

Abstract
Tumor-associated macrophages (TAMs) represent the M2-like phenotype with potent immunosuppressive activity, and play a pro-tumor role in pancreatic ductal adenocarcinoma (PDAC) biology. In this study, we investigated the role of the insulin-like growth factor binding protein 2 (IGFBP2) as a determinant of TAM polarity. Clinical data revealed that the levels of IGFBP2 correlated with M2 TAMs accumulation and disease progression in human PDAC. In vivo mouse model experiments showed that IGFBP2 promoted immunosuppressive microenvironment and tumor growth in a macrophage dependent manner. Bioinformatics analysis of PDAC transcriptome revealed a significant association between IGFBP2 expression and M2 macrophage polarization and signal transducer and activator of transcription 3 (STAT3) activation. Mechanistic investigations demonstrated that IGFBP2 augmented the expression and secretion of IL-10 through STAT3 activation in PDAC cells, which induced TAM polarization toward an M2 phenotype. IGFBP2-polarized M2 macrophages significantly increased Tregs infiltration and impaired antitumor T-cell immunity in mouse model. Thus, our investigations have illuminated the IGFBP2 signaling pathway that contributes to the macrophage-based immunosuppressive microenvironment in PDAC, suggesting that blocking IGFBP2 axis constitutes a potential treatment strategy to reset TAM polarization toward an antitumor state in PDAC.
Keywords: IGFBP2, TAM, alternative polarization, PDAC, STAT3
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is considered the most lethal malignancy owing to its aggressive nature [1]. Due to the lack of effective means for early diagnosis, most PDAC patients present with either locally advanced disease or distant metastasis and are therefore not suitable for radical resection [2,3]. As systemic treatments have brought only modest survival benefits, many studies have focused on the unique biological characteristics of PDAC to define new therapeutic targets [4].
A hallmark of PDAC is an extremely stroma-rich tumor microenvironment (TME), a great part of which is composed of extracellular matrix (ECM) [5-7]. The cellular components of the stroma, particularly infiltrating immune cells, are critically involved in nearly all aspects of PDAC biology and contribute to an immunosuppressive TME [8]. Macrophages, as differentiated leukocytes and being responsible for homeostasis, display an unusual plasticity. Guided by environmental signals, macrophages may polarize to M1 (classically activated or inflammatory) or M2 (alternatively activated or anti-inflammatory) phenotypes, thus exerting antagonistic functions. Furthermore, fully polarized macrophages can repolarize and transform reciprocally in response to a changing TME [9-11]. Tumor-associated macrophages (TAMs) mainly represent the M2-like phenotype, with potent immunosuppressive activity. As the major tumor-infiltrating immune cells of PDAC, M2 TAMs play a pro-tumor role in carcinogenesis, angiogenesis, metastasis, and therapeutic resistance [12-13]. Elevated infiltration of M2 TAMs has been associated with poor prognosis in PDAC [14,15]. Unfortunately, current immunotherapies have had limited efficacy in patients with PDAC [16]. Reprogramming TAMs could be a promising strategy to ameliorate the immunosuppressive TME and prevent the progression of PDAC.
Insulin-like growth factor binding protein 2 (IGFBP2) acts as a pleiotropic oncogene in various malignancies including PDAC [17-24]. IGFBP2 levels are increased in the serum and pancreatic juice of PDAC patients and IGFBP2 has been proposed as a potential biomarker in PDAC [25-28]. The involvement of IGFBP2 in immune microenvironment has been suggested by the observation that IGFBP2 correlated with a decreasing of CD8+ T cells and CD19+ B cells and increasing of CD163+ M2 macrophages in glioblastoma, but which is lacking establish the potential mechanism [29]. Our recent study revealed that IGFBP2 is a driver of epithelial-to-mesenchymal transition and metastasis through the inflammation and immunity-related NF-κB pathway [24]. We also reported that IGFBP2 potentiates signal transducer and activator of transcription 3 (STAT3) transactivation activities to augment downstream COX-2 expression, which plays a role in immunosuppressive TME formation [30]. Furthermore, IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma [31].
Persistent activation of STAT3 has been demonstrated to be a crucial mediator in governing pancreatic carcinogenesis, acinar-ductal-metaplasia (ADM) development, tumor progression, and inhibition of apoptosis and inflammatory response [32-38]. STAT3 also plays a critical role in the interplay between the TME and the immune system and contributes to tumor immune evasion [39]. Activated STAT3 in tumor cells induces the overexpression of particular cytokines, such as IL-6 and IL-10, which subsequently activate STAT3 in various immune cell types, including TAMs. Increased activation of STAT3 in these immune cell subsets triggers multiple immunosuppressive mechanisms and leads to the impairment of antitumor immunity [40-44].
In this study, we illustrate the role of IGFBP2 as a determinant of TAM polarity. IGFBP2 augmented the expression and secretion of IL-10 through STAT3 activation in PDAC cells, which induced TAM polarization toward an M2 phenotype. Thus IGFBP2-polarized M2 macrophages significantly increased Tregs infiltration and impaired antitumor T-cell immunity. Thus, our investigations have illuminated the IGFBP2 signaling pathway that contributes to the macrophage-based immunosuppressive microenvironment in PDAC.
2. Materials and methods
2.1. Patients and tissue samples
The use of patient-derived material was approved by the institutional research ethics committee at Tianjin Medical University General Hospital (TMUGH). Tumor samples were obtained from 96 patients who underwent surgical resection at the hospital between 2009 and 2018 and had a histological diagnosis of PDAC. Following informed consent, the patient’s demographic and clinical characteristics were recorded and analyzed. Consecutive sections of formalin-fixed, paraffin embedded tumor samples were subjected to IHC for IGFBP2, CD163, FOXP3, Ki-67, and phospho-(Y705)-STAT3 (Santa Cruz Biotechnology). The results were scored by two pathologists who were blinded to the clinicopathologic data. Fresh PDAC patient surgical samples from TMUGH were processed into single-cell suspensions with 1 mg/ml collagenase, 2.5 U/ml hyaluronidase and 0.1 mg/ml DNase and were subjected to flow cytometry for cell surface markers with antibodies to human CD4, CD25, FOXP3, CD8, CD45, CD68, and CD163 (BD Biosciences). For each tumor, the mirror image tumor sample was also collected and processed to FFPE samples for measuring the IGFBP2 expression by IHC.
2.2. Patient-derived xenograft cell lines and cell culture
Human PDAC PDX cell lines MDA-PATC53 (high endogenous IGFBP2) and MDA-PATC148 (low endogenous IGFBP2) (all KRas G12D) were established as previously reported and cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 50:50 medium supplemented with 10% fetal bovine serum (FBS) and incubated in a 5% CO2 atmosphere at 37°C [45]. IGFBP2 stimulation experiments were performed by using recombinant IGFBP2 (Abcam) with cells starved of serum overnight. The mouse PDAC cell line Panc02 was purchased from ATCC (Rockville) and authenticated using short tandem repeat profiling.
2.3. Orthotopic PDAC mouse model
Female C57BL/6 and BALB/c nude mice (4-week-old) were maintained under specific pathogen free conditions and animal experiments were conducted under a protocol approved by the Ethics Committee of Tianjin Medical University General Hospital in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals. To generate stable mouse IGFBP2 overexpression cell line (Panc02-IGFBP2) and control cell line (Panc02-EV), Panc02 cells were infected with mouse IGFBP2 or control lentiviral particles (GeneCopoeia) using polybrene, followed by puromycin selection for 3 weeks. Panc02-EV or Panc02-IGFBP2 cells (2×106) were injected into the pancreas of C57BL/6 and BALB/c nude mice to establish orthotopic PDAC mouse models. The BALB/c nude mice were sacrificed and the xenograft tumors were harvested and weighed four weeks later. The C57BL/6 mice were received administration of PBS or clodronate liposomes (Clo) from the first day for 2 weeks. Two weeks after the administration, the mice were sacrificed and the xenograft tumors were harvested and weighed. The tumor nodules in liver were also counted. The tumors were dissociated into single-cell suspensions with 1 mg/ml collagenase, 2.5 U/ml hyaluronidase and 0.1 mg/ml DNase and processed for flow cytometry analysis using antibodies to identify mouse M1 TAMs (F4/80+/CD206−), M2 TAMs (F4/80+/CD206+), Tregs (CD4+CD25+FOXP3+), cytotoxic T cells (CD8+/CD45+), and apoptotic T cells (Annexin V+/CD8+) (BD Biosciences). IFN-γ in mouse tumor tissue was measured by Enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s procedure (R&D Systems).
2.4. Lentivirus, plasmids, and siRNA
To generate stable human IGFBP2 overexpression cell lines and control cell line, MDA-PATC148 cells were infected with human IGFBP2 or control lentiviral particles (GeneCopoeia) using polybrene, followed by puromycin selection for 3 weeks.
To generate the Y705-mutated STAT3 sequence, nucleotide sequences coding amino acid residues Y705 of STAT3 (NM_139276.2) were mutated to F705 using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies). Human wild-type and mutated STAT3 cDNAs were cloned into the pcDNA3.1 plasmid (Invitrogen) and verified by DNA sequencing. MDA-PATC148 cells were transfected with plasmids using FuGENE HD (Promega), followed by G418 selection for 3 weeks.
Predesigned siRNAs were obtained from Sigma-Aldrich. MDA-PATC53 cells (high endogenous IGFBP2) were transfected with siRNA for IGFBP2 (SASI_Hs02_00302878, SASI_Hs01_00039595), IL-10 (SASI_Hs01_00060266, SASI_Hs01_00060268), STAT3 (SASI_Hs01_00061860, SASI_Hs01_00061861), or universal negative control #1 using Lipofectamine RNAiMAX (Invitrogen). MDA-PATC148 cells (low endogenous IGFBP2) were similarly transfected with siRNA for STAT3 or negative control #1. Total RNA or protein was isolated 48 h after transfection and analyzed by qRT-PCR or Western blotting.
2.5. PDAC PDX cell-conditioned medium (cell-CM)
IGFBP2 overexpression or knockdown, STAT3 overexpression, knockdown or inhibition, IL-10 overexpression, knockdown or neutralization, or negative control PDAC PDX cells were incubated in complete medium for 24 h and the cell-CM was collected and centrifuged for 20 min at 2,000 x g at 4°C. Supernatants were transferred and centrifuged an additional 20 min at 10,000 x g at 4°C to remove cellular debris. Cell-CM supernatants were measured for IL-10 concentrations by ELISA (R&D Systems) and used as a medium supplement for macrophage polarization.
2.6. Macrophage Studies
2.6.1. Macrophage polarization
Human PBMCs were isolated from healthy blood donors (Gulf Coast Blood Center) by density gradient centrifugation with Histopaque-1077 (Sigma-Aldrich). CD14+ monocytes were purified from PBMCs using CD14 microbeads (Miltenyi Biotech) and cultured in RPMI 1640 medium supplemented with 20% heat-inactivated FBS (Sigma-Aldrich) and 50 ng/ml GM-CSF (Peprotech) for 5 days to differentiate into nonpolarized M0 macrophages.
M0 macrophages on day 5 were differentiated using M1 medium (50 ng/ml GM-CSF, 50 ng/ml IFN-γ, 20 ng/ml TNF-α, and 100 ng/ml lipopolysaccharide), M2 medium (100 ng/ml M-CSF, 10 ng/ml IL-10, and 10 ng/ml TGF-β; Peprotech), or PDX cell line cell-CM for 48 h. Polarized macrophages on day 7 were subjected to flow cytometry for cell surface markers with antibodies to human MHC-II, CD80, CD86, CD163, CD204, and CD206 (BD Biosciences). Polarized macrophages and their conditioned medium were also used for the following studies.
2.6.2. Phagocytosis assay
Macrophages polarized by various treatment conditions were incubated with FITC-labeled Escherichia coli BioParticles (Thermo Fisher Scientific) for 2 h with 5% CO2 atmosphere at 37°C. After removing the BioParticle loading suspension, trypan blue suspension was added and incubated for 1 min to quench any membrane bound probe. Relative phagocytosis was quantified as fluorescence intensity measured using a microplate spectrophotometer. Percent phagocytosis levels relative to the positive control (M1 macrophage) were calculated for each experimental condition.
2.6.3. Nitric oxide (NO) production
NO production by experimentally polarized macrophages was measured using the Griess Reagent System (Promega). The Griess reagent was added to supernatant medium of different polarized macrophages and the mixture was incubated 10 min at room temperature in the dark. The absorbance at 540 nm was measured using a microplate spectrophotometer and NO concentration was determined with sodium nitrite as a standard.
2.6.4. Arg1 activity
Arg1 activity was measured following previously described methods [46]. Briefly, macrophages were lysed, the supernatant combined 1:1 with 10 mM MnCl2 (Sigma-Aldrich) and heated at 56°C for 7 min to activate the Arg1. The incubation was continued at 37°C for 2 h with 50 μmol of L-arginine to allow hydrolysis of L-arginine by Arg1. Levels of the byproduct metabolite urea were determined by incubation with α-isonitrosopropiophenone substrate and absorbance measured at 540 nm, with Arg1 urea production calculated in samples from a urea standard curve.
2.6.5. VEGF, TGFβ and IL-10 secretion
Macrophages polarized by various treatment conditions were rinsed with PBS and cultured in RPMI 1640 medium supplemented with 20% heat-inactivated FBS for a further 24 h. The amount of VEGF, TGFβ, and IL-10 secreted in supernatants was quantified by ELISA according to the manufacturer’s procedure (R&D Systems).
2.7. RNA isolation and qRT-PCR
Total RNA was extracted from cells with a mirVana miRNA Isolation Kit (Ambion). Reverse-transcription was performed with Superscript II Reverse Transcriptase (Invitrogen). TaqMan Gene Expression Assays (Applied Biosciences) were used to detect and quantify human MHCII, CD80, CD86, NOS2, IL1B, TNFA, CD163, CD204, CD206, ARG1, IL10, TGFB1, and STAT3. Relative expression was normalized to the endogenous control GAPDH using the 2 −ΔΔCt method. Experiments were carried out in triplicate.
2.8. Western blotting
Cells were harvested and subjected to lysis in RIPA Buffer (Thermo Fisher Scientific) with 1:100 Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The whole-cell lysate from each sample was separated by electrophoresis on a 10% polyacrylamide gel, blotted onto nitrocellulose membranes, and probed with primary antibodies against IGFBP2, cMYC, Bcl-xL, COX2, cyclin D1, iNOS, Arg1, p65, phospho-(Ser536)-p65, STAT3, phospho-(Y703)-STAT3, EGRF, and phospho-(Y1068)-EGRF (Santa Cruz Biotechnology). Proteins were detected by HRP-conjugated secondary antibodies and visualized using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Cellular fractionation was performed by using the NE-PER Nuclear and cytoplasmic Extraction Kit (Pierce Biotechnology) according to the manufacturer’s protocol. Densitometric analysis of immunoblot bands were quantified using the ImageJ software (US National Institutes of Health).
2.9. Ingenuity pathway and gene set enrichment analyses
The PDAC Australia (PDAC-AU) cohort from the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org) consists of 461 samples from 391 patient donors, of which 91 samples have available RNA-seq data and survival information (Supplemental Table S1). These patients include 47 males, 43 females and 1 without gender information, and their ages are between 36 and 86. These 91 patients were categorized into three groups based on IGFBP2 expression levels: high- (top 20%), low- (bottom 20%) and intermediate-IGFBP2 (the other 60%).
Differentially expressed genes between the high- and low-IGFBP2 groups were identified by the DESeq2 method [47]. Then the Ingenuity Pathway Analysis software (IPA; Ingenuity Systems Inc.) was used to identify the enriched pathways associated with high or low expression of IGFBP2. We also calculated the correlation of gene expression between IGFBP2 and all other genes and got the top correlated genes based on the correlation coefficients. With the IGFBP2-correlated genes, we further identified the STAT3 signaling pathway as top enriched using the Gene set enrichment analysis (GSEA).
2.10. Statistical analysis
Data are expressed as the mean ± SD and analyzed using Student’s t test for two groups or analysis of variance (ANOVA) for multiple groups. P values < 0.05 were statistically significant and levels are indicated in figures by asterisks (*P<0.05, **P<0.01). SPSS 17.0 software (SPSS Inc.) and R software were used for statistical analyses.
3. Results
3.1. High IGFBP2 levels correlate with disease progression in human PDAC
To identify the role of IGFBP2 in the progression of PDAC, we first evaluated the association between levels of IGFBP2 and tumor proliferation rates in a cohort of 96 patients from TMUGH. Immunohistochemical (IHC) staining revealed that IGFBP2 was abundant in human PDAC tumor cells but rarely present in adjacent normal pancreatic cells (Fig. 1A). We used Ki-67 antibody for IHC staining and observed a positive correlation between IGFBP2 protein levels and the percentage of Ki-67 positive PDAC cells (Fig. 1B, C). Furthermore, clinicopathological analyses showed that IGFBP2 overexpression in tumors was significantly correlated with T (primary tumor size) stage. The patients with high IGFBP2 (++/+++) showed larger tumor volumes and more advanced T stage than those with low IGFBP2 (−/+) (Fig. 1D). Kaplan-Meier analysis indicated that patients with tumors expressing high levels of IGFBP2 protein had significantly shorter relapse-free survival (RFS) (P=0.001; Fig. 1E) and overall survival (OS) (P=0.002; Fig. 1F), suggesting that IGFBP2 is associated with poor prognosis in PDAC patients.
Fig. 1. High IGFBP2 levels correlate with disease progression in human PDAC.
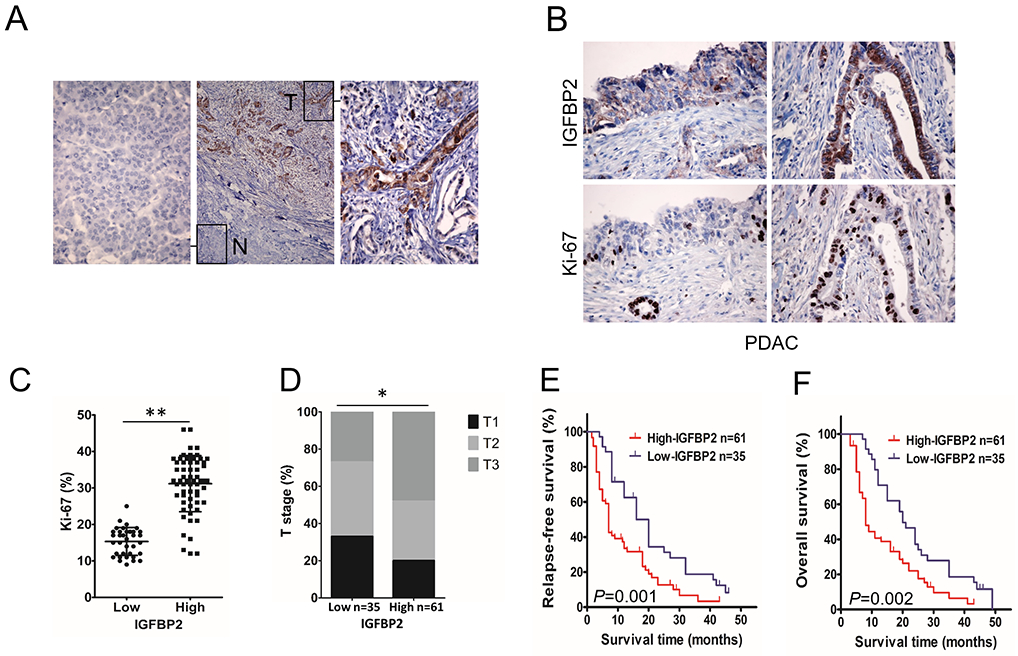
(A) Representative micrographs showing IGFBP2 expression in PDAC (T) compared to adjacent normal pancreatic tissues (N). Magnification: 10× and 400×. (B) IGFBP2 and Ki-67 IHC in tumors from high (right) and low (left) IGFBP2-expressing patient groups. Magnification: 400×. (C) Percentage of mitotic Ki-67 tumor cells (n=96; **P<0.01 by Student’s t-test). (D) The distribution of T stage (primary tumor size) between IGFBP2 high and low expression groups (n=96, *P<0.05 by χ2 test. T: T1, maximum tumor diameter ≤2 cm; T2, maximum tumor diameter >2 cm but ≤4 cm; T3, maximum tumor diameter >4 cm.). Kaplan-Meier curves comparing the relapse-free (E), and overall (F) survival rates of PDAC patients with opposing IGFBP2-expression levels (n=96; P=0.001 and P=0.002, respectively, by log-rank test).
3.2. High IGFBP2 levels correlate with accumulation of M2 TAMs in the human PDAC microenvironment
To explore the relationship between IGFBP2 levels and accumulation of immune cells in the human PDAC microenvironment, we analyzed gene expression data in the PDAC Australia (PDAC-AU) cohort from the International Cancer Genome Consortium (ICGC) (Supplemental Table S1). Of the 91 samples, we selected PDAC patients representing the top- and bottom-20th percentile of IGFBP2 gene expression for comparison to maximally differentiate the representative tumor microenvironments for high verses low IGFBP2 tumors. Notably, patients in the top-20th percentile of IGFBP2 gene expression showed significantly increased average expression of M2 macrophage markers (P=0.031) and Treg markers (P=0.00013; Fig. 2A). These data indicated that high IGFBP2 expression levels in PDAC correlate to accumulation of M2 macrophages and Tregs.
Fig. 2. High IGFBP2 levels correlate with M2 TAMs accumulation in human PDAC microenvironment.
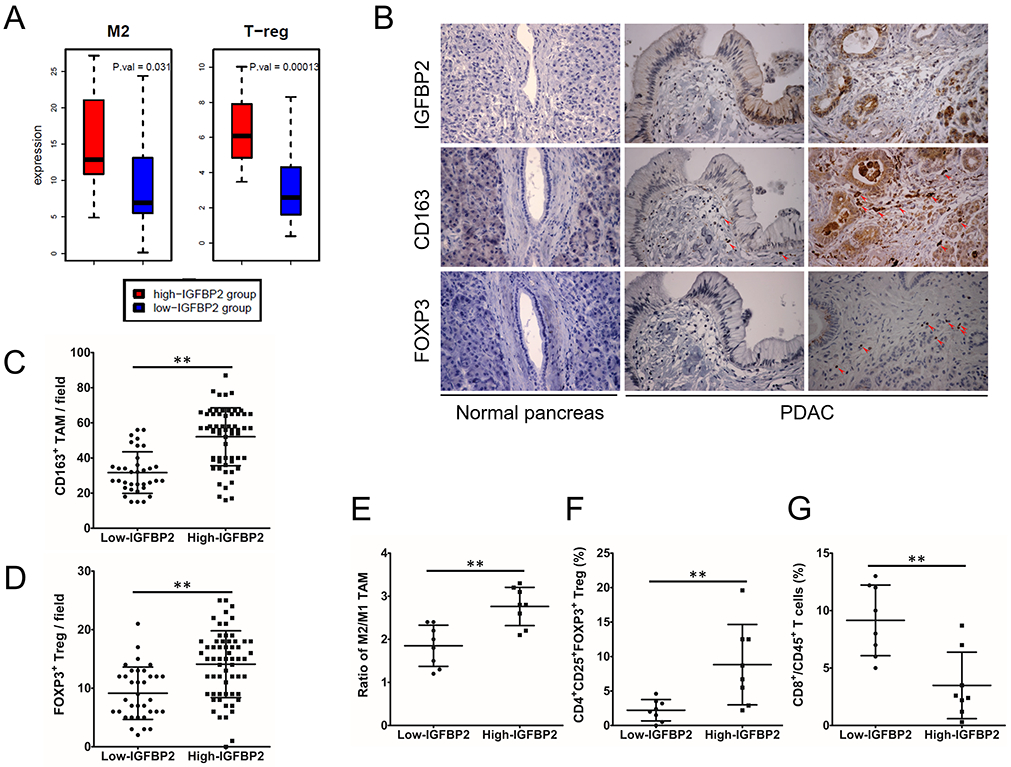
(A) Expression of M2 TAM an Treg markers in high- and low-IGFBP2 groups in patient samples from the ICGC PDAC-AU cohort (n=91; P=0.031 and P=0.00013 by Wilcoxon rank sum test). (B) IGFBP2, CD163 and FOXP3 expression in PDACs and normal pancreatic tissues. Magnification: 400×. Red arrows indicate CD163+ cells as M2 TAMs and FOXP3+ cells as Tregs. (C) Counts of M2 TAMs in PDAC microenvironment with low and high expression of IGFBP2. (D) Counts of Tregs in PDAC microenvironment with low and high expression of IGFBP2 (n=96; **P<0.01 by Student’s t-test). (E) Flow cytometry analysis of M2/M1 TAMs ratio in PDAC tissues from fresh surgical samples with low and high expression of IGFBP2 (CD68+CD163− cells as M1 TAMs and CD68+CD163+ cells as M2 TAMs). Flow cytometry analysis of CD4+CD25+FOXP3+ Tregs (F) and CD8+CD45+ T cells (G) in PDAC tissues from the same samples (n=16; **P<0.01 by Student’s t-test). Data represent the mean ± SD of at least 3 independent experiments.
To validate our findings in an independent patient group, we used IHC to survey numbers of putative CD163+ M2 TAMs and FOXP3+ Tregs in sections from the same cohort of 96 PDAC samples from TMUGH. We found that PDAC tissues with high IGFBP2 showed significantly greater numbers of M2 TAMs and Tregs than those with low IGFBP2 (Fig. 2B-D).
To explore the relationship between IGFBP2 levels and accumulation of immune cells in the human PDAC microenvironment, flow cytometry analysis was performed on 16 fresh tissue samples of human PDAC (8 each of IGFBP2 low and high cases characterized for IGFBP2 levels by IHC using the mirror image PDAC samples). We evaluated CD68+CD163− M1 TAMs, CD68+CD163+ M2 TAMs, CD4+CD25+FOXP3+ Tregs and CD8+CD45+ cytotoxic T cells. Samples from patients with high IGFBP2 levels showed significantly higher ratios of M2/M1 TAMs (Fig. 2E) and higher percentages of Tregs (Fig. 2F), which was consistent with our IHC results from the larger cohort (Fig. 2B-D). In addition, the percentage of cytotoxic T cells was significantly lower in samples with high IGFBP2 levels (Fig. 2G). These data suggest that high IGFBP2 expression in PDAC correlates with an immunosuppressive TME.
3.3. IGFBP2 promotes PDAC progression in a macrophage dependent manner
To investigate whether IGFBP2 promotes immunosuppressive cell accumulation and disease progression in vivo, we first established IGFBP2-overexpressing (Panc02-IGFBP2) and empty vector-transfected control (Panc02-EV) cell lines with Panc02 murine pancreatic cell line (Supplementary Fig. S1A). Then, an orthotopic PDAC mouse model was established in C57BL/6 mice (n=10 per group). Twenty-eight days following cell implantation, mice were sacrificed, and tumors were harvested (Supplementary Fig. S1B). Panc02-IGFBP2 group showed a significantly larger tumor size, larger tumor weight, and more liver metastases than Panc02-EV group (Supplementary Fig. S1C-E). Panc02-IGFBP2 group also contributed to significantly reduced OS in an independent cohort of mice (n=20 per group) (P = 0.008; Supplementary Fig. S1F). Flow cytometry analysis was performed on cell suspensions from the fresh mouse tumor tissues to interrogate the relationship between IGFBP2 levels and immune cell infiltration in the tumor microenvironment. Panc02-IGFBP2 group showed a significantly higher M2 (F4/80+/CD206+) / M1 (F4/80+/CD206−) TAMs ratio, a greater percentage of Tregs (CD4+CD25+FOXP3+), a lower percentage of cytotoxic T cells (CD8+CD45+), and a greater percentage of apoptotic cytotoxic T cells than measured in the Panc02-EV group (Supplementary Fig. S1G-J). These data suggested that IGFBP2 promotes immunosuppressive cell accumulation and disease progression in PDAC.
Next, we investigate whether immune cells contribute to IGFBP2 promoted PDAC progression, we established an orthotopic mouse model with Panc02-IGFBP2 and Panc02-EV cells in BALB/c nude mice (n=10 per group). Twenty-eight days later, mice were sacrificed, and tumors were harvested (Fig. 3A). The two groups didn’t show significantly differences in tumor size and tumor weight (Fig. 3B, C), which suggesting that the tumor promoting effect of IGFBP2 appears to be ascribed to the immunosuppressive TME.
Fig. 3. IGFBP2 promotes PDAC progression in a macrophage dependent manner.

(A) BALB/c nude mice were implanted with IGFBP2-overexpressing (IGFBP2) or empty vector-transfected control (EV) Panc02 cells to establish orthotropic PDAC model. After 28 days, mice were sacrificed and tumors were harvested. The tumor weight (B) and tumor size (C) were measured (n=10; NS, not statistically significant by Student’s t-test). (D) Orthotropic PDAC-bearing C57BL/6 mice we treated with Clo or PBS for 14 days. After 28 days, mice were sacrificed and tumors were harvested. The tumor weight (E) and liver tumor nodules (F) were measured. (G) M2/M1 TAMs ratio in PDAC microenvironment from both groups of mice (F4/80+/CD206− cells as M1 TAMs and F4/80+/CD206+ cells as M2 TAMs) were analyzed by flow cytometry. CD4+CD25+FOXP3+ Tregs (H), CD8+CD45+ T cells (I) and apoptotic CD8+ T cells (J) were also analyzed by flow cytometry. (K) The production of IFN-γ by cytotoxic T in PDAC microenvironment from both groups of C57BL/6 mice were analyzed by ILISA (n=10; **P<0.01 NS, not statistically significant by Student’s t-test). Data represent the mean ± SD of at least 3 independent experiments.
Furthermore, we studied the potential mechanisms behind the described IGFBP2-mediated the immunosuppressive TME in PDAC. TAMs are widely distributed in tumor microenvironment and mainly polarized to M2-like phenotype with potent immunosuppressive activity. They not only promote tumor angiogenesis and metastasis but also suppress antitumor immune response. To determine whether macrophages are involved in IGFBP2-mediated immunosuppression and tumor promotion, we treated orthotopic tumor-bearing mice with Clo or PBS (Fig. 3D). We found that macrophage depletion by Clo significantly disrupted the promotion of IGFBP2 on tumor growth and metastases (Fig. 3E, F), as well as the immunosuppression TME (Fig. 3G-K). Macrophage depletion effectively nullified the differences in percentage of Tregs (CD4+CD25+FOXP3+), cytotoxic T cells (CD8+CD45+), apoptotic cytotoxic T cells and the production of IFN-γ by cytotoxic T cells between Panc02-EV and Panc02-IGFBP2 groups. Together, these mouse model results serve to demonstrate that IGFBP2 initiates a macrophage-dependent pathway that promotes PDAC progression.
3.4. IGFBP2 modifies macrophage polarization to an immunosuppressive phenotype
To explore the impact of IGFBP2 on TAM polarization, we returned to public data in the ICGC PDAC-AU cohort, in this instance to validate the functional relationships observed for IGFBP2 in M2 macrophage polarization. As previously noted, of the 91 patient samples from the PDAC-AU cohort, we focused our analysis based on levels of gene expression comparing high-IGFBP2 (top 20th percentile) to low-IGFBP2 (bottom 20th percentile) to maximize representative tumor microenvironments. By comparing the high- to low-IGFBP2 groups, we discovered 543 genes upregulated and 503 genes downregulated in the high-IGFBP2 group (P<0.05 and ∣log2(FC)∣>1.5). These differential gene expression patterns were then submitted to Ingenuity pathway analysis (IPA) software to identify the associated pathways. Upregulated genes were enriched in the canonical pathways associated with LXR/RXR activation, FXR/RXR activation, atherosclerosis signaling, IL-12 signaling, macrophage production (Fig. 4A, red). These pathways have been reported to support activation of M2 macrophage polarization [48,49]. Downregulated genes in the high-IGFBP2 group were associated with cell cycle and metabolic function such as G2/M DNA damage checkpoint regulation, mitochondrial dysfunction, oxidative phosphorylation, etc. (Fig. 4A, blue). These pathways are associated with the prevention of macrophage polarization to the M2 phenotype [50-52]. As a result, we speculated that IGFBP2 could modify macrophage polarization to M2 phenotype.
Fig. 4. IGFBP2 modifies macrophage polarization to an immunosuppressive phenotype.

(A) IPA of data in the ICGC PDAC-AU cohort shows decreased and increased biological functions in the high-IGFBP2 group compared to the low-IGFBP2 group. The top 15 pathways with the largest negative log P values associated with higher (red bars) or lower (blue bars) expressed genes are shown. (B) Experimental scheme for human macrophage differentiation and polarization. Human monocytes are differentiated into nonpolarized M0 macrophages and then polarized into M1 or M2 macrophages by conditioned medium (CM) from PDAC cell lines differentially expressing IGFBP2. (C) The expression levels of M1 and M2 macrophage surface markers for polarized macrophages were detected by flow cytometry. (D) The expression levels of M1 and M2 macrophage phenotypic genes for polarized macrophages were detected by qRT-PCR and normalized to the endogenous control GAPDH. (E-J) Macrophage phagocytosis, nitric oxide (NO) production, IL-10, VEGF, TGF-β expression, and Arg1 activity measurements were established to characterize the functions of the IGFBP2 experimentally polarized macrophages (**P<0.01 by ANOVA for PATC53 CM or by Student’s t-test for PATC148 CM). Data represent the mean ± SD of at least 3 independent experiments.
Then we utilized two patient-derived PDAC cell lines that differentially expressed endogenous IGFBP2. IGFBP2 was silenced in MDA-PATC53 cells (high endogenous IGFBP2 expression) with two different pools of siRNA (siR-BP2-1 and siR-BP2-2). Alternatively, we overexpressed IGFBP2 in MDA-PATC148 cells (low endogenous IGFBP2 expression) using lentiviral infection to create a stably overexpressing line (MDA-PATC148 BP2). Conditioned medium from these cell lines (cell-CM) was collected to drive M0 macrophage polarization. The scheme of macrophage differentiation and polarization is summarized in Fig. 4B.
Phenotypic genes were used to define the M1 macrophages (HLA-DRA, CD80, CD86, NOS2, IL1B, and TNFA) and the M2 macrophages (CD163, CD204, CD206, ARG1, IL10, and TGFB1). When non-polarized M0 macrophages were treated with the cell-CM from the IGFBP2 knockdown group (MDA-PATC53 siR-BP2-1 and −2) and compared to high endogenous IGFBP2 MDA-PATC53 siR-ctrl, most of the classically activated M1 genes were upregulated and all the M2 genes were downregulated (Fig. 4C, top). Reciprocally, cell-CM from the IGFBP2 overexpression cells (MDA-PATC148 BP2) promoted alternative M2 gene expression panel markers and suppressed most of the M1 gene expression levels compared to low endogenous cell-CM from MDA-PATC148 EV (Fig. 4C, bottom). With classic cell surface markers to define M1 (MHC-II, CD80 and CD86) and M2 (CD163, CD204 and CD206) macrophages, we confirmed that the cell-CM from the high IGFBP2 expression cells (MDA-PATC53 siR-ctrl endogenous; MDA-PATC148 BP2 overexpression) promotes the alternative M2 macrophage phenotypes (Fig. 4D).
Furthermore, functional studies showed that when macrophages were differentiated in cell-CM from high IGFBP2 MDA-PATC148 BP2 cells and compared to low endogenous IGFBP2 MDA-PATC148 EV cells, they displayed a shift toward cancer promoting immunosuppression; including reduced phagocytosis and nitric oxide (NO) production, higher IL-10, VEGF, TGF-β expression and Arg1 activity (Fig. 4E-J). Complementarily, when macrophages were polarized in cell-CM where high endogenous IGFBP2 levels were knocked down (MDA-PATC53 siR-BP2-1 and −2) the reverse change in functionality, toward cancer suppressive activity, was observed.
3.5. IGFBP2 promotes STAT3 activation and IL-10 production in human PDAC cells
Next, we investigated the molecular mechanism through which IGFBP2 modulates TAM polarization. To this end, we focused on STAT3 signaling pathway, because STAT3 activation within tumor infiltrating immune cells governs multiple immunosuppressive mechanisms in malignancies, including macrophage polarization to the M2 phenotype. We analyzed the RNA-Seq data from ICGC PDAC-AU patient samples and performed gene set enrichment analysis (GSEA). STAT3-activated genes were significantly correlated with IGFBP2 (Fig. 5A), suggesting that IGFBP2 expression is strongly correlated with STAT3 activation. For optimal representation of the tumor microenvironment, we again compared the top- and the bottom-20th percentile of IGFBP2 expression groups and determined that STAT3 expression levels were significantly higher in IGFBP2 high patients (Fig. 5B). Meanwhile, IGFBP2 expression levels were significantly higher in STAT3 high patients (Fig. 5C).
Fig. 5. IGFBP2 promotes STAT3 activation and IL-10 production in human PDAC cells.

(A) GSEA demonstrated enrichment for STAT3 target genes based on correlation with IGFBP2 expression in the ICGC PDAC-AU database. The top of the panel shows the enrichment score (ES) for genes associated with STAT3 signaling pathway targets. The black lines indicate where the STAT3 target genes appear in the ranked gene list. The bottom of the panel shows the ranking scores (correlation of all genes associated with the STAT3 signaling pathway targets with IGFBP2). (B) Expression of STAT3 in high- and low-IGFBP2 groups in patient samples from the ICGC PDAC-AU cohort (n=91; P=0.005 by Wilcoxon rank sum test). (C) Expression of IGFBP2 in high- and low-STAT3 groups in patient samples from the ICGC PDAC-AU cohort (n=91; P=0.006, by Wilcoxon rank sum test). (D) IGFBP2 and pSTAT3 expression by IHC in patient PDAC and normal pancreatic tissues. Magnification: 400×. (E) Western blot analysis of MDA-PATC148 cells starved of serum overnight and then stimulated with exogenous IGFBP2 protein at the indicated dosages (0, 50, 100, 200, 400 ng/ml) for 60 min (left panel). Western blot analysis of MDA-PATC148 cells starved of serum overnight and then stimulated with exogenous IGFBP2 (100 ng/ml) for the indicated time points (0, 10, 15, 30, 60 min) (right panel). (F) Western blot analysis of MDA-PATC53 cells transfected with siRNA negative control (siR-ctrl) or one of two different IGFBP2 siRNAs (siR-BP2-1 and siR-BP2-2) for 72 hours to deplete IGFBP2 (left panel). Western blot analysis of MDA-PATC148 cells 72 hours after infection with negative control (EV) or human IGFBP2 (BP2) lentiviral particles to overexpress IGFBP2 (right panel). (G) Expression of cytokines reported to be downstream genes of STAT3 measured in these cells were detected by qRT-PCR and normalized to the endogenous control GAPDH. (H) The secretion of IL-10 was detected by ELISA of culture medium (**P<0.01 by ANOVA for PATC53 or by Student’s t-test for PTAC148). Data represent the mean ± SD of at least 3 independent experiments.
We inferred that IGFBP2 might mediate immunosuppressive macrophage polarization differentiation through STAT3 activation. We first evaluated levels of phosphorylated STAT3 (pSTAT3) in sections from TMUGH cohort of 96 PDAC patient samples characterized for IGFBP2 levels by IHC. We observed that increased IGFBP2 positively correlated with pSTAT3 immunostaining levels (Fig. 5D).
To investigate whether IGFBP2 is the upstream stimulator of the STAT3 signaling pathway in human PDAC cells, we stimulated MDA-PATC148 cells, which had been serum-starved overnight, with increasing amounts of exogenous IGFBP2 protein. Western blotting indicated an increasing expression of phosphorylated STAT3 (pSTAT3) in parallel with IGFBP2 uptake into the cell (Figure 5E). The expression of STAT3 transcriptional targets c-MYC, Bcl-xL, COX2, and cyclin D1 also increased in response to IGFBP2 stimulation. Next, we performed a time-course experiment in which MDA-PATC148 cells were stimulated with exogenous IGFBP2 after overnight serum starvation. Western blotting revealed phosphorylation of STAT3 and the expression of STAT3 transcriptional targets c-MYC, Bcl-xL, COX2, and cyclin D1 started from 10 min following addition of exogenous IGFBP2.
We also compared STAT3 activation and the expression of STAT3 downstream genes in MDA-PATC53 (high endogenous IGFBP2) siR-ctrl to IGFBP2 depleted MDA-PATC53-siR-BP2-1 and −2 cells. IGFBP2 knockdown decreased expression of pSTAT3 while total STAT3 levels were equivalent among groups, so did the STAT3 downstream genes. Corresponding results were observed by IGFBP2 overexpression in MDA-PATC148 BP cells, which showed increased pSTAT3 levels and upregulation of STAT3 downstream genes expression compared to low endogenous IGFBP2 MDA-PATC148 EV cells (Fig. 5F).
To explore potential targets involved in the regulation of macrophage polarization, a panel of cytokines reported to be downstream of STAT3 signaling and involved in the process of macrophage polarization were analyzed by qRT-PCR and ELISA. Notably, both mRNA expression and secreted levels of IL-10 protein were significantly decreased when endogenous IGFBP2 was depleted by siRNA in MDA-PATC53 cells. On the other hand, IL10 expression and IL-10 secretion were significantly upregulated after overexpression of IGFBP2 in MDA-PATC148 cells by lentiviral infection (Fig. 5G, H).
In our previous research, we reported that IGFBP2 also activated the NF-κB pathway in PDAC [24]. In order to determine whether IGFBP2 modified macrophage polarization through NF-κB pathway, we knocked down p65 in both PATC53 and PATC148-BP2 PDAC cells by two different pools of siRNAs (Supplementary Fig. S2A). However, we did not observe changes in the expression and secretion of IL-10 (Supplementary Fig. S2B), and the conditioned medium from these cell lines (cell-CM) did not affect the TAM polarization (Supplementary Fig. S2C).
3.6. IGFBP2 modifies macrophage polarization by promoting IL-10 production in human PDAC cells
To investigate the impact of tumor IL-10 on the polarization of macrophages, we carried out gain-of-function and loss-of-function studies. First, we depleted IL-10 by siRNA (siR-IL10-1 and siR-IL10-2) or neutralizing antibody in the MDA-PATC53 cells. Differentiation toward M1 signature markers was observed when M0 macrophages were cultured with the cell-CM from IL-10 depleted cells (Fig. 6A). The results of functional tests indicated that the macrophages polarized in IL-10-depleted PDAC cell-CM displayed increased phagocytosis and NO production, decreased IL-10, VEGF, TGF-β expression and Arg1 activity (Fig. 6B-G). These results revealed that IL-10 inhibition by siRNA or neutralized antibody abolished the capability of PDAC cells to generate the immunosuppressive M2 macrophages.
Fig. 6. IGFBP2 modifies macrophage polarization by promoting IL-10 production in human PDAC cells.
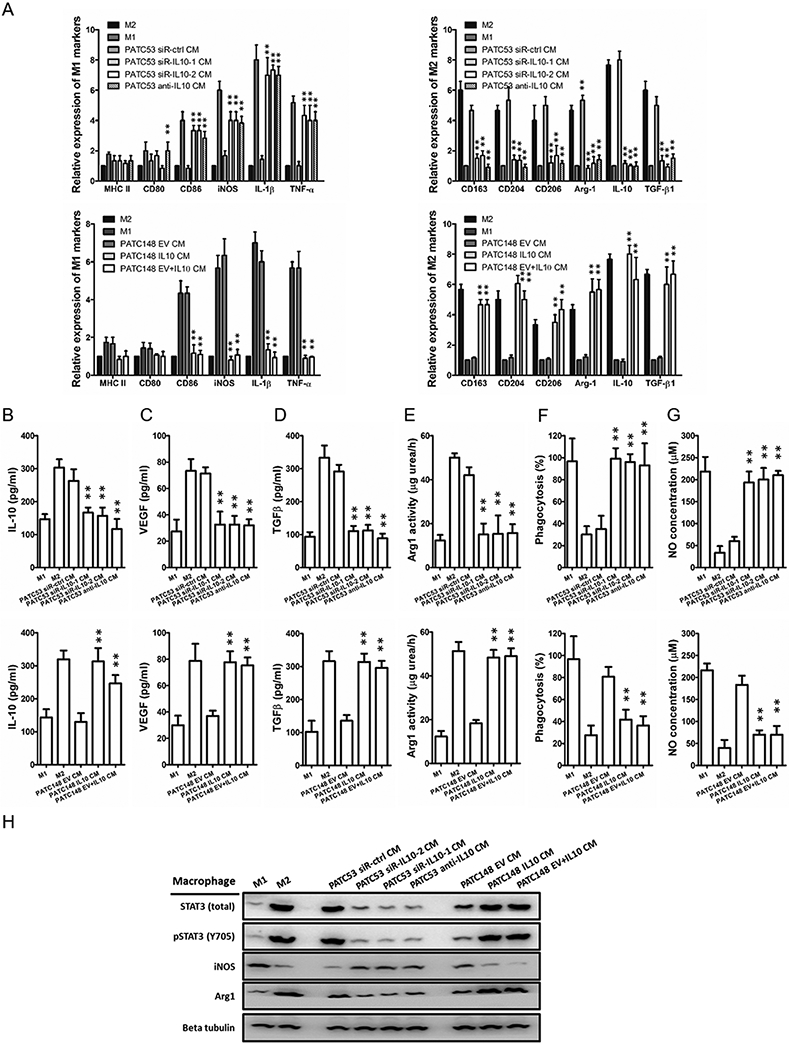
Human monocytes were differentiated into nonpolarized M0 macrophages and then polarized into M1 or M2 macrophages by conditioned medium (CM) from PDAC cell lines differentially expressing IL-10. (A) The expression levels of M1 and M2 macrophage phenotypic genes for IL-10 experimentally polarized macrophages were detected by qRT-PCR and normalized to the endogenous control GAPDH. (B-G) Macrophage phagocytosis, nitric oxide (NO) production, IL-10, VEGF, TGF-β expression, and Arg1 activity measurements were established to characterize the functions of the IL-10 experimentally polarized macrophages. (H) Western blot analysis of STAT3, pSTAT3, iNOS, and Arg1 expression in IL-10 experimentally polarized macrophages (**P<0.01 by ANOVA for PATC53 CM or by Student’s t-test for PATC148 CM). Data represent the mean ± SD of at least 3 independent experiments.
Alternatively, we overexpressed IL-10 in MDA-PATC148 cells using lentiviral infection to create a stably overexpressing line (MDA-PATC148 IL10). Conditioned medium from MDA-PATC148 EV supplement with or without IL10 or MDA-PATC148 IL10 was collected to drive M0 macrophage polarization. M2 signature markers were observed when M0 macrophages were cultured with the high IL-10 expressing cell-CM. The results of functional tests indicated that the macrophages polarized in IL-10-high PDAC cell-CM displayed an immunosuppressive phenotype. Polarized macrophages displayed decreased phagocytosis and NO production, increased IL-10, VEGF, TGF-β expression and Arg1 activity.
In addition, the protein expression of iNOS was remarkably upregulated and the expression of Arg1 was downregulated in macrophages polarized in IL-10-depleted PDAC cell-CM. Reciprocally, the protein expression of iNOS was downregulated and the expression of Arg1 was upregulated in IL-10-conditioned macrophages. We also evaluated the immunostaining levels of total and phosphorylated STAT3 (pSTAT3) in polarized macrophages. Western blotting indicated significant higher expression levels of total and phosphorylated STAT3 (pSTAT3) in M2 and M2-like macrophages compared with M1 and M1-like macrophages (Fig. 6H).
3.7. IGFBP2 promotes IL-10 secretion in human PDAC cells in a STAT3-dependent manner
To confirm that the IGFBP2-induced IL-10 secretion is mediated by activation of STAT3 in PDAC cells, we first treated MDA-PATC53 cells with a selective STAT3 inhibitor, Stattic, which inhibits activation, dimerization, and nuclear translocation of STAT3. Following the decline of phosphorylated STAT3 expression, the expression of some STAT3 downstream genes c-MYC, Bcl-xL, COX2, and cyclin D1 decreased in both dose-dependent and time-dependent manners, so did IL-10. (Fig. 7A-D). Then we silenced STAT3 by siRNA (siR-STAT3-1 and siR-STAT3-2) in MDA-PATC53 cells or BP2 overexpression MDA-PATC148 (MDA-PATC148 BP2) cells. Western blotting indicated the expression levels of total and phosphorylated STAT3 (pSTAT3) and some STAT3 downstream genes c-MYC, Bcl-xL, COX2, and cyclin D1 significant decreased (Fig. 7E). QRT-PCR and ELISA also established significantly decreased IL10 expression and IL-10 secretion (Fig. 7F, G). Then we established MDA-PATC148 cells stably overexpressing wild-type or Y705-mutant STAT3. Overexpression of wild-type STAT3 significantly increased the expression of total and phosphorylated STAT3 and some STAT3 downstream genes, including IL-10, while overexpression of mutated STAT3 only increased total STAT3 (Fig. 7H, I).
Fig. 7. IGFBP2 promotes IL-10 secretion in human PDAC cells in a STAT3-dependent manner.

(A-D) MDA-PATC53 cells were treated with the indicated concentrations of the STAT3 inhibitor Stattic for the indicated times. Whole-cell extracts were subjected to immunoblotting for STAT3, pSTAT3 and STAT3 downstream genes, including cMYC, Bcl-xL, COX2 and cyclin D1. The expression levels of IL10 were detected by qRT-PCR and normalized to the endogenous control GAPDH. The secretion of IL-10 was detected by ELISA. (E-G) High endogenous IGFBP2 MDA-PATC53 cells or IGFBP2-overexpressing PATC148 BP2 were transfected with siRNA negative control (siR-ctrl) or one of two different STAT3 siRNAs (siR-STAT3-1 and siR-STAT3-2) to knock down STAT3. (H-I) Low endogenous IGFBP2 MDA-PATC148 cells were transfected with wild-type or Y705-mutated STAT3 plasmid and analyzed by immunoblotting qRT-PCR and ELISA as described above. Human monocytes were differentiated into nonpolarized M0 macrophages and then polarized into M1 or M2 macrophages by conditioned medium (CM) from PDAC cell lines differentially expressing STAT3. (J) The expression levels of M1 and M2 macrophage phenotypic genes for STAT3 experimentally polarized macrophages were detected by qRT-PCR and normalized to the endogenous control GAPDH. (K-P) Macrophage phagocytosis, nitric oxide (NO) production, IL-10, VEGF, TGF-β expression, and Arg1 activity measurements were established to characterize the functions of the IL-10 experimentally polarized macrophages (**P<0.01 by ANOVA for PATC53 CM or by Student’s t-test for PATC148 CM). Data represent the mean ± SD of at least 3 independent experiments.
To confirm that the IGFBP2-induced immune suppressive microenvironment is mediated by activation of STAT3 in PDAC cells, we first silenced STAT3 by siRNA (siR-STAT3-1 and siR-STAT3-2) or STAT3 inhibitor, Stattic, in MDA-PATC53 cells. QRT-PCR established that the expression of M1 signature markers were increased when M0 macrophages were cultured with the cell-CM from STAT3 silenced cells (Fig. 7J). The results of functional tests indicated that the macrophages polarized in STAT3-depleted PDAC cell-CM displayed increased phagocytosis and NO production, decreased IL-10, VEGF, TGF-β expression and Arg1 activity (Fig. 7K-P). We also established MDA-PATC148 cells stably overexpressing wild-type or Y705-mutant STAT3. Overexpression of wild-type STAT3 significantly increased M2 markers expression, while overexpression of mutated STAT3 did not. The functional tests revealed that the macrophages polarized in wild-type STAT3 overexpressing PDAC cell-CM displayed decreased phagocytosis and NO production, increased IL-10, VEGF, TGF-β expression and Arg1 activity. These data demonstrated that IGFBP2 perform its reprogramming function by augmenting the expression and secretion of IL-10 through activation of the STAT3 pathway in PDAC cells.
3.8. IGFBP2 activates STAT3 by facilitating EGFR nuclear accumulation and activation in human PDAC cells
To explore the mechanism of how IGFBP2 activates STAT3 and promotes IL-10 expression and secretion, gain-of-function and loss-of-function studies were carried out. In our prior publication, we have shown that IGFBP2 augmented the nuclear accumulation of EGFR and activated the nuclear EGFR signaling pathway to potentiate STAT3 transactivation activities in human glioma and melanoma [30,31]. We hypothesize that IGFBP2 could uses the same mechanism in PDAC in activating STAT3 and IL-10 expression. The MDA-PATC148 cells (with low endogenous IGFBP2) were transfected with an IGFBP2 overexpression vector or empty vector to establish stable cell lines. MDA-PATC148 BP2 and MDA-PATC148 EV cells were fractionated into cytoplasmic and nuclear fractions to detect IGFBP2, EGFR, pEGFR, STAT3, and pSTAT3 by immunoblotting. Our results showed that IGFBP2 co-localized with EGFR in both the cytoplasm and nucleus, especially in the nucleus. Higher IGFBP2 levels and increases in pEGFR (Y1068) and pSTAT3 (Y705) were observed in the nuclear fractions from MDA-PATC148 BP2 cells than those from MDA-PATC148 EV cells (Fig. 8A). In contrast, IGFBP2 was knocked down in MDA-PATC53 cells using two different pools of siRNAs, and Western blot analysis was performed on the fractionated cell lysates. IGFBP2 knockdown led to impaired EGFR nuclear localization along with a decreased nuclear pEGFR and pSTAT3 (Fig. 8B).
Fig. 8. IGFBP2 activates STAT3 by facilitating EGFR nuclear accumulation and activation in human PDAC cells.

(A) Immunoblot analysis of cytoplasmic and nuclear fractions of MDA-PATC148.EV and MDA-PATC148.BP2 cells. Beta-tubulin represents a loading control for the cytoplasmic fraction, and PARP represents a loading control for the nuclear fraction. Densitometric analysis represented by the bar graph demonstrates the percentage of cytoplasmic or nuclear EGFR. (B) Immunoblot analysis comparing cytoplasmic and nuclear fractions of MDA-PATC53 cells depleted of IGFBP2 via two independent pools of IGFBP2 siRNA (siR-BP2-1, siR-BP2-2) to cells transfected with negative control siRNA (siR-ctrl). (C) Immunoblot analysis comparing cytoplasmic and nuclear fractions of MDA-PATC53 cells inhibited EGFR activation by erlotinib or treated with placebo. (D) Immunoblot analysis comparing cytoplasmic and nuclear fractions of MDA-PATC53 cells depleted of EGFR via two independent pools of EGFR siRNA (siR-EGFR-1, siR-EGFR-2) to cells transfected with negative control siRNA (siR-ctrl). (E) Schematic of the proposed mechanism of IGFBP2 in promoting tumor progression by inducing alternative polarization of macrophages in PDAC through the STAT3 pathway.
To further investigate the role of EGFR in this pathway, we inhibited EGFR activation by erlotinib, a specific EGFR inhibitor, in MDA-PATC53 cells. Decreased activation of EGFR and STAT3 in the nuclei was detected (Fig. 8C). Moreover, we knocked down EGFR by siRNAs. The expression of nuclear EGFR decreased along with decreased expression of pEGFR and pSTAT3 (Fig. 8D).
Taken together, these results suggest that IGFBP2 facilitated the nuclear accumulation and activation of EGFR in PDAC, which promoted STAT3 transcriptional activity, resulting in increased expression and secretion of STAT3 downstream mediator IL-10. Furthermore, IL-10 in TME modified TAM polarization towards an immunosuppressive phenotype. The proposed mechanism is summarized in Fig. 8E.
4. Discussion
Previous studies established the pleiotropic oncogenic role of IGFBP2 in various malignancies including PDAC. In the present study, clinical and experimental evidence highlight the tumor promotion effect of IGFBP2 through its immune-regulating function. We provide clear evidence that IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in PDAC through the STAT3 pathway.
In the current study, we found that patients whose tumor exhibited a high level of IGFBP2 had a more immunosuppressive TME and that is represented by greater accumulation of M2 TAMs, which was associated with poor prognosis in PDAC patients. The results of bioinformatics analysis form public database also supported this conclusion. We further validated whether IGFBP2 promoted disease progression by inducing an immunosuppressive TME by establishing an orthotopic PDAC nude mouse model. The tumor promoting effect of IGFBP2 was exhausted in immunodeficient mice. To determine whether macrophages are involved in IGFBP2-mediated tumor promoting effect, we depleted macrophages in mouse model with Clo. As expected, we found that specific depletion of macrophage disrupted the tumor promoting effect of IGFBP2, suggesting that macrophage were attributed to the IGFBP2-mediated immunosuppression and tumor promotion in PDAC. The mouse model also showed a significantly higher M2/M1 TAMs ratio in IGFBP2 overexpressing group compared with control group, indicating that there are more immune suppressive M2 macrophages infiltrating in the TME of IGFBP2 overexpressing mice. In vitro experiment, IGFBP2 showed potent function to educate macrophages polarizing toward immuno-suppressive M2 phenotype. We further demonstrated that IGFBP2 perform its reprogramming function by augmenting the expression and secretion of IL-10 through activation of the STAT3 pathway in PDAC cells. The fundamental contribution of this study to the characterization of IGFBP2 as an oncogene is the demonstration that IGFBP2 functions as an immunoregulator by driving repolarization of M2 TAMs in the PDAC microenvironment. Our investigations have illuminated the IGFBP2 signaling pathway that contributes to the macrophage-based immunosuppressive microenvironment in PDAC, suggesting that blocking IGFBP2 axis constitutes potential immunotherapeutic alleys to reset TAM polarization toward an antitumor state in PDAC.
STAT3 has been demonstrated to be a key mediator in driving immune escape, thus promoting tumor progression [41]. STAT3 activation within immune cells governs the recruitment and the activation of immunosuppressive cells, such as Tregs, Th17 cells, MDSCs, and M2 macrophages and thus maintaining an immunosuppressive microenvironment. STAT3 activation in TAMs drives polarization toward the M2 phenotype and augments their immunosuppressive activities. TAMs are shown to be predominantly STAT3-positive M2 macrophages in many cancers and predict a poor prognosis for those patients. STAT3 activation in cancer cells also regulates TAM polarization. STAT3 activation of melanoma cells correlated with infiltration of TAMs and tumor growth. Inhibiting STAT3 activation in tumor cells triggers macrophage production of NO and leads to macrophage-mediated, nitrite-dependent cytostatic activity against tumor cells. The STAT3- mediated communication between cancer cells and TAMs is mediated by numerous extracellular stimuli, such as growth factors and cytokines, including IL-10, IL-6, GM-CSF, G-CSF, VEGF, and IFN.
Numerous regulators have been reported to mediate the activation of STAT3. In glioma and melanoma, it has been shown that IGFBP2 promotes transportation of activated EGFR into nucleus where it forms a complex with STAT3 that activates STAT3 signaling pathway and its target genes including IL-10 [30,31]. Similar mechanism is likely operational in PDAC based on the observation of this study showing association of IGFBP2 and activated STAT3 and its downstream mediator IL-10.
In this study, we found that IGFBP2 facilitated the nuclear accumulation of EGFR, which promoted STAT3 transcriptional activity, resulting in increased IL-10, which is known to be a STAT3 downstream gene. Increased expression of IL-10 by PDAC cells functions as a bridge in the tumor microenvironment and drives the activation of STAT3 in tumor infiltrating-immune cells. STAT3-activated TAMs polarize towards the M2 phenotype and overexpress IL-10 to maintain an immunosuppressive microenvironment and promote tumor progression. IL-10 transcribed by STAT3 in turn activates the STAT3 pathways and maintains a stable feedforward loop between tumor cells and immunosuppressive cells. Thus, STAT3 functions as a central actor for inflammation-induced cancer such as PDAC, and IGFBP2 acts to initiate this feedforward loop.
In this study, we have demonstrated that IGFBP2 promotes PDAC progression by inducing alternative polarization of macrophages, and this function is mediated directly through activation of the STAT3 pathway, which promotes expression of IL-10 in the microenvironment. These results provided a mechanistic characterization of IGFBP2-induced repolarization of macrophages, and thus implicate IGFBP2 targeting as a potentially effective way to overcome immunotherapy resistance in human PDAC.
Supplementary Material
Supplemental Figure S1. IGFBP2 promotes PDAC progression in a macrophage dependent manner.
(A) The IGFBP2-overexpressing (Panc02-IGFBP2) and empty vector-transfected control (Panc02-EV) cell lines were established with Panc02 murine pancreatic cell line. Whole-cell extracts were subjected to immunoblotting for IGFBP2. (B) Experimental scheme for orthotropic PDAC model in C57BL/6 mice. Mice were divided into two groups and implanted with IGFBP2-overexpressing (IGFBP2) or empty vector-transfected control (EV) Panc02 cells. (C) After 28 days, mice were sacrificed and tumors were harvested. The tumor weight (D) and liver tumor nodules (E) were measured (n=10; **P<0.01 by Student’s t-test). (F) The same orthotropic pancreatic carcinoma model was repeated, and the overall survival rates were analyzed by Kaplan-Meier curves (n=20 per group; P=0.008 by log-rank test). (G) Flow cytometry analysis of M2/M1 TAMs ratio in PDAC microenvironment from both groups of C57BL/6 mice (F4/80+/CD206− cells as M1 TAMs and F4/80+/CD206+ cells as M2 TAMs). Flow cytometry analysis of CD4+CD25+FOXP3+ Tregs (H), CD8+CD45+ T cells (I) and apoptotic CD8+ T cells (J) in PDAC microenvironment from both groups of C57BL/6 mice (n=10 per group; **P<0.01 by Student’s t-test). Data represent the mean ± SD of at least 3 independent experiments.
Supplemental Figure S2. IGFBP2 promotes IL-10 secretion in human PDAC cells in a non-NF-κB-dependent manner.
(A) High endogenous IGFBP2 MDA-PATC53 cells or IGFBP2-overexpressing PATC148 BP2 were transfected with siRNA negative control (siR-ctrl) or one of two different p65 siRNAs (siR-p65-1 and siR-p65-2) to knock down p65. Whole-cell extracts were subjected to immunoblotting for p65 and p-p65. (B) The expression levels of IL10 were detected by qRT-PCR and normalized to the endogenous control GAPDH. The secretion of IL-10 was detected by ELISA. (C) Human monocytes were differentiated into nonpolarized M0 macrophages and then polarized into M1 or M2 macrophages by conditioned medium (CM) from PDAC cell lines differentially expressing p65. The expression levels of M1 and M2 macrophage phenotypic genes for p65 experimentally polarized macrophages were detected by qRT-PCR and normalized to the endogenous control GAPDH. (NS, not statistically significant by ANOVA). Data represent the mean ± SD of at least 3 independent experiments.
Supplemental Table S1. Information for the 91 patients from the ICGC PADC-AU cohort, and their expression levels (FPKM) of relative genes.
Highlights.
High IGFBP2 expression correlated with M2 TAMs accumulation and disease progression in human PDAC.
IGFBP2 augmented the expression and secretion of IL-10 through STAT3 activation in PDAC cells, which induced TAM polarization toward an M2 phenotype. IGFBP2-polarized M2 macrophages significantly increased Tregs infiltration and impaired antitumor T-cell immunity in PDAC.
Blocking IGFBP2 axis constitutes a potential treatment strategy to reset TAM polarization toward an antitumor state in PDAC.
Acknowledgments
We thank Limei Hu and David Cogdell at MD Anderson Cancer for their technical support. We thank Dr. Annette Hastie and Dr. Ping-Chieh Chou at Wake Forest Baptist Medical Center for their critical reviews of this manuscript. This study was partially supported by the U.S. National Institutes of Health/National Cancer Institute under awards P30CA012197 to Wake Forest University Health Sciences. This work was also supported by a grant from the National Foundation for Cancer Research (to W.Z.), the Skip Viragh Family Foundation (to J.F.), National Natural Science Foundation of China (81702410), Natural Science Foundation of Tianjin (17JCQNJC11100) and Natural Science Foundation of Tianjin Medical University (2016KYZQ02) (to L.S.). W.Z. was supported by the Hanes and Willis Family Endowed Professorship in Cancer and a Fellowship from the National Foundation for Cancer Research. L.S. was supported by a Postdoctoral Fellowship from the Tianjin Medical University General Hospital and Comprehensive Cancer Center of Wake Forest Baptist Medical Center.
Footnotes
Ethics approval and consent to participate
The present study was performed in accordance with the Declaration of Helsinki. Approval for the use of human subjects was obtained from the research ethics committee of Tianjin Medical University General Hospital, and informed consent was obtained from each individual enrolled in this study. All animal experiments were approved by the Animal Ethics Committee of Tianjin Medical University General Hospital.
Consent for publication
We obtained consent to publish this paper from all the participants of this study.
Conflicts of Interest
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018. January;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- [2].Neoptolemos JP. Adjuvant treatment of pancreatic cancer. Eur J Cancer. 2011. September;47 Suppl 3:S378–80. [DOI] [PubMed] [Google Scholar]
- [3].Barugola G, Partelli S, Marcucci S, Sartori N, Capelli P, Bassi C, Pederzoli P, Falconi M. Resectable pancreatic cancer: who really benefits from resection? Ann Surg Oncol. 2009. December;16(12):3316–22. [DOI] [PubMed] [Google Scholar]
- [4].Vaccaro V, Sperduti I, Milella M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011. August 25;365(8):768–9 [DOI] [PubMed] [Google Scholar]
- [5].Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA. Stromal biology and therapy in pancreatic cancer. Gut. 2011. June;60(6):861–8. [DOI] [PubMed] [Google Scholar]
- [6].Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013. June;144(6):1210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013. May 15;73(10):3007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012. August 15;18(16):4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med. 2010. March 11;362(10):875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008. December;8(12):958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, Capretti G, Mineri R, Todoric J, Zerbi A, Allavena P, Mantovani A, Marchesi F. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2016. October;65(10):1710–20. [DOI] [PubMed] [Google Scholar]
- [12].Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006. January 27;124(2):263–6. [DOI] [PubMed] [Google Scholar]
- [13].Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006. December 1;66(23):11238–46. [DOI] [PubMed] [Google Scholar]
- [14].Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim KH, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Goedegebuure SP, Linehan DC. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016. May;17(5):651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim YS, Engle D, Campbell F, Palmer D, Ko JH, Tuveson DA, Hirsch E, Mielgo A, Schmid MC. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016. May;18(5):549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015. August;14(8):561–84. [DOI] [PubMed] [Google Scholar]
- [17].Moore LM, Holmes KM, Smith SM, Wu Y, Tchougounova E, Uhrbom L, Sawaya R, Bruner JM, Fuller GN, Zhang W. IGFBP2 is a candidate biomarker for Ink4a-Arf status and a therapeutic target for high-grade gliomas. Proc Natl Acad Sci U S A. 2009. September 29;106(39):16675–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mehrian-Shai R, Chen CD, Shi T, Horvath S, Nelson SF, Reichardt JK, Sawyers CL. Insulin growth factor-binding protein 2 is a candidate biomarker for PTEN status and PI3K/Akt pathway activation in glioblastoma and prostate cancer. Proc Natl Acad Sci U S A. 2007. March 27;104(13):5563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Holmes KM, Annala M, Chua CY, Dunlap SM, Liu Y, Hugen N, Moore LM, Cogdell D, Hu L, Nykter M, Hess K, Fuller GN, Zhang W. Insulin-like growth factor-binding protein 2-driven glioma progression is prevented by blocking a clinically significant integrin, integrin-linked kinase, and NF-κB network. Proc Natl Acad Sci U S A. 2012. February 28;109(9):3475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Das SK, Bhutia SK, Azab B, Kegelman TP, Peachy L, Santhekadur PK, Dasgupta S, Dash R, Dent P, Grant S, Emdad L, Pellecchia M, Sarkar D, Fisher PB. MDA-9/syntenin and IGFBP-2 promote angiogenesis in human melanoma. Cancer Res. 2013. January 15;73(2):844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kang Z, Yu Y, Zhu YJ, Davis S, Walker R, Meltzer PS, Helman LJ, Cao L. Downregulation of IGFBP2 is associated with resistance to IGF1R therapy in rhabdomyosarcoma. Oncogene. 2014. December 11;33(50):5697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ben-Shmuel A, Shvab A, Gavert N, Brabletz T, Ben-Ze'ev A. Global analysis of L1-transcriptomes identified IGFBP-2 as a target of ezrin and NF-κB signaling that promotes colon cancer progression. Oncogene. 2013. July 4;32(27):3220–30. [DOI] [PubMed] [Google Scholar]
- [23].So AI, Levitt RJ, Eigl B, Fazli L, Muramaki M, Leung S, Cheang MC, Nielsen TO, Gleave M, Pollak M. Insulin-like growth factor binding protein-2 is a novel therapeutic target associated with breast cancer. Clin Cancer Res. 2008. November 1;14(21):6944–54. [DOI] [PubMed] [Google Scholar]
- [24].Gao S, Sun Y, Zhang X, Hu L, Liu Y, Chua CY, Phillips LM, Ren H, Fleming JB, Wang H, Chiao PJ, Hao J, Zhang W. IGFBP2 Activates the NF-κB Pathway to Drive Epithelial-Mesenchymal Transition and Invasive Character in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2016. November 15;76(22):6543–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McCaffery I, Tudor Y, Deng H, Tang R, Suzuki S, Badola S, Kindler HL, Fuchs CS, Loh E, Patterson SD, Chen L, Gansert JL. Putative predictive biomarkers of survival in patients with metastatic pancreatic adenocarcinoma treated with gemcitabine and ganitumab, an IGF1R inhibitor. Clin Cancer Res. 2013. August 1;19(15):4282–9. [DOI] [PubMed] [Google Scholar]
- [26].Kendrick ZW, Firpo MA, Repko RC, Scaife CL, Adler DG, Boucher KM, Mulvihill SJ. Serum IGFBP2 and MSLN as diagnostic and prognostic biomarkers for pancreatic cancer. HPB (Oxford). 2014. July;16(7):670–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ansari D, Aronsson L, Sasor A, Welinder C, Rezeli M, Marko-Varga G, Andersson R. The role of quantitative mass spectrometry in the discovery of pancreatic cancer biomarkers for translational science. J Transl Med. 2014. April 5;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen R, Brentnall TA, Pan S, Cooke K, Moyes KW, Lane Z, Crispin DA, Goodlett DR, Aebersold R, Bronner MP. Quantitative proteomics analysis reveals that proteins differentially expressed in chronic pancreatitis are also frequently involved in pancreatic cancer. Mol Cell Proteomics. 2007. August;6(8):1331–42. [DOI] [PubMed] [Google Scholar]
- [29].Liu Y, Song C, Shen F, Zhang J, Song SW. IGFBP2 promotes immunosuppression associated with its mesenchymal induction and FcγRIIB phosphorylation in glioblastoma. PLoS One. 2019. September 27;14(9):e0222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chua CY, Liu Y, Granberg KJ, Hu L, Haapasalo H, Annala MJ, Cogdell DE, Verploegen M, Moore LM, Fuller GN, Nykter M, Cavenee WK, Zhang W. IGFBP2 potentiates nuclear EGFR-STAT3 signaling. Oncogene. 2016. February 11;35(6):738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li T, Zhang C, Zhao G, Zhang X, Hao M, Hassan S, Zhang M, Zheng H, Yang D, Liu L, Mehraein-Ghomi F, Bai X, Chen K, Zhang W, Yang J. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020. May 1;477:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scholz A, Heinze S, Detjen KM, Peters M, Welzel M, Hauff P, Schirner M, Wiedenmann B, Rosewicz S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology. 2003. September;125(3):891–905. [DOI] [PubMed] [Google Scholar]
- [33].Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003. January 23;22(3):319–29. [DOI] [PubMed] [Google Scholar]
- [34].Nagaraj NS, Washington MK, Merchant NB. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin Cancer Res. 2011. February 1;17(3):483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nam S, Wen W, Schroeder A, Herrmann A, Yu H, Cheng X, Merz KH, Eisenbrand G, Li H, Yuan YC, Jove R. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol. 2013. June;7(3):369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka TA, Yamamoto K, Kato K, Nakamura Y, Akira S, Takeda K, Kajimoto Y, Yamasaki Y, Sandgren EP, Kawaguchi Y, Wright CV, Fujitani Y. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006. June 1;20(11):1435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011. July 15;71(14):5020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Toyonaga T, Nakano K, Nagano M, Zhao G, Yamaguchi K, Kuroki S, Eguchi T, Chijiiwa K, Tsuneyoshi M, Tanaka M. Blockade of constitutively activated Janus kinase/signal transducer and activator of transcription-3 pathway inhibits growth of human pancreatic cancer. Cancer Lett. 2003. November 10;201(1):107–16. [DOI] [PubMed] [Google Scholar]
- [39].Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007. January;7(1):41–51. [DOI] [PubMed] [Google Scholar]
- [40].Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005. October 15;65(20):9525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006. September 1; 108(5): 1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kong LY, Abou-Ghazal MK, Wei J, Chakraborty A, Sun W, Qiao W, Fuller GN, Fokt I, Grimm EA, Schmittling RJ, Archer GE Jr, Sampson JH, Priebe W, Heimberger AB. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. 2008. September 15;14(18):5759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004. January 1;172(1):464–74. [DOI] [PubMed] [Google Scholar]
- [44].Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005. December;11(12):1314–21. [DOI] [PubMed] [Google Scholar]
- [45].Kang Y, Zhang R, Suzuki R, Li SQ, Roife D, Truty MJ, Chatterjee D, Thomas RM, Cardwell J, Wang Y, Wang H, Katz MH, Fleming JB. Two-dimensional culture of human pancreatic adenocarcinoma cells results in an irreversible transition from epithelial to mesenchymal phenotype. Lab Invest. 2015. February;95(2):207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Leblond MM, Gérault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, Valable S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology. 2015. June 5;5(1):e1056442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kimura T, Nada S, Takegahara N, Okuno T, Nojima S, Kang S, Ito D, Morimoto K, Hosokawa T, Hayama Y, Mitsui Y, Sakurai N, Sarashina-Kida H, Nishide M, Maeda Y, Takamatsu H, Okuzaki D, Yamada M, Okada M, Kumanogoh A. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat Commun. 2016. October 12;7:13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roszer T, Menéndez-Gutiérrez MP, Lefterova MI, Alameda D, Núñez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011. January 1;186(1):621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016. January 11;213(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Giampietri C, Petrungaro S, Conti S, Facchiano A, Filippini A, Ziparo E. Cancer Microenvironment and Endoplasmic Reticulum Stress Response. Mediators Inflamm. 2015;2015:417281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MC, Ahmed M, Hoeksema MA, de Vos AF, de Winther MP. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016. October 11;17(3):684–696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. IGFBP2 promotes PDAC progression in a macrophage dependent manner.
(A) The IGFBP2-overexpressing (Panc02-IGFBP2) and empty vector-transfected control (Panc02-EV) cell lines were established with Panc02 murine pancreatic cell line. Whole-cell extracts were subjected to immunoblotting for IGFBP2. (B) Experimental scheme for orthotropic PDAC model in C57BL/6 mice. Mice were divided into two groups and implanted with IGFBP2-overexpressing (IGFBP2) or empty vector-transfected control (EV) Panc02 cells. (C) After 28 days, mice were sacrificed and tumors were harvested. The tumor weight (D) and liver tumor nodules (E) were measured (n=10; **P<0.01 by Student’s t-test). (F) The same orthotropic pancreatic carcinoma model was repeated, and the overall survival rates were analyzed by Kaplan-Meier curves (n=20 per group; P=0.008 by log-rank test). (G) Flow cytometry analysis of M2/M1 TAMs ratio in PDAC microenvironment from both groups of C57BL/6 mice (F4/80+/CD206− cells as M1 TAMs and F4/80+/CD206+ cells as M2 TAMs). Flow cytometry analysis of CD4+CD25+FOXP3+ Tregs (H), CD8+CD45+ T cells (I) and apoptotic CD8+ T cells (J) in PDAC microenvironment from both groups of C57BL/6 mice (n=10 per group; **P<0.01 by Student’s t-test). Data represent the mean ± SD of at least 3 independent experiments.
Supplemental Figure S2. IGFBP2 promotes IL-10 secretion in human PDAC cells in a non-NF-κB-dependent manner.
(A) High endogenous IGFBP2 MDA-PATC53 cells or IGFBP2-overexpressing PATC148 BP2 were transfected with siRNA negative control (siR-ctrl) or one of two different p65 siRNAs (siR-p65-1 and siR-p65-2) to knock down p65. Whole-cell extracts were subjected to immunoblotting for p65 and p-p65. (B) The expression levels of IL10 were detected by qRT-PCR and normalized to the endogenous control GAPDH. The secretion of IL-10 was detected by ELISA. (C) Human monocytes were differentiated into nonpolarized M0 macrophages and then polarized into M1 or M2 macrophages by conditioned medium (CM) from PDAC cell lines differentially expressing p65. The expression levels of M1 and M2 macrophage phenotypic genes for p65 experimentally polarized macrophages were detected by qRT-PCR and normalized to the endogenous control GAPDH. (NS, not statistically significant by ANOVA). Data represent the mean ± SD of at least 3 independent experiments.
Supplemental Table S1. Information for the 91 patients from the ICGC PADC-AU cohort, and their expression levels (FPKM) of relative genes.