

Abstract
Background:
Chronic visceral pain is persistent pain emanating from thoracic, pelvic or abdominal origin that is poorly localized with regard to the specific organ affected. The prevalence can range up to 25% in the adult population as chronic visceral pain is a common feature of many visceral disorders which may or may not be accompanied by distinct structural or histological abnormalities within the visceral organs. Mounting evidence suggest that changes in epigenetic mechanisms are involved in the top-down or bottom-up sensitization of pain pathways and the development of chronic pain. Epigenetic changes can lead to long-term alterations in gene expression profiles of neurons and consequently alter functionality of peripheral neurons, dorsal root ganglia (DRG), spinal cord, and brain neurons. However, epigenetic modifications are dynamic and thus detrimental changes may be reversible. Hence, external factors/therapeutic interventions may be capable of modulating the epigenome and restore normal gene expression for extended periods of time.
Purpose:
The goal of this review is to highlight the latest discoveries made towards understanding the epigenetic mechanisms that are involved in the development or maintenance of chronic visceral pain. Furthermore, this review will provide evidence supporting that targeting these epigenetic mechanisms may represent a novel approach to treat chronic visceral pain.
Keywords: Acetylation, Methylation, Gastrointestinal, Brain, Spinal Cord, Stress
1. Introduction
Chronic pain is defined as pain lasting longer than three months after the resolution or in absence of an injury, and is experienced by patients as a marked reduction in the threshold required to induce pain. In patients suffering from chronic pain an innocuous stimulus can cause pain (allodynia) and/or noxious stimulus can trigger an amplified response (hyperalgesia).1 Chronic pain is estimated to affect 100 million adults in the United States. Chronic pain significantly reduces a patient’s quality of life, but also increases the risk of developing mental health disorders such as anxiety and depression. The combined financial cost to society of chronic pain due to health care and loss of productivity is enormous and ranges up to a staggering 600 billion in the United States alone.2 This review will focus on chronic visceral pain, which is persistent pain emanating from thoracic, pelvic or abdominal origin that is poorly localized with regard to the specific organ affected. The prevalence of visceral pain is higher in women than in men and can range up to 25% in the adult population.3 This high prevalence can be explained by the fact that chronic visceral pain is a common feature of many visceral disorders which may or may not be accompanied by distinct structural or histological abnormalities within the visceral organ. Pain disorders with concurrent mucosal inflammation describe inflammatory bowel disease (IBD), pancreatitis, bladder pain syndrome/interstitial cystitis (BPS/IC), and gynecological pain; whereas pain disorders without distinct structural or histological abnormalities include functional dyspepsia, functional chest pain, functional heartburn, functional dysphagia, irritable bowel syndrome (IBS), centrally mediated abdominal pain syndrome (CAPS), narcotic bowel syndrome (NBS), and functional anorectal pain (Figure 1).4 Reliable therapies that relieve chronic visceral pain represents a major unmet medical need, and such therapies would greatly improve the patient’s quality of life and reduce the burden to society of managing these patients. The efficacy of current therapeutics to treat visceral pain such as opioids and non-steroidal anti-inflammatory drugs is far from optimal and can produce unwanted and serious side-effects.5 Despite extensive research on the neurobiological mechanisms of chronic visceral pain, our understanding of the underlying molecular pathways is incomplete. This knowledge gap hampers the highly desirable and anticipated development of new visceral pain-relieving medications. Chronic pain develops when neuronal networks become ‘sensitized’. This process starts when the balance of neurotransmitters, receptors and other molecules in neurons is disturbed. In this way, the cellular functionality of peripheral neurons, dorsal root ganglia (DRG), spinal cord, and brain neurons can be altered.1,6 Mounting evidence suggests that epigenetic modifications play an important role in sensitization and the development of chronic pain.7–10 Epigenetic modifications are described as stable alterations in gene expression that arise during development and cell proliferation. However, epigenetic modifications are dynamic and can also be influenced by external environmental factors. In this way, external factors are capable of modulating gene expression for extended periods of time following a stimulus through changes in the epigenome. The importance of epigenetic modifications in neurons was first highlighted in synaptic plasticity during memory formation.10–14 Interestingly, the epigenetic changes that occur during memory formation are similar to those that occur during the neuronal sensitization process. Epigenome alterations form an attractive molecular mechanism for sensitization, as these changes can lead to long-term changes in neuronal gene expression of the neurotransmitters, receptors, and other molecules involved in sensitization. Once established, these epigenetic changes that occur during sensitization may also explain chronic pain on a molecular level. In this review, we summarize the latest discoveries made towards understanding the epigenetic mechanisms that are involved in chronic visceral pain. Furthermore, we argue that targeting these epigenetic mechanisms may become a novel treatment for chronic visceral pain. As more and more drugs targeting the epigenome become available, we can start targeting the underlying causes of chronic visceral pain, instead of treating the symptoms with traditional analgesics.
Figure 1: Common Visceral Pain Disorders.
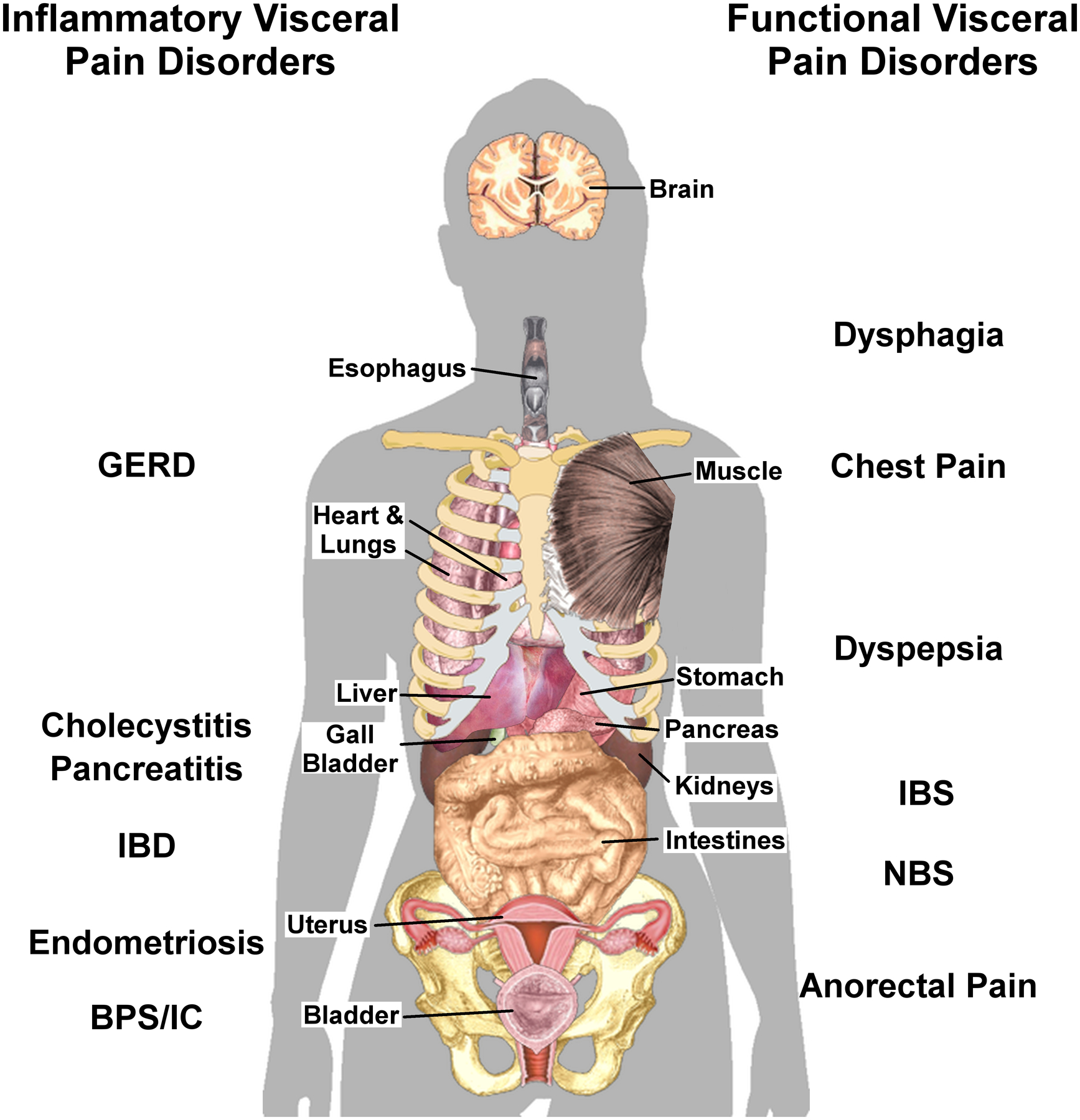
Left side: Visceral pain disorders associates with overt tissue inflammation, which may or may not be due to an infection. Right side: Visceral pain disorders with no identified pathological marker. Visceral pain is typically diffuse and may be referred to somatic structures (muscle, skin, joints) within the same or adjacent dermatomes or to other visceral organs. The location of selected organs is approximate. The name of the disorder has been illustrated near the affected organ. GERD, gastroesophageal reflux disease; IBD, inflammatory bowel disease; BPS, bladder pain syndrome; IC, interstitial cystitis; IBS, irritable bowel syndrome; NBS, narcotic bowel syndrome. This composite image was assembled from individual public domain images available from Wikimedia Commons.
2. The development of chronic visceral pain – neuronal sensitization from bottom up or top down
2.1. The acute pain pathway
Visceral organs are innervated by nociceptive neurons with cell bodies in the DRG. The free nerve endings of these neurons contain multiple receptor types to detect potential harmful changes in pH, stretch, temperature, or specific chemicals mediators such as those released in response to chronic stress.15–17 The acute pain pathway is activated when noxious signals are registered in the periphery. These signals are transmitted to the dorsal horn of the spinal cord, where the first synapse in the pain pathway is located. Usually visceral afferents synapse at multiple spinal levels, which causes a diffuse localization of the initial noxious signal.18 The dorsal horn of the spinal cord is an important location in the pain pathway as here the pain signal can be modulated by local inhibitory interneurons or descending projections from the brainstem, before sending the signal to the brain.6,19 The final synapse occurs in the thalamus, which acts as the primary hub in the central pain matrix.20 From the thalamus, the signal is dispersed to several different brain regions. Distributing the signal to the amygdala, anterior cingulate cortex, hippocampus and nucleus accumbens, process the emotional component of pain.21,22 The signal is sent to the somatosensory cortex for the localization of the pain and other regions such as the prefrontal cortex, cingulate and parietal cortex to define the quality and magnitude of the pain signal.23 The underlying mechanism that drives the transition from acute to chronic pain is not well understood. However, evidence suggests that the acute to chronic pain transition requires certain adaptations in the aforementioned pain pathways.24 In chronic visceral pain, the pain pathway can become detrimentally modulated when sensitization of primary (peripheral afferent), secondary (spinal) or tertiary (brainstem/thalamic) neurons occurs. Interestingly, sensitization can occur through a bottom-up or top down mechanism.
2.2. Sensitization from bottom-up mechanisms
Under normal circumstances, tissue injury or the release of local inflammatory mediators (chemokines, cytokines, histamine, proteases, prostaglandins, serotonin, corticotropin-releasing hormone (CRH)) trigger peripheral neurons to release neuromodulators (calcitonin gene related peptide (CGRP), nitric oxide (NO), substance P) that stimulate nerve activity.25,26 Sensitization starts when there is prolonged exposure to the initial stimulus and release of neuromodulators that activate second messenger signaling cascades, the phosphorylation and/or altered expression of certain receptors (cation channels), which may affect the cellular pathways that ensure proper neuronal function. In this way, the action potential threshold of the affected neuron can be lowered or the number of action potentials fired upon reaching threshold can be increased.1 In the bottom-up scenario, sensitization usually starts at the level of the primary peripheral neurons. As a result, the enhanced neuronal excitability of the primary nociceptive neuron will lead to increased neurotransmitter and neuromodulator release in the dorsal horn of the spinal cord after stimulation. On the secondary level, the increased and/or prolonged input from primary afferent neurons will increase the number of Ca2+ channels in dorsal horn neurons. Altering the Ca2+ permeability of dorsal horn neurons will lower the action potential threshold of these neurons. As a consequence, these neurons can fire action potentials with less input from the primary neurons. Another potential pathway leading to dorsal horn sensitization is through dysfunction of inhibitory interneurons. These neurons receive signaling input either directly from the sensitized primary neuron or indirectly via descending neurons from the brain.27,28 Increased peripheral excitability and/or decreased inhibitory tone can lead to remodeling and persistent excitation of second order neurons and thus contribute to chronic pain.29 Sensitization can also happen at the tertiary level. Increased neuronal afferent stimulation can cause remodeling in the brain regions of the central pain matrix. Enhanced signaling from the thalamus will subsequently sensitize the integration nuclei (amygdala, hippocampus, insula, or cingulate), which can also alter activation thresholds in these neurons. When sensitization has occurred in these regions, previously innocuous stimuli will not only be perceived as noxious, but also trigger the negative emotional response to chronic pain.
2.3. Sensitization from top-down mechanisms
It is known that stress and negative emotions can enhance the perception of nociception in the absence of an overt peripheral injury.30–32 This happens through the direct sensitization of the central pain matrix. The importance of this top-down mechanism for visceral pain is illustrated by epidemiological evidence showing correlations between chronic visceral pain and stress, anxiety and depression.33,34 Evidence from experimental models has shown that persistent stress can facilitate pain perception and sensitize pain pathways, promoting chronic visceral pain disorders.35,36 An important mediator of this pathway is the hypothalamus-pituitary-adrenal (HPA) axis. Stress can initiate the HPA axis, by activating the paraventricular nucleus of the hypothalamus (PVN) to secrete CRH into to hypophyseal portal circulation. CRH binds to CRH1 in the anterior pituitary gland, which stimulates the release of adrenocorticotropic hormone (ACTH) in the systemic circulation. In turn, ACTH binds in the adrenal cortex and cause the de novo synthesis and release of cortisol in humans and corticosterone in rats in the systemic circulation (CORT). Circulating CORT has a dual role, depending on the receptors and regions where it binds. When CORT binds to its high-affinity mineralocorticoid receptor (MR) and low-affinity glucocorticoid receptor (GR) in the hippocampus, PVN and cortical regions, it will trigger signaling cascades that induce a negative feedback loop in order to terminate the stress response. However, when CORT binds the same receptors in the amygdala, it increases CRH expression which opposes the feedback inhibition and facilitates the stress axis37–41. The central nucleus of the amygdala (CeA) plays an important role in visceral pain as this region integrates viscerosensory signaling with neuroendocrine and autonomic responses to stressors through the expression of MR, GR and CRH.42–44 The importance of CeA signaling has been illustrated by directly stereotaxically implanting CORT-containing micropellets bilaterally on the dorsal margin of the CeA of healthy rats. Elevated amygdala CORT induced visceral hypersensitivity, through CORT-mediated decreases in GR and increases in CRH within the CeA. Simultaneous infusions of GR agonists or CRH antagonists in the CeA reversed the CORT-induced visceral hypersensitivity. The same effects on visceral sensitivity and GR/CRH expression were found in rats after they underwent repeated water avoidance stress (WAS).43 Through neuronal remodeling and unbalancing GR/CRH signaling in the CeA, WAS induces visceral hypersensitivity to colonic distension in rodents45,46. HPA axis dysfunction can exacerbate visceral pain during stress.47 Interestingly these effects persisted long after the CORT pellets were depleted or the repeated WAS procedure was discontinued.45,48 Acute and chronic stress can alter the signaling in the central pain matrix, especially through HPA axis dysregulation and altered CeA activity/signaling. This can lead to exacerbation of pain perception and/or diminishment of anti-nociceptive and anti-stress signaling.49 These mechanisms have been elucidated in patients with irritable bowel syndrome (IBS) where brain imaging studies revealed increased activity in the amygdala after colorectal stimulation.50 Furthermore, these patients had an abnormal HPA axis reactivity to CRH challenge and an overall increase in CORT secretion, which is indicative of HPA axis dysregulation, and likely contributed to the chronic visceral pain in these patients.51,52
3. Epigenetic changes
Epigenetic changes refer to stable alterations in (long-term) gene expression potential resulting from developmental or environmental signals. These alterations are the result of cellular mechanisms that integrate external signals by structurally adapting chromosomal regions to register, signal, or perpetuate altered activity states or by modulating transcript levels.53 Epigenetic marks are essential for the structural and functional adaptation of chromosomal regions, making those regions more or less accessible for gene transcription. In this way, gene expression is modulated without interfering with the base pair sequence of the DNA itself. Structurally, epigenetic marks play an indispensable role in DNA condensation. By interacting with numerous other proteins, these marks allow the packing of an extremely long DNA strand into an extremely small nucleus. 140 base pairs of DNA, wrapped around a histone (H) octamer, forms a nucleosome. The central histone octamer is formed by two copies of each of the ‘core histones’ H2A, H2B, H3 and H4.54 The N-terminal histone tail protrudes from the nucleosome and is available for post-translational modifications which can attract DNA-condensation or DNA-unwrapping enzymes. Nucleosomes are further condensed in chromatin, which can switch between two forms: the “open” euchromatin and the “closed” heterochromatin. In euchromatin, coding DNA sequences are poised for transcriptional initiation and enhancer activity when they are detached from the nucleosomes. In contrast heterochromatin is a highly compact state where genes are silenced.55 Functionally, the precise interplay between transcription initiation factors and specific epigenetic marks allow the unwrapping of DNA at specific locations, where gene transcription needs to be initiated, without having to unwrap the whole chromatin structure. The multitude of these dynamic epigenetic “marks” are known as the epigenome. Interestingly, changing the epigenome does not always require the presence of chronic or prolonged stimuli. For instance, in the process of memory formation, short-lived external stimuli can also alter epigenetic marks causing persistent changes in gene expression.56–58 Here we will briefly discuss the mechanism by which DNA methylation, histone (de)acetylation and non-coding RNA’s can induce changes in long-term gene expression.
3.1. Histone modifications
As stated above, DNA is wrapped around a central histone octamer, forming nucleosomes. The protruding N-terminal histone tail is available for post-translational modifications: lysine acetylation, serine/threonine phosphorylation, lysine/arginine methylation, ubiquitination and ADP-ribosylation (Figure 2A).59 Of all these modifications, histone (de)acetylation and (de)methylation are the best studied. Histone acetyltransferases (HAT) can add acetyl groups to the lysine residues (K) of the histone tail. Acetylated histones are usually associated with increased gene expression.60 In contrast, histone deacetylases (HDAC) can remove acetyl groups and stabilize the positive charge of the lysine side chains to enhance the binding of DNA to the histones. Hence, deacetylation usually prevents binding of transcription factors on the DNA and is associated with reduced gene expression.61 The activity of histone methyltransferases and demethylases determine the degree of histone side chain methylation. This modification also influences gene expression, but is not as straightforward as histone acetylation, because the effect on gene expression depends on the number of added methyl groups and the particular lysine residue methylated. For instance, mono-methylation of H3K9, H3K27 and H4K20 is associated with transcriptional activation whereas di- or tri-methylation are associated with transcriptional repression. Methylation of H3K36 and H3K79 are associated with active transcriptional elongation, but also repress transcriptional from cryptic promoters in gene bodies.62–65
Figure 2: Epigenetic Modifications.
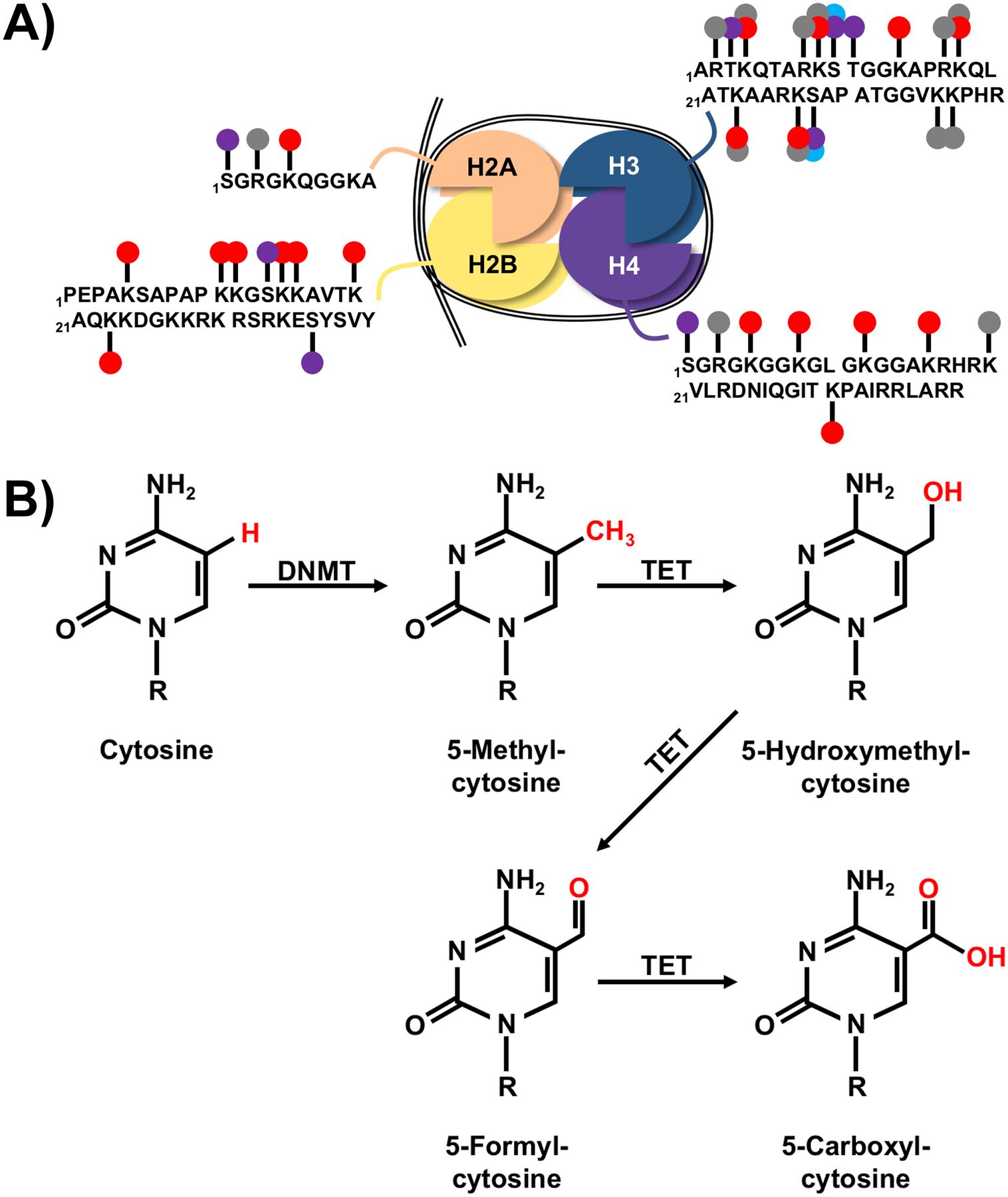
A) Nucleosomes are composed of an octamer of histones H2A, H2B, H3, and H4 (two dimers of 2A/2B and two dimers of H3/H4) with 146 base pairs of DNA wrapped around the complex (drawn as black double line). The H1 histone may also be attached as a linker to stabilize the DNA (not shown). Several epigenetically regulated modifications to the n-terminal tails of the histones influence DNA accessibility to transcription machinery including acetylation (red circles), methylation (gray circles), phosphorylation (purple circles), or ADP ribosylation (blue circles). The single letter abbreviation was used for each amino acid, with spaces after every ten, and subscript numbers indicating location. Multiple circles on a single amino acid represent alternative modifications. N-terminal sequences were based on consensus sequences for human histones at uniport.org: H2A - P0C0S8, H2B - P62807, H3 - P68431, H4 – P62805. B) DNA methyl transferases (DNMT) target cytosine residues immediately upstream of guanosine residues (CpG sites) within DNA to form 5-methylcytosine (5mC), which typically represses transcription. Ten-eleven-translocation (TET) proteins participate in demethylation by converting 5mC to 5-hydroxymethylcytosine (5hmC), which has also been demonstrated to participate in epigenetic regulation of gene expression. Further oxidation by TET proteins changes 5hmC to 5-formylcytosine (5fC) and subsequently 5-carboxylcytosine (5caC), which can then be converted back to cytosine (not pictured). The role of 5fC and 5caC in modulation of gene expression is uncertain due to the recent development of tools that can distinguish between 5mC, 5hmC, 5fC, and 5caC. For each structure, R is the sugar (deoxyribose)-phosphate group that forms the DNA nucleotide.
Histone modifications are dynamic, reversible and interconnected. The location, presence and combination of particular histone modifications form docking sites for other proteins. This “histone code” will recruit specific chromatin-associated and epigenetic proteins that will determine whether or not the DNA is accessible for transcription, repair, and replication.54,62,66 Also, this code can be read by transcription factors that will ultimately initiate gene expression activity.67 For example, histone deacetylation and methylation of certain lysine residues can recruit heterochromatin-associated proteins, which on their turn interact with DNA methyltransferases. As a result, histone modifications can result in DNA methylation and through interacting with the docked heterochromatin-associated proteins favor the condensation of methylated DNA into heterochromatin.68,69 The opposite can happen when HATs are recruited and heterochromatin is transitioned into euchromatin to allow gene expression. Acetylation of certain histone tail residues forms a docking site for other proteins that are involved in chromatin unwrapping. Certain attracted chromatin adapters possess HAT abilities that will add more negatively charged acetyl group to the histone tails. In this way, the histone tail’s positive charge is neutralized, subsequently inducing the relaxation of the chromatin structure and facilitates the binding of the transcriptional machinery. The highly dynamic nature of histone modifications means that it is regarded as a more transient cellular modification that could quickly promote or silence gene expression in response to changes in environmental stimuli, in contrast to DNA methylation, which is regarded as more stable.56,70
3.2. DNA methylation
DNA methylation is the process in which methyl groups are added to cytosine at the 5’ carbon position of the pyrimidine ring by DNA methyltransferases (DNMT). In mammals, methylation occurs predominantly on cytosines that are followed be a guanosine (CpG sites).71 DNMT3A and DNMT3B are responsible for de novo methylation, which occurs in response to environmental cues and is an important mechanism during development and cellular differentiation.72 These DNMTs are also responsible for the DNA methylation after the cell has fully differentiated. In order to maintain this cellular state, DNA methylation patterns are copied from the mother strand to the daughter strand during cell division by DNMT1. In most cases DNA methylation leads to gene silencing, as the added methyl groups structurally interfere with the binding capacity of transcription factors, thus preventing the initiation of gene transcription.73 However, under certain conditions DNA methylation can lead to gene activation. In the case of the human telomerase reverse transcriptase gene, hypomethylation was associated with gene silencing. In addition to DNA methylation, DNA hydroxymethylation, wherein oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) is catalyzed by ten-eleven-translocation (TET) proteins, has been identified in recent years as an important epigenetic process. In contrast to DNA methylation, hydroxymethylation often colocalizes with euchromatin and thus is associated with enhanced gene expression.74–77 5hmC is also an intermediary step in the demethylation process and has been shown to be involved in a myriad of other processes such as cellular differentiation, neuronal development, aging, and DNA strand separation (Figure 2B).78–82 Just like the ‘histone code’, the location and amount of methylated CpG’s can have profound effects on gene expression and chromatin structure. Methyl groups can serve as functional docking sites for proteins with a methyl-CpG binding domain. The ‘methylation code’ determines the outcome of these proteins docking. The proteins can recruit other transcriptional co-repressors such as HDACs (and initiate chromatin remodeling), but also facilitate binding of transcriptional co-activators.73,83,84 For instance, methyl-CpG-binding protein 2 (MeCP2) acts as a transcriptional repressor, by recruiting histone methyltransferases that methylate histone lysine residues associated with transcriptional repression.85 However, MeCP2 can also interact with transcriptional enhancer such as CREB1, promoting transcriptional initiation.86 Through the recruitment of additional repressor or activator proteins, de novo DNA methylation can have a profound effect on long-term gene expression and chromatin structure. As mentioned above, histone modifications and DNA methylation are intertwined and can work together in gene silencing and heterochromatin formation by recruiting histone decacetylases.87,88
3.3. Non-coding RNAs
Non-coding RNA plays an important role in cellular differentiation and development by regulating gene expression and chromatin structure.89 There are two broad classes of non-coding RNAs (ncRNA): short and long ncRNA. Short ncRNAs are RNA strands shorter than 200 nucleotides and thus microRNA, short interference RNA and PIWI-interacting RNA are part of this group. On the other hand, long ncRNAs are longer than 200 nucleotides. ncRNA mediate histone modifications and can recruit DNA methyltransferases, inducing de novo DNA methylation and transcriptional silencing.90
The best studied ncRNA are miRNAs, which are short (~19–25 nucleotides) non-coding RNA molecules that are generated from miRNA genes. The initial miRNA transcript is processed (via primary miRNA and precursor miRNA intermediates) into mature double stranded miRNA by the endonucleases ribonuclease 3 (Drosha) and Dicer. The guide strand from the double-stranded miRNA will be sequestered into the miRNA-induced silencing complex. This complex exerts control over the miRNA binding to its target mRNA. Perfect complementary binding between the miRNA and the target mRNA will lead to mRNA degradation, whereas incomplete binding inhibits further mRNA processing. Both mechanisms lead to the inhibition of target gene expression.91 Due to their short length, a miRNA can bind and regulate the expression of multiple target genes. In addition, each target gene can be regulated by a number of miRNAs. miRNAs are essential for the fine-tuning of gene expression and play important roles in developmental, physiological, and pathophysiological processes.92
3.4. Techniques to Measure Epigenetic Modifications
Many standard biochemical and molecular biological techniques have been developed to measure the post-translational modifications to histones (acetylation, methylation, phosphorylation, ADP ribosylation) or the differential methylation of DNA (5mC, 5hmC, 5fC, 5caC) (Figure 2) that form the basis of epigenetic changes to the genome. While we refer the reader to recent reviews focused on the different methodologies for detecting epigenetic modifications to histones93,94, DNA95, or non-coding RNA96, we will briefly provide a summary of the common techniques with a comparison of the strengths and weaknesses of each assay.
Targeted analysis of histone modifications, which will measure the amount or type of post-translational modification, can be accomplished with biochemical techniques, such as mass spectrometry or enzymatic assays, or through standard molecular biological techniques, such as western blot or immunofluorescence. The gold-standard for measuring histone-DNA interactions is chromatin immunoprecipitation (ChIP) that is used to provide insight about how the histone modification modifies the epigenetic regulation of a transcript. For well characterized histone modifications (acetylation, methylation), there are multiple vendors that provide kits and/or selective antibodies to analyze global expression within the cell or tissue of interest. A weakness of using the commercial kits is that nuclear protein extraction should be performed to enrich the sample for histones, rather than total protein extraction, which will limit the overall protein available for analysis from an individual sample and could require pooling samples across biologic replicates (when using cell culture or tissues from animal models) as typical commercial assays report a range of sensitivity from 1–500 ng for the target protein(s). A strength of the commercial kits is that the antibodies and reagents have been validated, and once a sufficient histone sample has been obtained, multiple modifications can be analyzed simultaneously using standard laboratory equipment. Alternatively, mass spectrometry may generally require a smaller amount of protein (depending on the purity of the sample) and can provide very specific information about the type of modification on the histone, but the cost of the specialized equipment typically requires a core-facility to provide the analysis service and there can be multiple purification steps to achieve a suitable sample for analysis.97–99 There are also kits (typically colorimetric) that measure the activity of acetyltransferases, methyltransferases, deacetylases, or demethylases using 96-well plates and standard plate readers. These assays provide a relatively high throughput (24 or more samples depending on the number of technical replicates) for minimal cost (most assays are $300-$500). The same technical weaknesses occur as with other commercial kits with generating enough sample to test duplicate/triplicate replicates in the assay; however, by testing for activity of broad classes of histone modifying enzymes, these assays can be useful when evaluating novel therapeutics or for assessing potential epigenetic regulation in specific diseases, such as cancer.100,101 Caution must also be used with interpretation of the results of the enzymatic assays as global changes in activity of the enzyme may or may not affect the expression of your gene of interest. ChIP is used to evaluate the association of histone modifications with specific DNA sequences. Typical protocols require several hundred-thousand to millions of cells, 5–50 mg of tissue, or 1–15 μg of purified chromatin, but some variants have been developed that use smaller samples. Standard ChIP protocols will cross-link proteins that are directly interacting with the DNA, and ChIP-ChIP assays can be performed to detect DNA regions with multiple bound proteins (i.e. a histone and a transcription factor); however, additional proteins that associate with the primary protein (i.e. a histone acetyltransferase that modifies the histone but does not interact directly with the DNA) may not be detected. Sequencing or PCR amplification identify the target DNA sequence that was cross-linked to the protein of interest. Bisulfite sequencing can also be used to evaluate the methylation status of the bound DNA.102
Multiple techniques can be used to assess DNA methylation at either specific regions of interest or the whole genome, and recent reviews have provided detailed information on the strengths and weaknesses of those assays.95,103 Briefly, to broadly analyze global methylation, there are methylation sensitive restriction enzymes (typically unable to cleave DNA when a methylated base is present) that can be used to digest DNA, and comparing sequencing information against restriction enzyme digestion that is insensitive to methylation can produce a map of the methylated sites within the sample region. An alternative strategy that can reduce costs is to enrich the sample for methylated regions by using methylcytosine antibodies or other methyl binding proteins to capture methylated DNA before analysis. For detecting single base differences in methylation, the gold standard is bisulfite sequencing that can be combined with methylation sensitive arrays, used with whole-genome sequencing, or with the more targeted approach of reduced representation bisulfite sequencing. In general, the major considerations with choosing an appropriate method is the amount of DNA within the sample (whole genome analysis methods require 100 ng – 10 μg of DNA) and the funds available to perform the assay and data interpretation (targeted assays can cost as little as $100 per sample, while whole genome analysis can exceed $5000 per sample). Additionally, specific assays are necessary to distinguish 5hmC from 5mC (such as oxidative bisulfite sequencing or TET-assisted bisulfite sequencing).104
4. Epigenetic mechanisms in visceral pain
4.1. Evidence pointing towards epigenetic regulation
Neurons survive for decades and therefore exhibit a remarkably low regeneration rate. Epigenetic regulation is a mechanism through which neurons maintain their specific activities throughout their lifespan. However, neuronal epigenetic regulation can be influenced by environmental changes such as physical and/or psychological stress. Hence, considering this knowledge, environmentally induced epigenetic changes in neurons were hypothesized to be a valuable underlying mechanism in the context of pain sensitization and chronic visceral pain. This idea was supported by the following observations in animal models of adult stress. In our laboratory, we investigated the effects of (chronic) stress on colonic sensitivity. We observed that bilateral implantation of a CORT micropellet on the dorsal margin of the CeA in rats lead to the development of visceral hypersensitivity that persisted for a prolonged time either after the implanted CORT-micropellets had fully dissolved. We also observed a similar persistent visceral hypersensitivity in rats exposed to WAS for 1 hr /day for 7 days. Animal models of early life adversity also clearly illustrate this principle. In these models, rats exposed to prenatal or neonatal stress exhibit visceral hypersensitivity in adulthood.105 Animal models of maternal separation, limited nesting and odor associated learning demonstrate visceral hypersensitivity in adulthood, long after the initial stressor in neonatal life has been removed.106–108 Compared to undisturbed littermates, animals that received neonatal stress had long-lasting changes in gene expression, underlying and inducing visceral hypersensitivity. The longevity of these changes is best explained through stress-induced detrimental changes in epigenetic regulation. An overview of the epigenetic changes involved in visceral hypersensitivity is presented in Table 1. These epigenetic changes will be discussed in detail below.
Table 1.
Summary of epigenetic changes involved in visceral hypersensitivity and the effects of pharmacological treatment in rodent models.
| Location | Epigenetic modification | Species | Sex | Model | Effect | Downstream gene expression | Pharmacology to reverse visceral hypersensitivity | Ref |
|---|---|---|---|---|---|---|---|---|
| Amygdala | DNA methylation | Rat | Male | Repeated WAS | ↑GR methylation, ↓CRH methylation | ↓GR, ↑CRH | HDAC inhibitor TSA i.c.v. | 109 |
| Histone acetylation | Rat | Male | CORT implant | ↓GR promotor acetylation | ↓GR | HDAC inhibitor TSA intra-amygdala | 110 | |
| Spinal cord | miRNA | Rat | Female | Neonatal cystitis | ↑miR181a & miR181b | ↓GABAAα−1 | / | 111 |
| Rat | Female | Neonatal cystitis | ↑miR-92–3p | ↓KCC2, ↓VGAT | miR-92 lentivirus i.t. | 112 | ||
| Histone acetylation | Rat | Both | Prenatal + adult HeICS | ↑H3 acetylation @BDNF promoter | ↑BDNF | BDNF siRNA i.t. HAT inhibitor curcumin p.o. HAT inhibitor anacardic acid i.t. |
105 | |
| Rat | Male | Neonatal + adult TNBS | ↑H3K9 & H3K12 acetylation @BDNF promoter | ↑BDNF | HAT inhibitor garcinol i.t. | 113 | ||
| Rat | Female | Forced swim test | ↓Acetylation mGlu2/3 promoter | ↓mGlu2/3 | HDAC inhibitor SAHA i.t. | 114 | ||
| Rat | Male | Maternal separation | ↓Global H4K12 acetylation | / | HDAC inhibitor SAHA i.p. | 119 | ||
| DRG | miRNA | Rat | Male | TNBS | ↓miR-199a | ↑TRPV1 | mir-199a lentivirus i.p. | 120 |
| DNA methylation | Rat | Male | Repeated WAS | ↑GR, CNR1 methylation | ↓GR, ↓CNR1 | DNMT1 siRNA i.t. | 121 | |
| Histone acetylation | Rat | Male | Repeated WAS | ↑TRPV1 acetylation | ↑TRPV1 | / | 121 |
Downward arrows indicate a decrease whereas upward arrows indicate an increase in epigenetic modifications or gene expression levels. / indicates not examined in the study. Pharmacological inhibitors were infused directly into the targeted area. DRG, dorsal root ganglia; miRNA, microRNA; WAS, water avoidance stress; CORT, corticosterone; HeICS, heterotypic intermittent chronic stress; TNBS, trinitrobenzenesulfonic acid; GR, glucocorticoid receptor; CRH, corticotropin-releasing hormone; H3, histone 3; BDNF, brain derived neurotrophic factor; H3K9, lysine 9 on histone 3; H3K12, lysine 12 on histone 3; mGlu2/3, group II metabotropic glutamate receptor; H4K12, lysine 12 on histone 4; CNR1, cannabinoid receptor 1; TRPV1, transient receptor potential cation channel subfamily V member 1; GABAAα−1, gamma-aminobutyric acid type A receptor alpha1 subunit; KCC2, Solute carrier family 12 member 5; VGAT, vesicular GABA transporter; HDAC, histone deacetylase; TSA, trichostatin A; HAT, histone acetyltransferase; SAHA, suberoylanilide hydroxamic acid; DNMT1, DNA methyltransferase 1; siRNA, small interfering RNA
4.2. Supraspinal epigenetic mechanisms responsible for visceral hypersensitivity
In our laboratory, we reported that DNA methylation patterns of key genes involved in the HPA axis were changed in the CeA of male Fisher rats exposed to 7 days of repeated WAS. Chronic stress increased the methylation of several CpG sites in the 17 promoter of GR, whereas the methylation at CpG dinucleotide 6 of CRH was decreased. These changes in DNA methylation were associated with concomitant decreases in GR and increases in CRH gene expression in the CeA of these animals, and resulted in heightened responses to visceral stimuli.109 As GR signaling can directly inhibit CRH expression, the decreased GR expression might have been directly responsible for the increased CRH expression. Interestingly, direct intracerebroventricular infusion of the HDAC inhibitor, trichostatin A (TSA), directly preceding WAS, reversed the colonic hypersensitivity indicating that central changes in histone acetylation were involved in stress-induced colonic hypersensitivity.109 In a subsequent study from our team, in rats with elevated amygdala CORT direct infusion of HDAC inhibitors into the CeA of male rats reversed colonic hypersensitivity. On a molecular level, CORT implantation reduced H3K9 acetylation at the GR promotor level. This deacetylation may have been caused by Sirtuin-6, a HDAC that was bound on the histones at the GR promoter. Sirtuin-6 may have decreased histone acetylation and consequently induced gene silencing. Not surprisingly, TSA infusion prevented Sirtuin-6 binding to the GR histones, restored GR expression and prevented visceral hypersensitivity.110 Both studies from our laboratory illustrate the importance of epigenetic mechanisms in the CeA-mediated induction of visceral pain. Interestingly, infusions of a HDAC inhibitor reversed both DNA methylation and histone deacetylation. This may imply that histone deacetylation precedes changes in DNA methylation in the CeA. The number of acetyl groups on the histone tails determines which other epigenetic regulators are attracted to the DNA. Hence, fewer acetylated residues may favor DNMTs. To date, no studies have been published that investigated miRNA-mediated regulation of GR and CRH in the CeA of stressed animals and their potential influence on visceral pain. These studies represent our future area of investigation.
4.3. Epigenetic mechanisms in the spinal cord of visceral hypersensitive animals
An example of bottom-up sensitization is the animal model for neonatal cystitis in which neonates receive an intravesicular injection of zymosan in the bladder. Often this neonatal challenge is combined with a second challenge in adulthood to trigger an increased response. Using this experimental model upregulation of miR-181a and miR-181b in the spinal cord has been observed.111 Both miRNAs have multiple complementary binding sites in the 3’ UTR region of GABAA receptors and induced long-term downregulation of GABAAa-1 in the adult animals. As GABAA receptors are important inhibitors of the pain signaling pathways, spinal inhibition of GABAergic neurotransmission may have unmasked excitatory pathways that resulted in visceral hypersensitivity.111 In a subsequent study of the same group, it was reported that other miRNAs, affecting different components of the GABA inhibitory pathway, were also upregulated after neonatally induced cystitis (and adult re-challenge). For instance, the upregulation of miR-92–3p caused the downregulation of KCC2 and VGAT in the lumbosacral spinal cord. Loss of these two ion transport channels interfered with GABA-mediated inhibition of pain signaling.112 Administration of a miR-92–3p sponge (a miRNA inhibitor) to the spinal cord of these animals, reversed visceral hypersensitivity, highlighting the importance of miR-mediated mechanisms.112 Detrimental effects of prenatal, neonatal and adult stress (top-down mechanism) on spinal cord neurons have been described by other investigators. When pregnant Sprague-Dawley rats underwent heterotypic intermittent chronic stress (HeICS), offspring were prone to visceral hypersensitivity after adult re-challenge. Visceral hypersensitivity in these animals was mediated by the increased pro-nociceptive BDNF expression in the spinal cord. The combination of prenatal HeICS and adult re-challenge, decreased HDAC1 binding to the histones at the BDNF promoter, while at the same time increasing H3 acetylation levels and RNA Pol II binding. Hence, BDNF expression in the spinal cord of these animals was upregulated, resulting in increased visceral nociception. Subsequent administration of BDNF antagonists or the HAT inhibitor curcumin suppressed BDNF upregulation in the spinal cord and decreased visceral hypersensitivity.105 The importance of the pro-nociceptive BDNF pathway was further investigated in the neonatal induced cystitis/adult re-challenge model. In these animals, increases in BDNF were observed in the spinal cord and in the DRG projecting to the colon. On a molecular level, the HAT CREB binding protein was recruited to the BDNF promotor, increasing H3K9 and H3K12 acetylation levels, providing a docking opportunity for RNA Pol II and inducing subsequent BDNF expression. As in the previously discussed model, spinal administration of the HAT inhibitor garcinol blocked the upregulation of BDNF and prevented colonic hypersensitivity in these animals.113 Cao et al. showed that a forced swim test (a model of subchronic adult stress) can induce colonic hypersensitivity in adult rats. However, pretreating these rats with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA or vorinistat) increased the expression of mGlu2/3 receptors. These receptors have analgesic effects in the spinal cord and their upregulation attenuated colonic hypersensitivity.114 Interestingly, the mGlu2 receptor is also important in the estrogen-induced visceral hypersensitivity. 17b-estradiol-induced colonic hypersensitivity was reversed by SAHA administration, due to increases in histone acetylation at the mGlu2 promoter.115
Early life adversity is associated with the development of visceral hypersensitivity. Evidence stems from epidemiological studies in humans and animal models that early life adversity is a risk factor for the development of chronic visceral hypersensitivity.116,117 A widely used animal model for early life adversity is maternal separation, as it can trigger the development of gastrointestinal disorders and psychological disorders.118 The profound influence of maternal separation on the epigenetic mechanisms in the spinal cord was illustrated by Moloney at al. (2015). Maternal separation of Sprague-Dawley rats lead to visceral hypersensitivity and a marked global decrease in H4K12 acetylation in the spinal cord with the administration of the HDAC inhibitor SAHA in adult animals normalizing acetylation levels and ameliorated colonic hypersensitivity.119
4.4. Epigenetic mechanisms in the dorsal root ganglia of visceral hypersensitive animals
Intracolonic infusion of trinitrobenzenesulfonic acid (TNBS) can be used for bottom-up sensitization and thus cause colonic hypersensitivity in adult rats both during and following recovery from the acute colonic inflammation. Zhou et al. showed that in TNBS-induced visceral hypersensitivity, TRPV1 was upregulated in the colon and DRG of these animals. TRPV1 is a non-selective ligand-gated cation channel that is expressed on peripheral primary afferent sensory neurons. TNBS induced a decrease in colonic miR-199a, a regulator of TRPV1 expression, thus upregulating TRPV1 and increasing colonic sensitivity. These effects were reversed by intraperitoneal injection of lentiviral miR-199a precursors in TNBS-treated animals. miR-199a decreased TRPV1 expression in the colon and DRG with a concomitant increase in colonic response thresholds.120
Chronic adult stress is also known to induce epigenetic changes (contributing to sensitization) of DRG neurons. Exposure to repetitive WAS increased CpG methylation at the GR promoter in nociceptive neuronal cell bodies within L6-S2 DRG, consequently decreasing GR mRNA and protein expression.121 The decrease in GR had profound effects on the anti-nociceptive properties of the endocannabinoid system, through the concomitant decrease in CNR1 expression, as GR acts as one of its transcriptional activators.121 The epigenetic effects of chronic stress were not limited to increases in DNA methylation at the GR promoter. Chronic stress also increased methylation at the Cnr1 promoter and histone acetylation at the TRPV1 promoter.121 The latter mechanism was mediated by the HAT EP300, that was bound to the TRPV1 promoter causing increased H3 acetylation and consequently upregulating TRPV1 expression. The simultaneous decrease of GR/CNR1, which has TRPV1-inhibitory properties, and increase in TRPV1 contributed to colonic hypersensitivity.121 The intrathecal administration of DNMT1-siRNA prevented colonic hypersensitivity by reversing the increases in DNA methylation at the GR and CNR1 promoter levels, but had no effect on TRPV1 methylation or acetylation levels. However, as GR expression was restored, GR could prevent the upregulation of EP300 and thus prevent downstream TRPV1 promoter H3 acetylation. Interestingly, these changes in anti-/pro-nociceptive gene expression were only observed in L6-S2 DRG that innervate the pelvic organs, but were absent in the L4-L5 DRG that innervate the lower extremities.121
4.5. Evidence for epigenetic changes in patients suffering from visceral pain
The number of studies investigating epigenetic changes in patients with visceral pain is limited. However, some studies have indicated that these mechanisms may play a role in diseases that are characterized by visceral pain, such as irritable bowel syndrome or interstitial cystitis. When comparing the miRNA expression profiles between colon biopsies from patients suffering from IBS-D with visceral pain and healthy controls, Zhou et al. reported a decrease in miR-199 in these patients, which correlated with TRPV1 expression and visceral pain scores.120 In a different study, it was found that miR-29a is upregulated in these patients.122 Comparing the genome-wide methylation patterns of peripheral blood mononuclear blood cells from IBS patients and healthy controls, revealed that 133 positions were differentially methylated. These affected genes were associated with the glutathione metabolism related to oxidative stress and neuropeptide hormone activity.123 A genome-wide methylation study on voided urine from patients with interstitial cystitis revealed genes downstream of the MAPK pathways were differentially methylated when compared to healthy controls.124 These studies highlight the epigenetic changes in the target tissues of patients. Changes in miRNA expression or DNA methylation pattern in these tissues can be used as valuable biomarkers for identifying patients at risk for functional visceral pain disorders that typically lack a single diagnostic test. Although an investigation of the epigenetic changes in the CNS of patients with chronic visceral pain is currently impossible we speculate that the epigenetic mechanisms elucidated in animal models may aid in our overall understanding of the underlying epigenetic reprogramming leading to chronic visceral pain in patients.
5. Epigenetic treatment approach for visceral pain
Although epigenetic marks are long-lasting and persistent in nature, they can be further modified or even reversed at a later time. These epigenetic marks are catalyzed and maintained by epigenetic modifier enzymes. In principle, this reversible nature of the epigenetic marks and the potential for the inhibition of these specific epigenetic enzymes, holds potential for the development of epigenetic therapeutics of the treatment of visceral pain.125 From the preclinical models discussed above, we are starting to understand the basic epigenetic mechanisms that underlie chronic visceral pain, which is critical for the development of drugs acting to modulate epigenetic reprogramming.126 In contrast, our knowledge of the therapeutic potential of the epigenetic effects of classic and new drugs on chronic visceral pain is still in its infancy. Classical analgesia such as non-steroidal anti-inflammatory drugs (NSAIDs) and opioids have epigenetic-altering properties, but these drugs are seldom used for this purpose. Aspirin, celecoxib, and sulindac are known to modulate miRNAs, suppress or reverse DNA methylation, and increase histone acetylation of several growth genes in human gastric mucosa, rat colon, and cultured human carcinoma cells.127 As a consequence, long-term administration of these COX-inhibitors could have therapeutically relevant effects on the epigenome of patients with chronic visceral pain. However, these reports were contradicted by one study that did not find any changes in global DNA methylation in blood cells of women on long-term NSAID treatment.128 To date, we know little about the effects if NSAIDs on selective DNA methylation or other epigenetic markers of specific pain-related genes. Recent data suggests that the mechanisms through which opioids induce opioid tolerance, addiction and hyperalgesia involve miRNA dysregulation and histone and DNA methylation of specific opioid receptor genes.129–131 However, due to their undesirable side effects of opioids it is unlikely that the ability of opioids to affect epigenetic programming will be employed to treat pain.
As a proof of concept, we and others have shown the beneficial influence of HDAC inhibition in the central nervous system on visceral hypersensitivity. TSA and SAHA infusions in the brain or spinal cord reversed psychological stress-induced visceral hypersensitivity. In one of our studies, we even managed to identify an HDAC involved in chronic visceral pain. TSA administration inhibited the HDAC Sirtuin-6, among other HDACs, and prevented WAS-induced visceral hypersensitivity. As there exist 5 classes of HDACs, more research is needed to elucidate which other HDACs are involved in the epigenetic pathology of chronic visceral pain. In the aforementioned pre-clinical studies, pharmacological modulation of histone acetylation and DNA methylation had profound effects on nociceptive responses. HAT/HDAC and DNMT inhibitors have obtained FDA approval and are currently being used in the clinic for the treatment of various malignancies or have advanced sufficiently in clinical trials to be considered as potential treatment options.132–135 In contrast, to date there are no miRNA modulating drug therapies that have obtained FDA approval. At this point miRNA therapies are entering the first stages of clinical trials.136,137 Hence, we will focus on the potential use of clinically available epigenetic drugs. Valproic acid, vorinostat and entinostat are HDAC inhibitors approved for clinical treatments. To date, most clinical trials focus on the treatment potential in cancer, and no clinical studies on the efficacy of HDAC inhibitors for modulating visceral pain have been conducted. However, there are indications of their usefulness in pain modulation.138,139 For instance, oral administration of the HDAC inhibitor givinostat reduced pain symptoms of systemic-onset juvenile idiopathic arthritis.140 Moreover, the intravenous administration of the HDAC inhibitor romidepsin improved bone pain in certain patient populations.141 This evidence supports the case that the effects of HDAC inhibitors could be investigated in chronic visceral pain. Furthermore, valproic acid is currently available for treating neurological conditions such as epilepsy and bipolar disorder. 5-azacytidine and decitabine are clinically approved DNMT inhibitors for the treatment of certain cancers.142 Both drugs are analogs to the nucleoside cytidine and are incorporated into the DNA in order to exert their effects on DNMTs. Once incorporated in the DNA, 5-azacytidine and decitabine respectively reversible and irreversibly bind to DNMTs, leading to the depletion of these enzymes and inducing DNA hypomethylation.143,144 Using these two DNMT has several drawbacks. As cell division is the most opportune and efficient time point for these drugs to get incorporated in the DNA, these drugs work well in (rapidly) diving cells.145,146 5-azacytidine can also be incorporated in RNA while decitabine activity is S-phase specific. Moreover, decitabine is rapidly degraded by cytosine deaminase and does not bind to proteins and is thus rapidly excreted from the body.147 Due to these problems, these drugs require high dosing to achieve therapeutic levels for malignancy treatment and even at these levels, the drugs induce unwanted side effects,148 and questions remain whether a non-toxic dose is even possible.149
The availability of epigenetic drugs may open up possibilities to use these drugs for the treatment of chronic visceral pain. However, targeting these pain-related pathways in the central nervous system faces a few unique challenges. First, little is known about the efficacy, especially from the DNMT inhibitors, in slowly or non-dividing cells such as neurons. New inhibitors are being developed specifically for non-dividing cells, which would render them interesting candidates for the modulation of chronic visceral pain. Second, the current generation of epigenetic drugs target the whole epigenome, don’t discriminate between particular HATs/HDACs/DNMTs and don’t distinguish between physiological and pathological epigenetic marks.150 New drugs with higher cellular or protein specificity are being developed and/or tested. For instance, class I and class II HDAC inhibitors have been identified as valid anticancer targets and are being tested in clinical studies.151 However, the potential uncoordinated changes in a number of signaling cascades can induce severe, unpredictable and long-lasting epigenetic dysregulation. Questions remain whether lowering the administered dose could sufficiently reverse subtle pathological DNA methylation or histones acetylation levels, with only minor interference in physiologically relevant epigenetic processes. In this light, it is possible that the dose of the HDAC inhibitor givinostat required to treat systemic-onset juvenile idiopathic arthritis is lower than the dose required to treat chronic myeloproliferative neoplasms or multiple myeloma.152,153 This might indicate that in certain cases non-transformed cells are more susceptible to epigenetic treatment. However, it is unclear whether these working concentrations in the periphery would also have an effect in the central nervous system, which brings us to the third hurdle to overcome. In order to modulate epigenetic pathways in the central nervous system these drugs will need to be able to cross the blood-brain or blood-spinal cord barrier. Certain viral vectors, nanoparticles, exosomes and various other techniques are being used to deliver drugs directly across the blood-brain and blood-spinal cord barrier or prolong their time at these barriers to facilitate their uptake in the central nervous system through other uptake methods, for instance receptor-mediated uptake or passive diffusion.154–160 Just like other drugs, epigenetic drugs could be packaged in lipophilic substances or hydrophilic agents and potentially cross the blood-brain/blood-spinal cord barrier. Despite these improvements in delivery methods, high doses and/or long-term administration of the drug will be required for the drug to reach the central nervous system and exert its effects. Hence, it cannot be excluded that these drugs induce systemic (side) effects as the current generation of epigenetic drugs wasn’t designed to target specific cell types. Fourth, when these drugs have crossed the blood brain barrier, they’ll need to reach the specific areas involved in chronic visceral pain. It is unlikely that in the near future drugs will target only a specific region of the brain, without affecting other regions. However, improvements have been made in brain implants and brain surgery, so it would be possible to directly deliver epigenetic drugs to the area of interest, increasing treatment efficacy and diminishing the potential side effects. Epigenetic mechanisms are also involved in the peripheral pathways of chronic visceral pain. Targeting the central pathways with the current available drugs is extremely challenging. However, targeting the peripheral components of the pain pathway would also be beneficial for patients. Restoration of the epigenetic marks in the neurons at the level of the visceral organs could potentially ameliorate allodynia or pain perception by reducing the input signaling in the pain pathway.
In summary, while still in the early days of discovery, new therapeutics targeting epigenetic processes have the potential to revolutionize the treatment of chronic visceral pain disorders. Chronic visceral pain is a significant health care burden due to the poor efficacy of current therapeutics, in part due to the lack of knowledge of the underlying mechanisms. Epigenetic processes regulating gene expression could be responsible for the transition from acute to chronic visceral pain with differences in methylation, acetylation, or miRNA expression occurring from bottom-up or top-down mechanisms. Specific histone tail acetylation, DNA methylation and miRNA expression patterns can cause long-term changes in expression of ion channels and/or receptors that alter neuronal excitability, promoting chronic visceral pain. However, our understanding of the epigenetic component in visceral pain is still advancing at a rapid pace. There are numerous histone tail modifications, such as histone tail methylation, whose role in visceral pain have yet to be investigated. With ever improving research techniques, it’s only a matter of time before the role of other epigenetic mechanisms will be revealed. Environmental cues, such as childhood or adult stressors or colonic inflammation promote epigenetic modification of peripheral, spinal, and/or central neuronal pathways to induce hypersensitivity, as demonstrated by multiple preclinical models. A growing body of clinical evidence has also demonstrated changes in DNA methylation patterns and miRNA expression associated with chronic visceral pain. Interestingly, there is no uniform relationship between environmental factors and epigenetic outcomes, in fact here may be a dual role of histone acetylation and deacetylation in the development or reduction of visceral pain. For instance, early life stress increases global histone tail acetylation, whereas adult stress has the opposite effect. How these global changes in acetylation specifically affect genes relevant for visceral pain also remains to be studied. A context-specific mechanism would require two completely different treatment therapies. Therefore, personalized pharmacological treatment will be necessary. A person with visceral pain, originating from adult stress, could be helped with HDAC inhibitors, whereas a person with visceral pain, originating from pre- or neonatal stress could only be helped with HAT inhibitors. Although the current generation of drugs lacks sufficient tissue or pathway specificity, the development of new classes of selective compounds and/or targeted delivery systems will open the door to new therapies targeting epigenetic processes to treat chronic visceral pain.
Acknowledgments
TL and BGVM wrote the first draft of the manuscript. COL and ACJ revised the manuscript. TL, COL, and ACJ prepared the figures and table. TL and BGVM revised the final version of the manuscript.
Funding
BGVM is a Senior Research Career Scientist for the Department of Veterans Affairs (IK6 BX003610). ACJ is a Career Development Award recipient from the Department of Veterans Affairs (IK2 BX003630).
Abbreviations
- 5caC
5-carboxylcytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcyosine
- ACTH
adrenocorticotropic hormone
- BDNF
brain derived neurotrophic factor
- BPS
bladder pain syndrome
- CAPS
centrally mediated abdominal pain
- CeA
central nucleus of the amygdala
- CGRP
calcitonin gene related peptide
- ChIP
chromatin immunoprecipitation
- CNR1
cannabinoid receptor 1
- CNS
central nervous system
- CORT
cortisol or corticosterone
- CpG
cytosine-guanosine dinucleotides
- CREB
cAMP response element binding protein
- CRH
corticotropin-releasing hormone
- CRH1
CRH receptor type 1
- DNMT
DNA methyltransferase
- DRG
dorsal root ganglia
- GABAAα−1
gamma-aminobutyric acid type A receptor alpha1 subunit
- GERD
gastroesophageal reflux disease
- GR
glucocorticoid receptor
- H
histone
- H3
histone 3
- H3K12
histone 3, lysine 12
- H3K9
histone 3, lysine 9
- H4K12
histone 4, lysine 12
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HeICS
heterotypic intermittent chronic stress
- HPA
hypothalamus-pituitary-adrenal
- IBD
inflammatory bowel disease
- IBS
irritable bowel syndrome
- IC
interstitial cystitis
- K
lysine
- KCC2
Solute carrier family 12 member 5
- MeCP2
methyl-CpG binding protein 2
- mGlu2/3
group II metabotropic glutamate receptor
- miRNA
microRNA
- MR
mineralocorticoid receptor
- mRNA
messenger RNA
- NBS
narcotic bowel syndrome
- ncRNA
non-coding RNA
- NO
nitric oxide
- NSAID
non-steroidal anti-inflammatory
- Pol
polymerase
- PVN
paraventricular nucleus of the hypothalamus
- SAHA
suberoylanilide hydroxamic acid
- siRNA
small interfering RNA
- TET
ten-eleven translocation protein
- TNBS
trinitrobenzenesulfonic acid
- TRPV1
transient receptor potential cation channel subfamily V member 1
- TSA
trichostatin A
- VGAT
vesicular GABA transporter
- WAS
water avoidance stress
Footnotes
Disclosures
The authors declare no conflicts of interest. The views expressed in this review article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
References
- 1.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. [DOI] [PubMed] [Google Scholar]
- 2.Policy. IoMBoHS. Relieving pain in America A Blueprint for Transforming Prevention, Care, Education and Research. The National Academies Press; Washington; D.C. 2011. [PubMed] [Google Scholar]
- 3.Collett B Visceral pain: the importance of pain management services. Br J Pain. 2013;7(1):6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drossman DA, Hasler WL. Rome IV-Functional GI Disorders: Disorders of Gut-Brain Interaction. Gastroenterology. 2016;150(6):1257–1261. [DOI] [PubMed] [Google Scholar]
- 5.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuner R Central mechanisms of pathological pain. Nat Med. 2010;16(11):1258–1266. [DOI] [PubMed] [Google Scholar]
- 7.Mauck M, Van de Ven T, Shaw AD. Epigenetics of chronic pain after thoracic surgery. Curr Opin Anaesthesiol. 2014;27(1):1–5. [DOI] [PubMed] [Google Scholar]
- 8.Rahn EJ, Guzman-Karlsson MC, David Sweatt J. Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: implications for pain and memory. Neurobiol Learn Mem. 2013;105:133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone LS, Szyf M. The emerging field of pain epigenetics. Pain. 2013;154(1):1–2. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Stefano GB, Kream RM. Epigenetic modification of DRG neuronal gene expression subsequent to nerve injury: etiological contribution to complex regional pain syndromes (Part I). Med Sci Monit. 2014;20:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bali P, Im HI, Kenny PJ. Methylation, memory and addiction. Epigenetics. 2011;6(6):671–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng J, Zhou Y, Campbell SL, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13(4):423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer JM, Kochinke K, Oortveld MA, et al. Epigenetic regulation of learning and memory by Drosophila EHMT/G9a. PLoS Biol. 2011;9(1):e1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20(2):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Million M, Wang L, Wang Y, et al. CRF2 receptor activation prevents colorectal distension induced visceral pain and spinal ERK1/2 phosphorylation in rats. Gut. 2006;55(2):172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochoa-Cortes F, Guerrero-Alba R, Valdez-Morales EE, et al. Chronic stress mediators act synergistically on colonic nociceptive mouse dorsal root ganglia neurons to increase excitability. Neurogastroenterol Motil. 2014;26(3):334–345. [DOI] [PubMed] [Google Scholar]
- 17.Vanner S, Greenwood-Van Meerveld B, Mawe G, et al. Fundamentals of Neurogastroenterology: Basic Science. Gastroenterology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz ES, Gebhart GF. Visceral pain. Curr Top Behav Neurosci. 2014;20:171–197. [DOI] [PubMed] [Google Scholar]
- 19.Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res Rev. 2009;60(1):214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morton DL, Sandhu JS, Jones AK. Brain imaging of pain: state of the art. J Pain Res. 2016;9:613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14(7):502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilder-Smith CH. The balancing act: endogenous modulation of pain in functional gastrointestinal disorders. Gut. 2011;60(11):1589–1599. [DOI] [PubMed] [Google Scholar]
- 23.Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32(12):611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arroyo-Novoa CM, Figueroa-Ramos MI, Miaskowski C, Padilla G, Stotts N, Puntillo KA. Acute wound pain: gaining a better understanding. Adv Skin Wound Care. 2009;22(8):373–380; quiz 381–372. [DOI] [PubMed] [Google Scholar]
- 26.Widgerow AD, Kalaria S. Pain mediators and wound healing--establishing the connection. Burns. 2012;38(7):951–959. [DOI] [PubMed] [Google Scholar]
- 27.Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron. 2014;82(3):522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeilhofer HU, Wildner H, Yevenes GE. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev. 2012;92(1):193–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaggi AS, Singh N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res. 2011;1381:187–201. [DOI] [PubMed] [Google Scholar]
- 30.Lampe A, Doering S, Rumpold G, et al. Chronic pain syndromes and their relation to childhood abuse and stressful life events. J Psychosom Res. 2003;54(4):361–367. [DOI] [PubMed] [Google Scholar]
- 31.Maizels M, Aurora S, Heinricher M. Beyond neurovascular: migraine as a dysfunctional neurolimbic pain network. Headache. 2012;52(10):1553–1565. [DOI] [PubMed] [Google Scholar]
- 32.Scarinci IC, McDonald-Haile J, Bradley LA, Richter JE. Altered pain perception and psychosocial features among women with gastrointestinal disorders and history of abuse: a preliminary model. Am J Med. 1994;97(2):108–118. [DOI] [PubMed] [Google Scholar]
- 33.Drossman DA, Chang L, Bellamy N, et al. Severity in irritable bowel syndrome: a Rome Foundation Working Team report. Am J Gastroenterol. 2011;106(10):1749–1759; quiz 1760. [DOI] [PubMed] [Google Scholar]
- 34.Hooten WM. Chronic Pain and Mental Health Disorders: Shared Neural Mechanisms, Epidemiology, and Treatment. Mayo Clin Proc. 2016;91(7):955–970. [DOI] [PubMed] [Google Scholar]
- 35.Mayer EA. The neurobiology of stress and gastrointestinal disease. Gut. 2000;47(6):861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray CD, Flynn J, Ratcliffe L, Jacyna MR, Kamm MA, Emmanuel AV. Effect of acute physical and psychological stress on gut autonomic innervation in irritable bowel syndrome. Gastroenterology. 2004;127(6):1695–1703. [DOI] [PubMed] [Google Scholar]
- 37.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20(2):78–84. [DOI] [PubMed] [Google Scholar]
- 38.Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505–2511. [DOI] [PubMed] [Google Scholar]
- 39.Sapolsky RM, McEwen BS, Rainbow TC. Quantitative autoradiography of [3H]corticosterone receptors in rat brain. Brain Res. 1983;271(2):331–334. [DOI] [PubMed] [Google Scholar]
- 40.Schulkin J, Gold PW, McEwen BS. Induction of corticotropin-releasing hormone gene expression by glucocorticoids: implication for understanding the states of fear and anxiety and allostatic load. Psychoneuroendocrinology. 1998;23(3):219–243. [DOI] [PubMed] [Google Scholar]
- 41.Shepard JD, Barron KW, Myers DA. Corticosterone delivery to the amygdala increases corticotropin-releasing factor mRNA in the central amygdaloid nucleus and anxiety-like behavior. Brain Res. 2000;861(2):288–295. [DOI] [PubMed] [Google Scholar]
- 42.Johnson AC, Greenwood-Van Meerveld B. Knockdown of steroid receptors in the central nucleus of the amygdala induces heightened pain behaviors in the rat. Neuropharmacology. 2015;93:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson AC, Tran L, Greenwood-Van Meerveld B. Knockdown of corticotropin-releasing factor in the central amygdala reverses persistent viscerosomatic hyperalgesia. Transl Psychiatry. 2015;5:e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myers B, Greenwood-Van Meerveld B. Divergent effects of amygdala glucocorticoid and mineralocorticoid receptors in the regulation of visceral and somatic pain. American journal of physiology Gastrointestinal and liver physiology. 2010;298(2):G295–303. [DOI] [PubMed] [Google Scholar]
- 45.Bradesi S, Schwetz I, Ennes HS, et al. Repeated exposure to water avoidance stress in rats: a new model for sustained visceral hyperalgesia. American journal of physiology Gastrointestinal and liver physiology. 2005;289(1):G42–53. [DOI] [PubMed] [Google Scholar]
- 46.Myers B, Greenwood-Van Meerveld B. Differential involvement of amygdala corticosteroid receptors in visceral hyperalgesia following acute or repeated stress. American journal of physiology Gastrointestinal and liver physiology. 2012;302(2):G260–266. [DOI] [PubMed] [Google Scholar]
- 47.Blanchard EB, Lackner JM, Jaccard J, et al. The role of stress in symptom exacerbation among IBS patients. J Psychosom Res. 2008;64(2):119–128. [DOI] [PubMed] [Google Scholar]
- 48.Myers B, Greenwood-Van Meerveld B. Elevated corticosterone in the amygdala leads to persistent increases in anxiety-like behavior and pain sensitivity. Behav Brain Res. 2010;214(2):465–469. [DOI] [PubMed] [Google Scholar]
- 49.Price JL. Prefrontal cortical networks related to visceral function and mood. Ann N Y Acad Sci. 1999;877:383–396. [DOI] [PubMed] [Google Scholar]
- 50.Guleria A, Karyampudi A, Singh R, et al. Mapping of Brain Activations to Rectal Balloon Distension Stimuli in Male Patients with Irritable Bowel Syndrome Using Functional Magnetic Resonance Imaging. J Neurogastroenterol Motil. 2017;23(3):415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang L, Sundaresh S, Elliott J, et al. Dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis in irritable bowel syndrome. Neurogastroenterol Motil. 2009;21(2):149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukudo S, Nomura T, Hongo M. Impact of corticotropin-releasing hormone on gastrointestinal motility and adrenocorticotropic hormone in normal controls and patients with irritable bowel syndrome. Gut. 1998;42(6):845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bird A Perceptions of epigenetics. Nature. 2007;447(7143):396–398. [DOI] [PubMed] [Google Scholar]
- 54.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839(8):627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108(4):439–451. [DOI] [PubMed] [Google Scholar]
- 56.Guan JS, Xie H, Ding X. The role of epigenetic regulation in learning and memory. Exp Neurol. 2015;268:30–36. [DOI] [PubMed] [Google Scholar]
- 57.Halder R, Hennion M, Vidal RO, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19(1):102–110. [DOI] [PubMed] [Google Scholar]
- 58.Rudenko A, Tsai LH. Epigenetic modifications in the nervous system and their impact upon cognitive impairments. Neuropharmacology. 2014;80:70–82. [DOI] [PubMed] [Google Scholar]
- 59.Geranton SM, Tochiki KK. Could targeting epigenetic processes relieve chronic pain states? Curr Opin Support Palliat Care. 2015;9(2):138–146. [DOI] [PubMed] [Google Scholar]
- 60.Petty E, Pillus L. Balancing chromatin remodeling and histone modifications in transcription. Trends in genetics : TIG. 2013;29(11):621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–5552. [DOI] [PubMed] [Google Scholar]
- 62.Kouzarides T Chromatin modifications and their function. Cell. 2007;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 63.Kim SK, Jung I, Lee H, et al. Human histone H3K79 methyltransferase DOT1L protein [corrected] binds actively transcribing RNA polymerase II to regulate gene expression. J Biol Chem. 2012;287(47):39698–39709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25(8):3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krogan NJ, Kim M, Tong A, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23(12):4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–266. [DOI] [PubMed] [Google Scholar]
- 67.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. [DOI] [PubMed] [Google Scholar]
- 68.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–113. [DOI] [PubMed] [Google Scholar]
- 69.Peters AH, Mermoud JE, O’Carroll D, et al. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet. 2002;30(1):77–80. [DOI] [PubMed] [Google Scholar]
- 70.Hart AK, Fioravante D, Liu RY, Phares GA, Cleary LJ, Byrne JH. Serotonin-mediated synapsin expression is necessary for long-term facilitation of the Aplysia sensorimotor synapse. J Neurosci. 2011;31(50):18401–18411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zamudio N, Bourc’his D. Transposable elements in the mammalian germline: a comfortable niche or a deadly trap? Heredity (Edinb). 2010;105(1):92–104. [DOI] [PubMed] [Google Scholar]
- 72.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. [DOI] [PubMed] [Google Scholar]
- 73.Lubin FD, Gupta S, Parrish RR, Grissom NM, Davis RL. Epigenetic mechanisms: critical contributors to long-term memory formation. Neuroscientist. 2011;17(6):616–632. [DOI] [PubMed] [Google Scholar]
- 74.Chen H, Dzitoyeva S, Manev H. Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor Neurol Neurosci. 2012;30(3):237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39(12):5015–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubiura M, Okano M, Kimura H, Kawamura F, Tada M. Chromosome-wide regulation of euchromatin-specific 5mC to 5hmC conversion in mouse ES cells and female human somatic cells. Chromosome Res. 2012;20(7):837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chouliaras L, van den Hove DL, Kenis G, et al. Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr Alzheimer Res. 2012;9(5):536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chouliaras L, van den Hove DL, Kenis G, et al. Prevention of age-related changes in hippocampal levels of 5-methylcytidine by caloric restriction. Neurobiol Aging. 2012;33(8):1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hashimoto H, Liu Y, Upadhyay AK, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40(11):4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Severin PM, Zou X, Schulten K, Gaub HE. Effects of cytosine hydroxymethylation on DNA strand separation. Biophys J. 2013;104(1):208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mifsud KR, Gutierrez-Mecinas M, Trollope AF, Collins A, Saunderson EA, Reul JM. Epigenetic mechanisms in stress and adaptation. Brain Behav Immun. 2011;25(7):1305–1315. [DOI] [PubMed] [Google Scholar]
- 84.Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10(4):631–647. [PubMed] [Google Scholar]
- 85.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278(6):4035–4040. [DOI] [PubMed] [Google Scholar]
- 86.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31(9):2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21(10):1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dey BK, Mueller AC, Dutta A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription. 2014;5(4):e944014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24(20):2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Preall JB, Sontheimer EJ. RNAi: RISC gets loaded. Cell. 2005;123(4):543–545. [DOI] [PubMed] [Google Scholar]
- 92.Friedman RF KK; Burge CB; Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moradian A, Kalli A, Sweredoski MJ, Hess S. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics. 2014;14(4–5):489–497. [DOI] [PubMed] [Google Scholar]
- 94.Saavedra F, Marty-Lombardi S, Loyola A. Characterization of Posttranslational Modifications on Histone Variants. Methods Mol Biol. 2018;1832:21–49. [DOI] [PubMed] [Google Scholar]
- 95.Kurdyukov S, Bullock M. DNA Methylation Analysis: Choosing the Right Method. Biology (Basel). 2016;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kashi K, Henderson L, Bonetti A, Carninci P. Discovery and functional analysis of lncRNAs: Methodologies to investigate an uncharacterized transcriptome. Biochim Biophys Acta. 2016;1859(1):3–15. [DOI] [PubMed] [Google Scholar]
- 97.Zhang G, Ueberheide BM, Waldemarson S, et al. Protein quantitation using mass spectrometry. Methods Mol Biol. 2010;673:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gundry RL, White MY, Murray CI, et al. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol. 2009;Chapter 10:Unit10 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karch KR, Denizio JE, Black BE, Garcia BA. Identification and interrogation of combinatorial histone modifications. Front Genet. 2013;4:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gul S Epigenetic assays for chemical biology and drug discovery. Clin Epigenetics. 2017;9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeAngelis JT, Farrington WJ, Tollefsbol TO. An overview of epigenetic assays. Mol Biotechnol. 2008;38(2):179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rodriguez-Ubreva J, Ballestar E. Chromatin immunoprecipitation. Methods Mol Biol. 2014;1094:309–318. [DOI] [PubMed] [Google Scholar]
- 103.Yong WS, Hsu FM, Chen PY. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Field SF, Beraldi D, Bachman M, Stewart SK, Beck S, Balasubramanian S. Accurate measurement of 5-methylcytosine and 5-hydroxymethylcytosine in human cerebellum DNA by oxidative bisulfite on an array (OxBS-array). PLoS One. 2015;10(2):e0118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winston JH, Li Q, Sarna SK. Chronic prenatal stress epigenetically modifies spinal cord BDNF expression to induce sex-specific visceral hypersensitivity in offspring. Neurogastroenterol Motil. 2014;26(5):715–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prusator DK, Greenwood-Van Meerveld B. Gender specific effects of neonatal limited nesting on viscerosomatic sensitivity and anxiety-like behavior in adult rats. Neurogastroenterol Motil. 2015;27(1):72–81. [DOI] [PubMed] [Google Scholar]
- 107.Prusator DK, Greenwood-Van Meerveld B. Sex-related differences in pain behaviors following three early life stress paradigms. Biol Sex Differ. 2016;7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yi L, Zhang H, Sun H, et al. Maternal Separation Induced Visceral Hypersensitivity from Childhood to Adulthood. J Neurogastroenterol Motil. 2017;23(2):306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tran L, Chaloner A, Sawalha AH, Greenwood Van-Meerveld B. Importance of epigenetic mechanisms in visceral pain induced by chronic water avoidance stress. Psychoneuroendocrinology. 2013;38(6):898–906. [DOI] [PubMed] [Google Scholar]
- 110.Tran L, Schulkin J, Ligon CO, Greenwood-Van Meerveld B. Epigenetic modulation of chronic anxiety and pain by histone deacetylation. Mol Psychiatry. 2015;20(10):1219–1231. [DOI] [PubMed] [Google Scholar]
- 111.Sengupta JN, Pochiraju S, Kannampalli P, et al. MicroRNA-mediated GABA Aalpha-1 receptor subunit down-regulation in adult spinal cord following neonatal cystitis-induced chronic visceral pain in rats. Pain. 2013;154(1):59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang J, Yu J, Kannampalli P, et al. MicroRNA-mediated downregulation of potassium-chloride-cotransporter and vesicular gamma-aminobutyric acid transporter expression in spinal cord contributes to neonatal cystitis-induced visceral pain in rats. Pain. 2017;158(12):2461–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aguirre JE, Winston JH, Sarna SK. Neonatal immune challenge followed by adult immune challenge induces epigenetic-susceptibility to aggravated visceral hypersensitivity. Neurogastroenterol Motil. 2017;29(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cao DY, Bai G, Ji Y, Karpowicz JM, Traub RJ. EXPRESS: Histone hyperacetylation modulates spinal type II metabotropic glutamate receptor alleviating stress-induced visceral hypersensitivity in female rats. Mol Pain. 2016;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao DY, Bai G, Ji Y, Traub RJ. Epigenetic upregulation of metabotropic glutamate receptor 2 in the spinal cord attenuates oestrogen-induced visceral hypersensitivity. Gut. 2015;64(12):1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bradford K, Shih W, Videlock EJ, et al. Association between early adverse life events and irritable bowel syndrome. Clin Gastroenterol Hepatol. 2012;10(4):385–390 e381–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Drossman DA, Leserman J, Nachman G, et al. Sexual and physical abuse in women with functional or organic gastrointestinal disorders. Ann Intern Med. 1990;113(11):828–833. [DOI] [PubMed] [Google Scholar]
- 118.Gareau MG, Jury J, Perdue MH. Neonatal maternal separation of rat pups results in abnormal cholinergic regulation of epithelial permeability. American journal of physiology Gastrointestinal and liver physiology. 2007;293(1):G198–203. [DOI] [PubMed] [Google Scholar]
- 119.Moloney RD, Stilling RM, Dinan TG, Cryan JF. Early-life stress-induced visceral hypersensitivity and anxiety behavior is reversed by histone deacetylase inhibition. Neurogastroenterol Motil. 2015;27(12):1831–1836. [DOI] [PubMed] [Google Scholar]
- 120.Zhou Q, Yang L, Larson S, et al. Decreased miR-199 augments visceral pain in patients with IBS through translational upregulation of TRPV1. Gut. 2016;65(5):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology. 2015;148(1):148–157 e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haberle J, Gorg B, Rutsch F, et al. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med. 2005;353(18):1926–1933. [DOI] [PubMed] [Google Scholar]
- 123.Mahurkar S, Polytarchou C, Iliopoulos D, Pothoulakis C, Mayer EA, Chang L. Genome-wide DNA methylation profiling of peripheral blood mononuclear cells in irritable bowel syndrome. Neurogastroenterol Motil. 2016;28(3):410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bradley MS, Burke EE, Grenier C, Amundsen CL, Murphy SK, Siddiqui NY. A genome-scale DNA methylation study in women with interstitial cystitis/bladder pain syndrome. Neurourol Urodyn. 2018;37(4):1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–263. [DOI] [PubMed] [Google Scholar]
- 126.Buchheit T, Van de Ven T, Shaw A. Epigenetics and the transition from acute to chronic pain. Pain Med. 2012;13(11):1474–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yiannakopoulou E Targeting epigenetic mechanisms and microRNAs by aspirin and other non steroidal anti-inflammatory agents--implications for cancer treatment and chemoprevention. Cell Oncol (Dordr). 2014;37(3):167–178. [DOI] [PubMed] [Google Scholar]
- 128.Wilson LE, Kim S, Xu Z, Harlid S, Sandler DP, Taylor JA. Non-Steroidal Anti-Inflammatory Drug Use and Genomic DNA Methylation in Blood. PLoS One. 2015;10(9):e0138920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Doehring A, Oertel BG, Sittl R, Lotsch J. Chronic opioid use is associated with increased DNA methylation correlating with increased clinical pain. Pain. 2013;154(1):15–23. [DOI] [PubMed] [Google Scholar]
- 130.Hwang CK, Wagley Y, Law PY, Wei LN, Loh HH. MicroRNAs in opioid pharmacology. J Neuroimmune Pharmacol. 2012;7(4):808–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sun H, Maze I, Dietz DM, et al. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci. 2012;32(48):17454–17464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buckstein R, Yee K, Wells RA, Canadian Consortium on Evidence-based Care in MDS. 5-Azacytidine in myelodysplastic syndromes: a clinical practice guideline. Cancer Treat Rev. 2011;37(2):160–167. [DOI] [PubMed] [Google Scholar]
- 133.Friday BB, Anderson SK, Buckner J, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14(2):215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Greenberg PL, Garcia-Manero G, Moore M, et al. A randomized controlled trial of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving decitabine. Leuk Lymphoma. 2013;54(2):321–328. [DOI] [PubMed] [Google Scholar]
- 135.Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121(23):4655–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol Ther Nucleic Acids. 2017;8:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Titze-de-Almeida R, David C, Titze-de-Almeida SS. The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res. 2017;34(7):1339–1363. [DOI] [PubMed] [Google Scholar]
- 138.Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today. 2014;19(5):654–660. [DOI] [PubMed] [Google Scholar]
- 139.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–5318. [DOI] [PubMed] [Google Scholar]
- 140.Vojinovic J, Damjanov N, D’Urzo C, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011;63(5):1452–1458. [DOI] [PubMed] [Google Scholar]
- 141.Niesvizky R, Ely S, Mark T, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117(2):336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics. 2015;7:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. [DOI] [PubMed] [Google Scholar]
- 144.Nguyen CT, Weisenberger DJ, Velicescu M, et al. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2’-deoxycytidine. Cancer Res. 2002;62(22):6456–6461. [PubMed] [Google Scholar]
- 145.Brunner N, Bronzert D, Vindelov LL, Rygaard K, Spang-Thomsen M, Lippman ME. Effect on growth and cell cycle kinetics of estradiol and tamoxifen on MCF-7 human breast cancer cells grown in vitro and in nude mice. Cancer Res. 1989;49(6):1515–1520. [PubMed] [Google Scholar]
- 146.Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I-II study on 5-aza-2’-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs. 1997;8(4):358–368. [DOI] [PubMed] [Google Scholar]
- 147.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008;112(11):2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hollenbach PW, Nguyen AN, Brady H, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5(2):e9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ritchie EK. Safety and efficacy of azacitidine in the treatment of elderly patients with myelodysplastic syndrome. Clin Interv Aging. 2012;7:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. [DOI] [PubMed] [Google Scholar]
- 151.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–5468. [DOI] [PubMed] [Google Scholar]
- 152.Galli M, Salmoiraghi S, Golay J, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89(2):185–190. [DOI] [PubMed] [Google Scholar]
- 153.Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150(4):446–455. [DOI] [PubMed] [Google Scholar]
- 154.Georgieva JV, Hoekstra D, Zuhorn IS. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood-Brain Barrier. Pharmaceutics. 2014;6(4):557–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gray SJ, Woodard KT, Samulski RJ. Viral vectors and delivery strategies for CNS gene therapy. Ther Deliv. 2010;1(4):517–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–530. [DOI] [PubMed] [Google Scholar]
- 157.Masserini M Nanoparticles for brain drug delivery. ISRN Biochem. 2013;2013:238428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Peura L, Malmioja K, Huttunen K, et al. Design, synthesis and brain uptake of LAT1-targeted amino acid prodrugs of dopamine. Pharm Res. 2013;30(10):2523–2537. [DOI] [PubMed] [Google Scholar]
- 160.Song Q, Song H, Xu J, et al. Biomimetic ApoE-Reconstituted High Density Lipoprotein Nanocarrier for Blood-Brain Barrier Penetration and Amyloid Beta-Targeting Drug Delivery. Mol Pharm. 2016;13(11):3976–3987. [DOI] [PubMed] [Google Scholar]