

Abstract
INTRODUCTION:
Variability exists in the disease trajectories of Alzheimer’s disease (AD) patients. We performed a genome-wide association study to examine rate-of-cognitive-decline (ROD) in AD patients.
METHODS:
We tested for interactions between genetic variants and time since diagnosis to predict the ROD of a composite cognitive score in 3,946 AD cases and performed pathway analysis on the top genes.
RESULTS:
Suggestive associations (p<1.0×10−6) were observed on chromosome 15 in DNA polymerase-γ (rs3176205, p=1.11×10−7), chromosome 7 (rs60465337, p=4.06×10−7) in contactin-associated protein-2, in RP11–384F7.1 on chromosome 3 (rs28853947, p=5.93×10−7), and family with sequence similarity 214 member-A on chromosome 15 (rs2899492, p=5.94×10−7), and intergenic regions on chromosomes 16 (rs4949142, p=4.02×10−7), and 4 (rs1304013, p=7.73×10−7). Significant pathways involving neuronal development and function, apoptosis, memory, and inflammation were identified.
DISCUSSION:
Pathways related to AD, intelligence, and neurological function determine AD progression, while previously identified AD risk variants, including APOE, do not have a major impact.
1. Introduction
Alzheimer ‘s disease (AD) is characterized by progressive cognitive deterioration and substantial variability exists in the cognitive trajectories of affected individuals. Several studies have examined factors associated with cognitive decline in non-demented adults1–9, conversion from mild cognitive impairment (MCI) to AD10–18 and rate of decline (ROD) after AD diagnosis19–26. Several non-genetic determinants of decline, including lifestyle factors, biomarkers and biometric variables, and co-morbid diagnoses have also been reported.27–37,26,38.
Several genetic and epigenetic factors that potentially contribute to ROD in AD cases have also been identified. Expression levels of leucine rich repeat and fibronectin type III domain containing 2 (LRFN2)39, beta-nerve growth factor (β-NGF) and its receptors40, myocyte-enhancer factor 2C (MEF2C)41, and inositol polyphosphate-5-phosphatase (INPP5D)42 have been associated with ROD in brains of people with AD. Epigenetic protein dysregulation has also been implicated in AD progression43,44. A recent study identified 519 proteins whose abundance were associated with cognitive trajectory in adults without dementia at baseline. These proteins were enriched in pathways related to neuronal mitochondrial activities synaptic abundance, inflammation, and apoptosis45. Two studies have examined the role of AD risk genes in cognitive decline and found specific SNPs and polygenic risk scores predict ROD in older, non-demented individuals46,47. A different study also found that the AD risk variants or polygenic risk scores do not affect ROD in AD individuals26. Genetic association studies identified two SNPs in astrocytic water channel aquaporin-4 (AQP4) that predict ROD48, and a genome-wide association study (GWAS) identified variants in six genes that interacted with AD diagnosis (vs MCI) to predict longitudinal cognitive change49. We previously conducted a GWAS for ROD using AD cases from the Alzheimer’s Disease Neuroimaging Initiative (ADNI, N=303) and cases from the Religious Orders Study and Rush Memory and Aging Project (ROS/MAP, N=323) as replication, and identified significant associations with variants in several genes, including F-spondin (SPON1), whose product binds the central domain of the amyloid precursor protein (APP)50. We present here the results from a GWAS of ROD in an expanded sample of 3,946 AD cases of European ancestry and discuss methodological challenges related to analysis of cognitive data and interaction tests (SNP genotype x time with AD) using longitudinal data.
2. Methods
2.1. Composition of the Data
Eleven cohorts with longitudinal cognitive data and genome-wide SNP data were available for study: ADNI51 (PMID: 23932184), ROS/MAP52,53 the Three City Study (3C)54, AddNeuroMed (ANM)55–57, Myriad Flurizan phase III clinical trial58, National Alzheimer’s Coordinating Centers (NACC)59, Pfizer60, Lilly Semagacestat phase III clinical trial61, Washington University in St. Louis (WashU)26, the Adult Changes in Thought study (ACT)62, and the Washington Heights Inwood Community Aging Project (WHICAP)63. Details about the design, recruitment, and genotyping methods for each cohort are provided in supplemental materials.
2.2. Imputation and Quality Control
Following genotype chip quality control (removal of low call rate SNPs and individuals, individuals with excess heterozygosity, or ambiguous sex) each dataset was phased and imputed to the 1,000 Genomes Project (phase 1 integrated release 3, March 2012)64using SHAPEIT/IMPUTE265,66 or MaCH/Minimac67,68software. All reference population haplotypes were used for the imputation. Rare variants (MAF < 2%) and those with an r2 <0.70 were excluded from further analyses. In the mega-analysis, variants were excluded if they were missing or poorly imputed in > 30% of all samples. King Robust69 was used to generate a kinship coefficient for each pair of individuals using a set of genotyped SNPs common to each cohort (N = 41,625 after LD pruning) using a merged dataset from all eleven cohorts. The member of each related or duplicate pair (kinship coefficient ≥0.1) with the shortest amount of follow-up time was removed. Individuals were assigned ancestry using K-means clustering implemented in R, where K=3 based on the three reference populations (Eur, Afr, Asn) in the 1000 Genomes populations. Individuals were assigned to the cluster. The member of each related pair (kinship coefficient ≥0.1) with the shortest amount of follow-up time was removed. Individuals were assigned ancestry using K-means (K=3) clustering with the 1000 Genomes populations (Eur, Afr, Asn) whose centroid was nearest across the first 10 PCs. Those samples that didn’t cluster with Eur reference population were removed from downstream analysis. Subsequent PC analysis was conducted within cohort and also in the combined sample.
2.3. Composite Cognitive Score
Methods for combining cognitive tests in each cohort and harmonizing them across cohorts to produce a composite indicator of general cognitive performance (GCP) are published elsewhere70. Each study administered at least two and as many as 21 cognitive tests (see68). Briefly, we used item response theory (IRT) methods to derive a measure of general cognitive performance. We first identified common tests across studies (i.e., anchor tests) and tests that were not common. Anchor tests serve to anchor the cognitive metric across studies so that a unit difference in the underlying factor score has the same meaning across study71. Next, we estimated a confirmatory factor analysis, consistent with a graded-response IRT model72,73, of all tests across all studies and time. This approach allows items to be weighted differently, by accommodating different factor loadings. Items also provide measurement at different locations or points along the general cognitive trait depending on how well respondents do on the tests.
2.4. Association Tests
Association tests were performed using two regression-based repeated measures methods. In one approach, linear regression models were solved with generalized estimating equations (GEE) assuming an autoregressive correlation structure with GCP as the outcome. To assess rate of decline rather than levels of cognitive performance, models included a term for the interaction between SNP allele dosage and time since AD diagnosis as the predictor of interest. This construct tests whether SNP genotype modifies the effect of duration of illness on cognitive performance. All models were adjusted for age, sex, ancestry principal components (computed within cohorts for cohort specific analyses and in the total sample for the mega-analyses), the main effects of SNP and time since diagnosis, and squared and cubic terms for time since AD diagnosis which account for any non-linear effects of time since diagnosis on GCP. Analyses were conducted within cohort and in the total sample through fixed effects inverse variance meta-analysis. In another approach, analyzed the total sample using linear mixed effects models including the same interaction term and covariates with random intercepts for individual and cohort. All association tests were performed using Universal Genome Analyst (Koesterer, Ryan. Universal Genome Analyst (uga). https://github.com/rmkoesterer/uga. DOI: 10.5281/zenodo.578712.), which parallelizes tests within the R packages GEEpack (https://CRAN.R-project.org/package=geepack) and LME4 (https://github.com/lme4/lme4/). We limited analyses to cognitive tests performed during the first two years of post-diagnosis follow-up. The top variants were further tested for association with GCP after adjusting for years of education.
2.5. Functional Annotation of Variants
We assessed regulatory potential for genic and intergenic SNPs using the online databases Genotype-Tissue Expression project (GTEx, http://www.gtexportal.org/home/) and BRAINEAC (www.braineac.org) to identify any eQTLs among the top SNPs. All SNPs were annotated using SNPeff which uses data from ENCODE and other sources to assign SNPs to promoter regions, CpG islands, DNAase hypersensitivity sites, and quantifies cross-species conservation and the impact of coding mutations74.
2.6. Pathway Analysis
Genes containing variants with p-values < 1×10−4 (N=334) in at least two models tested were included in an Ingenuity pathway analysis (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis). Only SNPs within introns, exons, and 3’ and 5’ UTRs (according to SNPeff annotation) were considered.
3. Results
After QC, 3,946 AD cases were available for analysis. Table 1 shows characteristics of each cohort, including the mean age at baseline, length of follow-up, and change in GCP during the study period, which we limited to the first two years of follow-up. The interaction terms between time with AD and age (p=0.0001), sex (p=0.02), and education status (p=3.5×10−5) were significantly associated with GCP.
Table 1.
Demographic information by study cohort
| Cohort | Age first visit, μ (SD) | N Males/N Females | Follow-up years, μ (SD) | Change in GCP*, μ (SD) | Change in MMSE* μ (SD) | λ† GEE‡ | λ LME§ |
|---|---|---|---|---|---|---|---|
| 3C | 77.9(5.6) | 139/216 | 6.5(2.5) | 2.8(4.5) | 2.1(3.6) | 1.06 | 1.36 |
| ACT | 78.2(6.1) | 68/106 | 7.7(2.8) | 4.1(5.4) | 1.40(.9) | 1.45 | 1.16 |
| AddNeuroMed | 77.3(6.8) | 110/179 | 1.2(1.0) | 1.5(4.9) | 1.9(3.9) | 1.15 | 3.25 |
| ADNI | 75.7(6.4) | 157/115 | 2.7 (1.4) | 4.8 (5.1) | 2.4(4.6) | 1.11 | 3.18 |
| Flurizan | 74.6(8.2) | 548/533 | 1.2(0.6) | 2.2(7.1) | 3.6(5.0) | 1.05 | 2.26 |
| NACC | 78.3(7.9) | 271/268 | 2.2(1.7) | 6.2(6.2) | 1.8(3.8) | 1.10 | 2.14 |
| Pfizer | 75.7(5.0) | 69/92 | 1.5(2.5) | 2.5(4.3) | 2.0(3.6) | 1.06 | 2.31 |
| ROS/MAP1 | 82.4(6.5) | 120/262 | 7.9(4.5) | 4.5(7.0) | 1.1(3.5) | 1.08 | 3.23 |
| ROS/MAP2 | 82.6(7.7) | 24/55 | 1.4(.5) | 3.5(5.8) | 0.9(3.3) | 1.28 | 2.94 |
| Semagacestat | 74.1(7.7) | 152/155 | 1.3(.5) | 1.5(4.9) | 2.6(4.4) | 1.08 | 10.37 |
| WashU | 74.2(7.5) | 110/129 | 5.7(4.9) | 2.9(4.9) | 1.5(3.3) | 1.14 | 5.37 |
| WHICAP | 82.3(7.9) | 20/48 | 5.0(3.5) | 1.1(6.1) | 1.7(3.8) | 3.42 | 1.63 |
during the first two years of follow up post AD diagnosis,
genomic inflation factor,
Generalized Estimating Equations,
Linear Mixed Effects
3.1. Inflation
Significant inflation (λ) of the genome-wide interaction term test statistics was observed in several cohorts using both LME and GEE. The λ was moderately correlated with sample size. We attempted several approaches to reduce λ, including computing p-values using F and t distributions, setting the degrees of freedom equal to the sample size minus the number of variables in the model, using the Boss R package75, including terms for time with AD squared and cubed, and limiting the follow-up time to two years post diagnosis. Ultimately, none of these steps eliminated inflation, and in three cohorts λ remained above 1.2 (ACT, ROS/MAP2, WHICAP) although inflation was reduced in models including non-linear time with AD terms and limiting to two years of follow-up. Consequently, we corrected all test statistics for the cohort-specific λ and conducted the meta-analyses with and without cohorts with λ > 1.2. Supplementary figures 1–3 show the λ-corrected quintile-quintile plots for both the GEE (meta-analysis with (supplementary figure 1) and without (supplementary figure 2) ACT, ROS/MAP2, WHICAP, and LME (mega-analysis including all cohorts, supplementary figure 3).
3.3. Association Results
Although multiple SNPs and structural variants in several independent regions were significantly associated with ROD in the GEE or LME models including all cohorts, none of these showed evidence in both GEE and LME models nor were they robust to the exclusion of the three cohorts with λ > 1.2. Using these stringent criteria, no SNPs showed genome-wide significance for associations with ROD. Table 2 shows variants (trimmed for LD) with p-values < 1×10−4 in all three models, as well as any gene(s) whose expression was predicted to be significantly altered by the SNP according to the Genotype-Tissue Expression (GTEx) database (https://gtexportal.org/home/)76. Suggestive associations (p<1.0×10−6) were observed in a large region on chromosome 15 spanning several genes including DNA polymerase-γ (POLG) (rs3176205, pLME=1.11×10−7, figure 1), on chromosome 7 (rs60465337, pGEE=4.06×10−7, figure 2) in an intron of contactin-associated protein 2 (CNTNAP2), in the lincRNA RP11–384F7.1 gene region on chromosome 3 (rs28853947, pGEE=5.93×10−7), and family with sequence similarity 214 member A (FAM214A) on chromosome 15 (rs2899492, pGEE=5.94×10−7); and in intergenic regions on chromosomes 16 (rs4949142, pGEE=4.02×10−7), and 4 (rs1304013, pGEE=7.73×10−7). A variant in SPON1 was associated with ROD at (rs200230690, pGEE=2.36×10−5). Fourteen of the SNPs in Table 2 are significant eQTLs, including several predicted to affect POLG expression.
Table 2.
Results with p<1×10−4 in all models tested
| Chr | SNP | A1 * | A2 † | Freq ‡ | Gene | eQTLs § | Beta¶ | PGEE | PGEEλ # | PLME | Direction** |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs61776523 | A | G | 0.86 | Y_RNA | - | −0.51 | 5.99E-05 | 5.97E-05 | 6.28E-05 | +----x----++ |
| 2 | rs62146087 | A | G | 0.62 | CD8B | CD8B, PLGLB1 | 0.40 | 1.96E-05 | 2.47E-05 | 7.49E-05 | +++++++-+x-+ |
| 2 | rs10930401 | T | C | 0.88 | intergenic | METTL5 | −0.49 | 7.00E-05 | 6.85E-05 | 7.81E-05 | -------+--x- |
| 2 | rs11683533 | G | A | 0.63 | intergenic | - | −0.45 | 2.09E-06 | 6.50E-06 | 9.10E-05 | ---------+-- |
| 3 | rs1447793 | G | A | 0.95 | ROBO2 | - | 0.89 | 5.73E-05 | 1.19E-05 | 2.22E-05 | +x+++++x-+xx |
| 3 | rs79279449 | T | C | 0.84 | LSAMP | - | −0.48 | 2.54E-05 | 8.21E-05 | 1.77E-05 | -+--+------x |
| 3 | rs28853947 | C | T | 0.70 | RP11–384F7.1 | - | −0.45 | 2.29E-06 | 5.93E-07 | 8.59E-05 | --------+++x |
| 3 | rs4857800 | T | A | 0.65 | intergenic | - | −0.40 | 7.90E-06 | 1.03E-06 | 8.39E-05 | -----+----++ |
| 4 | rs12501599 | G | C | 0.78 | CLNK | ZNF518B | 0.46 | 1.34E-05 | 7.35E-05 | 2.42E-05 | ++++-+++++++ |
| 4 | rs2168075 | A | G | 0.45 | CCSER1 | CCSER1 | −0.37 | 4.62E-05 | 8.89E-05 | 9.38E-05 | --------+-+- |
| 4 | rs1304013 | C | T | 0.73 | intergenic | - | 0.50 | 4.73E-07 | 8.82E-06 | 2.48E-05 | -++++++x-+++ |
| 6 | rs9393409 | A | G | 0.36 | intergenic | - | −0.39 | 2.70E-05 | 1.81E-05 | 9.17E-05 | -----x++---+ |
| 6 | rs9380681 | T | C | 0.70 | intergenic | - | 0.46 | 1.40E-06 | 1.80E-06 | 3.09E-05 | ++++-++x-+-+ |
| 6 | rs45604140 | C | G | 0.90 | PTK7 | DNPH1, KLHDC3 | −0.54 | 6.07E-05 | 1.98E-05 | 7.15E-06 | -+----+---++ |
| 6 | rs4897203 | T | C | 0.09 | TRDN | - | 0.74 | 1.34E-06 | 2.14E-06 | 9.56E-07 | +++++++-+-++ |
| 7 | rs7806833 | T | G | 0.22 | SCIN | - | −0.46 | 4.99E-05 | 5.64E-05 | 4.44E-05 | ----------+- |
| 7 | rs39437 | G | C | 0.16 | OSBPL3-CYCS | - | 0.51 | 4.68E-06 | 2.15E-06 | 9.70E-05 | ++++++++-++- |
| 7 | rs17150563 | T | C | 0.72 | intergenic | HIBADH, TAX1BP1 | 0.40 | 6.08E-05 | 2.13E-05 | 3.61E-05 | ++-++++x-+-+ |
| 7 | rs7792776 | G | A | 0.87 | intergenic | - | −0.55 | 1.08E-05 | 1.68E-05 | 2.07E-05 | ----------+- |
| 7 | rs6959165 | A | G | 0.45 | HECW1 | - | 0.36 | 2.96E-05 | 1.82E-05 | 1.64E-05 | +++++++x--++ |
| 7 | rs60465337 | C | T | 0.97 | CNTNAP2 | - | 1.03 | 8.59E-07 | 4.06E-07 | 2.86E-05 | ++++++++++x- |
| 8 | rs16877878 | A | G | 0.96 | RP11–566H8.3 | - | 1.18 | 1.13E-05 | 9.35E-05 | 1.33E-05 | x+++++x++++x |
| 10 | rs182768834 | G | A | 0.95 | intergenic | - | −0.87 | 1.60E-06 | 9.96E-06 | 6.31E-05 | ----------x- |
| 11 | rs61897000 | G | A | 0.66 | CHRDL2 | XRRA1 | −0.36 | 7.25E-05 | 1.69E-05 | 6.44E-05 | --+-----+-++ |
| 12 | rs7301894 | A | G | 0.44 | ANO2 | - | −0.34 | 8.59E-05 | 6.21E-05 | 7.54E-05 | -------x+--+ |
| 12 | rs10785192 | T | A | 0.07 | RP11–585P4.5 | RP11–585P4.5, GLIPR1L2 | −0.69 | 2.23E-05 | 5.99E-05 | 1.69E-06 | --+--------x |
| 12 | rs660322 | G | A | 0.24 | TMEM132D | - | 0.53 | 1.07E-05 | 9.44E-06 | 1.14E-05 | x+-+++++-xx+ |
| 15 | rs2899492 | C | T | 0.16 | FAM214A | ARPP19 | 0.62 | 5.94E-07 | 9.70E-07 | 1.37E-05 | ++-++++x+xx+ |
| 15 | rs8041705 | T | C | 0.56 | HMGB1P8 | - | −0.41 | 1.76E-05 | 2.66E-05 | 2.22E-05 | -----x---x+- |
| 15 | rs12324317 | T | C | 0.61 | RLBP1 | RLBP1, POLG | −0.40 | 1.80E-05 | 2.11E-05 | 1.91E-05 | ----+-+x+-+- |
| 15 | rs9788714 | G | A | 0.62 | RLBP1-FANCI | POLG | −0.42 | 2.50E-06 | 1.70E-06 | 1.70E-06 | -----++-+-+x |
| 15 | rs2238301 | A | G | 0.61 | FANCI | POLG | −0.46 | 3.30E-07 | 1.59E-07 | 3.15E-07 | ------+-+-+x |
| 15 | rs3176205 | T | C | 0.61 | POLG | POLG | −0.46 | 2.75E-07 | 1.49E-07 | 1.11E-07 | -----++-+-+x |
| 16 | rs4949142 | A | G | 0.85 | intergenic | - | −0.60 | 5.83E-07 | 4.02E-07 | 7.82E-06 | -------x--x+ |
| 16 | rs12448088 | G | C | 0.40 | PLCG2 | - | −0.41 | 2.91E-05 | 2.36E-05 | 1.10E-05 | +---x----x-+ |
| 17 | rs2071194 | C | A | 0.36 | EVPL | TEN1 | 0.41 | 1.44E-05 | 3.84E-05 | 6.67E-05 | +++++++x-+++ |
effect allele,
other allele,
frequency of effect allele,
Expression Quantitative Trait Locus: genes differentially expressed by SNP genotype according to GTEx database,
beta from GEE model including all cohorts,
p-value from GEE model excluding cohorts with λ>1.2,
effect direction in individual cohorts from the GEE model including all cohorts. The order of the symbols is Pfizer, 3C, ADNI, Flurizan, NACC, ROS/MAP1, Semagacestat, ADNeuroMed, ACT, WashU, WHICAP, ROS/MAP2.
Figure 1. Association results for the region containing DNA polymerase-γ on chromosome 15.
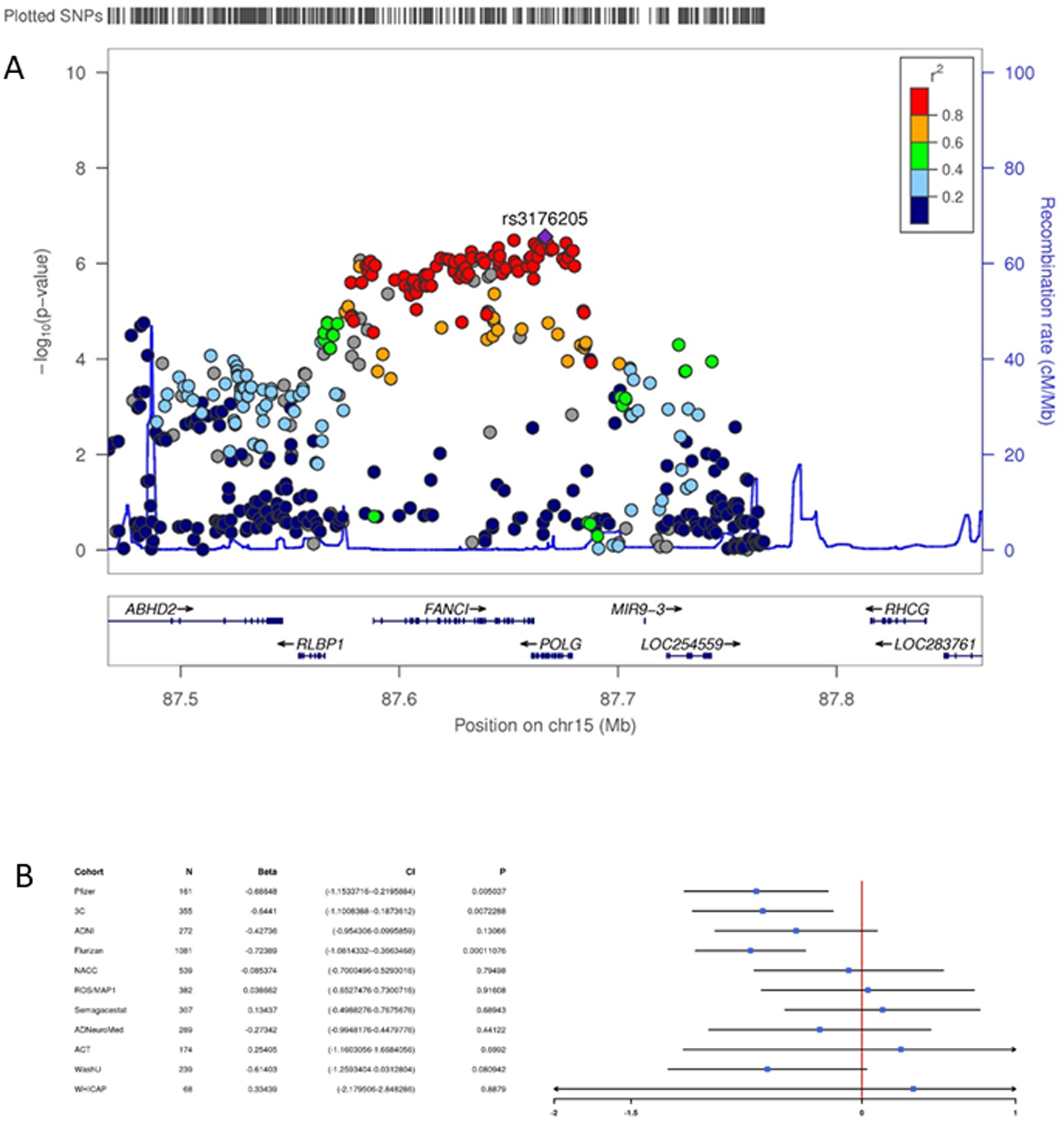
Regional Manhattan plot (A) and forest plot (B) showing the full generalized estimating equations model results for the region containing DNA polymerase-γ on chromosome 15. SNPs are color coded according to their linkage disequilibrium with the lead SNP in the region. The forest plot shows the beta and associated 95% confidence interval in each cohort.
Figure 2. Association results for contactin-associated protein 2 on chromosome 7.
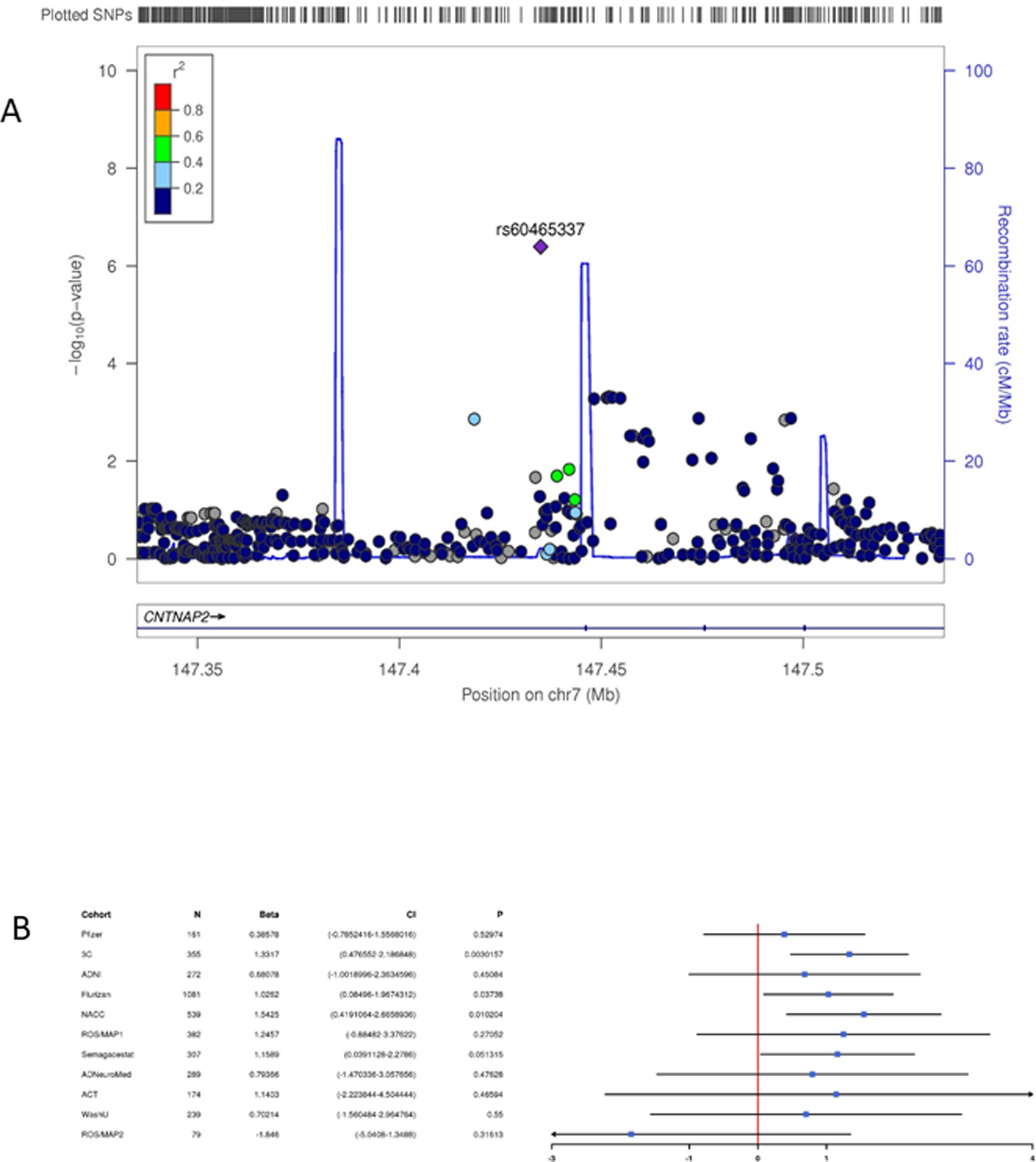
Regional Manhattan plot (A) and forest plot (B) showing the full generalized estimating equations model results for the region containing contactin-associated protein 2 on chromosome 7. SNPs are color coded according to their linkage disequilibrium with the lead SNP in the region. The forest plot shows the beta and associated 95% confidence interval in each cohort.
We also examined the top SNPs in 32 known AD risk genes77–79, and also the SNPs tagging the APOE ε2 and ε4 alleles, for association with ROD. After correcting for the number of SNPs tested, only rs1476679 in zinc finger CW-type and PWWP domain containing 1 (ZCWPW1, pLME=3.07×10−6, pGEE=3.9×10−4), was significantly associated with ROD. Notably, the minor allele (C) is protective for AD and associated with slower ROD77. Although the associations were observed with different variants, CNTNAP279 and phospholipase C gamma 2 (PLCG2)80 have also recently been implicated as AD risk genes.
3.4. Pathway Analysis
Among the genes that met criteria for inclusion in pathway analysis (Supplementary table 1), several have direct links to AD pathology, including amyloid beta precursor protein binding family A member 1 (APBA1), beta-secretase 1 (BACE1), paired immunoglobin like type 2 receptor alpha (PILRA), are in the same families as established AD risk genes such as ATP binding cassette subfamily A member 1 (ABCA1) and EPH receptor B1(EPHB1). Three genes are related to other neurodegenerative diseases. Synuclein alpha (SNCA), and parkin RBR E3 ubiquitin protein ligase (PRKN) are associated with Parkinsons disease risk, and HECT, C2 and WW domain containing E3 ubiquitin protein ligase 1 (HECW1) is associated with familial amyotrophic lateral sclerosis. Supplementary table 2 shows the individual SNP results for these genes. Subsets of these genes were significantly overrepresented in 56 canonical pathways (many of which are closely related and contain a largely overlapping set of genes) at Benjamini-Hochberg81 p-value < 0.05 (Supplementary table 3). Many of these pathways are related to neuronal development and function (Gαq signaling, ephrin signaling, synaptic long term depression, axonal guidance signaling), neuronal apoptosis (G beta gamma signaling, Huntington’s disease signaling, phospholipase C signaling), memory (CREB signaling in neurons, protein kinase A signaling), and inflammation and immunity (CXCR4 signaling, thrombin signaling). Similarly, a portion of these genes were significantly (FDR corrected p-value ≤ 0.05) overrepresented in 53 physiological systems (supplementary table 4) related to nervous system function, including development of neurons (p=7.03×10−9), neuritogenesis (p=9.02×10−8), morphology of the nervous system (p=9.08×10−8), neurite branching (p=1.64×10−7), neurotransmission (p=3.03×10−7), and synaptic transmission (p=3.61×10−7).
4. Discussion
We report results from a GWAS for ROD in the largest cohort of AD cases with longitudinal cognitive data assembled to date. We identified several suggestive associations in genes with no previous links to AD risk, as well as one study-wide significant association with ZCWPW1 in tests focused on previously established AD risk genes, and identified novel variants in CNTNAP2 and PLCG2. These newly implicated genes have roles in a diverse set of physiological pathways that have functions related to known AD processes and more generalized neural biology and development. Several of these pathways showed statistically significant enrichment of the top-ranked genes in the GWAS.
POLG is involved in proofreading during mitochondrial DNA (mtDNA) replication82. Mutations in the gene have been associated with multiple mitochondrial disorders including Alpers type mtDNA depletion syndrome83 and progressive external ophthalmoplegia84. Several animal studies have induced accelerated aging phenotypes by altering the function of POLG85,86, and the effects appear to be driven by increased neuronal apoptosis85. Given the well-established role of mitochondrial dysfunction in AD (reviewed in87–89) and the links between variants in this gene and aging phenotypes, this gene is a biologically compelling candidate for a ROD mediator. The top variant in the gene is a significant eQTL for POLG, suggesting its effects on ROD might be through increased expression.
CNTNAP2 encodes a neuronal member of the neurexin superfamily and is involved in neural-glia interactions and clustering of potassium channels in axons. It is expressed at high levels in the prefrontal and anterior temporal cortex, the dorsal thalamus, caudate, putamen, and amygdala, with enriched expression in circuits involved in higher cortical functions including language90. Variants have been associated with neurodevelopmental disorders including autism91–93, ADHD94, intellectual disabilities95 through multiple protein function and regulatory mechanisms. It is downregulated in the hippocampus of AD patients, possibly through increased expression of the transcription factor Storkhead box 1A (STOX1A)96. The variant we identified is intronic with no known regulatory effects.
There is evidence that several of the top-ranked genes have roles in the immune system and neuroinflammation, including (cytokine dependent hematopoietic cell linker) (CLNK)97, CD8b molecule (CD8B)98, and PLCG299. Hect, c2, and ww domains-containing e3 ubiquitin-protein ligase 1 (HECW1) binds to mutated superoxide dismutase 1 (SOD1) to produce Lewy body-like hyaline inclusions in ventral horn motor neurons in familial amyotrophic lateral sclerosis patients100.
The pathway analysis results highlight additional mechanisms affecting ROD, broadly implicating neuronal development, apoptosis, and synapse formation. The vast majority of the significant canonical pathways are linked by the involvement of genes encoding G protein coupled receptor (GPCR) subunits. GPCRs regulate many neurotransmitters in the brain and also directly influence the amyloid cascade by modulating α-, β-, and γ-secretase, proteolysis of the amyloid precursor protein (APP), and regulation of amyloid-β degradation101. The top pathway, Gαq signaling, is involved in axon growth and has been a drug target for multiple disorders, including a negative phase 2 clinical trial for AD102. The second ranked pathway, G beta gamma signaling, has also been studied in the context of AD and affects apoptosis103.The significant diseases and biological functions largely involve a different set of genes than those overrepresented in the canonical pathways and suggest roles for neural development and neurotransmission in ROD. CNTNAP2, APBA1, and BACE1 are all involved in the top functions, although it is unclear from these data whether these findings represent pre or post disease alterations.
Our findings also highlight genetic links between intelligence and AD-related pathways. A recent study identified 187 independent loci associated with intelligence from a meta-analysis of 248,482 non-demented subjects104. Of these loci, ten (APBA1 pmin=1.63×10−6, BANK1 pmin=4.30×10−5, KCNH5 pmin=5.42×10−5, NEGR1 pmin=3.09×10−7, PDE4D pmin=1.01×10−6, PTPRN2 pmin=1.59×10−5, RBFOX1 pmin=2.23×10−5, SGCZ pmin=3.91×10−6, SLC17A3 pmin=4.27×10−5, and ZCCHC4 pmin=2.34×10−5 were among our top-ranked genes for ROD measured in individuals after onset of AD symptoms. Each of these associations remained or increased in significance after adjusting for years of education, suggesting that the effects of these genes are not limited to general, pre-disease cognitive ability and may actively alter disease pathology. Of these genes, only APBA1 is known to be involved in AD pathology105,106. None of the top SNPs in these ten genes that were associated with ROD were tagged by the lead SNP associated with intelligence in104, making it impossible to determine whether the effect directions matched, but also suggesting the possibility that different causal variants within those genes may affect ROD and general intelligence. These results, combined with the significant ROD pathways we identified and the observation that ROD is associated with rs1476679 in ZCWPW1 only among known AD risk variants (although different variants in CNTNAP2 and PLCG2 were associated with ROD), suggest that post-diagnosis cognitive functioning may be determined more by genetic variation influencing general neural function and connectivity than by genes involved in the cascade of events leading to AD-related pathology.
In aggregate, these results suggest that like AD itself, cognitive decline is highly polygenic and controlled by a diverse set of pathways. The individual variant results suggest roles for mitochondrial dysfunction, neuron function, and immunity, while the pathway results implicate, GPRC-mediated amyloid-β and/or neurotransmitter processing neuronal development, pruning, and survival.
4.1. Strengths and Limitations
Several limitations to this work should be noted. The data are comprised of multiple, relatively small cohorts with different ascertainment schemes. This, combined with the inherently heterogeneous nature of AD presentation, symptom profile, and pathology, suggests that participants in this study may be at different stages of the disease and/or may represent multiple biologically distinct AD subtypes. The different sets of cognitive tests performed across cohorts may have limited our ability to detect true genetic associations with ROD, although our previous work demonstrated that the metric of the GCP composite factor is consistent across studies70. Finally, the longitudinal interaction tests we used were associated with inflation in the test statistics for both LME and GEE models and, consequently, our results may be less robust after a heavy correction for genomic control. However, we minimized this concern by excluding datasets showing high levels of inflation.
Despite these issues, several indicators suggest our findings are robust. First, the significance of the top results are commensurate with the sample size, and the effect sizes and directions are generally consistent across cohorts, with no single sample exerting an excessive effect on the overall association. The variants reported are also associated with ROD using two distinct regression-based approaches to modeling correlated data, and are robust even when the cohorts showing the greatest inflation are excluded. The top-ranked findings were observed generally with relatively common variants that were well imputed (r2 ≥ 0.8). In addition, evidence suggesting we identified genes in AD-relevant pathways, significant pathways related to neuronal function, and genes that are also significantly associated with cognitive performance more broadly suggest our analysis uncovered true determinants of ROD. Future directions include further expanding the sample and repeating the analyses using pre-diagnosis cognitive scores. Finally, our phenotype is a measure of global cognitive function and it is possible that additional genes contribute to specific domains of cognition (i.e. memory or executive function).
Supplementary Material
Research in context.
Systematic review:
The authors reviewed the literature using PubMed sources. While the genetics of cognitive decline has not been widely studied (except by our group), non-genetic factors influencing AD progression have been identified. The relevant citations are appropriately cited.
Interpretation:
Our findings point to novel variants and pathways affecting cognitive decline, and show a limited role for known AD risk variants. These results may inform the design and analysis of future clinical trials of AD drugs.
Future directions:
The manuscript outlines a framework for the generation and analysis of longitudinal cognitive scores which will be applied to larger samples and specific domains of cognitive function in order to confirm and expand these findings. Functional studies are necessary to determine whether the genes/pathways identified are suitable for drug targets.
Acknowledgements
The study was supported by NIH grants P30AG10161, R01AG15819, R01AG17917, K25AG055620 P30AG10161, R01AG15819, R01AG17917, R01AG042437, R01AG044546, P01AG003991, RF1AG053303, R01AG058501, U01AG058922, U01AG052411, R01AG05777, P50 AG05681, P01 AG03991, R01 AG067501 and P01 AG026276, Alzheimer’s Association grants NIRG-11-200110, BAND-14-338165, AARG-16-441560 and BFG-15-362540, and Young Investigator Award (Sherva/Green)
The Three City (3C) Study is conducted under a partnership agreement among the Institut National de la Santé et de la Recherche Médicale (INSERM), the University of Bordeaux, and Sanofi-Aventis. The Fondation pour la Recherche Médicale funded the preparation and initiation of the study. The 3C Study is also supported by the Caisse Nationale Maladie des Travailleurs Salariés, Direction Générale de la Santé, Mutuelle Générale de l’Education Nationale (MGEN), Institut de la Longévité, Conseils Régionaux of Aquitaine and Bourgogne, Fondation de France, and Ministry of Research-INSERM Programme ‘Cohortes et collections de données biologiques.’ Lille Génopole received an unconditional grant from Eisai. The Three-city biological bank was developed and maintained by the laboratory for genomic analysis LAG-BRC - Institut Pasteur de Lille.
References
- 1.Kaffashian S et al. Predicting cognitive decline: a dementia risk score vs. the Framingham vascular risk scores. Neurology 80, 1300–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prince M, Lewis G, Bird A, Blizard R & Mann A A longitudinal study of factors predicting change in cognitive test scores over time, in an older hypertensive population. Psychol Med 26, 555–68 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Formiga F et al. Factors predicting 2-year cognitive decline in nonagenarians without cognitive impairment at baseline: the NonaSantfeliu study. J Am Geriatr Soc 55, 1152–4 (2007). [DOI] [PubMed] [Google Scholar]
- 4.De Jager C, Blackwell AD, Budge MM & Sahakian BJ Predicting cognitive decline in healthy older adults. Am J Geriatr Psychiatry 13, 735–40 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Chodosh J, Reuben DB, Albert MS & Seeman TE Predicting cognitive impairment in high-functioning community-dwelling older persons: MacArthur Studies of Successful Aging. J Am Geriatr Soc 50, 1051–60 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Crowe M et al. Life-space and cognitive decline in a community-based sample of African American and Caucasian older adults. J Gerontol A Biol Sci Med Sci 63, 1241–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodard JL et al. Prediction of cognitive decline in healthy older adults using fMRI. J Alzheimers Dis 21, 871–85 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llado-Saz S, Atienza M & Cantero JL Increased levels of plasma amyloid-beta are related to cortical thinning and cognitive decline in cognitively normal elderly subjects. Neurobiol Aging 36, 2791–7 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Pankratz VS et al. Predicting the risk of mild cognitive impairment in the Mayo Clinic Study of Aging. Neurology 84, 1433–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adak S et al. Predicting the rate of cognitive decline in aging and early Alzheimer disease. Neurology 63, 108–14 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Landau SM et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 75, 230–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devanand DP et al. MRI hippocampal and entorhinal cortex mapping in predicting conversion to Alzheimer’s disease. Neuroimage 60, 1622–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Rodriguez E et al. Genetic risk score predicting accelerated progression from mild cognitive impairment to Alzheimer’s disease. J Neural Transm (Vienna) 120, 807–12 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Heister D et al. Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology 77, 1619–28 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H et al. Prognostic polypeptide blood plasma biomarkers of Alzheimer’s disease progression. J Alzheimers Dis 40, 659–66 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Moradi E et al. Machine learning framework for early MRI-based Alzheimer’s conversion prediction in MCI subjects. Neuroimage 104, 398–412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tosto G, Zimmerman ME, Carmichael OT, Brickman AM & Alzheimer’s Disease Neuroimaging I. Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol 71, 872–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellendt S et al. Predicting Stability of Mild Cognitive Impairment (MCI): Findings of a Community Based Sample. Curr Alzheimer Res 14, 608–619 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Treiber KA et al. Cognitive stimulation and cognitive and functional decline in Alzheimer’s disease: the cache county dementia progression study. J Gerontol B Psychol Sci Soc Sci 66, 416–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Small BJ, Viitanen M, Winblad B & Backman L Cognitive changes in very old persons with dementia: the influence of demographic, psychometric, and biological variables. J Clin Exp Neuropsychol 19, 245–60 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Capitani E, Cazzaniga R, Francescani A & Spinnler H Cognitive deterioration in Alzheimer’s disease: is the early course predictive of the later stages? Neurol Sci 25, 198–204 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Nagahama Y et al. Cerebral correlates of the progression rate of the cognitive decline in probable Alzheimer’s disease. Eur Neurol 50, 1–9 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Lopez OL et al. Predicting cognitive decline in Alzheimer’s disease: an integrated analysis. Alzheimers Dement 6, 431–9 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Mielke MM et al. Interaction between vascular factors and the APOE epsilon4 allele in predicting rate of progression in Alzheimer’s disease. J Alzheimers Dis 26, 127–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canevelli M et al. Predicting the Rate of Cognitive Decline in Alzheimer Disease: Data From the ICTUS Study. Alzheimer Dis Assoc Disord 30, 237–42 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Del-Aguila JL et al. Assessment of the Genetic Architecture of Alzheimer’s Disease Risk in Rate of Memory Decline. J Alzheimers Dis 62, 745–756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bleckwenn M et al. Impact of coronary heart disease on cognitive decline in Alzheimer’s disease: a prospective longitudinal cohort study in primary care. Br J Gen Pract 67, e111–e117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benedictus MR et al. Lower cerebral blood flow is associated with faster cognitive decline in Alzheimer’s disease. Eur Radiol 27, 1169–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eldholm RS et al. Progression of Alzheimer’s Disease: A Longitudinal Study in Norwegian Memory Clinics. J Alzheimers Dis 61, 1221–1232 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Farina N, Jerneren F, Turner C, Hart K & Tabet N Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Exp Gerontol 99, 146–150 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Reyes-Coronel C et al. Predicting rapid cognitive decline in Alzheimer’s disease patients using quantitative EEG markers and neuropsychological test scores. Conf Proc IEEE Eng Med Biol Soc 2016, 6078–6081 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Ferrari C et al. Alzheimer’s Disease Progression: Factors Influencing Cognitive Decline. J Alzheimers Dis 61, 785–791 (2018). [DOI] [PubMed] [Google Scholar]
- 33.De Vos A et al. The Cerebrospinal Fluid Neurogranin/BACE1 Ratio is a Potential Correlate of Cognitive Decline in Alzheimer’s Disease. J Alzheimers Dis 53, 1523–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanders C et al. Nutritional Status is Associated with Faster Cognitive Decline and Worse Functional Impairment in the Progression of Dementia: The Cache County Dementia Progression Study1. J Alzheimers Dis 52, 33–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drummond E et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol 133, 933–954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farina N, Rusted J & Tabet N The effect of exercise interventions on cognitive outcome in Alzheimer’s disease: a systematic review. Int Psychogeriatr 26, 9–18 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Woods B, Aguirre E, Spector AE & Orrell M Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev, CD005562 (2012). [DOI] [PubMed] [Google Scholar]
- 38.McDermott KL, McFall GP, Andrews SJ, Anstey KJ & Dixon RA Memory Resilience to Alzheimer’s Genetic Risk: Sex Effects in Predictor Profiles. J Gerontol B Psychol Sci Soc Sci 72, 937–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bereczki E et al. Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141, 582–595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crispoltoni L et al. Changes in Plasma beta-NGF and Its Receptors Expression on Peripheral Blood Monocytes During Alzheimer’s Disease Progression. J Alzheimers Dis 55, 1005–1017 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Sao T et al. MEF2C mRNA expression and cognitive function in Japanese patients with Alzheimer’s disease. Psychiatry Clin Neurosci 72, 160–167 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Yoshino Y et al. INPP5D mRNA Expression and Cognitive Decline in Japanese Alzheimer’s Disease Subjects. J Alzheimers Dis 58, 687–694 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Mahady L et al. Frontal Cortex Epigenetic Dysregulation During the Progression of Alzheimer’s Disease. J Alzheimers Dis 62, 115–131 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Yu L et al. Association Between Brain Gene Expression, DNA Methylation, and Alteration of Ex Vivo Magnetic Resonance Imaging Transverse Relaxation in Late-Life Cognitive Decline. JAMA Neurol 74, 1473–1480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wingo AP et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun 10, 1619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrews SJ, Das D, Anstey KJ & Easteal S Late Onset Alzheimer’s Disease Risk Variants in Cognitive Decline: The PATH Through Life Study. J Alzheimers Dis 57, 423–436 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Andrews SJ, Das D, Cherbuin N, Anstey KJ & Easteal S Association of genetic risk factors with cognitive decline: the PATH through life project. Neurobiol Aging 41, 150–158 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Burfeind KG et al. The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimers Dement (N Y) 3, 348–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee E et al. Single-nucleotide polymorphisms are associated with cognitive decline at Alzheimer’s disease conversion within mild cognitive impairment patients. Alzheimers Dement (Amst) 8, 86–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherva R et al. Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement 10, 45–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiner MW et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement 9, e111–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett DA et al. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res 9, 646–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett DA, Schneider JA, Arvanitakis Z & Wilson RS Overview and findings from the religious orders study. Curr Alzheimer Res 9, 628–45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Group CS Vascular factors and risk of dementia: design of the Three-City Study and baseline characteristics of the study population. Neuroepidemiology 22, 316–25 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Lovestone S et al. AddNeuroMed--the European collaboration for the discovery of novel biomarkers for Alzheimer’s disease. Ann N Y Acad Sci 1180, 36–46 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Lourdusamy A et al. Identification of cis-regulatory variation influencing protein abundance levels in human plasma. Hum Mol Genet 21, 3719–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voyle N et al. A Pathway Based Classification Method for Analyzing Gene Expression for Alzheimer’s Disease Diagnosis. J Alzheimers Dis 49, 659–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Green RC et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris JC et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 20, 210–6 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Jones RW et al. The Atorvastatin/Donepezil in Alzheimer’s Disease Study (LEADe): design and baseline characteristics. Alzheimers Dement 4, 145–53 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Doody RS et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369, 341–50 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Kukull WA et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 59, 1737–46 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Albert SM et al. Hospitalization and Alzheimer’s disease: results from a community-based study. J Gerontol A Biol Sci Med Sci 54, M267–71 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Genomes Project C et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howie BN, Donnelly P & Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delaneau O, Marchini J & Zagury JF A linear complexity phasing method for thousands of genomes. Nat Methods 9, 179–81 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Li Y, Willer CJ, Ding J, Scheet P & Abecasis GR MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34, 816–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Howie B, Fuchsberger C, Stephens M, Marchini J & Abecasis GR Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44, 955–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gross AL et al. Calibrating longitudinal cognition in Alzheimer’s disease across diverse test batteries and datasets. Neuroepidemiology 43, 194–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dorans NJ Principles and Practices of Test Score Equating. (ed. Moses TP) (ETS, Princeton, NJ, 2010). [Google Scholar]
- 72.Samejima F Estimation of Latent Ability Using a Response Pattern of Graded Scores. Psychometrika 34, 1–& (1969). [Google Scholar]
- 73.Takane Y & Deleeuw J On the Relationship between Item Response Theory and Factor-Analysis of Discretized Variables. Psychometrika 52, 393–408 (1987). [Google Scholar]
- 74.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voorman A, Rice K & Lumley T Fast computation for genome-wide association studies using boosted one-step statistics. Bioinformatics 28, 1818–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Consortium GT The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sims R et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Benjamini Y & Hochberg Y Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Statistical Methodology 57, 289–300 (1995). [Google Scholar]
- 82.Bogenhagen DF, Rousseau D & Burke S The layered structure of human mitochondrial DNA nucleoids. J Biol Chem 283, 3665–75 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Naviaux RK & Nguyen KV POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. Ann Neurol 58, 491 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Van Goethem G, Dermaut B, Lofgren A, Martin JJ & Van Broeckhoven C Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28, 211–2 (2001). [DOI] [PubMed] [Google Scholar]
- 85.Trifunovic A et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–23 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Kujoth GC et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–4 (2005). [DOI] [PubMed] [Google Scholar]
- 87.Macdonald R, Barnes K, Hastings C & Mortiboys H Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem Soc Trans 46, 891–909 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Van Giau V, An SSA & Hulme JP Mitochondrial therapeutic interventions in Alzheimer’s disease. J Neurol Sci 395, 62–70 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Onyango IG, Khan SM & Bennett JP Jr. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front Biosci (Landmark Ed) 22, 854–872 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Abrahams BS et al. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc Natl Acad Sci U S A 104, 17849–54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alarcon M et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet 82, 150–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arking DE et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 82, 160–4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bakkaloglu B et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet 82, 165–73 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elia J et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 15, 637–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mikhail FM et al. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A 155A, 2386–96 (2011). [DOI] [PubMed] [Google Scholar]
- 96.van Abel D et al. Direct downregulation of CNTNAP2 by STOX1A is associated with Alzheimer’s disease. J Alzheimers Dis 31, 793–800 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Cao MY, Davidson D, Yu J, Latour S & Veillette A Clnk, a novel SLP-76-related adaptor molecule expressed in cytokine-stimulated hemopoietic cells. J Exp Med 190, 1527–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnson P A human homolog of the mouse CD8 molecule, Lyt-3: genomic sequence and expression. Immunogenetics 26, 174–7 (1987). [DOI] [PubMed] [Google Scholar]
- 99.Kang JS et al. Cloning and functional analysis of the hematopoietic cell-specific phospholipase C(gamma)2 promoter. FEBS Lett 399, 14–20 (1996). [DOI] [PubMed] [Google Scholar]
- 100.Miyazaki K et al. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J Biol Chem 279, 11327–35 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Thathiah A & De Strooper B The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat Rev Neurosci 12, 73–87 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Lenz RA et al. Adaptive, dose-finding phase 2 trial evaluating the safety and efficacy of ABT-089 in mild to moderate Alzheimer disease. Alzheimer Dis Assoc Disord 29, 192–9 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Giambarella U et al. G protein betagamma complex-mediated apoptosis by familial Alzheimer’s disease mutant of APP. EMBO J 16, 4897–907 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hill WD et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol Psychiatry 24, 169–181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Saluja I, Paulson H, Gupta A & Turner RS X11alpha haploinsufficiency enhances Abeta amyloid deposition in Alzheimer’s disease transgenic mice. Neurobiol Dis 36, 162–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xie Z, Romano DM & Tanzi RE RNA interference-mediated silencing of X11alpha and X11beta attenuates amyloid beta-protein levels via differential effects on beta-amyloid precursor protein processing. J Biol Chem 280, 15413–21 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.