

Abstract
Background & Aims:
Excess and unresolved endoplasmic reticulum (ER) stress in intestinal epithelial cells (IECs) promotes intestinal inflammation. Activating transcription factor 6 (ATF6) is one of the signaling mediators of ER stress. We studied the pathways that regulate ATF6 and its role for inflammation in IECs.
Methods:
We performed an RNA interference screen, using 23,349 unique small interfering RNAs targeting 7783 genes and a luciferase reporter controlled by an ATF6-dependent ERSE (ER stress-response element) promoter, to identify proteins that activate or inhibit the ATF6 signaling pathway in HEK293 cells. To validate the screening results, intestinal epithelial cell lines (Caco-2 cells) were transfected with small interfering RNAs or with a plasmid overexpressing a constitutively active form of ATF6. Caco-2 cells with a CRISPR-mediated disruption of autophagy related 16 like 1 gene (ATG16L1) were used to study the effect of ATF6 on ER stress in autophagy-deficient cells. We also studied intestinal organoids derived from mice that overexpress constitutively active ATF6, from mice with deletion of the autophagy related 16 like 1 or X-Box binding protein 1 gene in IECs (Atg16l1∆IEC or Xbp1∆IEC, which both develop spontaneous ileitis), from patients with Crohn’s disease and healthy individuals (controls). Cells and organoids were incubated with tunicamycin to induce ER stress and/or chemical inhibitors of newly identified activator proteins of ATF6 signaling, and analyzed by real-time PCR and immunoblots. Atg16l1∆IEC and control (Atg16l1fl/fl) mice were given intraperitoneal injections of tunicamycin and were treated with chemical inhibitors of ATF6 activating proteins.
Results:
We identified and validated 15 suppressors and 7 activators of the ATF6 signaling pathway; activators included the regulatory subunit of casein kinase 2 (CSNK2B) and acyl-CoA synthetase long chain family member 1 (ACSL1). Knockdown or chemical inhibition of CSNK2B and ACSL1 in Caco-2 cells reduced activity of the ATF6-dependent ERSE reporter gene, diminished transcription of the ATF6 target genes HSP90B1 and HSPA5 and reduced NF-κB reporter gene activation upon tunicamycin stimulation. Atg16l1∆IEC and or Xbp1∆IEC organoids showed increased expression of ATF6 and its target genes. Inhibitors of ACSL1 or CSNK2B prevented activation of ATF6 and reduced CXCL1 and TNF expression in these organoids upon induction of ER stress with tunicamycin. Injection of mice with inhibitors of ACSL1 or CSNK2B significantly reduced tunicamycin-mediated intestinal inflammation and IEC death and expression of CXCL1 and TNF in Atg16l1∆IEC mice. Purified ileal IECs from patients with CD had higher levels of ATF6, CSNK2B, and HSPA5 mRNAs than controls; early-passage organoids from patients with active CD show increased levels of activated ATF6 protein, incubation of these organoids with inhibitors of ACSL1 or CSNK2B reduced transcription of ATF6 target genes, including TNF.
Conclusions:
Ileal IECs from patients with CD have higher levels of activated ATF6, which is regulated by CSNK2B and HSPA5. ATF6 increases expression of TNF and other inflammatory cytokines in response to ER stress in these cells and in organoids from Atg16l1∆IEC and Xbp1∆IEC mice. Strategies to inhibit the ATF6 signaling pathway might be developed for treatment of inflammatory bowel diseases.
Keywords: IBD, inflammation, signal transduction, gene expression
Graphical Abstract
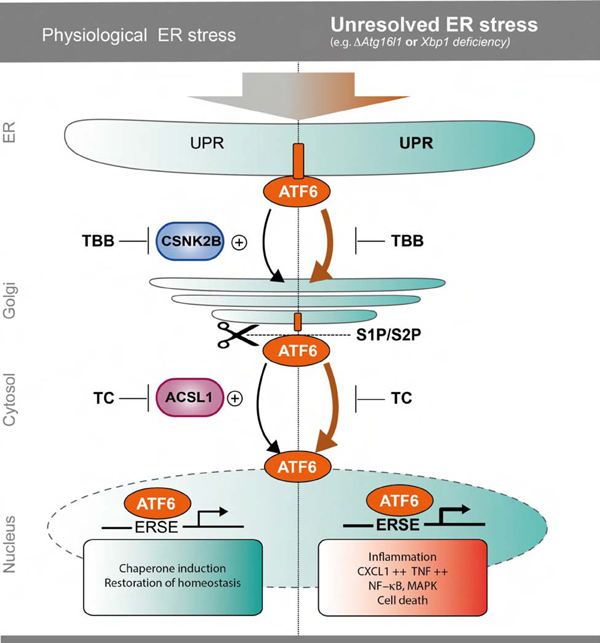
Introduction
The Endoplasmic Reticulum (ER) is a tightly controlled cellular compartment for synthesis and folding of secretory proteins. Accumulation of unfolded/misfolded proteins within the ER provokes an unfolded protein response (UPR) with the aim to reduce ER-stress and restore homeostasis. Unresolved ER-stress can lead to excessive UPR activation, which can be inflammatory and ultimately lead to programmed cell death1. Three main arms govern the ER-stress-induced response in mammalian cells, regulated by three key molecules, respectively: IRE1α (endoribonuclease inositol-requiring enzyme 1α), PERK (protein kinase RNA-like ER kinase) and ATF6α2, 3. These three proximal transmembrane sensors are activated by dissociation of the ER chaperone glucose regulated protein 78 (GRP78) in favor of binding to misfolded proteins. Unbound ATF6α translocates to the Golgi network, where it undergoes regulated intramembrane proteolysis mediated by the site 1 and site 2 protease (S1P/S2P). The released cytosolic N-terminal portion of ATF6α migrates to the nucleus and induces the expression of genes containing ER-stress response elements (ERSE-I and -II) e.g. GRP784. Recent findings indicate that activation of the UPR induces macroautophagy and that autophagy in turn is able to alleviate the UPR5–9. A strong link for an impaired UPR/autophagy crosstalk has been identified in the etiopathogenesis of inflammatory bowel disease (IBD), by both functional and genetic evidence7, 8, 10–12. Conditional deletion of XBP1 in the intestinal epithelium leads to paradoxical activation of ER-stress and a spontaneous enteritis in mice8. Likewise, mice with a conditional intestinal epithelial deletion of the Crohńs disease (CD) risk gene Autophagy related 16 like 1 (ATG16L1), a core component of the autophagic machinery, display signs of unresolved ER stress, impaired Paneth cell architecture and suffer from spontaneous, age-dependent onset of ileitis 13. The models share increased pro-inflammatory signals via NF-κB, high levels of TNFα secretion, increased necroptotic epithelial cell death and accumulation of IRE1α5, 13, 14, reflecting molecular alterations observed in IBD patients.
Surprisingly little is known about the executioners of the inflammatory signaling under conditions of hyperactivated ER-stress. Importantly, while the downstream transcriptional program induced by ATF6α signaling has been extensively studied in the context of ER homeostasis15, 16, our knowledge regarding the exact regulatory network of the ATF6 branch within the intestinal epithelium is still limited. We hypothesized that modulation of ATF6α function might counteract detrimental signals of aggravated ER-stress in IECs, specifically in conditions of genetically disturbed autophagy.
In this study, we set out to systematically understand modulators of ATF6α signaling using a stringent high-throughput RNAi screening. Among the validated hits, two upstream co-activators of ATF6α signaling were identified and further validated: Acyl-CoA Synthetase Long Chain Family Member 1 (ACSL1) and the Casein Kinase 2 Beta (CSNK2B). Using primary murine intestinal organoid cultures, we show that impairment of autophagy or unresolved ER stress in IECs results in the compensatory upregulation of the ATF6α branch, which we link to enhanced pro-inflammatory signaling and cytokine secretion, which could be restricted by inhibition of ACSL1 or CSNK2B in vitro and in vivo. Our findings point to upstream inhibition of ATF6α as a novel therapeutic strategy to overcome detrimental pro-inflammatory effects of failing autophagy and the UPR in IECs.
Materials and methods
Cell culture and reagents
Information on cell culture and reagents can be found in the supplement.
Cultivation of murine SI organoids
Crypts were isolated from mouse small intestine (SI) by EDTA-based Ca2+/Mg2+ chelation and intestinal organoids were cultivated as described by Sato et al.17.
Culture of human intestinal organoids
Human intestinal biopsy specimens were obtained from patients who underwent endoscopic examination. The study was approved by the Ethics Committee of the Medical Faculty, University Kiel (vote B231/98) and written informed consent was obtained from each patient prior to study related procedures. Isolation of the crypts and establishment of intestinal organoids were performed as described18. Organoids were passaged every 6–7 days. For all experiments, organoids were used at passage 3–5.
High-throughput RNAi screening procedure
23,349 unique siRNAs targeting 7,783 genes were screened using the Silencer Human Druggable Genome siRNA library V3 (Ambion, Austin, USA). HEK-293 cells were reverse transfected with either single siRNAs (Ambion; primary and secondary screen) or siRNA pools (siGENOME; SMARTpool; Dharmacon, Lafayette, USA; tertiary screen) complexed with siPORT Amine (Ambion) as described19.
Dual luciferase reporter assays
Plasmid-encoded ERSE-dependent firefly luciferase (pGL3-ERSE, 12 ng/well) and a constitutive thymidine-kinase driven Renilla luciferase were used in a dual luciferase assay (Clontech, 3 ng/well) to assay ATF6/ERSE activation. For quantification of the NF-κB promoter activity, a reporter system based on NF-κB responsive promoter elements driving expression of the Firefly luciferase (40 ng/well) was used 19. For all luciferase reporter assays, the fold change is depicted. For calculating the fold change, unstimulated WT controls were set to 1.
RNA extracts and quantitative RealTime PCR
Total RNA was isolated using the RNeasy kit (Qiagen). Total RNA was reverse-transcribed to cDNA using the Maxima H Minus First Strand cDNA Synthesis kit (Thermo Scientific). Quantitative RealTime PCR was performed using the TaqMan Gene Expression Master Mix (Applied Biosystems) and analyzed by the 7900HT Fast Real Time PCR System (Applied Biosystems). The following TaqMan assays (Applied Biosystems) were used: ACSL1(00960561), CSNK2B(00365635), ATF6(00232586), HSP90B1(00427665), DNAJC3(00534483), Hsp90b1(00441926), Hspa5(00517690), and Tnf(00443258). Relative transcript levels were determined using the indicated housekeeper and the standard curve method20.
Immunoblotting
Western blots were performed as described14. α-ATF6 antibody was purchased from Acris (SM7007P), α-GRP78 from Abcam, Cambridge, UK (ab21685), α-ATG16L1 from CST, Danvers, USA (#8089), GRP94 from CST (#2104), α-p100/p52 from CST (#52583), α-p-p65 from Abcam (ab86299), α-p65 from CST (#82429, α-p38 and α-p-p38 from CST (#9212 and #9211, respectively)
Histopathological analyses of murine small intestinal tissue
Histological scoring was performed in a blinded fashion. The histological score displays the combined score of inflammatory cell infiltration, cell death (TUNEL) and tissue damage as described elsewhere5.
Mice
Villin(V)-cre+; Xbp1fl/fl (Xbp1ΔIEC), Villin(V)-cre+; Atg16l1fl/fl (Atg161ΔIEC) 5 mice, backcrossed for at least six generations on a C57BL/6 background, were used at an age of 8–20 weeks. All mice were maintained in a specific pathogen-free facility. All experiments were performed in accordance with the guidelines for Animal Care of Kiel University and in conformity to national and international laws and policies and with appropriate permissions (acceptance no.:V242–7224.121–33).
In vivo treatment of mice
Atg16l1ΔIEC or Atg16l1fl/fl mice were treated with 1mg/kg bodyweight tunicamycin or DMSO i.p. for 24 h or 72h before being sacrificed. Groups of mice received 2.5 μg/g bodyweight TC or 40μg/g bodyweight CX-4945 i.p., respectively, at 0 h, 24 h, and 48 h post Tunicamycin injection (72h experiment) or once at 0h (24h experiment). This animal experiment was approved by the Animal Investigation Committee of the University Hospital Schleswig-Holstein (acceptance no.:V242–32647/2018 (59–7/18)).
Statistics
Statistical analysis was performed using the GraphPad Prism 5 software package (GraphPad Software Inc., La Jolla, USA). Unless otherwise stated, the Student’s unpaired t-test was performed. Data are shown as mean ± standard error of the mean (SEM). In case multiple groups were compared, the ANOVA with post hoc Tukey’s test was used for statistical analysis. A p-value of ≤ 0.05 was considered as significant (*). A p-value of ≤ 0.01 was considered as strongly significant (**) and p-value of ≤ 0.001 as highly significant (***). ****= p-value ≤ 0.0001.
Results
Identification and network analysis of ATF6α signaling modulators
To identify modulators of the ATF6α signaling pathway, we targeted 7,783 genes using a commercially available “druggable” genome siRNA library. The screen was performed in human embryonic kidney cells (HEK-293) transfected with siRNA and a luciferase reporter construct driven by an ATF6-specific ERSE cassette (Fig.1A)4. Luciferase activity was measured 24 h after stimulation with the ER-stress inducer tunicamycin (5 µg/ml), which inhibits N-glycosylation. Each transcript was targeted using three different siRNAs, resulting in a total number of 23,349 assays for ATF6 activation (Fig.1B). Genes with a normalized, averaged fold-induction higher than 2.0 or lower than −2.0 were considered as candidate genes. To validate the findings, the 157 genes (Table S1A) were rescreened using the same experimental setup. The remaining 104 candidate genes (Table S1B) were subjected to a third screen using pools of four independent siRNAs per transcript (Fig.1B). This stringent approach resulted in 22 hits (Table S1C), comprising 15 suppressors and 7 activators of ATF6α signaling (Fig.1B,C). A protein interaction network (STRING) analysis revealed an increased connectivity from the primary to the tertiary screen (primary screen: average local clustering coefficient 0.367, p-value 0.00147; third screen: 0.469 and 1.15×10−8, respectively).
Figure 1: Systematic siRNA screening reveals modulators of ATF6α activation.
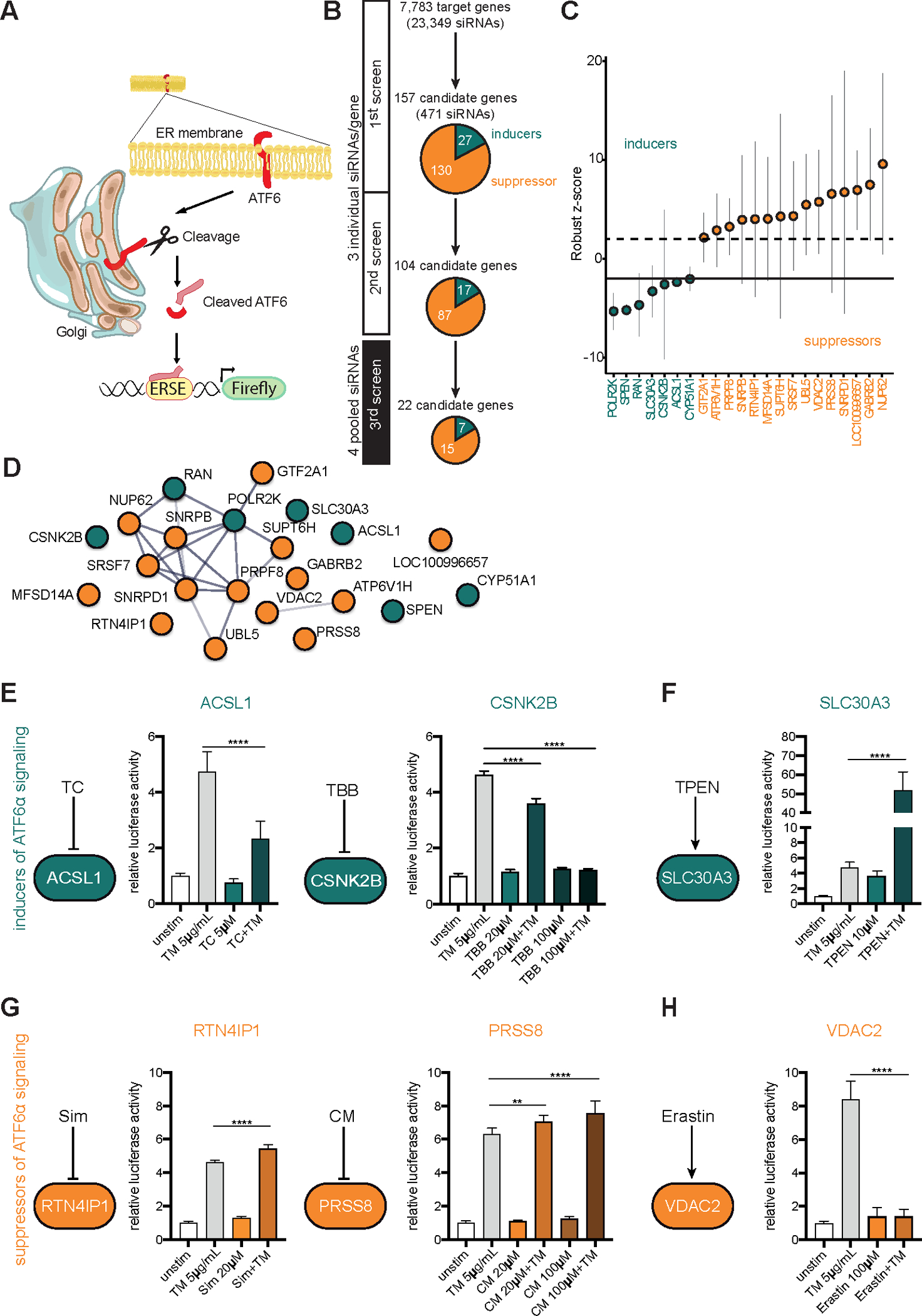
(A) Schematic representation of the screening approach. (B) Screening procedure and number of candidates at different screening stages. In the primary and secondary screens each gene was targeted with three individual siRNAs tested separately. Pools of four siRNAs were used for the third screen. Candidate genes with an inducing or repressing effect on ATF6 activation are indicated in green or orange, respectively (C) Final set of 22 candidates after the third screen. Bars depict mean and 95th confidence interval (3 replicates). (D) STRING Network of candidate genes (third screen). Only interactions with a confidence score >0.4 were considered. Inducers depicted in green, repressors shown in orange. (E–H) ERSE promoter activity quantified by dual luciferase reporter assay in HEK-293 cells. n=6. Cells were exposed to tunicamycin (5 µg/ml) and small molecule agents for 24 h. Depicted data representative of 3 independent experiments. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test. TC=Triacsin C; TBB=4,5,6,7-Tetrabromo-2-azabenzimidazole; TPEN=N,N,N′,N′-Tetrakis(2-pyridylmethyl)ethylenediamine; CM=Camostat mesylate; Sim=Simvastatin.
Validated siRNA-mediated cellular ER-stress regulation by selected individual candidates is phenocopied by chemical interference
From the regulatory network of 22 validated ATF6α signaling modulators, we selected 6 candidates for further functional characterization based on their (1) known biological function, (2) cellular localization (ER, Golgi, nucleus), (3) availability of specific inhibitors/inducers, (4) antibody availability and (5) available mouse models (Fig.S1B, Table S1D). To independently validate the siRNA-mediated effects, we used corresponding chemical inhibitors or inducers (Fig.1E–H). Direct inhibition of the identified ATF6α signaling inducers ACSL1 (Acyl-CoA Synthetase Long Chain Family Member 1) and CSNK2B (Casein Kinase 2β) using Triacsin C (TC) and 4,5,6,7-Tetrabromo-2-azabenzimidazole (TBB), respectively, significantly reduced ERSE promoter activity upon ER-stress induction (Fig.1E). Treatment of cells with N,N,N′,N′-Tetrakis (2-pyridylmethyl)ethylenediamine (TPEN), a Zn2+ chelator, known to increase the expression of the identified ATF6α inducer SLC30A321, elevated the activity of the ATF6α (Fig.1F). Inhibition of the serine protease 8 (PRSS8) activity by Camostat mesylate (CM) augmented ATF6α signaling (Fig.1G). Indirect inhibition of RTN4IP1 (Reticulon 4 Interacting Protein 1) signaling with Simvastatin (Sim), which blocks RhoA signaling, a downstream target of RTN4 (Reticulon 4)22 verified RTN4IP1 as a repressor of ATF6α signaling (Fig.1G). Treatment of cells with the VDAC2 (voltage dependent anion channel 2) binding small molecule Erastin, known to induce VDAC2 expression23, diminished ATF6α signaling and confirmed VDAC2 as repressor of this signaling branch (Fig.1H).
ACSL1 and CSNK2B act on distinct steps of ATF6α signaling in IECs
To further confirm the relevance of ACSL1 and CSNK2B in the intestinal epithelium, we silenced ACSL1 and CSNK2B in the Caco-2 cells using siRNA transfection (for knockdown efficiency, see Fig. S2A). This resulted in significantly reduced ERSE promoter activity (Fig.2A) and reduced mRNA levels of the canonical ATF6α-target gene HSP90B1 (GRP94) after tunicamycin stimulation (Fig.2B).
Figure 2: CSNK2B controls ATF6α signaling upstream of intramembrane cleavage.
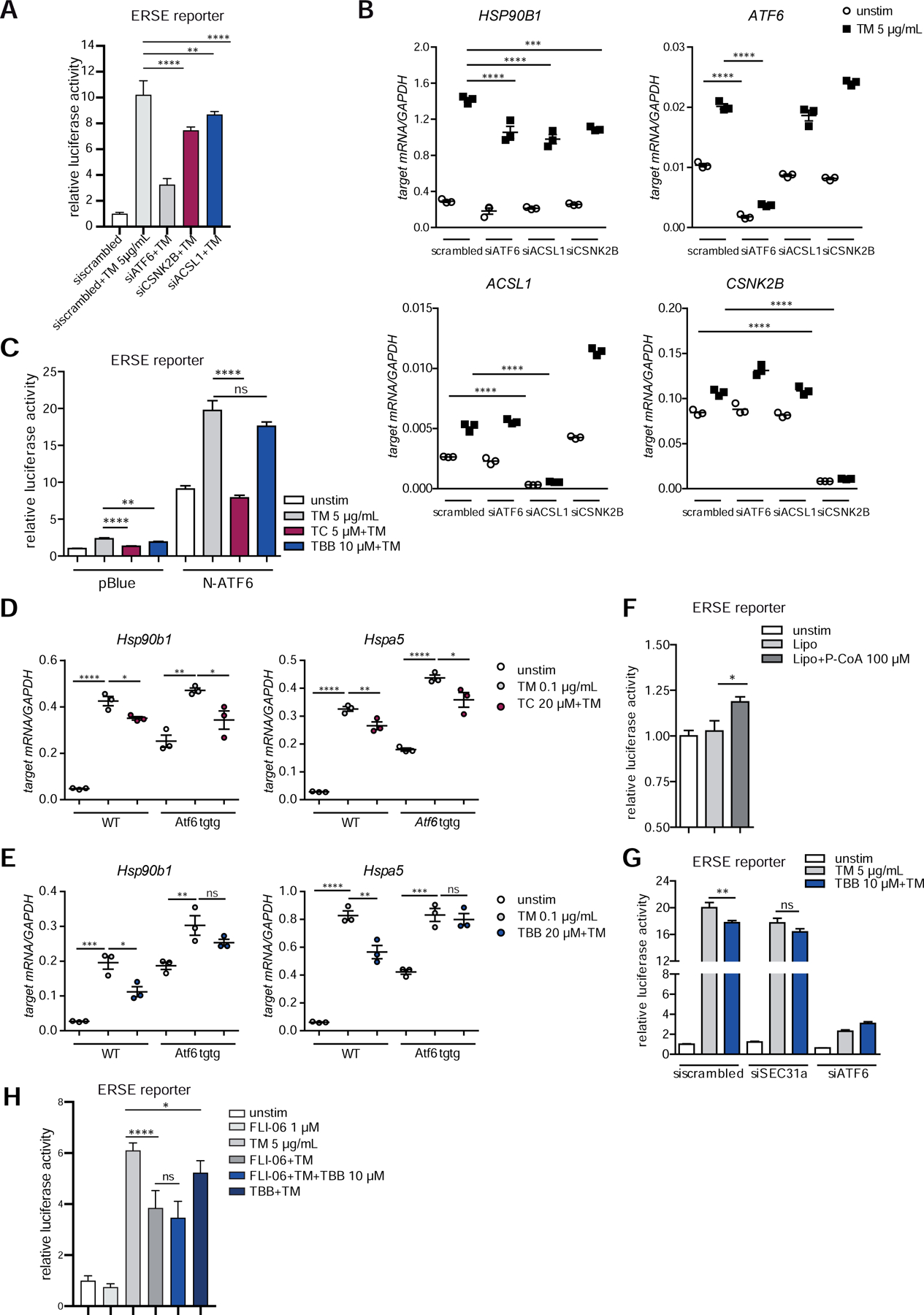
(A–B) siRNA-mediated knockdown of ACSL1 (siACSL1), CSNK2B (siCSNK2B) and ATF6α (siATF6) in Caco-2 cells. scrambled= non-targeting control siRNA. (A) ERSE promoter activity quantified by dual luciferase reporter assays. After 24 h, cells were stimulated with 5 µg/ml tunicamycin (TM) for additional 24 h. (B) mRNA levels of ATF6α target HSP90B1 were measured by qPCR (n=3) 24 h after TM stimulation. (C) Effects of TC and TBB treatment on ERSE promoter activity in Caco-2 cells quantified by dual luciferase reporter assays. Cells transfected either with N-ATF6α or with the empty plasmid (pBlue) and stimulated with tunicamycin and inhibitors (24 h). (D–E) Transcript levels of Hsp90b1 and Hspa5 in WT and Atf6α transgenic (Atf6 tgtg) SI organoids treated with tunicamycin (0.1 µg/ml) and TC (D) or TBB (E) for 24 h. (F) Caco-2 cells were stimulated with lipofectamine-complexed Palmitoyl coenzyme A (100 µM) or lipofectamine alone (Lipo) for 24 h and ERSE dual luciferase reporter activity was measured. (G) ERSE promoter activity in Caco-2 cells upon siRNA-mediated depletion of SEC31a (siSEC31a). (H) ER-Golgi transport was inhibited in Caco-2 cells with FLI-06 (1 µM) in presence or absence of tunicamycin and TBB, respectively. Cells stimulated for 24 h. ERSE promoter activity quantified by dual luciferase reporter assay. Shown data representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
To address the molecular mechanism how ACSL1 and CSNK2B act on ATF6α signaling, we first transfected Caco-2 cells either with a plasmid encoding the transcriptionally active N-terminal ATF6α fragment 24. In this model, influence of an inhibitor would point to an effect downstream of the S1/2P-dependent intramembrane proteolysis of ATF6α. We found that ACSL1 inhibition by TC repressed ERSE activation in cells overexpressing N-terminal ATF6α, whereas treatment with the CSNK2B inhibitor TBB did not inhibit ERSE-dependent reporter gene activity (Fig.2C). Similarly, in intestinal organoids derived from transgenic mice overexpressing the activated form of ATF6α (Villin-Cre::nAtf6α tg carrying a loxP-STOP-loxP cassette in front of the transgene, termed Atf6tgtg hereafter)24, TBB treatment did not diminish mRNA levels of Atf6α target genes (Hsp90b1, Hspa5) upon ER-stress induction by tunicamycin and at baseline. In contrast, inhibition of ACSL1 with TC resulted in reduced mRNA levels of Hsp90b1 (Grp94) and Hspa5 (Grp78) (Fig.2D,E). These results imply that ACSL1 mediates its co-activating effect on ATF6α signaling downstream of the cleavage event at the Golgi apparatus. Treatment of IECs with Palmitoyl coenzyme A, product of the enzymatic reaction catalyzed by ACSL1, caused increased ERSE reporter activity (Fig.2F) supporting the role of ACSL1 as inducer of ATF6α signaling. Importantly, the lack of effect of the CSNK2B inhibitor TBB on Atf6tgtg-induced signaling implies that CSNK2B regulates ATF6α signaling upstream of the intramembrane proteolysis. Koreishi et al.25 demonstrated previously that the casein kinase 2 (CK2), composed of CSNK2B and CSNK2A, phosphorylates the COPII constituent Sec31, thereby facilitating ER-Golgi trafficking. As it was shown that ATF6α trafficking is dependent on COPII vesicles26, we hypothesized that CSNK2B might be involved in the transport of ATF6α from the ER to the Golgi apparatus. Indeed, depletion of SEC31 by siRNA in IECs abolished the inhibitory effect of TBB on ERSE reporter gene activity (Fig.2G). In further support of these findings, inhibition of ER-Golgi trafficking by treatment with the dihydropyridine FLI-06 (1 µM)27 diminished the effect of CSNK2B inhibition on ERSE promoter activity (Fig.2H).
The ATF6α branch of the UPR is a critical modulator of ER-stress-induced pro-inflammatory signals in IECs
Unresolved ER-stress in IECs has emerged as an important mechanism favoring intestinal inflammation8. First, we examined the levels of pro-inflammatory cytokines in intestinal organoids derived from Atf6tgtg transgenic mice and littermate control mice. Atf6tgtg organoids displayed an elevation of transcript levels of Cxcl1 and Tnfα in the presence of tunicamycin, confirming a co-activating role of nATF6α (Fig.3A). Levels of the ER stress target gene Transcripts (Hspa5 and Hsp90b1) as well as the ATF6 transcript itself could also be synergistically increased by tunicamycin stimulation in Atf6tgtg transgenic organoids. We detected, both on mRNA and on protein level, altered expression of canonical and non-canonical NF-κB signaling components, indicated by increased levels of Rela and p-p65 levels and increased levels of Relb, Nfkb2, p100 and p52, respectively (Fig.S2B–C) already at baseline. This was accompanied by enhanced phosphorylation of p38 (Fig.S2C) in the Atf6tgtg organoids. In line with these findings, inhibition of NF-κB signaling using the aromatic diamine JSH-23, which blocks the nuclear translocation of NF-κB28, abolished the pro-inflammatory signature in organoids overexpressing the N-terminal ATF6 fragment illustrated by significantly reduced mRNA levels of both Cxcl1 and Tnfα upon ER-stress induction (Fig.3B). As ER-stress dependent activation of pro-inflammatory signals might involve autocrine release of TNFα5 we employed an anti-TNF neutralizing antibody (100 ng/ml) in Atf6tgtg organoids, which, however, only had mild effects on the mRNA levels of these pro-inflammatory cytokines and two NF-κB target genes (Birc2/3)29 (Fig.3B, Fig.S2D). Of note, whereas it has been shown that a ENU-induced hypomorphic mutation of the S1P gene renders mice susceptible to colitis30, pharmacological inhibition of S1P required for ATF6α cleavage at the Golgi complex with PF-429242 (10 µM) was able to inhibit tunicamycin-induced NF-κB reporter activation (Fig.3C). To further validate this finding, we performed siRNA knockdown of ATF6α, ACSL1 or CSNK2B in IECs and could confirm the co-activation effect of the endogenous ATF6 signaling module on NF-κB reporter gene activity upon ER-stress induction (Fig.3D). In agreement with these findings, stimulation with the ACSL1 product Palmitoyl coenzyme A caused increased NF-κB reporter gene activity (Fig.3E). Taken together, our results identify ATF6 as a critical regulator of pro-inflammatory signaling in IECs and suggests a functional interaction of NF-κB and ATF6 signaling under conditions of ER-stress.
Figure 3: ATF6α regulates NF-κB signaling upon ER-stress induction.
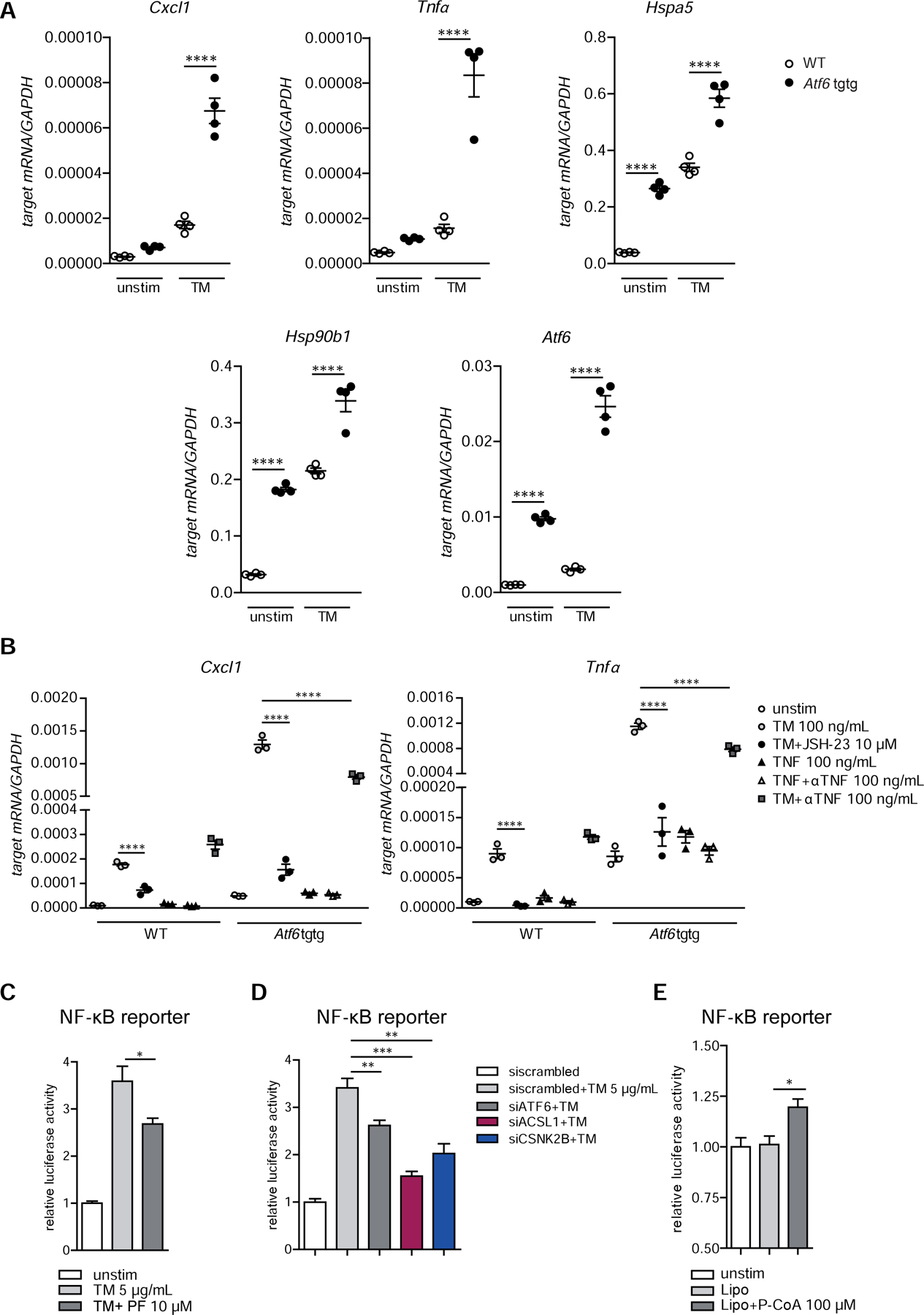
(A) Cxcl1, Tnfα, Hspa5 (Grp78), Hsp90b1 (Grp94), Atf6 transcript levels of in WT and Atf6α transgenic (Atf6 tgtg) SI organoids stimulated with tunicamycin (100 ng/ml, 24 h). (B) Cxcl1 and Tnfα mRNA levels in WT and Atf6 tgtg SI organoids stimulated for 24 h. (C–D) NF-κB promoter activity in Caco-2 cells upon (C) inhibition of S1P with PF-429242 (10 µM) or (D) siRNA-mediated depletion of ATF6α(siATF6), ACSL1(siACSL1) or CSNK2B(siCSNK2B). (E) NF-κB dual luciferase reporter assay in Caco-2 cells stimulated with lipofectamine-complexed Palmitoyl coenzyme A (100 µM) or lipofectamine alone (Lipo) for 24 h. Depicted data representative of 3 independent experiments. Statistical analysis was performed using one-way ANOVA together with post hoc tukey’s.
Inhibition of the ATF6α activators CSNK2B and ASCL1 attenuates the pro-inflammatory profile of genetically induced ER-stress: impact of ATF6α signaling on Xbp1- and Atg16l1-deficiency
We next turned our attention to a potential role of ATF6α and its upstream regulators for the execution of impaired, pro-inflammatory ER-stress responses observed in Xbp1- and Atg16l1deficient IECs5, 8, 13, 14. Indeed, Caco-2 cells carrying a genetic deletion of the autophagy gene ATG16L1 introduced by CRISPR-Cas914 (ΔATG16L1-Caco-2) exhibited increased ATF6α cleavage compared to their respective wild type comparators (WT-Caco-2) (Fig.4A,B). Additionally, the ERSE reporter assays revealed significantly increased activation of the ATF6α branch in ΔATG16L1 cells compared to WT (Fig.4C), both at baseline and upon further ER-stress induction with tunicamycin (5 µg/ml). Likewise, Atg16l1-deficient small intestinal (SI) organoids showed an upregulation of ATF6 target genes compared to wild-type controls (Fig.S3A). Tunicamycin-induced ERSE reporter activity and target gene induction could be blocked by the cognate inhibitors TC (ACSL1) or TBB (CSNK2B), respectively (Fig.4C,D). In addition, cell viability of ΔATG16L1 cells upon ER-stress induction-assessed by MTS assay was significantly improved by treatment with the two ATF6 inhibitors (Fig.4E,F). Next, we sought to study the effect of inhibition of the ATF6α branch on the increased NF-κB signaling tone in autophagy-deficient IECs31. ΔATG16L1-Caco2 cells transfected with an NF-κB reporter plasmid showed increased activity upon tunicamycin stimulation compared to wild type cells (Fig.4G,H). Notably, treatment with TBB/TC again significantly reduced NF-κB reporter gene activity. Increased mRNA levels of Cxcl1 and Tnfα observed in the Atg16l1-deficient organoids were reduced upon treatment with the ACSL1 and the CSNK2B inhibitor, respectively (Fig.4I).
Figure 4: Reduction of the hyperactivation of the ATF6α branch in ATG16L1-deficient IECs alleviates levels of pro-inflammatory cytokines.
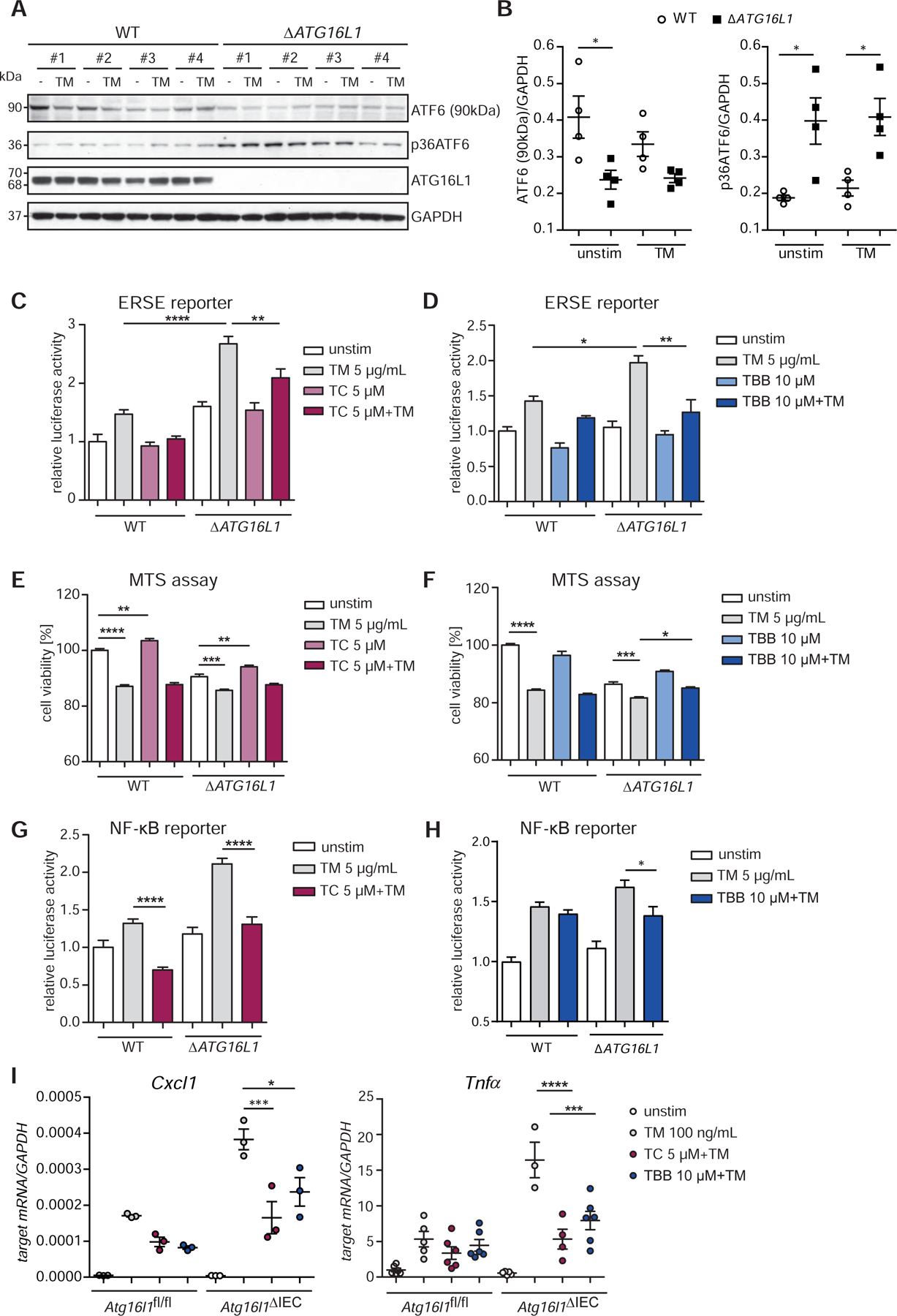
(A) Western blot analysis and quantification (B) of ΔATG16L1-Caco-2 and the WT cells. Cells stimulated with tunicamycin (5 µg/ml, 6 h). #1-#4 refers to 4 independent biological replicates derived from one CRISPR clone. (C) Effects of TC and (D) TBB on the ERSE promoter in Caco-2 cells measured by dual luciferase reporter assays. Cell viability of Caco-2 WT and ∆ATG16L1-deficient cells quantified by MTS assay in the presence of TC (E) and TBB (F) after tunicamycin stimulation (5 µg/ml, 24 h). (G–H) NF-κB Luciferase activity in Caco-2 WT and ATG16L1-deficient cells. Cells stimulated with tunicamycin (5 µg/ml, 24 h) in the presence or absence of (G) TC (5 µM) or (H) TBB (10 µM). (I) Cxcl1 and Tnfα transcript levels in SI organoids (Atg16l1fl/fl, Atg16l1∆IEC) treated with tunicamycin (100 ng/ml) and TC/TBB (24 h, n=3). Shown data representative of 3 independent experiments. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
We also found elevated activation of the ATF6α arm in the SI epithelial cell line MODE-K stably transduced with a short hairpin Xbp1 (shXbp1) lentiviral vector (Fig.5A–C, Fig.S3C)5. We detected enhanced mRNA levels of ATF6α targets (Hsp90b1, Hspa5) in Xbp1-deficient SI organoids (Fig.S3B). Inhibition of ATF6α signaling by treatment with TC or TBB alleviated ERSE promoter activity (Fig.5C,D) and improved cell viability upon ER-stress induction in MODE-K.iXbp1 (ΔXbp1) cells (Fig. 5E,F). Moreover, both augmented NF-κB activity (Fig.5G,H) in ΔXbp1 cells, and Cxcl1 and Tnfα levels in Xbp1-deficient organoids (Fig.5I) were reduced in the presence of the inhibitors upon tunicamycin treatment compared to controls.
Figure 5: Inhibition of the ATF6α branch in Xbp1-deficient IECs alleviates levels of pro-inflammatory cytokines.
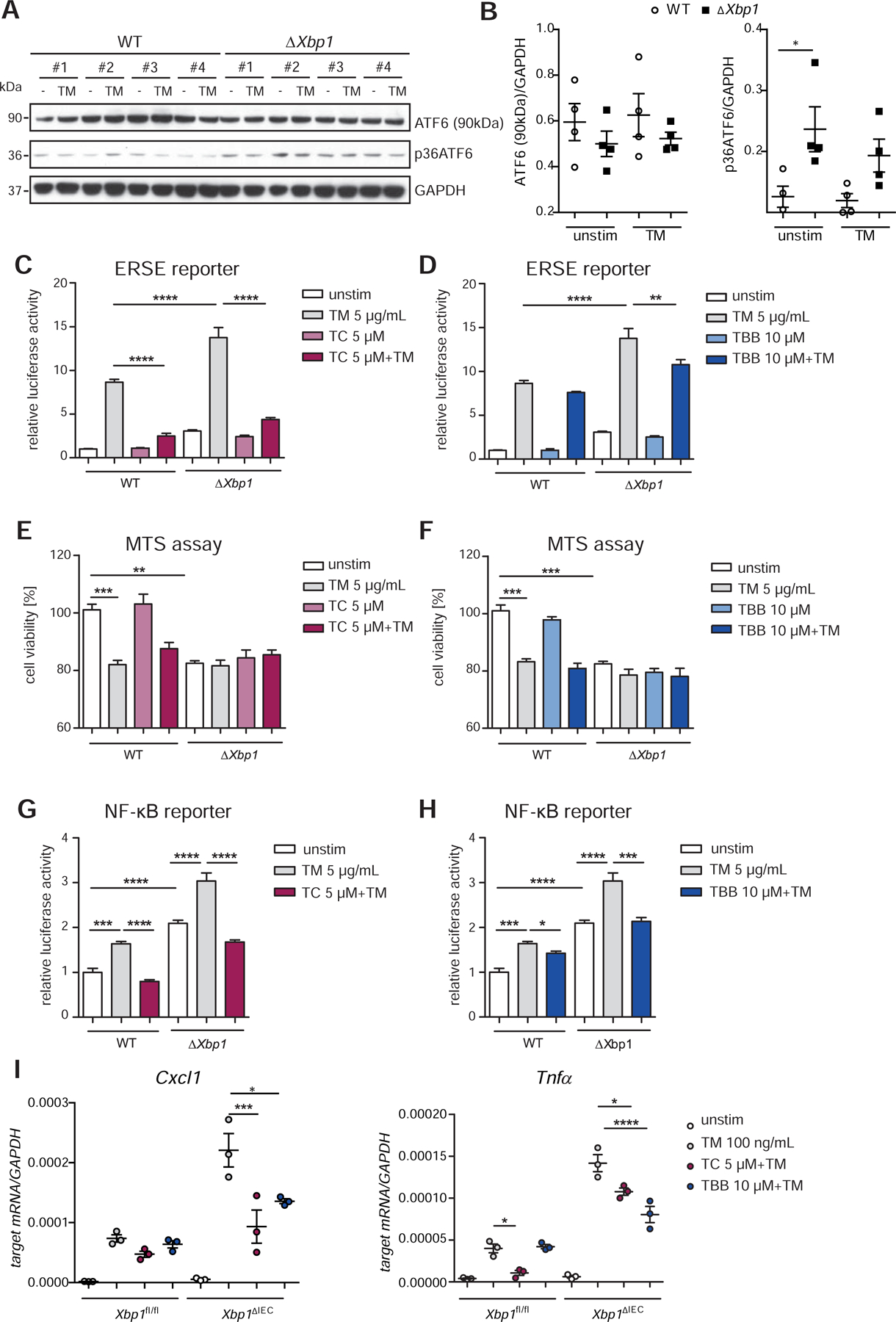
(A) Immunoblotting and quantification (B) of MODE-K cells stably transduced with a short hairpin Xbp1 lentiviral vector and the respective wild type control. Cells stimulated with tunicamycin (5 µg/ml, 6 h). #1-#4 refers to 4 independent biological replicates. (C–D) Activation of the ATF6α branch upon ER-stress induction (tunicamycin, 24 h, 5 μg/ml) quantified in the presence of (C) TC and (D) TBB in MODE-K.iXbp1 (ΔXbp1) and MODE-K.iCtrl (WT) cells by dual luciferase reporter assays. Effects of TC (E) and TBB (F) on cell viability quantified by MTS assay after exposure to tunicamycin (5 μg/ml, 24 h) in WT and ΔXbp1 cells. (G–H) NF-κB luciferase activity in WT and Xbp1-deficient Mode-K cells. Cells exposed to tunicamycin (5 μg/ml, 24 h) in the presence or absence of (G) TC (5 μM) or (H) TBB (10 μM). (I) qPCR of Cxcl1 and Tnfα of SI organoids (Xbp1fl/fl, Xbp1ΔIEC) treated with tunicamycin (100 ng/ml) and inhibitors (TC 5 μM; TBB 10 μM) for 24 h (n=3). Data shown is representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
We next assessed the contribution of ATF6α signaling to the ileitis phenotype of Atg16l1ΔIEC mice in a short-term (24 h, Fig.S5) and in a longer in vivo ER stress model, in which mice were followed up for 72 h (Fig.6). In both experiments, Atg16l1fl/fl and Atg16l1ΔIEC mice were injected intraperitoneally with a single dose of tunicamycin (1mg/kg bodyweight) at 0 h. To block ATF6α-mediated signaling, mice were simultaneously injected with either TC or CX-4945 (silmatasertib) at 0 h, and additionally at 24 h, and 48 h post Tunicamycin injection in case of the 72 h experiment (see Fig.6A and S5A for experimental design). Similar to TBB, CX-4945 is an ATP-competitive inhibitor of the CK2 and inhibited ATF6-mediated ERSE and NF-κB reporter gene activity in ΔATG16L1-Caco-2 cells (Fig.S3D–E). However, superior to TBB, CX-4945 is orally bioavailable32 and is currently tested in clinical trials for haematological and solid cancer treatment (ClinicalTrials.gov NCT01199718,NCT02128282,NCT00891280). Both TC and CX-4945 injections significantly attenuated tunicamycin-mediated body weight loss after 72 h in Atg16l1ΔIEC mice (Fig.6B, Fig.S4A). Moreover, we observed attenuated shortening of the small intestine (Fig.6C), reduced Cxcl1 protein levels in the serum (Fig.6D) of Atg16l1ΔIEC mice, and reduced mRNA levels of Tnfα and Ifit1 upon Tunicamycin injection in the presence of the tested inhibitors (Fig.S4B–C). Both transcripts are known to be induced by ER stress signals in Atg16l1ΔIEC mice14. Likewise, histological analysis demonstrated reduced levels of inflammation (Fig.6G) and reduced epithelial cell death as depicted by reduced numbers of TUNEL+ epithelial cells in Atg16l1-deficient IECs in the presence of the tested inhibitors (Fig.6E–F). Staining was concentrated at the bottom of the crypts and -in line with previous studies-marked Paneth cells as well as other epithelial cells14, 33. In line with these findings, already 24 h after Tunicamycin injection, TC and CX-4945 treatment resulted in reduced epithelial cell death in the absence of Atg16l1 as assessed by TUNEL staining (Fig.S5D,E), and reduced mRNA levels of Tnfα and Ifit1 (Fig.S5F).
Figure 6: Inhibition of the ATF6α branch mitigates ER-stress mediated inflammation and cell death in Atg16l1ΔIEC mice.
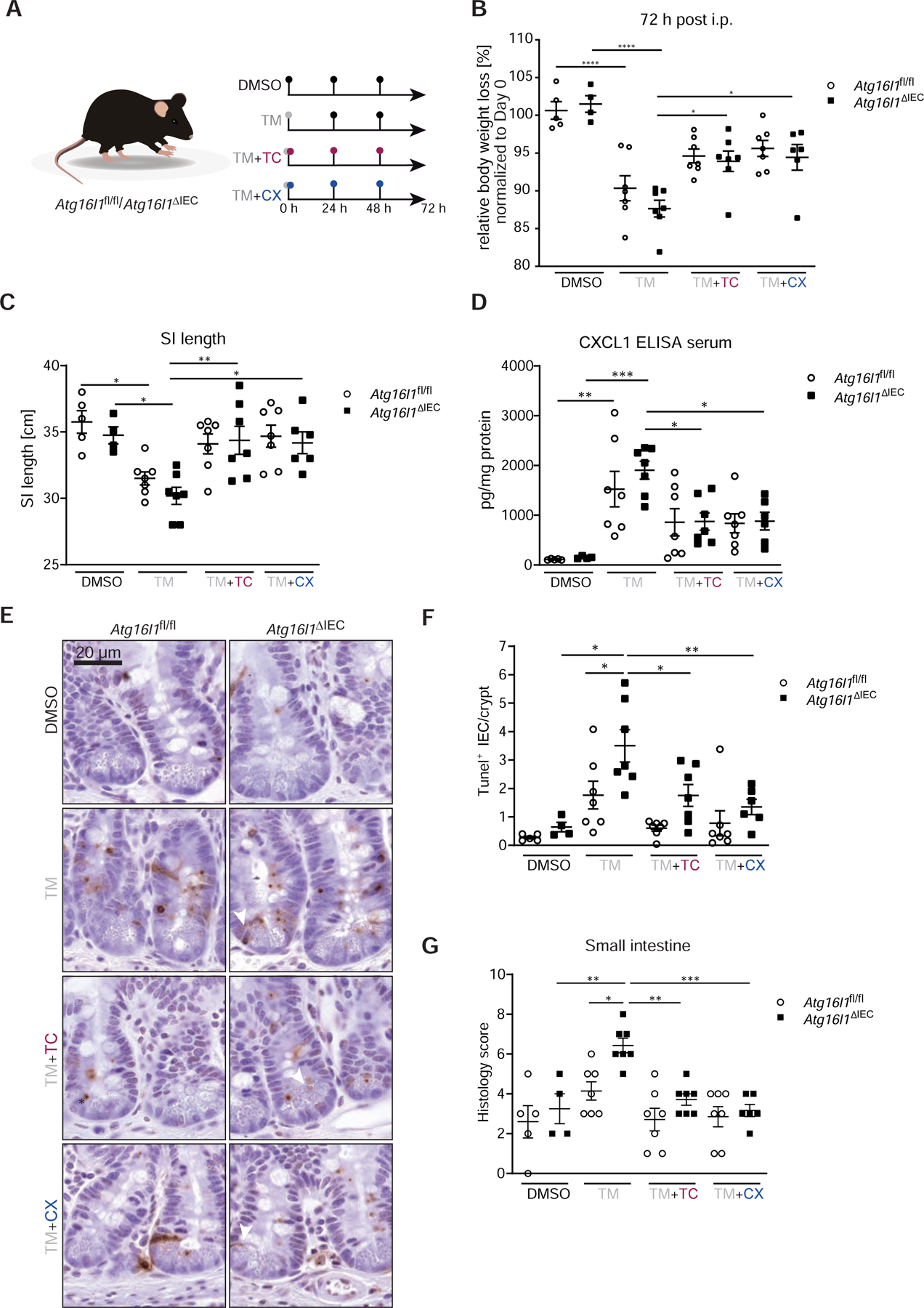
(A) Stimulation scheme of Atg16l1fl/fl and Atg16l1ΔIEC mice (n = 4–7). Mice were treated with 1 mg/kg bodyweight of tunicamycin i.p., when indicated mice additionally received either TC (2.5 µg/g bodyweight) or CX-4945 (40 µg/g bodyweight) at 0, 24, and 48 h. Control groups received DMSO. After 72 h mice were sacrificed. (B) Weight loss 72 h after injection. (C) SI length 72 h after injection. (D) CXCL1 concentration in serum quantified by ELISA. (E–F) TUNEL staining of SI sections with representative pictures (E, arrowheads denote TUNEL+ IECs outside of the Paneth cell/stem cell niche) and quantification (F). Bars=20 µm. A minimum of 50 crypts/intestine were assessed in each treatment group. (G) Histological evaluation of small intestinal sections. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
Limiting ATF6α signaling attenuates the pro-inflammatory profile in human organoids upon ER-stress induction: relevance for human IBD
We next assessed expression levels and activation of ATF6 in human IBD patients. Analysis of mRNA level in purified IECs from ileal biopsies revealed significantly higher expression of ATF6α and the ATF6 target HSPA5 in pediatric CD patients compared to healthy controls (Fig.7A, comparison to UC in Fig.S6B) 34. Likewise, CSNK2B, but not ACSL1 mRNA levels were upregulated (Fig. S6B). Using protein lysates of early-passage ileal human organoids from CD patients and healthy controls, we next demonstrated higher levels of the active p36 fragment of ATF6α in organoids derived from adult CD patients, which was more pronounced in organoids from inflamed tissue (Fig.7B, Fig.S6A, Table S2). Next, we analysed mRNA levels of ATF6 targets in human organoids generated from biopsies of CD patients and healthy controls. Upon Tunicamycin-mediated ER stress induction, we detected significantly increased levels of HSPA5, DNAJC3 and HSP90B1 in organoids derived from CD patients compared to healthy controls (Fig.7C). Subsequently, we investigated in human ileal organoids whether ATF6α signaling can be limited by ACSL1- or CSNK2B inhibition. Indeed, exposure to either TC (Fig.7D) or TBB (Fig.7E) resulted in significantly reduced expression of ATF6α targets upon exposure to tunicamycin. Importantly, inhibition of ATF6α signaling by TC or TBB, respectively, resulted in significantly lower mRNA expression of pro-inflammatory cytokines (IL8, TNFα) in human organoids exposed to tunicamycin (Fig.7F,G).
Figure 7: Limiting ATF6α signaling attenuates ER-stress mediated inflammation in human organoids.
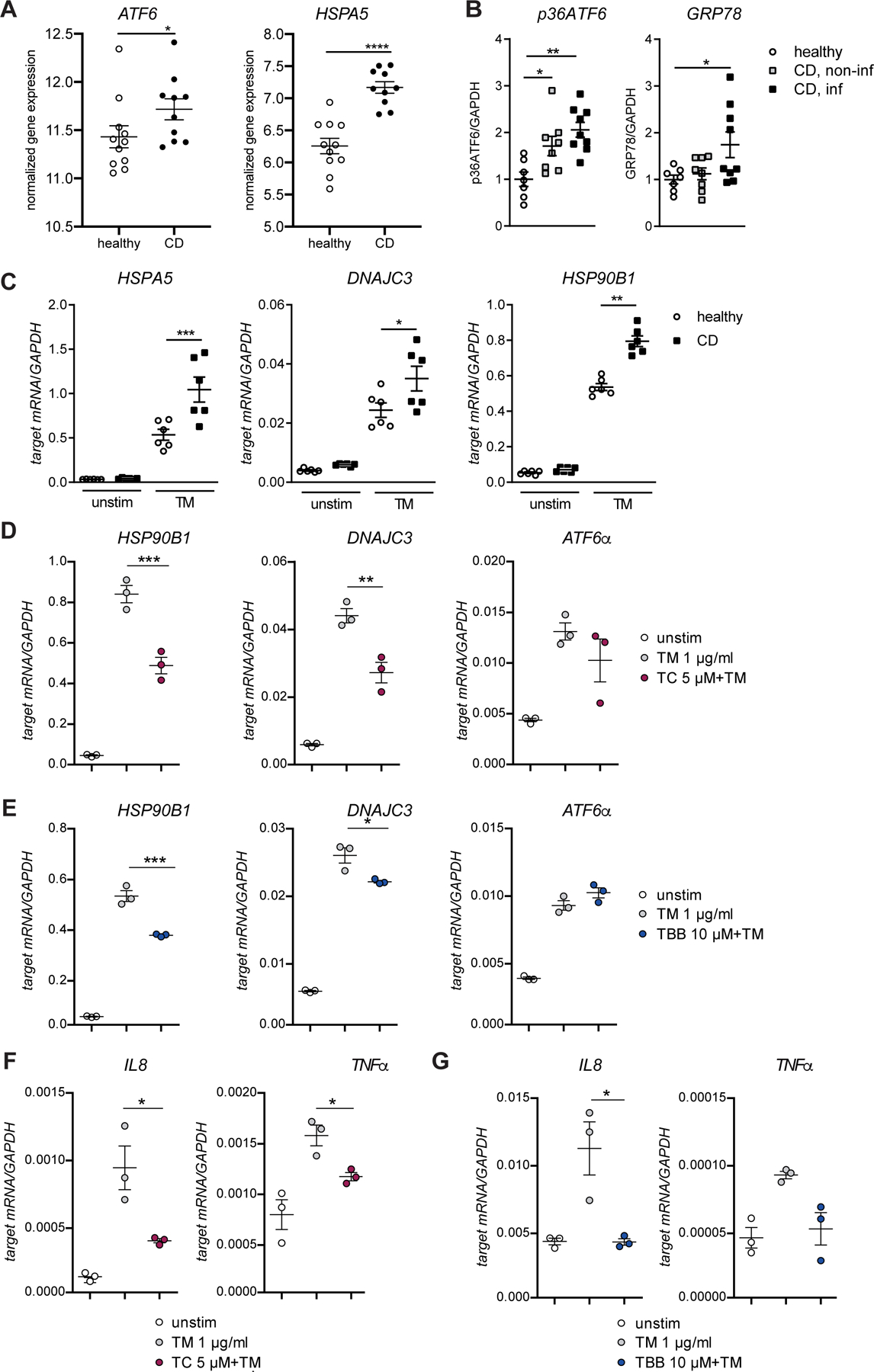
(A) Relative mRNA expression of ATF6α and HSPA5 in IECs from ileal biopsies from paediatric CD patients and healthy controls. (B) Quantification of protein levels of p36ATF6 and GRP78 derived from SI organoid lysates generated from healthy, CD non-inflamed, and CD inflamed tissue, respectively. (C) mRNA levels of HSPA5, DNAJC3, and HSP90B1 in human SI organoids from healthy controls and CD patients treated with tunicamycin (1 µg/ml; 24 h). (D) Transcript levels of HSP90B1 and DNAJC3 in human SI organoids treated with tunicamycin (1 µg/ml) and TC (D) or TBB (E) for 24 h. (F–G) IL8 and TNFα transcript levels in human SI organoids treated with tunicamycin (1 µg/ml) and inhibitor TC (F) or TBB (G), respectively (24 h, n=3). Depicted data representative of 3 independent experiments. Each data point represents one organoid line derived from an individual CD patient. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test or Mann-Whitney test (for pair comparisons).
Discussion
In this study, we identified regulators of ATF6α signaling using a stringent siRNA screening approach. Among the 22 validated upstream regulators of ATF6α, ACSL1 and CSNK2B were further analyzed. ACSL1 (acyl-CoA synthetase long-chain family member 1) is a 78-kDa intrinsic membrane protein that mediates the conversion of fatty acids (FAs) to acyl-CoAs. Importantly, ACSL1 localizes to the ER and to mitochondria-associated membranes. The other identified ATF6α co-activator CSNK2B encodes the regulatory subunit of the CK2, which is a tetrameric serine/threonine-selective protein kinase composed of two catalytic subunits and two regulatory subunits. CK2 is localized in the ER and the Golgi complex35. Several studies have described a modulatory function of the CK2 on the UPR36, 37. However, to our knowledge, none of these studies have provided a mechanistic link between the ATF6α branch of the UPR and CSNK2B. Using transgenic organoids overexpressing a constitutively active ATF6α form, which mimics the S1P-cleaved protein, we show that the two targets act either downstream (ACSL1) or upstream (CK2) of the cleavage event. It is important to note that pharmacological inhibition of the upstream ATF6α regulators did not completely abolish all tunicamycin-mediated ER stress effects. This could be due to incomplete abrogation of ATF6α signals by the inhibitors, but also due to the extensive crosstalk between the three UPR branches1, 38, which should be carefully considered when targeting the UPR for therapeutic benefit.
Several studies have revealed an intensive crosstalk between unresolved ER-stress, failing autophagy and pro-inflammatory signaling in IECs in the context of IBD 5, 8, 13, 14, 39. In Xbp1- and Atg16l1-deficient IECs, increased TNF-dependent NF-κB signaling and spontaneous intestinal inflammation in vivo are observed and have been attributed to elevated IRE1α levels 5, 8, 13. In this context, our study reveals a significantly increased activation of the ATF6α branch in cells lacking the autophagy gene Atg16l1 or the UPR gene Xbp1. Importantly, inhibition of ATF6α upstream signaling using the small molecule inhibitors TC/TBB was able to diminish the observed hyper-inflammatory phenotype of Atg16l1- and Xbp1-deficient cells5, 13. Interestingly, direct activation of ATF6α by active IRE1 has been proposed40 and our data suggests a role of ATF6α and its upstream regulators for the execution of impaired, pro-inflammatory ER-stress responses in IECs. It has been proposed that ATF6α induces the phosphorylation of AKT to finally activate NF-κB signaling41, 42, but engagement of other pro-inflammatory signaling events, e.g. the activation of p38MAPK have also been reported during ER stress43. As MAPK activation has been shown to shift the balance of NF-κB signals in IECs from an anti-apoptotic to a pro-inflammatory function44, such additional signals could be important for the effector function of ATF6 itself or might be modulated by downstream effectors of ATF6α.
In line with a co-activating role of ATF6 on the NF-κB pathway, we demonstrate an up-regulation of components of the NF-κB machinery (NF-κB2 coding for p100 and its processed form p52) and increased p65 as well as p38MAPK phosphorylation in organoids overexpressing the active N-terminal ATF6α fragment already under steady state conditions. It must be noted that this model only incompletely reflects the physiological situation of normal ATF6 activation as the organoids face a constant stimulation by the transgene-encoded transcription factor, however under additional stimulation with the ER stress inducer tunicamycin, synergistic induction of Cxcl1 and Tnfa mRNA levels was still observed. The induction of the two transcripts could be blocked by an inhibitor of the nuclear translocation of p6528, whereas it was only partially inhibited by application of anti-TNF antibodies arguing against a main role of autocrine TNF release in this system.
We have shown that homozygous Atf6tgtg mice develop spontaneous colon adenomas at 12 weeks of age24. Increased pro-inflammatory cytokine mRNA levels in this model were only detected at late stages of tumour development in whole colon tissue (>20 weeks) supporting the hypothesis that ATF6α activation alone is not sufficient to generate IBD-like tissue pathology, but additional signals must be present. As tunicamycin inhibits N-linked protein glycosylation and thereby activates additional ER stress signaling at the level of all three UPR branches 45, which includes IRE1-dependent signaling, the results suggest that the ATF6α branch serves as a co-activating executioner towards a hyper-inflammatory phenotype. Pharmacological inhibition of the two ATF6α upstream activators CSNK2B and ASCL1 upon tunicamycin application in vivo was able to block ER stress-induced epithelial cell death and signs of mucosal inflammation in the small intestine. The protective effect was more pronounced in mice lacking Atg16l1 in the intestinal epithelium, which are prone to develop ileal inflammation dependent on IRE1 and the TNFR1-NF-κB axis5, 13 and to TNF dependent necroptosis33. Still, it remains an interesting question whether and how ATF6α acts upon the autophagic flux of IECs under autophagy-proficient conditions.
These observations in the small intestine are important for human IBD, as the role of ATF6α signaling in intestinal inflammation has only been shown in colonic IECs from UC patients, where increased cleavage of ATF6α and augmented expression of the ATF6α targets GRP78 and GRP94 were demonstrated12. Increased ATF6α expression itself was recently suggested as a marker for precancerous dysplasia in colitis-associated colorectal cancer46. Here, we show that increased mRNA levels and activation of the ATF6 branch are present in small intestinal epithelial cells from CD patients. This activatability is maintained -at least during early passages- in patient-derived small intestinal organoids, arguing for a sustained deregulation of this pathway in this disease condition. It is important to note that, although the hypomorphic ATG16L1T300A variant is a risk factor for ileal CD47, it seems unlikely that small intestinal ATF6 hyper-activation can be explained by genetics only. ER stress rather should be regarded as a central hub integrating signals on the state of the cell48. As such it is influenced by a variety of environmental factors, such as diet49, microbiota50 or proliferative signals14, 51 that act on IECs as a barrier constituent. The individual life history of exposure to such stressors may exceed the epithelium’s resilience leading to unresolved ER stress and ATF6 activation as a pro-inflammatory signal, whereby hypomorphic ATG16L1T300A may be an important determinant of the threshold.
Our findings suggest that engagement of the ATF6α branch may represent an executioner mechanism of pro-inflammatory ER stress signals, particularly in IECs with defective autophagy or exaggerated UPR signaling. We demonstrate the presence of activated ATF6 signaling as a characteristic feature of small intestinal IECs isolated from CD patients. Importantly, we show that inhibition of the ATF6 branch is able to mitigate the pro-inflammatory signature of ER stress induction in human small intestinal organoids. Interfering with the ATF6α pathway targeting the upstream inducers ACSL1 and CSNK2B, respectively, might thus represent a novel therapeutic approach in intestinal inflammation.
Supplementary Material
(A) Network of candidate genes after the primary screen based on STRING. Only interactions with a confidence score >0.4 were considered. p=0.00147. (B) Schematic cellular localization of the 6 ATF6α modulators subjected to further functional studies. ATF6 signaling inducers are depicted in green, inhibitors shown in orange.
(A) knockdown efficiency for ATF6, ACSL1 and CSNK2B in Caco-2 cells was calculated (related to Fig. 2B). Samples treated with the non-targeting siRNA (siscrambled, unstimulated) were set to 1. (B) Transcript levels of NF-κB signaling components in small intestinal organoids treated with tunicamycin (TM 100ng/ml) for 24 h. (C) Immunoblotting of protein lysates collected from WT and Atf6 tgtg small intestinal organoids stimulated with Tunicamycin (TM, 100ng/ml). Relative p-p65, p52 and p-p38 protein levels were quantified. The average value of the unstimulated WT controls was set to 1. n=3 biological replicates. (D) cIAP1/Birc2 and cIAP2/Birc3 mRNA levels in WT and Atf6 tgtg SI organoids stimulated for 24 h. Related to Fig. 3B. Depicted data representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
Transcript levels of Atf6α target genes Hsp90b1 (Grp94) and Hspa5 (Grp78) in small intestinal organoids treated with tunicamycin (TM, 100ng/ml) for 24 h. (A) Atg16l1fl/fl and Atg16l1∆IEC small intestinal organoids. n=4 biological replicates. (B) Xbp1fl/fl and Xbp1ΔIEC small intestinal organoids. n=4 biological replicates. (C) Transcript levels of Xbp1 in Xbp1-deficient MODE-K cells (n=4). (D) ERSE promoter activity and (E) NF-κB luciferase activity in Caco-2 cells upon exposure to tunicamycin (5 μg/ml) for 24 h in the presence or absence of CX-4945 (3 μM) as indicated. Shown data representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
(A) Stimulation scheme of Atg16l1fl/fl and Atg16l1ΔIEC mice (n = 5/6). Mice were treated with 1 mg/kg bodyweight of tunicamycin i.p, when indicated mice additionally received either TC (2.5 µg/g bodyweight) or CX-4945 (40 µg/g bodyweight). Control groups received DMSO. After 24 h mice were sacrificed. (B) Weight loss 24 h after injection. (C) CXCL1 concentration in SI crypt lysates detected via ELISA. (D-E) Tunel staining of SI sections with representative pictures (D) and quantification (E). A minimum of 50 crypts/intestine were assessed in each treatment group. Bars=20 µm. Arrowheads show apoptotic Paneth cells. (F) Transcript levels of Tnfα and Ifit1 in SI crypt lysates of treated Atg16l1fl/fl and Atg16l1ΔIEC mice after 24 h. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
Mice received 1 mg/kg bodyweight of tunicamycin i.p, when indicated mice were additionally treated with either TC (2.5 µg/g bodyweight) or CX-4945 (40 µg/g bodyweight) at 0, 24, and 48 h. Control groups received DMSO. Mice were sacrificed after 72 h (n = 4/7). (A) Body weight loss. (B) Transcript levels of Tnfα and Ifit1 in SI crypt lysates of Atg16l1fl/fl and Atg16l1ΔIEC mice after 72 h of treatment. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
(A) Immunoblotting of protein levels of p36ATF6 and GRP78 derived from SI organoid lysates generated from healthy (n =7), CD non-inflamed (n = 8), and CD inflamed tissue (n = 9). Related to Fig. 7B. (B) Relative mRNA expression of ATF6, HSPA5, ACSL1 and CSNK2B in IECs from ileal biopsies from paediatric IBD patients (CD, n=10 and UC, n=11) and healthy controls (n=11) (data obtained from Howell et al., 2018, Gastroenterology). Differential expression calculated between control and diseased using DE-Seq2. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
(A) Gene list of identified ATF6 signaling modulators in the primary RNAi screen performed in HEK-293 cells. (B) Gene list of identified AFT6 signaling modulators in the second RNAi screen. (C) Gene list and (D) criteria selection of identified AFT6 signaling modulators in the third RNAi screen.
Details on patients that provided biopsies for generating human small intestinal organoids.
What you need to know:
Background and Context:
Endoplasmic reticulum stress in intestinal epithelial cells promotes intestinal inflammation. Activating transcription factor 6 (ATF6) is one of the cellular mediators of the ER stress response.
New Findings:
Ileal epithelial cells from patients with CD have higher levels of activated ATF6, which is regulated by CSNK2B and ACSL1. ATF6 increases expression of tumor necrosis factor and other inflammatory cytokines in response to endoplasmic reticulum stress in intestinal cells.
Limitations:
This study was performed in cell lines, tissues from patients, and mice; further studies of this pathway are needed in humans.
Impact:
Strategies to inhibit ATF6 signaling pathway might be developed for treatment of inflammatory bowel diseases.
Lay Summary:
This study identified a gene that is activated in stressed intestinal cells, resulting in production of factors that promote intestinal inflammation.
Grant support:
This work was supported by DFG Excellence Clusters Inflammation at Interfaces and Precision Medicine in Inflammation (RTFIII) (P.R.); the DFG Research Training Group 1743 (P.R.), the CRC877 B9 project (P.R.), H2020 SYSCID Contract 733100 (P.R.), the SH Excellence Chair program (P.R.); the Wellcome Trust Investigator award 106260/Z/14/Z (A.K.), European Research Council under the Horizon 2020 ERC CoG agreement n° 648889 (A.K.); Cambridge Biomedical Research Centre (A.K.) and the National Institutes of Health (grants DK044319, DK051362, DK053056, and DK088199) (R.S.B.) and grant to the Harvard Digestive Diseases Center DK034854 (R.S.B). D.H. is supported by the DFG CRC 1335 project P11.
Abbreviations used in this paper:
- ACSL1
Acyl-CoA Synthetase Long Chain Family Member 1
- ATF6
activating transcription factor 6
- Atg16l1
autophagy related 16 like 1
- CSNK2B
casein kinase 2 beta
- ER
endoplasmic reticulum
- ERSE
ER-stress response elements
- GRP78
glucose-regulated protein 78 kDa
- IBD
inflammatory bowel disease
- IECs
intestinal epithelial cells
- IRE1
endoribonuclease inositol-requiring enzyme 1
- NF-κB
nuclear factor kappaB
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- S1P/S2P
site 1/2 protease
- UC
ulcerative colitis
- UPR
unfolded protein response
- Xbp1
X-box binding protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors disclose no conflicts.
References
- 1.Grootjans J, Kaser A, Kaufman RJ, et al. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 2016;16:469–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haze K, Yoshida H, Yanagi H, et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999;10:3787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999;397:271–4. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida H, Haze K, Yanagi H, et al. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 1998;273:33741–9. [DOI] [PubMed] [Google Scholar]
- 5.Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013;503:272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006;4:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deuring JJ, Fuhler GM, Konstantinov SR, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014;63:1081–91. [DOI] [PubMed] [Google Scholar]
- 8.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008;134:743–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006;26:9220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 2008;5:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology 2007;132:190–207. [DOI] [PubMed] [Google Scholar]
- 12.Treton X, Pedruzzi E, Cazals-Hatem D, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology 2011;141:1024–35. [DOI] [PubMed] [Google Scholar]
- 13.Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J Exp Med 2017;214:401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aden K, Tran F, Ito G, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp Med 2018. [DOI] [PMC free article] [PubMed]
- 15.Wu J, Rutkowski DT, Dubois M, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 2007;13:351–64. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 2007;13:365–76. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–5. [DOI] [PubMed] [Google Scholar]
- 18.Fujii S, Suzuki K, Kawamoto A, et al. PGE2 is a direct and robust mediator of anion/fluid secretion by human intestinal epithelial cells. Sci Rep 2016;6:36795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipinski S, Grabe N, Jacobs G, et al. RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A 2012;109:21426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 21.Smidt K, Jessen N, Petersen AB, et al. SLC30A3 responds to glucose- and zinc variations in beta-cells and is critical for insulin production and in vivo glucose-metabolism during beta-cell stress. PLoS One 2009;4:e5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci 2010;11:799–811. [DOI] [PubMed] [Google Scholar]
- 23.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman OI, Lobner EM, Bierwirth S, et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology 2018. [DOI] [PubMed]
- 25.Koreishi M, Yu S, Oda M, et al. CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS One 2013;8:e54382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci U S A 2009;106:17775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer A, Mentrup T, Kleizen B, et al. Small molecules intercept Notch signaling and the early secretory pathway. Nat Chem Biol 2013;9:731–8. [DOI] [PubMed] [Google Scholar]
- 28.Shin HM, Kim MH, Kim BH, et al. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett 2004;571:50–4. [DOI] [PubMed] [Google Scholar]
- 29.Till A, Rosenstiel P, Krippner-Heidenreich A, et al. The Met-196 -> Arg variation of human tumor necrosis factor receptor 2 (TNFR2) affects TNF-alpha-induced apoptosis by impaired NF-kappaB signaling and target gene expression. J Biol Chem 2005;280:5994–6004. [DOI] [PubMed] [Google Scholar]
- 30.Brandl K, Rutschmann S, Li X, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U S A 2009;106:3300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Zhang L, Joo D, et al. NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siddiqui-Jain A, Drygin D, Streiner N, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 2010;70:10288–98. [DOI] [PubMed] [Google Scholar]
- 33.Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med 2017;214:3687–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howell KJ, Kraiczy J, Nayak KM, et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells From Pediatric Patients With Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate With Outcome. Gastroenterology 2018;154:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faust M, Jung M, Gunther J, et al. Localization of individual subunits of protein kinase CK2 to the endoplasmic reticulum and to the Golgi apparatus. Mol Cell Biochem 2001;227:73–80. [PubMed] [Google Scholar]
- 36.Hosoi T, Korematsu K, Horie N, et al. Inhibition of casein kinase 2 modulates XBP1-GRP78 arm of unfolded protein responses in cultured glial cells. PLoS One 2012;7:e40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manni S, Brancalion A, Tubi LQ, et al. Protein kinase CK2 protects multiple myeloma cells from ER stress-induced apoptosis and from the cytotoxic effect of HSP90 inhibition through regulation of the unfolded protein response. Clin Cancer Res 2012;18:1888–900. [DOI] [PubMed] [Google Scholar]
- 38.Haze K, Okada T, Yoshida H, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J 2001;355:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diamanti MA, Gupta J, Bennecke M, et al. IKKalpha controls ATG16L1 degradation to prevent ER stress during inflammation. J Exp Med 2017;214:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Shen J, Arenzana N, et al. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem 2000;275:27013–20. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima S, Hiramatsu N, Hayakawa K, et al. Selective abrogation of BiP/GRP78 blunts activation of NF-kappaB through the ATF6 branch of the UPR: involvement of C/EBPbeta and mTOR-dependent dephosphorylation of Akt. Mol Cell Biol 2011;31:1710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamazaki H, Hiramatsu N, Hayakawa K, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 2009;183:1480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hung JH, Su IJ, Lei HY, et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem 2004;279:46384–92. [DOI] [PubMed] [Google Scholar]
- 44.Guma M, Stepniak D, Shaked H, et al. Constitutive intestinal NF-kappaB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med 2011;208:1889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger E, Haller D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun 2011;409:610–5. [DOI] [PubMed] [Google Scholar]
- 46.Hanaoka M, Ishikawa T, Ishiguro M, et al. Expression of ATF6 as a marker of pre-cancerous atypical change in ulcerative colitis-associated colorectal cancer: a potential role in the management of dysplasia. J Gastroenterol 2018;53:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duraes C, Machado JC, Portela F, et al. Phenotype-genotype profiles in Crohn’s disease predicted by genetic markers in autophagy-related genes (GOIA study II). Inflamm Bowel Dis 2013;19:230–9. [DOI] [PubMed] [Google Scholar]
- 48.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010;140:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nezami BG, Mwangi SM, Lee JE, et al. MicroRNA 375 mediates palmitate-induced enteric neuronal damage and high-fat diet-induced delayed intestinal transit in mice. Gastroenterology 2014;146:473–83 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hodin CM, Verdam FJ, Grootjans J, et al. Reduced Paneth cell antimicrobial protein levels correlate with activation of the unfolded protein response in the gut of obese individuals. J Pathol 2011;225:276–84. [DOI] [PubMed] [Google Scholar]
- 51.Powell N, Pantazi E, Pavlidis P, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 2020;69:578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Network of candidate genes after the primary screen based on STRING. Only interactions with a confidence score >0.4 were considered. p=0.00147. (B) Schematic cellular localization of the 6 ATF6α modulators subjected to further functional studies. ATF6 signaling inducers are depicted in green, inhibitors shown in orange.
(A) knockdown efficiency for ATF6, ACSL1 and CSNK2B in Caco-2 cells was calculated (related to Fig. 2B). Samples treated with the non-targeting siRNA (siscrambled, unstimulated) were set to 1. (B) Transcript levels of NF-κB signaling components in small intestinal organoids treated with tunicamycin (TM 100ng/ml) for 24 h. (C) Immunoblotting of protein lysates collected from WT and Atf6 tgtg small intestinal organoids stimulated with Tunicamycin (TM, 100ng/ml). Relative p-p65, p52 and p-p38 protein levels were quantified. The average value of the unstimulated WT controls was set to 1. n=3 biological replicates. (D) cIAP1/Birc2 and cIAP2/Birc3 mRNA levels in WT and Atf6 tgtg SI organoids stimulated for 24 h. Related to Fig. 3B. Depicted data representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
Transcript levels of Atf6α target genes Hsp90b1 (Grp94) and Hspa5 (Grp78) in small intestinal organoids treated with tunicamycin (TM, 100ng/ml) for 24 h. (A) Atg16l1fl/fl and Atg16l1∆IEC small intestinal organoids. n=4 biological replicates. (B) Xbp1fl/fl and Xbp1ΔIEC small intestinal organoids. n=4 biological replicates. (C) Transcript levels of Xbp1 in Xbp1-deficient MODE-K cells (n=4). (D) ERSE promoter activity and (E) NF-κB luciferase activity in Caco-2 cells upon exposure to tunicamycin (5 μg/ml) for 24 h in the presence or absence of CX-4945 (3 μM) as indicated. Shown data representative of 3 independent experiments. For statistical analysis, one-way ANOVA together with Tukey post hoc test was performed.
(A) Stimulation scheme of Atg16l1fl/fl and Atg16l1ΔIEC mice (n = 5/6). Mice were treated with 1 mg/kg bodyweight of tunicamycin i.p, when indicated mice additionally received either TC (2.5 µg/g bodyweight) or CX-4945 (40 µg/g bodyweight). Control groups received DMSO. After 24 h mice were sacrificed. (B) Weight loss 24 h after injection. (C) CXCL1 concentration in SI crypt lysates detected via ELISA. (D-E) Tunel staining of SI sections with representative pictures (D) and quantification (E). A minimum of 50 crypts/intestine were assessed in each treatment group. Bars=20 µm. Arrowheads show apoptotic Paneth cells. (F) Transcript levels of Tnfα and Ifit1 in SI crypt lysates of treated Atg16l1fl/fl and Atg16l1ΔIEC mice after 24 h. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
Mice received 1 mg/kg bodyweight of tunicamycin i.p, when indicated mice were additionally treated with either TC (2.5 µg/g bodyweight) or CX-4945 (40 µg/g bodyweight) at 0, 24, and 48 h. Control groups received DMSO. Mice were sacrificed after 72 h (n = 4/7). (A) Body weight loss. (B) Transcript levels of Tnfα and Ifit1 in SI crypt lysates of Atg16l1fl/fl and Atg16l1ΔIEC mice after 72 h of treatment. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
(A) Immunoblotting of protein levels of p36ATF6 and GRP78 derived from SI organoid lysates generated from healthy (n =7), CD non-inflamed (n = 8), and CD inflamed tissue (n = 9). Related to Fig. 7B. (B) Relative mRNA expression of ATF6, HSPA5, ACSL1 and CSNK2B in IECs from ileal biopsies from paediatric IBD patients (CD, n=10 and UC, n=11) and healthy controls (n=11) (data obtained from Howell et al., 2018, Gastroenterology). Differential expression calculated between control and diseased using DE-Seq2. Statistical analysis was performed using one-way ANOVA together with Tukey post hoc test.
(A) Gene list of identified ATF6 signaling modulators in the primary RNAi screen performed in HEK-293 cells. (B) Gene list of identified AFT6 signaling modulators in the second RNAi screen. (C) Gene list and (D) criteria selection of identified AFT6 signaling modulators in the third RNAi screen.
Details on patients that provided biopsies for generating human small intestinal organoids.