

Abstract
The sequences of SARS-CoV-2 spike (S) from Saudi Arabia along with SARS-CoV and bat SARS-like CoVs were obtained. Positive selection analysis and secondary structure investigation of spike sequences were performed. Adaptive molecular evolution was observed in SARS-CoV-2 displayed by positive selection pressure at N-terminal domain (NTD; codons 41, 163, 174 and 218), Receptor binding domain (RBD; codons 378 and 404) and S1/S2 Cleavage site (codon 690). Furthermore, the spike protein secondary structure depicted by the homo-trimer structure showed a high similarity between Saudi SARS-CoV-2 isolate and the parental strain (bat SL-COVZC45). Despite the high similarity depicted in the spike sequence model alignment, it displayed a significant difference when each chain was treated solely owing to 7 motif differences in the three composing chains. In addition, SARS-CoV-2 S trimer model uncovered the presence of N-acetyl glucosamine ligands. Eventually, 3C-like proteinase cleavage site was observed in S2 domain could be used as a site for drug discovery. Genetics and molecular evolutionary facts are useful for assessment of evolution, host adaptation and epidemic patterns ultimately helpful for adaptation of control strategies.
Keywords: SARS-CoV-2, Saudi Arabia, Spike, Selection pressure, Secondary structure, Cleavage site
1. Introduction
Novel Coronavirus is termed as Severe Acute Respiratory Syndrome (SARS-CoV-2) originated from Hubei, china in December 2019 and a vast spread was observed throughout the world (Wang et al., 2020). This infectious disease is named as COVID-19 and human to human transmission has been established (Chanet al., 2020). The disease symptoms depicted in SARS-CoV-2 infections were found similar to the infections caused by SARS coronavirus (SARS-CoV) in 2003 (Peiriset al., 2004). Coronaviruses are enveloped, non-segmented, positive sense single stranded RNA viruses with genome size of 26 kb to 32 kb, responsible for respiratory diseases in various animals as well as human beings. Human coronaviruses like SARS-CoV, MERS-CoV and SARS-CoV-2 are zoonotic pathogens (Chenet al., 2020). Previous sequence analyses showed high percentage of similarity among SARS-CoV-2, SARS-CoV and Bat corona viruses (Luet al., 2020). In addition, coronaviruses are found in a wide range of animal hosts like bats, camels, cats, mice and dogs (Tennantet al., 1993).
Once the COVID-19 causing agent was identified in January 2020, SARS-CoV-2 nucleotides sequence alignments were performed to clarify the origin and the incident virus evolution as well as the prediction of any probable intermediate host. Coronaviruses contain mainly four types of structural and nearly 16 types of non-structural proteins. Spike protein is a principal structural protein that mediates recognition and attachment of coronaviruses to the host cell receptor termed as angiotensin-converting enzyme 2 (Li et al., 2020, Donnelly et al., 2004). SARS-CoV-2 genome analysis depicted a similarity index of 79.5% with SARS-CoV and 96% resemblance with bat coronavirus (Chen et al., 2020). Sequence alignments of coronaviruses provide information with respect to the genetic characteristics of different viruses and sequence dependent data can be employed for accurate diagnostic purposes of etiological agents and adaptation of effective control measures. Super spreading of COVID-19 has been reported globally and high incidence rates are recorded in multiple regions of the world (World Health Organization, 2020). The growing number of infections over the times may result in emergence of several different types of variants due to mutations and recombination. So, genome sequence tracking and characterization is important to analyze different variants. Emergence of SARS-CoV-2 as global pandemic not only affected the human health but also drastically affected the global economy. Furthermore, analysis of mutations occurring in the S sequence representing the most frequently variable region of SARS-CoV-2 sequences will help us to understand their high inter-human transmissibility and evolution patterns of corona viruses. The data generated will be helpful for development of effective strategies to deal with existing and future epidemics.
2. Methods
2.1. Sequences gathering
The GISAID Epiflu Database comprises a COVID-19 related page (https://www.epicov.org/), where COVID-19 genome sequences are accessible. The current research was intended to compare Saudi Arabia SARS-CoV-2 spike sequences to that of the previously occurring SARS-CoV and bat-like SARS CoV. Thus, only three submitted sequences of Saudi Arabia SARS-CoV-2 were used. In addition, 7 bat SARS-COV sequences collected from 2011 to 2017 and two human SARS-CoV sequences were added from NCBI GenBank. Accession number, location and collection dates are shown in table 1.
Table 1.
List of genome sequences used in phylogenetic analysis.
| Accession Number |
Sequence name | Data Source |
Strain location | Date of collection |
|---|---|---|---|---|
| EPI_ISL_416432 | hCoV-19/Saudi Arabia/KAIMRC- Alghoribi/2020 | GISAID | Riyadh/Saudi Arabia | 3/7/2020 |
| EPI_ISL_416521 | hCoV-19/Saudi Arabia/SCDC-3321/2020 |
GISAID | Riyadh/Saudi Arabia |
3/10/2020 |
| EPI_ISL_416522 | hCoV-19/Saudi Arabia/SCDC-3324/2020 |
GISAID | Riyadh/Saudi Arabia |
3/10/2020 |
| AVP78031.1 | bat-SL-CoVZC45 | NCBI | Zhoushan city/Zhejiang province/China |
2/2017 |
| AID16716.1 | bat-SL-CoV_Longquan- 140 |
NCBI | Guizhou province/China |
2012 |
| ATO98218.1 | bat-SL-CoV_Rs7327 | NCBI | Yunnan Province/China |
10/24/2014 |
| ATO98145.1 | bat-SL-CoV_Rf4092 | NCBI | Yunnan Province/China |
9/18/2012 |
| ATO98108.1 | bat-SL-CoV_As6526 | NCBI | Yunnan Province/China |
5/12/2014 |
| AGZ48806.1 | bat-SL-CoV_RsSHC014 | NCBI | Yunnan Province/China |
4/17/2011 |
| AKZ19087.1 | bat-SL-CoV_YNLF_34C | NCBI | China | 5/23/2013 |
| AY278487.3 | Hu-SARS-CoV_BJ02 | NCBI | China | 6/5/2003 |
| AAP51227.1 | Hu-SARS-CoV_GD01 | NCBI | China | 6/5/2003 |
2.2. Single likelihood ancestor counting (SLAC)
To investigate this adaptive evolution, SLAC method was used to ensure the selection pressures acting on the COVID-19 S gene. Global non-synonymous to synonymous (dN/dS) rate ratios were estimated with the SLAC method (Kosakovsky Pond and Frost, 2005) in Datamonkey (http://www.datamonkey.org). Briefly, the alignment sequences were uploaded to a full automated workflow-dependent Datamonkey that recognized codons and lineages under selection even when recombinant sequences existed. Duplicated sequences were removed prior to analysis execution (Spielman et al., 2019).
2.3. Secondary structure investigation of spike protein
To comprehend the COVID-19 structure, comprising any possible deviation from a previously emerging related SARS-CoV as well as the detection of distinctive residues orientation that could be involved in target binding, the COVID-19 spike glycoprotein homo-trimer structure (target) was modeled via SWISS-MODEL (https://swissmodel.expasy.org/) (Waterhouse et al., 2018) against the structure of bat-SL-COVZC45 (template). Moreover, the obtained model for both was used for alignment and comparison using CLC Main Workbench V20.0 (QIAGEN).
2.4. Investigation of 3C-like proteinase cleavage sites
Coronaviruses have 3C or 3C-like proteases (3Cpro or 3CLpro, respectively), that comprise a standard chymotrypsin-like fold and a catalytic triad with a Cys residue as a nucleophile (12). These 3Cpro or 3CLpro, which contain conserved principle sites, may act as attractive targets for the design of essential antivirals for multiple viruses in the supercluster. Thus, 3C-like proteinase cleavage sites were investigated using a server (https://services.healthtech.dtu.dk/) that includes NetCorona service. NetCorona 1.0 predicts coronavirus 3C-like proteinase (or protease) cleavage sites using artificial neural networks on amino acid sequences (Kiemer et al., 2004).
3. Results
To investigate this apparent transition further, the single likelihood ancestor counting (SLAC) method was implemented to compare the selection pressures acting on the SARS-C0V-2 Spike sequence. It was observed that selection pressure was positive (dN/dS = 3.3981), occurring at 7 codons with p value threshold of 0.1 at codons 41, 163, 174, 218, 378, 404 and 690 and all sites were parsimony informative where minimum of two dissimilar nucleotides existed at least twice (Table 2). Four of them were at the N-terminal domain (NTD) of S1 subunit causing non-conservative missense mutation, involving F41C (p = 0.041), N163Y (p = 0.022), F174I (p = 0.032) and H218P (p = 0.083) owing to substitution mutation that mostly occurred by transversion (T > G, A > T and A > C at sites 41, 163 and 218, respectively), however a single transition mutation occurred at 174 (T > C). Moreover, two positive selection occurred at the receptor-binding domain (RBD) of S1 subunit leading to a single non-conservative missense mutation (C378L, p = 0.092) and a silent mutation (L404L, p = 0.054). However, the last positive selection pressure occurred at the S1/S2 cleavage site resulting in a conservative missense mutation (S690C, p = 0.098) owing to a AGC > GCC transition substitution at the first nucleotide (Fig. 1).
Table 2.
Sites showing signatures of positive selection in S as determined by dN/dS analysis.
| Codon | Partition | S | N | dS | dN | Selection detected? |
|---|---|---|---|---|---|---|
| 41 | 1 | 0.000 | 8.000 | 0.000 | 3.970 | Pos. p = 0.041 |
| 163 | 1 | 0.000 | 11.000 | 0.000 | 6.022 | Pos. p = 0.022 |
| 174 | 1 | 0.000 | 9.000 | 0.000 | 4.428 | Pos. p = 0.032 |
| 218 | 1 | 0.000 | 7.000 | 0.000 | 3.355 | Pos. p = 0.083 |
| 378 | 1 | 0.000 | 6.000 | 0.000 | 2.976 | Pos. p = 0.092 |
| 404 | 1 | 0.000 | 11.000 | 0.000 | 5.224 | Pos. p = 0.054 |
| 690 | 1 | 0.000 | 6.000 | 0.000 | 2.948 | Pos. p = 0.098 |
Fig. 1.


Amino acid sequence alignment of Saudi Arabia SARS-CoV-2 isolates, bat SARS-like CoV isolates and SARS-CoV isolates. The red arrows represent the sites of positive selection. The blue arrows display the S1/S2 cleavage site.
To better understand the structure of COVID-19 spike, including the deviation from a previously emerging related SARS-like CoV that was observed to be a probable major parental strain as obtained from RDP analysis, hCoV-19/Saudi Arabia/KAIMRC-Alghoribi (used as the Saudi hCoV- 19 representative) and Bat-SL-COVZC45 (the major parent) spike sequences were translated and used for examining the best templates based on target-template alignment features. Consequently, the templates with the highest quality have then been selected for model building to construct the COVID-19 spike glycoprotein homo-trimer structure (Fig. 2A) as well as that of Bat-SL-COVZC45 (Fig. 2B). The model was found to consist of 3 Chains (A, B and C). Full model alignment (Fig. 2.C) have revealed a high similarity, however when the 3 chains were aligned and compared solely, it displayed a significant difference owing to two motif differences (amino acids 151–277 and 451–536) in Chain A (Fig. 2.D), two motif differences (amino acids 202–227, 361–448) in chain B (Fig. 2.E) and three motif differences (amino acids 148–170, 243–264 and 441–480) in chain C (Fig. 2F). Moreover, SWISS-model has displayed the presence of N-acetyl glucosamine ligands in chain A (Fig. 3A), Chain B (Fig. 3B) and for interaction with Chain B (Fig. 3C) as well as Chain C (Fig. 3D) of hCoV-19/Saudi Arabia/KAIMRC-Alghoribi (Fig. 3).
Fig. 2.


Spike glycoprotein homo-trimer structure modeling. SWISS model was used to get the best template and the fitting model (GQME = 0.75, QMEAN = -2.07, 0.94 coverage, 95% identity for hCoV-19/SA/KAIMRC-Alghoribi and GQME = 0.78, QMEAN = -3.87, 0.94 coverage and 76.73% identity for bat-ST-COVZC45) followed by importing the model to QIAGEN CLC Main Workbench V20.0 to obtain the final homo-trimer of (A) hCoV-19/SA/KAIMRC-Alghoribi and (B) bat-ST-COVZC45 and (C) models alignment. Three chains were observed for each of hCoV- 19 and bat-SL-CoV, thus each chain was aligned on basis of their amino acid sequence and their 3D structure, involving (D) Chain A, (E) Chain B and (F) Chain C.
Fig. 3.

N-acetyl glucosamine ligands linked to (A) Chain A, (B) Chain B, and interacting with (C) Chain B and (D) Chain C, resolved by using SWISS model.
Moreover, 3C-like proteinase cleavage site (TGRLQ^SLQTY) was recognized at amino acids 992–1002 in the spike glycoprotein of SARS-CoV-2 (Fig. 4) that is needed for cell–cell fusion and consequently could be used as a target site for blocking to cease viral infection.
Fig. 4.
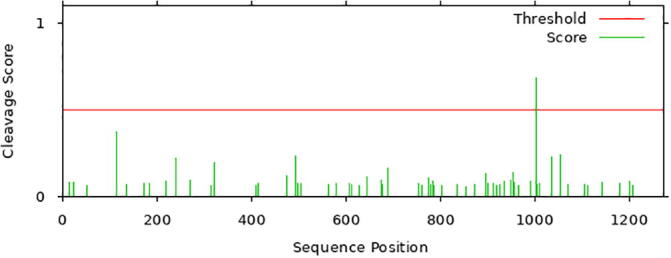
Predicted 3C-like proteinase cleavage site in SARS-CoV-2 Spike glycoprotein.
4. Discussion
Our knowledge is limited about SARS-COV-2 regarding basic and intermediate host species, evolution and genetic variation in relation to other corona viruses like MERS-COV and SARS-COV. Genetic map analysis of these corona viruses can help us to understand the evolutionary process and may be helpful to understand and control the ongoing epidemic of SARS-COV-2.
The virus is spreading globally and with increased number of infections, virus evolutionary rate should be considered with significant priority. Further studies are needed from more closely related animal and human viruses and especially the divergence in host tropism. The S protein mediates both receptor binding and membrane fusion (Li, 2016) and is crucial for defining host tropism and transmissibility patterns (Luet al., 2015). Therefore, the current study was focused on inspection of S sequences rather than the whole genome. Positively selected sites were found to be located in NTD and RBD of SARS-CoV-2 (Tagliamonte et al., 2020). The current study observed positive selection at NTD, RBD and S1/S2 cleavage site of Saudi SARS-CoV-2 isolates which approves the previous study. These positive selection residues and most especially in RBD that was observed in our study to result in non-conservative missense mutation could lead to higher receptor affinity and consequently higher infectivity and broader transmission pattern as shown in previous literature (Islam et al., 2020, Kaushal et al., 2020, Wrapp et al., 2020). For instance, the U.K SARS-CoV-2 variant B.1.1.7 was depicted to have novel 17 mutations including a notable mutation in the RBD resulting in being 70–80% more transmissible (Kupferschmidt, 2021, Leung et al., 2020).
Moreover, substitution mutation was observed in S1/S2 cleavage site that was recognized as a conserved region elsewhere (Zhanget al., 2020). It has a key function in defining virus infectivity and host range owing to significant cleavage by furin and other proteases (Andersenet al., 2020). Follis, et al observed that furin cleavage site insertion at the S1/S2 junction boosts cell–cell fusion without influencing virus entry (Folliset al., 2006). Furthermore, effectual MERS-CoV S protein cleavage empowers bat MERS-like coronaviruses to infect humans (Menacheryet al., 2020). Likely, low-pathogenicity avian influenza viruses are converted into highly pathogenic forms upon attainment of polybasic cleavage sites in hemagglutinin, by insertion or recombination (Alexander and Brown, 2009). In addition, these above described positively selected sites were parsimony informative which may have affected the resultant protein. Ghosh and Chakraborty detected 28 parsimony-informative sites out of 119 variable regions in 82 SARS-CoV2 genomes resulting in 79 variable sites occurred in the yielded proteins (Ghosh and Chakraborty, 2020).
Moreover, the spike protein secondary structure depicted by the homo-trimer structure showed a high similarity between Saudi SARS-CoV-2 isolate and the major parental strain (bat SL- COVZC45). However, on the individual chain sub-level, there was an observed difference due to 7 motif differences in the 3 chains composing the homo-trimer structure. Notably, the homo- trimer structure of SARS-CoV-2 displays an open conformation of S1 domains, which allows better interaction with target host proteins, which is in line with a recent study (Vankadari and Wilce, 2020). Since this homo-trimer glycoprotein is surface-exposed and consequently facilitates viral entry, it is considered as a crucial target for the neutralizing antibodies (Abs) upon infection. Therefore, S trimers are intensively decorated with N-linked glycans that is essential for carrying out appropriate folding (Wallset al., 2020) and for moderating accessibility to host proteases needed for entry and cell–cell fusion and neutralizing Abs (Walls et al., 2019, Xiong et al., 2018, Yang et al., 2015). This further approves the present study findings that showed the presence of N-acetyl glucosamine ligands in the three chains composing SARS-CoV-2 S trimer.
On the other hand, the current study determined a 3C-like proteinase cleavage site in S2 domain of spike glycoprotein which approves another study elsewhere (Vankadari and Wilce, 2020). Moreover, this observed 3C-like proteinase cleavage site can be used as a potential drug discovery site (Grum-Tokars et al., 2008) in the same manner as presently being suggested for SARS-CoV-2 (Rothan and Teoh, 2021). In this regard, boceprevir and calpain inhibitor XII were recently reported as 3CLpro selective inhibitors (Liuet al., 2021).
Conclusively, analysis of three Saudi hCoV-19 and seven representative Bat SARS-like CoVs, and 2 human SARS-CoV describes about the virus origin, selection pressures and receptor binding properties as well as possible target for drug discovery. Nonetheless, efficient strategies based on continuous screening of SARS-CoV-2 mutations and associated structural influence analysis could assist in earlier vaccines efficacy re-evaluation and development if needed (Mehmood et al., 2021, Plante et al., 2020). Genetics and molecular evolutionary facts are considered as useful tools to understand the epidemic pattern and transmission dynamics which are ultimately useful to adopt preventive measures.
CRediT authorship contribution statement
Islam Nour: Conceptualization, Formal analysis, Investigation, Methodology, Validation, Writing - original draft, Writing - review & editing. Ibrahim O. Alanazi: Conceptualization, Formal analysis, Investigation, Methodology, Validation, Writing - review & editing. Atif Hanif: Conceptualization, Funding acquisition, Writing - original draft, Writing - review & editing. Saleh Eifan: Conceptualization, Data curation, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
| Isolate ID. | Country. | Collection.date. | Isolate name. | Originating. Laboratory. |
Submitting. Laboratory. |
Authors. |
| EPI_ISL_416521. | Saudi Arabia. | 2020-03-10. | hCoV-19/Saudi Arabia/SCDC-3321/2020. | Public Health Laboratory. | Public Health Laboratory, Saudi CDC. | Albarrag, A. |
| EPI_ISL_416522. | Saudi Arabia. | 2020-03-10. | hCoV-19/Saudi Arabia/SCDC-3324/2020. | Public Health Laboratory Saudi CDC. | Public Health Laboratory Saudi CDC. | Albarrag, A. |
| EPI_ISL_416432. | Saudi Arabia. | 2020-03-07. | hCoV-19/Saudi.Arabia/KAIMRC- Alghoribi/2020. | Clinical Microbiology Lab. | Infectious Disease Research Department, King Abdullah International Medical Research Center (KAIMRC) | Majed Alghoribi, Sadeem Alhayli, Abdulrahman Alswaji, Liliane Okdah, Sameera Al Johani, Michel Doumith. |
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no. (RG-1441-492).
We gratefully acknowledge the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu™ Database on which this research is based. The list is detailed below.
Footnotes
Peer review under responsibility of King Saud University.
References
- Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395(10223):470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.W., Yuan S., Kok K.H., To K.K.W., Chu H., Yang J., Xing F., Liu J., Yip C.C.Y., Poon R.W.S., Tsoi H.W. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S.M., Guan Y., Yuen K.Y. Severe acute respiratory syndrome. Nat. Med. 2004;10(12):S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant B.J., Gaskell R.M., Jones R.C., Gaskell C.J. Studies on the epizootiology of canine coronavirus. Vet. Rec. 1993;132(1):7. doi: 10.1136/vr.132.1.7. [DOI] [PubMed] [Google Scholar]
- Donnelly C.A., Fisher M.C., Fraser C., Ghani A.C., Riley S., Ferguson N.M., Anderson R.M. Epidemiological and genetic analysis of severe acute respiratory syndrome. Lancet. Infect. Dis. 2004;4(11):672–683. doi: 10.1016/S1473-3099(04)01173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Qiao S., Zhang G. Analysis of angiotensin-converting enzyme 2 (ACE2) from different species sheds some light on cross-species receptor usage of a novel coronavirus 2019-nCoV. J. Infect. 2020;80(4):469–496. doi: 10.1016/j.jinf.2020.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Frost S.D. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22(5):1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Spielman S.J., Weaver S., Shank S.D., Magalis B.R., Li M., Pond S.L.K. Evolutionary Genomics. Humana; New York, NY: 2019. Evolution of viral genomes: interplay between selection, recombination, and other forces; pp. 427–468. [DOI] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., Lepore R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiemer L., Lund O., Brunak S., Blom N. Coronavirus 3CL pro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinf. 2004;5(1):72. doi: 10.1186/1471-2105-5-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annual Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G., Wang Q., Gao G.F. Bat-to-human: spike features determining ‘host jump’of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015;23(8):468–478. doi: 10.1016/j.tim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliamonte, M.S., Abid, N., Chillemi, G., Salemi, M. and Mavian, C., 2020. Re-insights into origin and adaptation of SARS-CoV-2. bioRxiv.
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M.R., Hoque M.N., Rahman M.S., Alam A.R.U., Akther M., Puspo J.A., Akter S., Sultana M., Crandall K.A., Hossain M.A. Genome-wide analysis of SARS-CoV- 2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020;10(1):1–9. doi: 10.1038/s41598-020-70812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N., Gupta Y., Goyal M., Khaiboullina S.F., Baranwal M., Verma S.C. Mutational frequencies of SARS-CoV-2 genome during the beginning months of the outbreak in USA. Pathogens. 2020;9(7):565. doi: 10.3390/pathogens9070565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K. Fast-spreading UK virus variant raises alarms. Science. 2021:9–10. doi: 10.1126/science.371.6524.9. [DOI] [PubMed] [Google Scholar]
- Leung, K., Shum, M.H., Leung, G.M., Lam, T.T. and Wu, J.T., 2020. Early empirical assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. medRxiv. [DOI] [PMC free article] [PubMed]
- Zhang T., Wu Q., Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020 doi: 10.1016/j.cub.2020.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26(4):450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis K.E., York J., Nunberg J.H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology. 2006;350(2):358–369. doi: 10.1016/j.virol.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Dinnon K.H., Yount B.L., McAnarney E.T., Gralinski L.E., Hale A., Graham R.L., Scobey T., Anthony S.J., Wang L., Graham B. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J. Virol. 2020;94(5) doi: 10.1128/JVI.01774-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander D.J., Brown I.H. History of highly pathogenic avian influenza. Revue scientifique et technique (Int. Office Epizoot.). 2009;28(1):19–38. doi: 10.20506/rst.28.1.1856. [DOI] [PubMed] [Google Scholar]
- Ghosh, S. and Chakraborty, S., 2020. Phylogenomics Analysis of SARS-CoV2 Genomes Reveals Distinct Selection Pressure on Different Viral Strains. BioMed research international, 2020. [DOI] [PMC free article] [PubMed]
- Vankadari N., Wilce J.A. Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020;9(1):601–604. doi: 10.1080/22221751.2020.1739565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020 doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Xiong X., Park Y.J., Tortorici M.A., Snijder J., Quispe J., Cameroni E., Gopal R., Dai M., Lanzavecchia A., Zambon M. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell. 2019;176(5):1026–1039. doi: 10.1016/j.cell.2018.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Tortorici M.A., Snijder J., Yoshioka C., Walls A.C., Li W., McGuire A.T., Rey F.A., Bosch B.J., Veesler D. Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J. Virol. 2018;92(4) doi: 10.1128/JVI.01628-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liu C., Du L., Jiang S., Shi Z., Baric R.S., Li F. Two mutations were critical for bat-to-human transmission of Middle East respiratory syndrome coronavirus. J. Virol. 2015;89(17):9119–9123. doi: 10.1128/JVI.01279-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133(1):63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan H.A., Teoh T.C. Cell-Based High-Throughput Screening Protocol for Discovering Antiviral Inhibitors Against SARS-COV-2 Main Protease (3CLpro) Mol. Biotechnol. 2021;19:1–9. doi: 10.1007/s12033-021-00299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Boland S., Scholle M.D., Bardiot D., Marchand A., Chaltin P., Blatt L.M., Beigelman L., Symons J.A., Raboisson P., Gurard-Levin Z.A. Dual inhibition of SARS-CoV-2 and human rhinovirus with protease inhibitors in clinical development. Antiviral Res. 2021 doi: 10.1016/j.antiviral.2021.105020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood, I., Ijaz, M., Ahmad, S., Ahmed, T., Bari, A., Abro, A., Allemailem, K.S., Almatroudi, A. and Tahir ul Qamar, M., 2021. SARS-CoV-2: An Update on Genomics, Risk Assessment, Potential Therapeutics and Vaccine Development. International Journal of Environmental Research and Public Health, 18(4), p.1626. [DOI] [PMC free article] [PubMed]
- Plante J.A., Liu Y., Liu J., Xia H., Johnson B.A., Lokugamage K.G., Zhang X., Muruato A.E., Zou J., Fontes-Garfias C.R., Mirchandani D. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020:1–6. doi: 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, 2020. Novel Coronavirus (2019-nCoV): situation report, 3.