Nedd8–ROC1–CUL1 (SI Appendix, Fig. S15). Note that CUL5 is the only major canonical cullin that was not tested in our studies; however, CUL5 is highly similar to CUL2. It is thus clear that 33-11 and KH-4-43 are less effective than 33 in inhibiting ubiquitination by the CUL1-based E3 CRL1/SCF. By contrast, KH-4-43/33-11 effectively inhibit ubiquitination by CRL4 (Fig. 2 and SI Appendix, Figs. S10 and S12). Combined with the binding data that show KH-4-43 and 33-11 bind to ROC1–CUL4A CTD 100- or 20-fold, respectively, more effectively than to ROC1–CUL1 CTD (Fig. 1 and SI Appendix, Fig. S7A), it can be concluded that KH-4-43/33-11 inhibit E3 CRL4 more specifically.
Compound 33 appears to be a more promiscuous inhibitor of E3 CRL as it inhibits ubiquitination by CRL1/SCF (SI Appendix, Figs. S2–S4) and by CRL4 (Fig. 2 D and E). These properties are consistent with the ability of 33 to bind to both ROC1–CUL4A CTD and ROC1–CUL1 CTD with a comparable Kd of ∼0.7 and 1.6 µM, respectively. It should be noted that we are mindful of the discrepancy between binding Kd measurement and ubiquitination inhibition assessment. For example, KH-4-43 shows a difference in Kd of two orders of magnitude in binding to ROC1–CUL4A CTD vs. ROC1–CUL1 CTD (Fig. 1). However, KH-4-43 inhibits ubiquitination by CRL4 and CRL1/SCF in the similar dose range of 10 and 30 µM (Fig. 2 B and C and SI Appendix, Figs. S13 and S14), respectively. Such discord is not understood at the present time. However, it should be remembered that, unlike binding experiments using only ligand and E3, the ubiquitination reactions employed in this work require many additional components. Thus, it may not be possible to compare binding affinity and E3 inhibition at the same molar scale.
Stabilization of E3 CRL4 Substrate CDT1.
Treatment of acute myeloid leukemia (AML) MV4-11 cells with KH-4-43/33-11 caused accumulation of the E3 CRL4 substrate CDT1 (12, 13), with KH-4-43 exhibiting more pronounced stabilization effects (Fig. 3A). CDT1 is a DNA replication initiation factor and its accumulation induces a DNA damage response to trigger apoptosis.
Fig. 3.

Treatment of cells with compounds KH-4-43 and 33-11 caused accumulation of the E3 CRL4 substrate CDT1. (A) KH-4-43 or 33-11 caused accumulation of CDT1 in AML MV4-11 cells in a dose-dependent manner. The abundance of CDT1 in cells treated with the compounds was analyzed by immunoblot as described in SI Appendix, Methods. The graph shows the quantification of three independent experiments with error bars indicating experimental variations. (B) Comparison of 33-11 with the Nedd8 inhibitor MLN4924 in MV4-11 cells. (C) Comparison of 33-11 and 33 in the ability to cause accumulation of CDT1 in AML MV4-11 and NB-4 cells. Loading controls: GAPDH, cyclophilin B, and tubulin.
33-11 caused accumulation of CDT1 to levels comparable to those observed with MLN4924 (Fig. 3B, lanes 1 to 3), which is a Nedd8 inhibitor that unselectively blocks all CRL activity (14). In contrast, 33-11 did not change the levels of p27 (Fig. 3B, lane 6), which is a substrate of E3 CRL1/SCFSkp2 (15) and was stabilized by MLN4924 (Fig. 3B, lane 5). These findings support the higher inhibitory activity of 33-11 toward E3 CRL4 over CRL1. Moreover, 33-11 was found to be more effective than 33 in causing stabilization of CDT1 in AML MV4-11 and NB-4 cells (Fig. 3C), consistent with the observation that 33-11 is more potent than 33 in CRL4 binding and inhibition (Fig. 1 and SI Appendix, Fig. S12). The effects of KH-4-43/33-11 on the stability of the CRL2 substrate Hif-α (16) and CRL3 substrate Nrf2 (17) cannot be examined in MV4-11, NB-4, or MOLT-4 cells due to the lack of expression in these cells.
Cytotoxicity by 33-11/KH-4-43 against a Subset of Tumor Cells.
To evaluate the cytotoxic effects of 33-11/KH-4-43, we employed flow cytometry with Annexin V staining to detect cells undergoing apoptosis (18) (Fig. 4A and SI Appendix, Fig. S17) and an ATP-based cell viability assay (Fig. 4B). The results showed a significant variation among the 36 tumor cell lines in response to the compounds. As shown, a subset of tumor lines was relatively sensitive to the treatment with 33-11/KH-4-43 including AML (NB4/MV4-11/THP-1/ML2), acute lymphoblastic leukemia (ALL; MOLT-4/CCRF-CEM), T lymphoma (Jurkat), pancreas (CAPAN-2 and K8082), as well as ovary (OVCAR-3), while a panel of breast, liver, and lung tumor cell lines was more resistant with EC50 >100 µM (Fig. 4A; EC50 is the concentration of an inhibiotr that gives half-maximal response in cell-based apoptotic or viability assays). Cytotoxicity was observed in both the ATP-based cell viability assay and Annexin V staining, despite the former assay appearing to be more sensitive. KH-4-43 was evidently more toxic than 33-11 in multiple cell lines tested, exhibiting EC50s of 1.8, 3.0, 3.9, and 4.8 µM in NB-4, MV4-11, OVCAR-3, and CAPAN-2 cells, respectively (Fig. 4B). Moreover, apoptosis was confirmed because the compound-induced effects were reversed by zVAD-fmkm (an apoptosis inhibitor). To assess the requirement of time exposure by 33-11/KH-4-43 for cytotoxicity, we performed washout experiments (Fig. 4C). The results showed that exposure between 6 and 24 h was required for either KH-4-43 or 33-11 to achieve maximal toxic effects on MV4-11 cells. These effects were similar to what was observed with the proteasome drug bortezomib (19). Together, these findings suggest that 33-11 and KH-4-43 are not generically toxic, as they have tumor-specific effects, indicating that this approach has clinical potential.
Fig. 4.

Cytotoxicity of compounds 33-11/KH-4-43 against a panel of tumor lines. (A) The ability of 33-11 to induce apoptosis was measured by Annexin V flow cytometry as described in SI Appendix, Methods. EC50/EC90 for each cell line is indicated. Note that EC50 or EC90 valued at 100 µM is the upper limit, representing those with EC50 or EC90 ≧100 µM. (B) Toxic response of a panel of cancer cells to 33-11/KH-4-43, as determined by viability assay (CellTiter-Glo). Note that mouse pancreatic cancer cell lines K8484 and DT8082 were isolated from KPC mice (38); the rest (34 in total) are all human cancer cell lines. (C) Washout. MV4-11 cells at 105 cells per milliliter were treated with compound at the concentrations indicated. At specified hours post compound treatment, the cells were washed once with fresh complete media without compound and allowed to grow for an additional 48 h followed by a viability test as in B. For B and C, the graph shows the quantification of three independent experiments with error bars indicating experimental variations.
33-11 Cytotoxicity in a Subset of Tumor Lines Was Correlated with Low CUL4 Abundance.
CUL4A and CUL4B are highly homologous except that CUL4B contains a distinct N-terminal extension that comprises a nuclear localization sequence, thereby shifting its cellular localization, making it a predominantly nuclear protein (20, 21). To determine if the cytotoxicity of 33-11 was related to the protein levels of CUL4A and CUL4B, we employed immunoblots to analyze their abundance in a panel of 10 tumor cell lines (Fig. 5A). The relative protein abundance is shown in Fig. 5B, revealing differences in protein levels of both CUL4A and CUL4B among the tumor cell lines. Using quantitative immunoblots with purified recombinant CUL4A and CUL4B proteins as standards, we have estimated the concentrations of both CUL4A and CUL4B in these 10 tumor lines as shown in SI Appendix, Table S1. A scatterplot of CUL4 protein level versus observed EC50 for 33-11 in the 10 cell lines reveals a trend indicating that CUL4 expression impacts sensitivity to 33-11 as indicated by the Spearman rank test, Rs = 0.797, P = 0.01, using a two-sided test (Fig. 5C). As shown, AML NB-4/MV4-11/ML-2, ALL MOLT-4, and HCT116/U2OS/H1299 appeared to fit into this regression line, suggesting that these tumor lines exhibit an inverse relationship between CUL4 abundance and cytotoxic response to 33-11. In all, these findings suggest that a subset of tumor cell lines 1) express CUL4 at strikingly low levels in comparison with others, by a factor as large as 70-fold, and 2) appear to respond to 33-11 for apoptosis at levels more profound than those with more abundant CUL4. Note that the difference in CUL4 abundance appears to be specific, because this subset of low-CUL4–expressing cells does not underexpress related cullin proteins (CUL1/CUL2/CUL3/CUL5) (SI Appendix, Fig. S18).
Fig. 5.

Low expression of CUL4A and CUL4B in a subset of tumor cells. (A) Immunoblot. The abundance of CUL4A or CUL4B in a panel of 10 tumor lines was analyzed by immunoblot. (B) Quantification. The graphs show the relative abundance of CUL4A (Left) or CUL4B (Right). The numbers of biological replicates are seven (MV4-11), four (NB-4), four (MOLT-4), two (ML-2), two (K562), three (THP1), three (U2OS), three (MDA-MB-231), three (HCT116), and two (H1299). The range of differences observed in these biological repeats is represented by the error bars. (B, Insets) Close-up views of the low-CUL4–expressing lines. (C) A scatterplot of CUL4 abundance vs. cytotoxic response to 33-11. The CUL4 concentration and 33-11 sensitivity (EC50) for the indicated cell lines are obtained from SI Appendix, Table S1.
However, it cannot be generalized that increased sensitivity to CUL4 inhibition is inversely correlated with CUL4 abundance in all tumor cell types, as AML THP-1 has high levels of CUL4 (∼1,125 fmol/mg) but is sensitive to 33-11 with an EC50 of 14 µM (Fig. 5B and SI Appendix, Table S1). In addition, both K8082 and OVCAR-3 were more sensitive to KH-4-43 than K8484 and SK-OV-3, respectively (Fig. 4B and SI Appendix, Fig. S17), but exhibited no significant difference in CUL4 abundance.
Taken together, these findings suggest that the low abundance of CUL4 protein might be correlated with its sensitivity to 33-11 in a tumor cell-specific manner. Note that the CUL4B protein is expressed at levels significantly higher than CUL4A in most tumor lines examined (SI Appendix, Table S1). In recently published proteomics studies (21, 22), CUL4B was also found more abundant than CUL4A.
Reducing CUL4 Levels Sensitized U2OS Cells to 33-11.
To evaluate whether decreased CUL4 expression sensitizes cells to 33-11, we used small interfering RNAs (siRNAs) targeting CUL4A, CUL4B, or both. U2OS cells expressing these siRNAs showed loss of CUL4A, CUL4B, or both as predicted (Fig. 6A). The siRNA-transfected cells were then treated with 33-11 and followed by flow cytometry analysis using Annexin V staining to quantify apoptosis. The results showed that combined depletion of both CUL4A and CUL4B enhanced the apoptotic response of the cells to 33-11 for apoptosis in a statistically significant manner (Fig. 6B; P = 0.0047). These findings suggest that at least in some tumor cell types, reducing CUL4 levels sensitized cells to 33-11 for apoptosis.
Fig. 6.

33-11 targets CRL4/CDT1 specifically. (A and B) Depletion of CUL4 sensitized U2OS cells to 33-11 for apoptosis. (A) Immunoblots confirmed depletion of CUL4A or CUL4B in U2OS cells treated with siRNAs. (B) siRNA-exposed U2OS cells were treated with 33-11, followed by Annexin V flow cytometry to quantify apoptosis. The graph represents three independent experiments with error bars indicating experimental variations. IC50 was calculated using SigmaPlot and a two-sided t test was performed using Microsoft Excel to determine a P value of 0.0047. (C and D) CDT1 knockdown diminishes cytotoxic effects of 33-11. An AML MV4-11 cell line was created to induce expression of shRNA by doxycycline to deplete CDT1. (C) Western analysis confirms depletion of CDT1 by DOX. HSP90/GAPDH/cyclophilin B are loading controls. (D) Apoptotic response with or without DOX. The Annexin V-positive apoptotic cells in response to 33-11 were reduced by DOX treatment. The graph incorporates two technical repeats.
Depletion of CDT1 Partially Overcomes 33-11–Induced Cytotoxicity.
Our findings suggest that aberrant accumulation of CDT1, as a result of inactivation of E3 CRL4 by 33-11, triggers apoptosis. If this hypothesis were true, small hairpin RNA (shRNA)-mediated CDT1 knockdown would reverse the cytotoxic effects by 33-11. To test this hypothesis, we have employed the Dharmacon SMARTvector Inducible Lentiviral shRNA System (GE) to create an inducible AML MV4-11 cell line to regulate the abundance of CDT1 by doxycycline (DOX). In the absence of compound, DOX treatment appeared to have little effect on CDT1 expression (Fig. 6C, lanes 1 and 2), presumably because this cell line expresses CDT1 protein at very low levels (also see Fig. 3). Consistent with previous observations (Fig. 3), 33-11 induced accumulation of CDT1 (Fig. 6C, lane 3). However, the 33-11–induced CDT1 accumulation was abolished by DOX treatment (Fig. 6C, lane 4). Under these conditions, the DOX-treated cells exhibited a significant decrease in 33-11–induced apoptosis as compared with the control (Fig. 6D). These findings further support the hypothesis that 33-11 is specifically impacting the cellular E3 CRL4/CDT1 pathway.
AML Xenograft Mouse Model.
We next determined if KH-4-43/33-11 possess antitumor activity in mice. To assess toxicity, female BALB/c nude mice (n = 3) received vehicle only (40 µL dimethyl sulfoxide; DMSO) or 50 or 100 mg/kg of 33-11 by intraperitoneal (IP) injection and were monitored for 7 d. Data showed no significant body weight loss by either vehicle alone or 33-11 at 50 mg/kg (SI Appendix, Fig. S19A), with the 33-11 100 mg/kg group showing >10% but <20% body weight loss. The results of necropsy revealed no adverse effects with either vehicle alone or the 33-11 50 mg/kg group. However, nonlethal abnormalities were observed in the 33-11 100 mg/kg group, including light red ascites, small intestine enlargement with yellow liquid, a yellow lump on the liver (4 × 3 mm), intestinal adhesion, and slight adhesion of abdominal viscera. These data reveal a preliminary no-adverse effect level at 50 mg/kg and thus limited structure-based (33-11) and target-based (E3 CRL4 inhibition) toxicity as well as a useful safety window (i.e., <50 mg/kg dose) for our therapeutic disease model approach.
AML MV4-11 contains receptor FLT3 internal tandem repeat domain insertions that lead to FLT3 constitutive activation and ligand-independent growth. FLT3 mutations are found in one-third of AML patients and MV4-11 has been used as a model for AML xenograft studies (23). We initiated studies to test antitumor activity in the mouse MV4-11 xenograft model with lead KH-4-43. MV4-11 tumors (n = 10 per group) were implanted in female BALB/c nude mice by inoculation subcutaneously at the right flank (107 MV4-11 cells). Treatment was initiated at day 19 when all mice had a tumor volume of ∼150 mm3. For a positive control, group 5 mice (G5) were treated with sorafenib (23) by daily oral administration at 3 mg/kg. For a negative control, G1 received vehicle only (40 µL DMSO) by IP injection. G2 and G3 were IP injected every 2 d (Q2D) with KH-4-43 at 25 or 50 mg/kg, respectively. G4 was IP injected daily (QD) with KH-4-43 at 50 mg/kg. Each treatment was for a total of 23 d of dosing. Animal body weight and tumor volume were measured twice a week. Given the guideline that >20% body weight loss is considered toxic, KH-4-43 at the tested dose levels is judged to be safe in the earlier dose range study (SI Appendix, Fig. S19B).
The changes of tumor volume and weight in G1 to G5 at the indicated time points after tumor inoculation were subjected to statistical analysis using two- and one-way ANOVA, respectively. Fig. 7 shows tumor volume changes among the five groups. G2 (KH-4-43, 25 mg/kg, Q2D), G3 (KH-4-43, 50 mg/kg, Q2D), G4 (KH-4-43, 50 mg/kg, QD), and G5 (sorafenib, 3 mg/kg, QD) exhibited tumor inhibition values of 18.60, 25.15, 38.90, and 46.02%, respectively. At day 42, G4 (P < 0.01) and G5 (P < 0.001) showed significant difference in tumor size in comparison with the vehicle control (G1). Consistent with this, G2, G3, and G4 exhibited reduction of tumor weight compared with the control (SI Appendix, Fig. S19C). End-of-study pharmacokinetics (PK) analyses detected KH-4-43 in plasma (SI Appendix, Fig. S19D) and in tumor (SI Appendix, Fig. S19E). In summary, KH-4-43 inhibited tumor growth in a dose-dependent manner and daily dosing at 50 mg/kg reached a statistically significant tumor growth inhibition effect in these MV4-11 xenografts. KH-4-43 exhibited improved antitumor efficacy in comparison with 33-11. While 33-11 showed a trend of tumor growth inhibition in an earlier AML MV4-11 xenograft model study, the inhibitory effects did not reach statistically significant levels. Moreover, KH-4-43 was measured in plasma with an area under the curve (AUC) over 24 h of ∼3.3 μM/h and slightly higher in the tumor of ∼4.7 μM/h of the MV4-11 xenograft mice, which is a sixfold higher AUC than that found in tumors of mice dosed with 33-11 (0.75 μM/h) (SI Appendix, Fig. S19 D and E for KH-4-43). Overall, the results of the mouse model studies help establish proof-of-concept evidence that compound KH-4-43 possesses modest antitumor activity in vivo.
Fig. 7.

Effects of KH-4-43 in the AML MV4-11 subcutaneous xenograft. AML MV4-11 subcutaneous xenograft experiments were performed as described in SI Appendix, Methods. Ten mice were used in each group (n = 10). The changes of tumor volume in G1 to G5 at the indicated time points after tumor inoculation were subjected to statistical analysis using two-way ANOVA.
Analog SAR, ADME/PK, and Other Properties.
Both commercial (21) and synthetic (56) analogs were used to develop a SAR at various positions of the E3 CRL inhibitor scaffold. The results of MST binding experiments have revealed that 33-7, a related analog without the 8-position hydroxyl group (-OH) of hit 33, is inactive in blocking ubiquitination, with drastically reduced binding activity to ROC1–CUL1 CTD (Kd decreases by >30-fold) (SI Appendix, Table S2). 33-3, lacking both the 7- and 8-hydroxyl groups, is completely inactive in both blocking ubiquitination and E3 binding (SI Appendix, Table S2). On the other hand, 33-2, an analog with the replacement of the 2-position trifluoromethyl (-CF3) group with a simple methyl (-CH3), exhibits a sevenfold drop in IC50 and an eightfold reduction of Kd (SI Appendix, Table S2). These results strongly suggest that 33’s hydroxyl group at the 8 position is essential, and its trifluoromethyl group at the 2 position is required for maximal inhibitory activity.
For further SAR studies, KH-4-43 and 33-11 were compared with their related structural analogs KH-4-119, KH-3-141, KH-3-115, MM-007, MM-008, and MM-009 (Table 1). KH-4-43 and 33-11 bound to ROC1–CUL4A (full length) with a Kd of 0.55 and 1.59 µM, respectively. The reason for lower binding with ROC1–CUL4A than ROC1–CUL4A CTD (Fig. 1 B and C) is not understood at this time. Note that ROC1–CUL4A possesses a free CUL4A N terminus, which binds to the DDB1–DCAF subcomplex in the context of a holoenzyme. It is possible that the unoccupied CUL4A N terminus may negatively impact the binding activity of KH-4-43/33-11 to ROC1–CUL4A. KH-4-119, a related analog with the 8-position methoxy group (-OMe; compare 8-OH of 33-11), has no detectable binding activity to ROC1–CUL4A (Table 1). In addition, KH-4-119 (7-OH, 8-OMe analog) shows very weak activity in blocking ubiquitination in vitro using a biochemical assay as described in SI Appendix, Fig. S12, and is incapable of inducing apoptosis in cancer cells using the Annexin V flow cytometry assay as described in Fig. 4A. Moreover, KH-3-115 lacking the pyrazole N-pendant phenyl group of 33-11 exhibits a >20-fold drop in Kd, and is weak in inhibiting ubiquitination and inactive in inducing apoptosis (Table 1). Finally, substitution of the pendant phenyl group with carbon chains of varying length (MM-007, MM-008, and MM-009, respectively) significantly weakened analogs in E3 binding/inhibition and apoptosis induction. These results confirm the importance of the hydroxyl group at the 8 position, and suggest that the pendant phenyl group is required for improved E3 binding, E3 inhibitory activity, and apoptosis.
Table 1.
33-11/KH-4-43 analogs and SAR studies
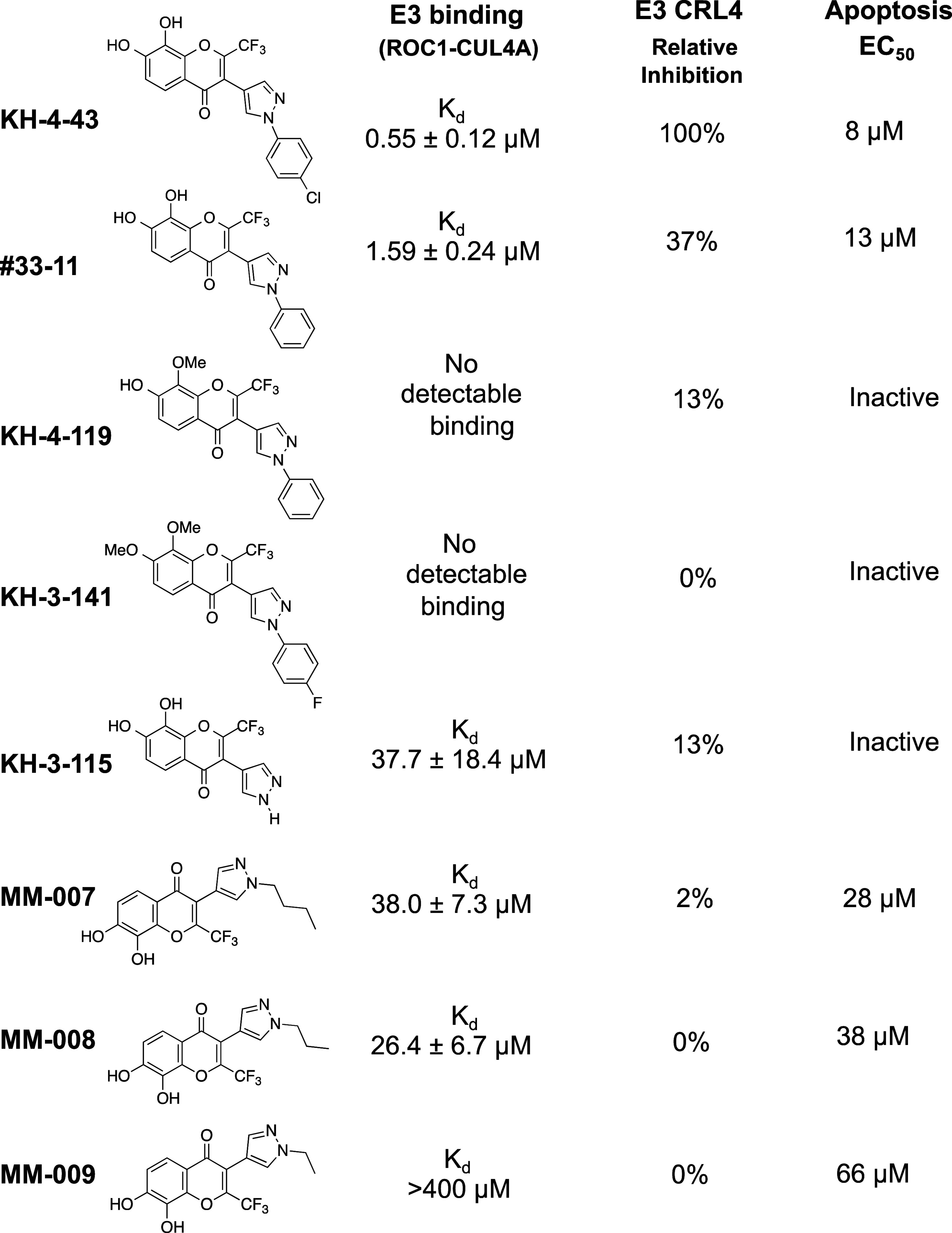 |
E3–ligand binding was determined using MST with the E3 complex ROC1–CUL4A as described in Fig. 1. E3 CRL4 inhibition was determined using the assay described in SI Appendix, Fig. S12. Values are expressed as relative inhibition in reference to KH-4-43 (100%). Apoptosis experiments were carried out as described in Fig. 4A. At 30 µM, KH-4-119, KH-3-141, and KH-3-115 were unable to induce any detectable level of apoptosis.
SI Appendix, Table S3 shows the results of in vitro ADME/DMPK (absorption, distribution, metabolism, and excretion/drug metabolism and pharmacokinetics) assays as well as IP PK in mice with 33-11/KH-4-43. Both 33-11 and KH-4-43 have many viable ADME properties, as well as some minor liabilities expected of early-stage lead compounds. For example, 33-11 has high plasma stability in three species and stability in rat and human liver microsomes. KH-4-43 shows excellent in vitro stability in mouse, rat, and human microsomes with low clearance (<10 h and long in vitro half-life >5 h) as a result of the 4-chloro substituent, which blocks a potential site of metabolism in the 4-phenyl position of 33-11 (see Fig. 1A for a chemical structure comparison). KH-4-43 is also highly permeable in the CACO-2 assay and is not a substrate of the PGP transporter (MDR1). KH-4-43 performed well in the in vivo mouse PK study when IP dosed at 100 mg/kg. KH-4-43 was well-tolerated and provided a Cmax of ∼35 µM with higher clearance in the first 3 h (α-phase), though it provided a sizable AUC (0 to 24 h of 19.2 µM/h), long terminal half-life of ∼9.6 h (β-phase elimination), and 24-h C-trough concentration of 89 nM. Taken together, 33-11/KH-4-43 have important properties for a new lead, but also reveal areas for improvement, especially on initial (α-phase) mouse PK clearance parameters.
33-11 and KH-4-43 both contain a catechol moiety (Fig. 1A), which might be deactivated rapidly in vivo by the enzyme catechol O-methyl transferase (COMT) (24). However, side-by-side comparison between 33-11 and tolcapone, a potent inhibitor of COMT (25), showed that at a range of substrate concentrations, 33-11 is 277- to 978-fold less active than tolcapone (SI Appendix, Table S3). These findings suggest that 33-11 is not a substrate of COMT.
The presence of the catechol moiety in 33-11/KH-4-43 (Fig. 1A) also raises perceived concerns about its stability due to redox cycling. However, our data have refuted these perceptions; ADME/PK analysis has shown that 33-11/KH-4-43 are stable in mouse, rat, and human microsomes and exhibit high plasma stability (SI Appendix, Table S3). In addition, we have observed antitumor activity by KH-4-43 in mouse xenografts (Fig. 7 and SI Appendix, Fig. S19) and detected this compound in both the plasma (SI Appendix, Fig. S19D) and the tumor (SI Appendix, Fig. S19E) of the experimental mice. Importantly, the results of SAR studies demonstrate the importance of not only the hydroxyl group at the 8 position but also the pendant pyrazole N-phenyl group required for improved E3 binding, E3 inhibitory activity, and apoptosis (Table 1). These findings are at odds with any hypothesis that the observed inhibition by KH-4-43/33-11/33 is mediated by nonspecific effects of the catechol group alone on E3 CRL. Specifically, KH-3-115, which possesses the catechol group, is inactive in apoptosis assays and weakly active in E3 binding and CRL4 assays (Table 1). Finally, the results of immobilization experiments (SI Appendix, Figs. S6 and S11) suggest that KH-4-43/33 act reversibly, which would be inconsistent with a possibility that these compounds exert inhibitory effects on E3 CRL4 by using the catechol group to mediate covalent interactions. There is a long history of chromones (contained in 33-11/KH-4-43) in drug discovery including several that are marketed drugs (26). Indeed, the chromone scaffold has been referred to as a “privileged structure” for drug discovery (27), indicating it is a useful starting scaffold to optimize (28). Of note, multiple approved chromone drugs include cromolyn, nedocromil, diosmin, and, more recently, alvocidib as well as flavoxate. There are also 17 FDA-approved catechol drugs (29). Taken together, our data demonstrate that 33-11/KH-4-43 are worthy starting points for further medicinal chemistry optimization.
Discussion
Scaffold against E3 CRL4.
According to the Structural Genomics Consortium, chemical probes are required to minimally have in vitro potency of the target protein at <100 nM, possess >30× selectivity relative to other sequence-related proteins of the same target family, and have demonstrated on-target effects at <1 μM (30). KH-4-43 appears close to these criteria because it has a binding Kd to E3 ROC1–CUL4A CTD or the highly related ROC1–CUL1 CTD at 83 nM or 9.4 µM, respectively (Fig. 1), which represents a difference of two orders of magnitude. KH-4-43 exhibits cytotoxicity in a subset of tumor cell lines with EC50 approaching ∼2 µM (Fig. 4). The results of RNA interference (RNAi)-mediated sensitization (CUL4 depletion; Fig. 6 A and B) and rescue (CDT1 depletion; Fig. 6 C and D) experiments suggest on-target effects by the KH-4-43–related analog 33-11. Note that lead 33-11 is not described in the chemical literature beyond its Chemical Abstracts Service number (431068-08-5), while KH-4-43 has not been previously reported.
Preliminary SAR study suggests that the 33-11/KH-4-43 scaffolds (Fig. 1A) are highly amenable to medicinal chemistry optimization with several substituents available for modification and building blocks that have been easily incorporated into the scaffold. The C-3 position likely contributes significantly to the binding specificity for E3 CRL4A. 33-11 differs from hit 33 (Fig. 1A) only at C-3 (N-phenyl pyrazole in 33-11 vs. phenyl ether in 33). Chromone substituent change from the aryl ether of 33 to the N-phenyl pyrazole group of 33-11 as the C-3 linker enhances the binding affinity by 20-fold (Fig. 1 and SI Appendix, Fig. S7A). SAR also has underscored the significance of the hydroxyl group at the 8 position and the pendant phenyl group (Table 1 and SI Appendix, Table S2). Future SAR optimization by exploring various phenyl substituents and replacement of pyrazole by other heteroaryl (or aryl) groups will lead to further increase of affinity for CRL4.
Ligand–Target Interactions and Specificity.
An effective drug typically exerts its effects via interactions with receptors, thereby initiating biochemical and physiological changes that characterize the drug’s response. The binding of compounds 33-11/KH-4-43 to E3 CUL4’s CTD (Fig. 1) presumably antagonizes E3’s ability to interact with an E2-conjugating enzyme(s), hence resulting in biochemical changes including inhibition of ubiquitination (Fig. 2 and SI Appendix, Figs. S10 and S12) and stabilization of E3 CRL4’s substrate CDT1 (Fig. 3). The physiological response by a subset of tumor cell lines to 33-11 and KH-4-43 is apoptosis (Fig. 4A and SI Appendix, Fig. S17). The cytotoxicity has been validated in the mouse tumor suppression in vivo model (Fig. 7 and SI Appendix, Fig. S19).
Despite conservation among cullins 1 to 5, KH-4-43/33-11 exhibit specificity in targeting E3 CRL4’s core ligase. First, biophysical experiments showed that KH-4-43/33-11 bind to the core ligase ROC1–CUL4A CTD with ∼100 to 200 nM affinity, 20- to 100-fold higher than the related ROC1–CUL1 CTD complex (Fig. 1). Second, biochemical assays showed that KH-4-43 is more potent than 33 in inhibiting the ubiquitination of CK1α by E3 CRL4CRBN (Fig. 2). Third, by contrast, KH-4-43 is less effective than 33 in inhibiting the ubiquitination of SCFβTrCP substrates IκBα (SI Appendix, Fig. S13) and β-catenin (SI Appendix, Fig. S14). 33-11 exhibits little inhibitory activity toward di-Ub synthesis reactions catalyzed by E3 subcomplexes including ROC1–CUL1 (SI Appendix, Fig. S8C) or ROC1–CUL2/ROC1–CUL3 (SI Appendix, Fig. S16). Fourth, cell-based experiments revealed that 33-11/KH-4-43 induce accumulation of the E3 CRL4 substrate CDT1 but not the E3 CRL1 substrate p27 (Fig. 3). Fifth, RNAi experiments showed that depletion of CUL4 sensitized U2OS cells for 33-11–induced apoptosis (Fig. 6 A and B) and knockdown of the E3 CRL4 substrate CDT1 partially overcame 33-11’s cytotoxicity (Fig. 6 C and D), providing physiological evidence that 33-11 targets the CRL4/CDT1 pathway specifically.
Thus, our data demonstrate that it is possible for a small-molecule agent to selectively target a specific cullin CTD. Despite a common globular CTD adopted by cullins 1 to 5, however, different cullin CTDs have divergent folds (31–34) and different total areas of interface with ROC1/Rbx1 that result in significant divergent orientation of the ROC1/Rbx1 RING domain among CRLs (33). It is therefore conceivable that a small molecule (such as 33-11/KH-4-43) may bind to a specific site within a cullin CTD that impacts the cullin–E2 interaction and/or alters the ROC1/Rbx1 orientation, leading to selective inhibition of ubiquitination.
E3 CRL4 Inhibitors Possess Tumor Inhibitory Potential.
The observed antitumor activity by our E3 CRL4 lead inhibitors is consistent with previous studies that have shown oncogenic potential of both CUL4A and CUL4B (35). In mouse tumor models, both overexpression and silencing approaches have established a tumor-promoting role for either CUL4A or CUL4B in multiple cancer types including lung, breast, colon, and hepatocellular carcinomas. In addition, a large body of investigations have linked CUL4 overexpression to human cancers including breast, ovary, stomach, colon, pancreas, lung, and bile ducts.
The most successful cancer therapies are chemical entities that preferentially target a protein or enzyme that carries a tumor-specific vulnerability, such as a mutation or other alteration that is specific to cancer cells and not found in normal host tissue. This is best exemplified by the clinical success of Gleevec, which is an inhibitor with exceptional affinity for the oncogenic, tumor-specific fusion kinase BCR-Abl that drives tumorigenesis in chronic myelogenous leukemia (36). In this study, we suggest a different type of vulnerability in a subset of tumor cells, which are characterized by low expression of the E3 component CUL4 (Fig. 5) and sensitivity to our CRL4 lead inhibitors 33-11/KH-4-43 (Fig. 4 and SI Appendix, Table S1). In a support of the key role played by CUL4 abundance in the inhibitors’ cytotoxic effects of certain tumor cells, we have demonstrated that reducing CUL4 levels by means of siRNA-mediated depletion sensitized U2OS cells to 33-11 treatment for apoptosis (Fig. 6 A and B). Moreover, lead inhibitors 33-11/KH-4-43 exhibited in vivo antitumor activity in AML MV4-11 xenograft mouse model studies (Fig. 7). The major impact of low target expression in enhancing druggability has been previously recognized as clinically significant. A well-known example, leukemic del(5q) myelodysplastic syndrome cells, are haploinsufficient for CK1α and these cells are sensitized to lenalidomide therapy that specifically targets CK1α for degradation (7). If validated, the low-target expression-driven drug sensitivity mechanism may be advantageous as compared with targeting amplified or mutated oncoproteins because targeting these oncogenic drivers frequently leads to selection events such as secondary mutations which result in drug resistance (37).
Methods
SI Appendix contains a methods section that describes in detail the experimental procedures used in this study, including HTS and hit validation and classification, chemical synthesis of analogs, protein expression and purification, in vitro ubiquitination assays, siRNA transfection, MST E3–ligand binding, compound treatment and immunoblot analysis, Annexin V flow cytometry, CellTiter viability assay, ADME and mouse IP PK studies, mouse tolerance/toxicity study, tumor mouse xenograft assay, and COMT assay.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants R01GM122751 (to Z.-Q.P.) and 1R01CA251425-01 (to Z.-Q.P. and R.J.D.) and R01GM074830 and R01GM130144 (to L.H.). We thank S. Mai and S.-H. Chen for preliminary mouse tumor studies; S. K. Athuluri-Divakar and E. P. Reddy for advice on MST binding experiments; S. Aaronson, E. P. Reddy, and P. Samir for providing tumor cell lines; Q. Yu for assistance in the early phase of this project; H. Y. Liu and R. J. Garippa (Memorial Sloan-Kettering Cancer Center Core Screening Facility) for expert assistance in HTS; and K. Kumar and C. Suebsuwong for assistance in preparing the manuscript concerning synthetic chemistry.
Footnotes
Competing interest statement: R.J.D., Z.-Q.P., K.W., K.Q.H., and M.M. are inventors on patent application 63/144,358 submitted by Icahn School of Medicine at Mount Sinai claiming inhibitors of Cullin-RING E3 ubiquitin ligase 4 (CRL4) to treat leukemia and other cancers.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2007328118/-/DCSupplemental.
Data Availability
All study data are included in the article and/or SI Appendix.
References
- 1.Petroski M. D., Deshaies R. J., Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Sarikas A., Hartmann T., Pan Z.-Q., The cullin protein family. Genome Biol. 12, 220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wertz I. E., Wang X., From discovery to bedside: Targeting the ubiquitin system. Cell Chem. Biol. 26, 156–177 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Plechanovová A., Jaffray E. G., Tatham M. H., Naismith J. H., Hay R. T., Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu K., et al., Suramin inhibits cullin-RING E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 113, E2011–E2018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seidel S. A., et al., Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krönke J., et al., Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petzold G., Fischer E. S., Thomä N. H., Structural basis of lenalidomide-induced CK1α degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Wu K., Kovacev J., Pan Z.-Q., Priming and extending: A UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol. Cell 37, 784–796 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu G., et al., UBE2G1 governs the destruction of cereblon neomorphic substrates. eLife 7, e40958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sievers Q. L., Gasser J. A., Cowley G. S., Fischer E. S., Ebert B. L., Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 132, 1293–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higa L. A., Mihaylov I. S., Banks D. P., Zheng J., Zhang H., Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 5, 1008–1015 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Hu J., McCall C. M., Ohta T., Xiong Y., Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6, 1003–1009 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Soucy T. A., et al., An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Skaar J. R., Pagan J. K., Pagano M., Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. 14, 369–381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamura T., et al., Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. U.S.A. 97, 10430–10435 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi A., et al., Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin S. J., Henry C. M., Distinguishing between apoptosis, necrosis, necroptosis and other cell death modalities. Methods 61, 87–89 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Brignole C., et al., Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J. Natl. Cancer Inst. 98, 1142–1157 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Zou Y., et al., Characterization of nuclear localization signal in the N terminus of CUL4B and its essential role in cyclin E degradation and cell cycle progression. J. Biol. Chem. 284, 33320–33332 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reichermeier K. M., et al., PIKES analysis reveals response to degraders and key regulatory mechanisms of the CRL4 network. Mol. Cell 77, 1092–1106.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Wang D., et al., A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auclair D., et al., Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia 21, 439–445 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Eisenhofer G., Kopin I. J., Goldstein D. S., Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 56, 331–349 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Antonini A., et al., COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): Relevance for motor and non-motor features. Neuropsychiatr. Dis. Treat. 4, 1–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva C. F. M., Pinto D. C. G. A., Silva A. M. S., Chromones: A promising ring system for new anti-inflammatory drugs. ChemMedChem 11, 2252–2260 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Keri R. S., Budagumpi S., Pai R. K., Balakrishna R. G., Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 78, 340–374 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Matos M. J., Vazquez-Rodriguez S., Uriarte E., Santana L., Borges F., Synthesis and pharmacological activities of non-flavonoid chromones: A patent review (from 2005 to 2015). Expert Opin. Ther. Pat. 25, 1285–1304 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Yang D.-P., Ji H.-F., Tang G.-Y., Ren W., Zhang H.-Y., How many drugs are catecholics. Molecules 12, 878–884 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arrowsmith C. H., et al., The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angers S., et al., Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Zheng N., et al., Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Cardote T. A. F., Gadd M. S., Ciulli A., Crystal structure of the Cul2-Rbx1-EloBC-VHL ubiquitin ligase complex. Structure 25, 901–911.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duda D. M., et al., Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell 134, 995–1006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng J., et al., The emerging role for cullin 4 family of E3 ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 1871, 138–159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ren R., Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 5, 172–183 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Orlando E., Aebersold D. M., Medová M., Zimmer Y., Oncogene addiction as a foundation of targeted cancer therapy: The paradigm of the MET receptor tyrosine kinase. Cancer Lett. 443, 189–202 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Olive K. P., et al., Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or SI Appendix.