

Abstract
Methylmercury (MeHg) is a global pollutant and potent neurotoxin. In humans, MeHg damages the central nervous system (CNS), causing irreversible neuronal shrinkage, and neuronal loss. Most chelators for clinical mercury detoxification are thiol-containing agents. N,N ‘bis-(2-mercaptoethyl) isophthalamide (NBMI) is a lipophilic thiol agent synthesized from natural chemicals. NBMI has high affinity for mercury, cadmium and lead, and can decrease their concentrations in polluted water. However, the efficacy of NBMI for MeHg toxicity has yet to be evaluated in intact animals. Here we used the nematode Caenorhabditis elegans (C. elegans) to test the efficacy of NBMI in attenuating MeHg toxicity in vivo in the whole organism. The results showed that NBMI reduced both the acute toxicity (125 μM MeHg, 1 h) and chronic (5 μM MeHg, 24 h) MeHg toxicity. Co-treatment with NBMI achieved maximal efficacy against MeHg toxicity, however delayed treatment 6 days after initiation of exposure was also effective at reducing neurotoxicity. Co-treatment of NBMI reduced the worms’ death rate, structural damage in DAergic neurons, and restored antioxidant response levels. While this study provides proof of principle for the therapeutic value of NBMI in MeHg toxicity, future studies are needed to address the cellular and molecular mechanisms and translatability of these effects to humans and other animals.
Keywords: Methylmercury, Chelation, Oxidative stress, DAergic neuron
Introduction
Recognition of the toxicity of organomercury dates back to the 1860s when dimethylmercury (DiMeHg) was firstly synthesized in a chemical laboratory (Nierenberg et al. 1998). In subsequent years, sporadic cases of organomercury poisoning were reported (Bakir et al. 1973; Clarkson 2002; Davis et al. 1994). The industrial use of mercury compounds, such as mercury sulfate as a catalyst to produce acetaldehyde, increased the levels of mercury in aquatic environments. The conversion of industrially-derived inorganic mercury into organomercury (mainly methylmercury, MeHg) and its biomagnification in the aquatic food chain, caused chronic and fetal MeHg poisoning (Minamata disease, MD) in Japan in the 1950s and 1960s (Ekino et al. 2007).
MD in adults reflects severe MeHg poisoning after chronic ingestion of fish-borne MeHg. Although some MD patients received supportive medical treatments, the prognosis of severe cases was poor, with 32.8% mortality in acute MD (Davies 1991). In early 1972, another outbreak of acute MeHg poisoning took place in an Iraqi rural area, where local farmers misused MeHg-coated grain seeds for food preparation (Bakir et al. 1973). Within several months, 6530 MeHg poisoning cases with 459 hospital deaths were reported (Bakir et al. 1973). Patients who received treatment either with 2,3-dirmecaptopropanol (dimercaprol), N-acetyl-d-L-penicillamine, and penicilamine in Iraq showed only a limited or no therapeutic benefit for severe or very severe cases of MeHg poisoning (Bakir et al. 1976; Clarkson et al. 1981). Furthermore, the removal of blood MeHg designated as percentage decline in blood MeHg level after treatment was not correlated with the prognosis (Bakir et al. 1976). It was also noted that the prognosis was correlated with the clinical stages. For severe or very severe cases, there was no clinical improvement even though blood MeHg levels were reduced, while clinical improvement was found in mild or moderate cases irrespective of the treatment (Bakir et al. 1976). This was also corroborated in a more recent DiMeHg poisoning case, establishing that the patient’s neurologic deterioration continued and persisted while receiving chelation therapy with 2,3-dimercaptosuccinic acid (DMSA) and after exhibiting a marked decrease in the body-burden of Hg (Nierenberg et al. 1998). Failure in the treatment of MeHg poisoning reveals that once severe brain damage starts, it cannot be reversed by lowering MeHg’s body burdens with the use of conventional thiol-containing medications.
The delayed neuropathological outcomes after severe intoxications with MeHg or DiMeHg suggest that the organomecurials first are distributed (and bind) to a great proportion of non-target or non-critical proteins. Subsequently, a small portion of the total burden of Hg are distributed to the brain, where they will interact with critical proteins and trigger a cascade of events that will results in neuronal cell death. The conventional thiol-containing medications possibly only remove Hg from the non-target proteins located outside the central nervous system. Thus, it is fundamental to search for new therapeutic agents that can remove Hg from deep compartments (i.e., the nervous system). With respect to animal-based experimental studies, chelating therapy has been reported to present beneficial effects in decreasing mercury body burden and preventing toxicity in MeHg-exposed animals, but such effects also depend on the degree of exposures and time-point when the antidotal therapy is initiated (Bridges et al. 2009; Carvalho et al. 2007).
Today, acute organomercury poisoning cases are rare. However, there is still a need to develop novel antidotes for organomercury poisoning, given that current treatments are ineffective for severe organomercury poisoning and the potential adverse health effect concern for those having elevated blood MeHg level by routinely ingesting marine foods (Myers et al. 2000; Silbernagel et al. 2011). The exposure to neurotoxic levels of MeHg via fish consumption is not uncommon (Silbernagel et al. 2011) and the recovery after stopping fish ingestion is very slow. Accordingly, the development of new chelating agent that could accelerate the neurological recovery after moderate exposure to MeHg can be useful. Recently, in a human study on the effect of N,N’bis-(2-mercaptoethyl) isophthalamide (NBMI) for mild chronic mercury (Hg) toxicity, it was shown that NBMI failed to lower the body burden of Hg, but had some beneficial effects in ameliorating clinical symptoms associated with Hg exposure (Schutzmeier et al. 2018).
NBMI is a lipophilic thiol agent synthesized from natural chemicals (Clarke et al. 2012b). The two components used to synthesize NBMI (dicarboxybenzoate and cysteamine) are derivatives of human food (fruit and meat) (Clarke et al. 2012b). Originally, NBMI was used as a complexing agent to eliminate metals in polluted water by Environmental Protection Agency (Clarke et al. 2012b). An in vitro study has shown that NBMI has a high affinity for Hg, cadmium, and lead. Polluted water treated with NBMI contained no detectable metals, including Hg and lead (Zaman et al. 2007), and the complex could not be leached out in various experimental conditions (Zaman et al. 2007), supporting the concept that NBMI may serve as a potential antidote for metal poisoning.
The model organism Caenorhabditis elegans (C. elegans) is being used to evaluate the efficacy and safety of new therapeutics (Xiong et al. 2017). Early findings on NBMI showed positive effects on mercury chloride-induced toxicity in rats (Clarke et al. 2012b), as well as in humans chronically exposed to Hg vapor (Schutzmeier et al. 2018). However, no studies have been performed to date to test the efficacy of NBMI for organomercury toxicity in vivo (Secor et al. 2011). In the current study, we used C. elegans to test the efficacy of NBMI in both acute and chronic toxicity MeHg paradigms, and its possible mechanisms associated with MeHg toxicity (transportation and detoxification). We found that, analogous to the irreversible nature of severe MeHg poisoning in human cases, the death rate of C. elegans could be prevented only upon co-treatment of NBMI with MeHg, while post-treatment NBMI had more limited beneficial effects. Nevertheless, co-treatment of NBMI with MeHg had greater protection than pre-treatment in a series of chronic toxicity tests. Altogether, our novel findings demonstrate the efficacy of NBMI in treating MeHg poisoning.
Materials and Methods
NBMI Treatment
Synchronized worms were treated with NBMI in NGM buffer. Briefly, newly hatched Larvae stage 1 worms were treated in 100 μl NGM buffer (3 g NaCl, 2.5 g peptone, 975 ml H2O, 1 ml cholesterol (5 mg ml – 1 in ethanol), 1 ml nystatin, 1 ml CaCl2 1 M, 1 ml MgSO4 1 M, 25 ml KH2PO4 pH 6). A stock solution of NBMI (1000×) was prepared with DMSO, which final concentration is 0.1% in the treatment buffer. For 1 h treatment, worms were treated with NGM without food. For 24 h treatment, worms were treated with dehydrated dead OP50 in NGM buffer (Ke and Aschner 2019).
C. elegans Strains
C. elegans were cultured at 20 °C in a thermostatic incubator. Standard recipes of worm growth medium (NGM) were used to make agar plates, which were seeded with OP50 bacteria. The strains used in this study were N2, CL2166 (dvIs19 [(pAF15) gst-4p::GFP::NLS] III), LD1171 (IdIs3[gcs-1p::GFP_rol-6(su1006)]), and OH7193 (otIs181 III;him-8(e1489) IV). All C. elegans strains were provided by the Caenorhabditis Genetic Center (University of Minnesota). To achieve worm synchronization, eggs were harvested by bleaching gravid worms and sucrose column separation of eggs. After 18 to 20 h, the newly hatched larvae stage 1 (L1) worms were treated.
Lethality Assay
Synchronized newly hatched worms were counted by sampling a 50 μl sample on a glass slide. The worms were pelleted by centrifugation at 609 g for 1 min. The concentration of worms was titrated using NGM buffer to the final concentration of 10 worms per microliter. In chronic treatment experiment, 92.5 μl worms were mixed with 5 μl dehydrated dead OP50 (Ke and Aschner 2019), and 2.5 μl 40× concentrated methylmercury chloride (MeHgCl) stock solution. In acute treatment experiment, 97.5 μl worms were mixed with 2.5 μl 40× concentrated MeHgCl stock solution. Thus, for each experiment, ~ 1000 worms were treated in 100 μl NGM buffer in a sterile 96-well plate (costar, corning). After treatment, worms were washed 3 times with M9 buffer, and transferred to OP50 seeded 35 mm plates. Twenty-four h later, 200–300 worms were counted for the calculation of death rate. The MeHg doses used to treat worms were based on the characterization of acute MeHg toxicity in a previous study (Ke and Aschner 2019), and they correspond to levels of exposure previously reported in humans (Aschner 2012).
Gene Expression Assay
Total RNA was extracted using TRIzol reagents (Ambion, Life Technologies, Grand Island, NY USA). Briefly, around 1000 worms in 50 μl ddH2O were mixed with 500 μl Trizol, and vortex 10 s for every 5 min. After three vortexes, add 100 μl chloroform, and centrifuge at 20,000 g for 15 min. Then, transfer a 50 μl of top aqueous solution (yields > 100 ng/μl) to the RNA binding column (NEB, Monarch RNA Cleanup Kit, Ipswich, MA, USA) to eliminate organic solvents and salts in the extraction solution. The quantity and quality of RNA were checked using NanoDrop 2000 spectrophotometer (Fisher, Wilmington, DE, USA). To assure high quality RNA used for reverse transcription and q-PCR, only samples that have a single curve peaked at 260 nm were used for gene expression experiment. Fresh RNA samples were reverse transcribed with High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). An aliquot of cDNA templates was quantified by Quantitative Real Time PCR with iCycler Thermal Cycler (BioRad, Hercules, CA, USA). The mRNA levels corresponding to each gene were normalized to the relatively stable expression gene ama-1. The relative mRNA levels were determined with the 2−△△CT method (Livak and Schmittgen 2001). Predesigned probes used in the current study are skn-1 (ID:Ce02407445_g1), sod-1 (ID:Ce02434432_g1), aat-1 (ID: Ce02458009_g1), aat-2 (ID:Ce02479487_g1), aat-3 (ID:Ce02492837_m1).
DAergic Neuron Morphology
Morphology of CEP DAergic neurons were scored as described in a previous study (Ke et al. 2020). Briefly, MeHg or NBMI treated worms were transferred to agar pads after three washes. The worms were paralyzed with 1 mM levamisole. Then, the morphology of DAergic neurons were checked under fluorescence microscope (Olympus BX41). C. elegans has eight (four pairs) DAergic neurons. Three pairs (2 ADEs, 4CEPs) are in the head region. CEP neurons have long dendrites extending to the tip of the mouth. The presence of bright puncta (thereafter named “puncta”) in CEP dendrites were checked for each worm.
ICP-MS
For ICP-MS (Agilent 8800 ICP-QQQ), around 20,000 worms per sample (n ≥ 3) were treated with MeHg or NBMI. After three washes with M9, worms were flash-frozen in liquid nitrogen for three times. Next, the samples were sonicated. After centrifugation, Hg content in the supernatant was measured. Briefly, the sample was digested in the microwave with 1.6 ml bidest H2O, 250 μl HNO3 suprapur and 250 μl HCl suprapur. Hg content was measured with the No gas mode ICP-MS. 0.01 μg/l Rhodium was used as internal standard. The calibration was prepared in 10% HNO3 suprapur and 10% HCl suprapur (concentration range: 1–300 ng/l). The washout solution contained 1 ppm Gold in 5% HNO3 and 5% HCl. An aliquot of supernatant was used to measure protein concentration (BCA protein assay). The content of Hg was calculated with dividing total Hg by total protein (ng Hg/mg protein).
Fluorescence Quantification
After treatment, worms were transferred to agar pads on glass slide, and paralyzed by treating 1 mM levamisole. Images were taken with Leica SP8 confocal microscope. Parameters for imaging were analogous for each treatment group. Quantification of fluorescence level was made with Fiji software. Mitotracker™ Red (Invitrogen) is a mitochondrion-selective probe that binds with thiol group containing molecules in mitochondria. After treatment, N2 worms were incubated with 10 μM Mitotracker™ Red for 1 h followed by 5 washes in M9. After 2 h in OP50 seeded plates, Mitotracker™ Red strained worms were transferred to agar pads for imaging process.
Statistical Analysis
Death rates of worms were analyzed with Chi-square test followed by multiple comparisons with Chi-square partition method. Quantitative data were shown as mean ± SD, and analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. Two-way ANOVA was used for data with a two-factor experimental design. Graphpad 7 (La Jolla, CA, USA) was used for statistical analysis. p < 0.05 was considered to be statistically significant.
Results
To test the efficacy of NBMI in attenuating MeHg’s toxicity, worms were treated with 10–1000 μM NBMI for 1 h, followed by exposure of MeHg in NBMI treatment buffer. The worms’ death rate was significantly inhibited by NBMI (Fig. 1a). However, the death rate was not inhibited by NBMI in worms treated with 10–1000 μM NBMI post MeHg exposure (Fig. 1a).
Fig. 1.
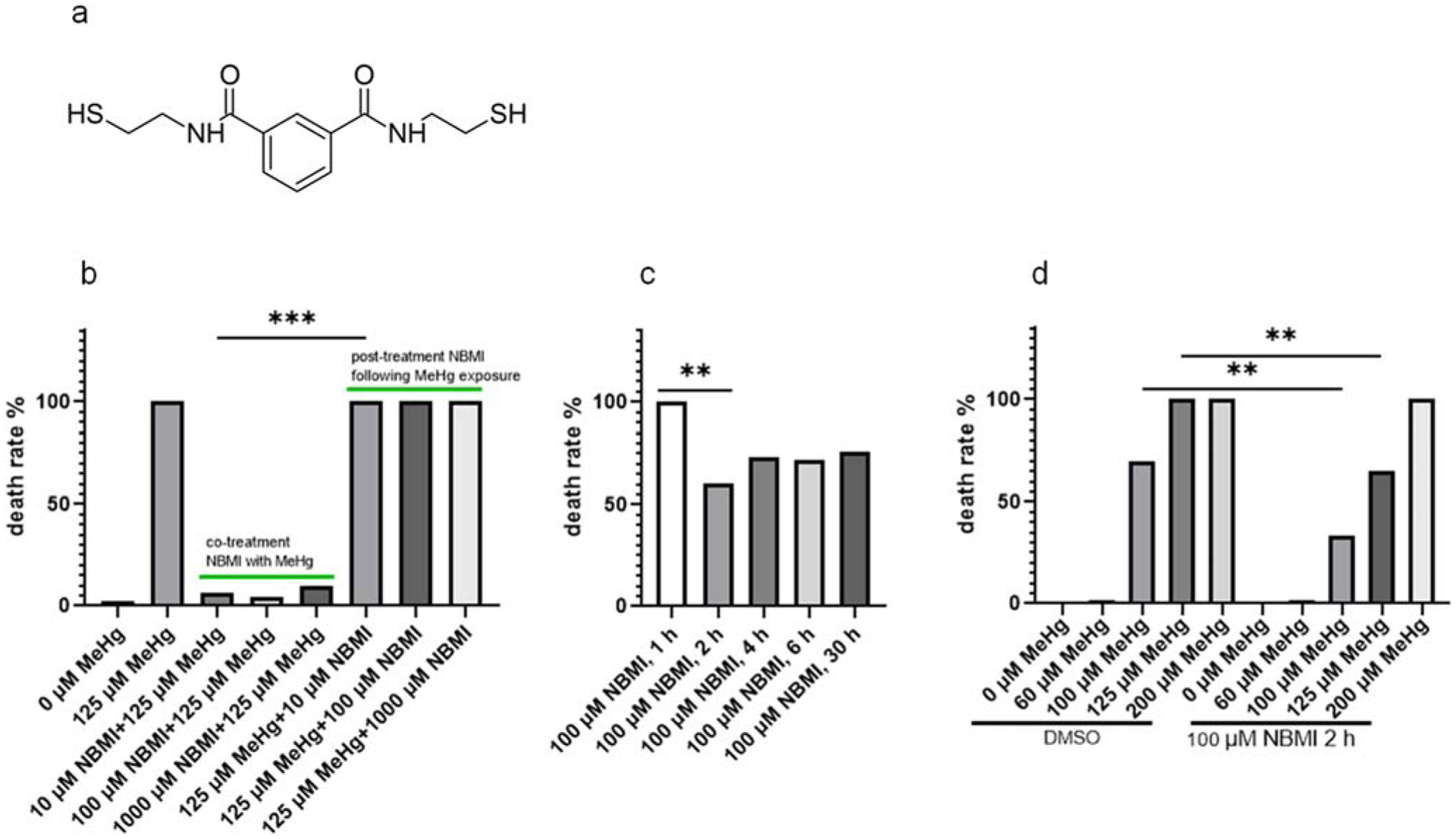
NBMI treatment prevents acute toxicity of MeHg. a Molecular formation and structure of NBMI. b Death rate of worms treated with MeHg and NBMI. For co-treatment NBMI: worms pre-treated with 10–1000 μM NBMI for 1 h, and co-treated with 125 μM MeHg for 1 h. For post-treatment NBMI: worms treated with 125 μM MeHg for 1 h, and post-treated with 10–1000 μM NBMI for 1 h. c Death rate of worms pre-treated with 100 μM NBMI for 0–30 h, and treated with 125 μM MeHg for 1 h in NBMI-free buffer (the medium was replaced before MeHg treatment). d Death rate of worms pre-treated with 100 μM NBMI for 2 h, and treated with 0–200 μM MeHg for 1 h in NBMI-free buffer (the medium was replaced before MeHg treatment). DMSO was used as the vehicle control, which final concentration is 0.1%. Death rate was calculated from 200 to 300 worms per sample. Comparisons of death rate were made with Chi-square test followed by multiple comparisons with Chi-square partition method. The horizontal bars represent a statistical significant difference between the groups with post-hoc multi-comparison test. *P < 0.05, **P < 0.01, and ***P < 0.001
The efficacy of 10 μM NBMI co-treatment in inhibiting the death rate in response to a lethal dose of MeHg (125 μM) suggests that NBMI has strong affinity for MeHg. The effect could result from the in vitro (within the treatment buffer) interaction between MeHg and NBMI. Thus, worms were pre-treated with NBMI, followed by MeHg exposure in NBMI-free buffer to minimize the in vitro interaction between MeHg and NBMI. In this test, the death rate was significantly reduced only upon NBMI treatment for a minimum of 2 h (Fig. 1b). No difference was noted when comparing death rate between 2 h, 4 h, 6 h, or 30 h pre-treatment with NBMI (Fig. 1b). In worms pre-treated with 100 μM NBMI for 2 h, the death rate was significantly reduced by NBMI in the 100 μM and 125 μM MeHg groups (Fig. 1c). No significant effect was observed in the 200 μM MeHg group (Fig. 1c). Based on the fact that post-treatment of NBMI has no effect on the death rate in worms exposed to a lethal dose of MeHg (Fig. 1a), while pre-treatment has a mild yet significant effect (Fig. 1c), it was concluded that co-treatment of NBMI with MeHg has a maximal protective effect.
Accordingly, the mild, but significant effect of NBMI pre-treatment on MeHg toxicity seems to invoke detoxifying mechanisms upon MeHg treatment in C. elegans. Aat-1, aat-2, and aat-3 are the three homologs of human lat-1 gene (Caito et al. 2013). It was hypothesized that these genes play a role in MeHg transport in worms (Caito et al. 2013). Thus, worms treated with a sublethal dose of MeHg (60 μM) were examined following pre-treatment with NBMI. Gene expression analysis revealed that, NBMI treatment alone had no effect on the expression of aat-1, aat-2, or aat-3 (Fig. 2a–2c). However, in worms treated with MeHg following NBMI pre-treatment, the expression of these genes (aat-1 and aat-2) significantly increased compared to other groups (Fig. 2a, b and c). However, the intra-worm content of Hg (ng Hg/mg protein) was not significantly changed between the MeHg and MeHg + NBMI groups (Fig. 2d).
Fig. 2.
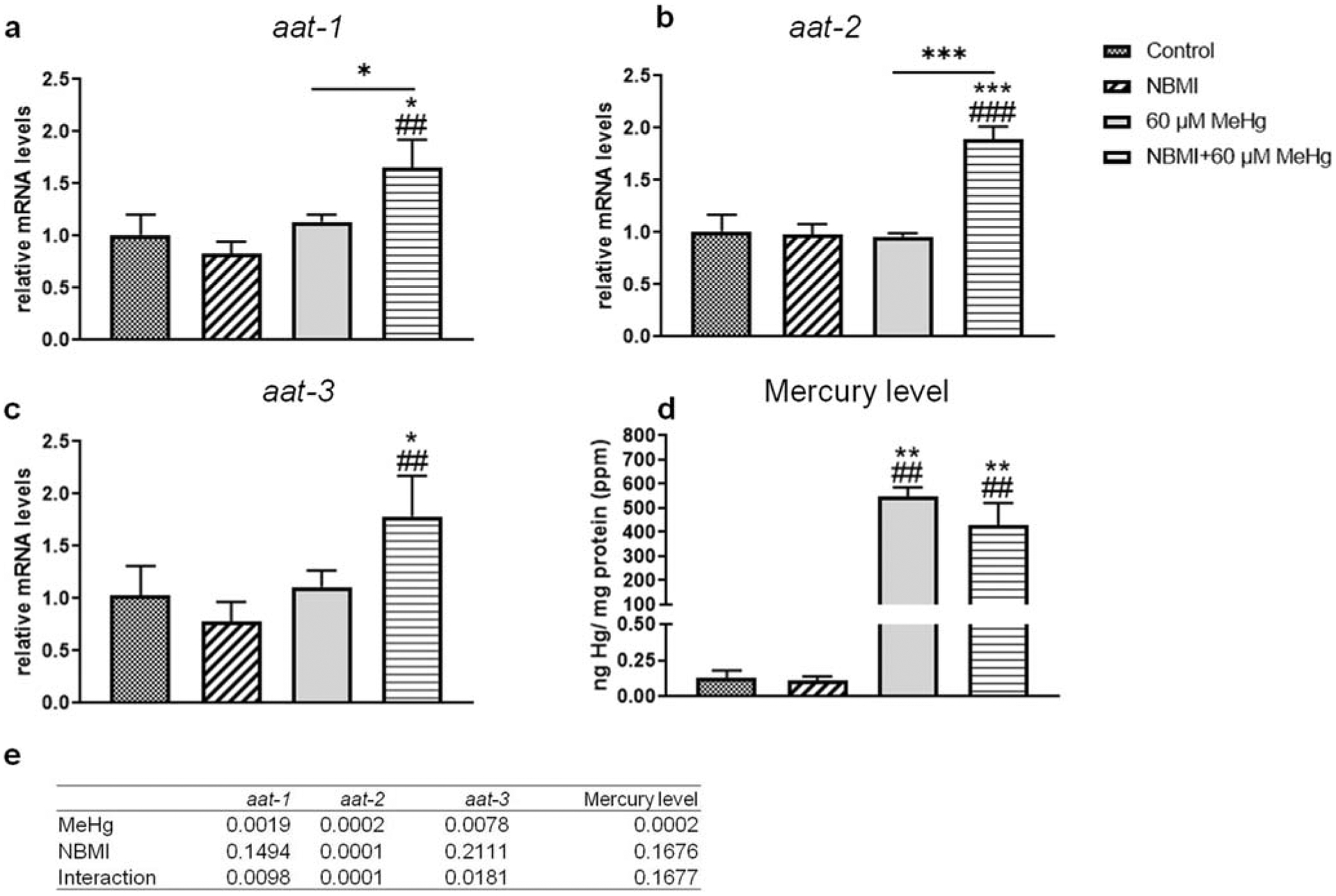
NBMI treatment does not affect intra-worm Hg level. a–c Expression of aat genes in worms treated with 60 μM MeHg or 100 μM NBMI, or pre-treated with 100 μM NBMI for 2 h, then treated 60 μM MeHg for 1 h in NBMI-free buffer. d Intra- worm Hg content of worms treated with MeHg or NBMI. Data are expressed as mean ± SD of at least three biological replicates. e p values of two-way ANOVA analysis. Comparisons were made by two-way ANOVA and Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared with untreated control. #P < 0.05, ##P < 0.01, and ###P < 0.001 as compared with the group with NBMI treatment alone. The horizontal bars represent a statistical significant difference between the groups
To further characterize putative mechanisms of NBMI’s protection, the expression level of skn-1 and sod-1 were compared in the various treatment groups (Fig. 3a, b). MeHg treatment significantly increased the expression of skn-1 (Fig. 3a) and sod-1 (Fig. 3b). However, NBMI pre-treatment failed to affect the expression level of these genes (Fig. 3a, b).
Fig. 3.
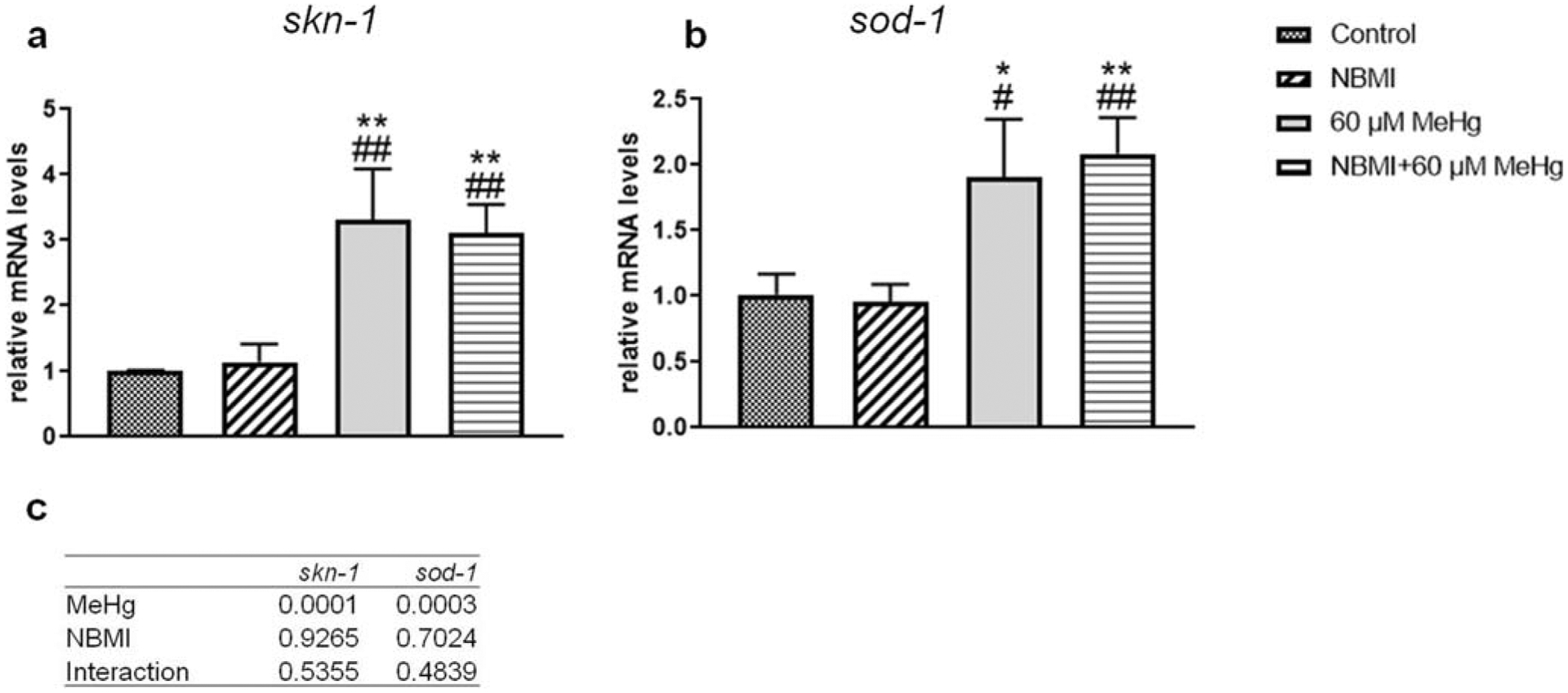
Expression of skn-1 and sod-1 is affected by MeHg but not NBMI. a,b Expression of skn-1, and sod-1 genes in worms treated with 60 μM MeHg or 100 μM NBMI, or pretreated with 100 μM NBMI for 2 h, and treated with 60 μM MeHg for 1 h in NBMI-free buffer. Data are expressed as mean ± SD of at least three biological replicates. c p values of two-way ANOVA analysis. Comparisons were made by two-way ANOVA and Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared with untreated control. #P < .05, ##P < .01, and ###P < .001 as compared with the group with NBMI treatment alone
To evaluate the effect of NBMI on the chronic MeHg toxicity, a chronic exposure model (5 μM MeHg for 10 days) was used to assess DAergic neuron morphology (Ke et al. 2020). In the absence of NBMI treatment, nearly 80% of the worms in the MeHg group showed bright, sphere-like shape puncta in the dendrites of CEP (DAergic) neurons (Fig. 4e, and f). Upon treatment with NBMI, the percentage of puncta-positive worms was significantly reduced to less than 60% (Fig. 4c). The average number of puncta in DAergic dendrites was also significantly reduced by NBMI (Fig. 4c). MeHg tended to increase the number of puncta in the soma of DAergic neurons (2 ADEs, 4 CEPs); however, it is not statistically different between the various groups (Fig. 4b).
Fig. 4.
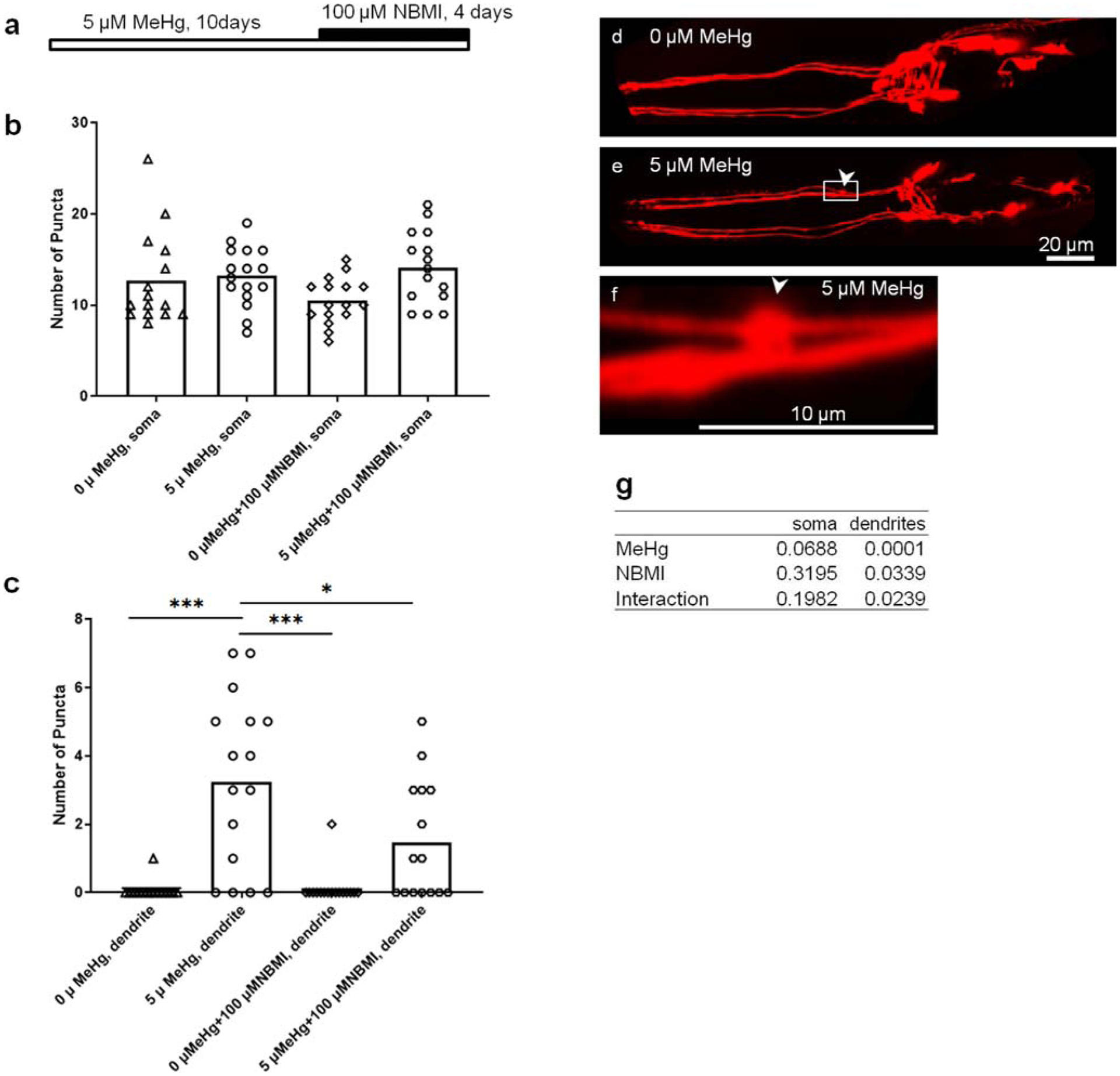
NBMI treatment inhibits MeHg- induced puncta formation in DAergic neurons. a Schematic representation of experimental design. b Puncta numbers in the soma of DAergic neurons of worms treated with MeHg or NBMI. c Puncta numbers in the dendrites of DAergic neurons of worms treated with MeHg or NBMI. d Representative image of mCherry-labeled DAergic neurons (dat-1::mCherry) in the head region of worms without MeHg treatment. e Representative image of mCherry-labeled DAergic neurons (dat-1::mCherry) in the head region of worms treated with 5 μM MeHg. f Magnification of d showing a punctae formed in the dendrite of CEP neuron. g p values of two-way ANOVA analysis. Comparisons were made by two-way ANOVA and Tukey’s multiple comparisons test. The horizontal bars represent a statistical significant difference between the groups. *P < 0.05, **P < 0.01, and ***P < 0.001
To test the effect of NBMI in reducing the pro-oxidant effects of MeHg, worms carrying transgenic reporter genes of antioxidant responses (gst-4 and gsc-1) were analyzed. Here, a relatively short exposure period (24 h) was used according to our previous study showing that the antioxidant response (gst-4 transcriptional level) is time-dependently associated with MeHg exposure (Ke et al. 2020). Treatment with 5 μM MeHg for 24 h induced greater than two-folds increase in gst-4 transcriptional level (Fig. 5). Co-treatment with 100 μM NBMI significantly reduced this event (p < 0.001, Fig. 5), but it remained significantly higher than in the untreated group (p = 0.044, Fig. 5). However, upon pre-treatment with 100 μM NBMI for 2 h, and treatment with MeHg in NMBI-free buffer, gst-4 level was statistically indistinguishable from the MeHg group. Unlike gst-4, MeHg induced a mild increase in the transcriptional level of gcs-1 (p = 0.024, Fig. 6). NBMI co-treatment fully inhibited gcs-1 level (Fig. 6), while pre-treatment had no effect (p = 0.135, Fig. 6).
Fig. 5.
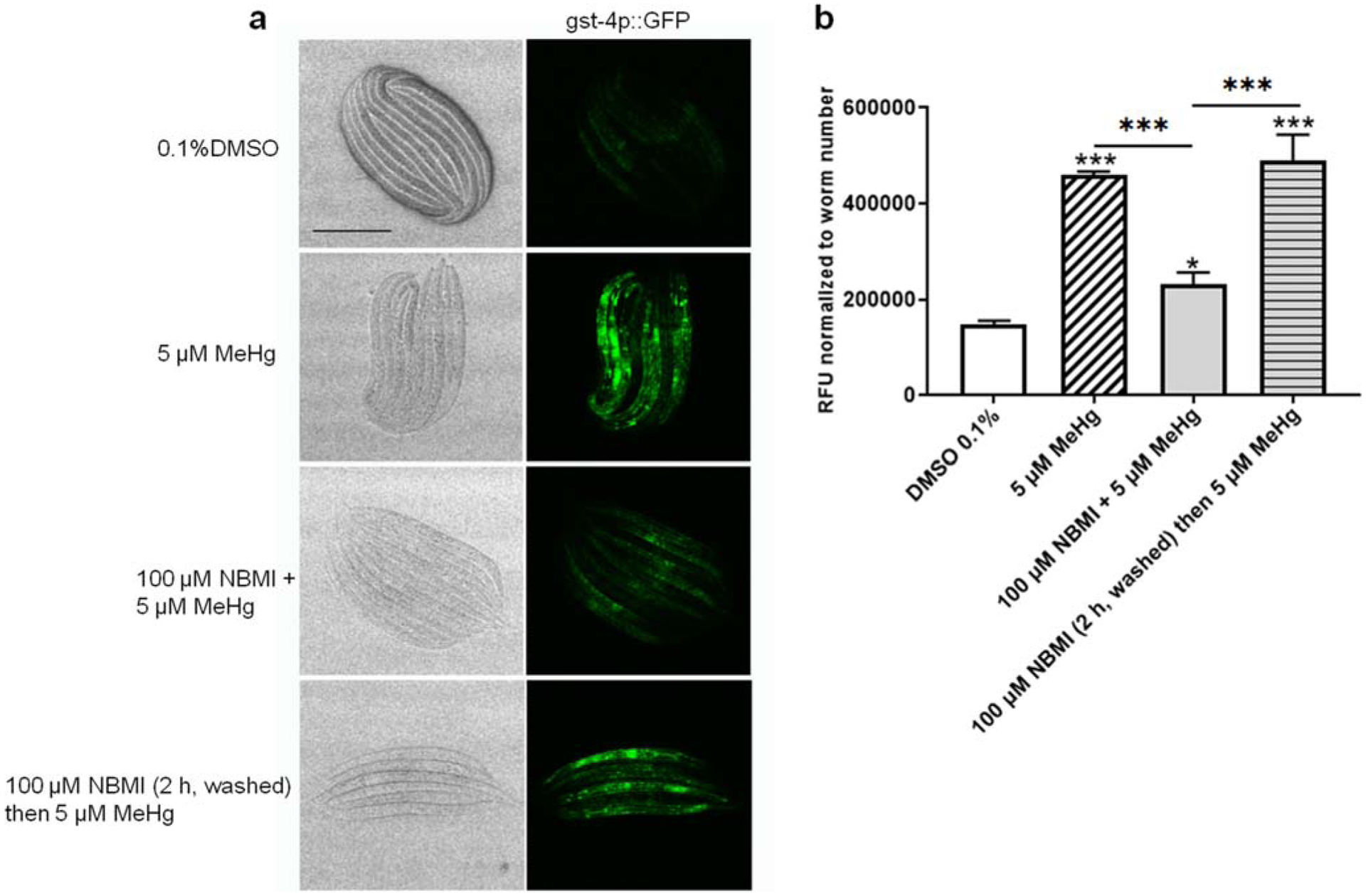
NBMI co-treatment inhibits MeHg-induced upregulation of gst-4. a Bright field and fluorescence images of worms carrying gst-4:: GFP. b Mean fluorescence level of worms treated with MeHg or NBMI. For each group, data were derived from at least 30 worms. Comparisons were made with one-way ANOVA and Tukey’s multiple comparisons test. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared with untreated control. The horizontal bars represent a statistical significant difference between the groups
Fig. 6.
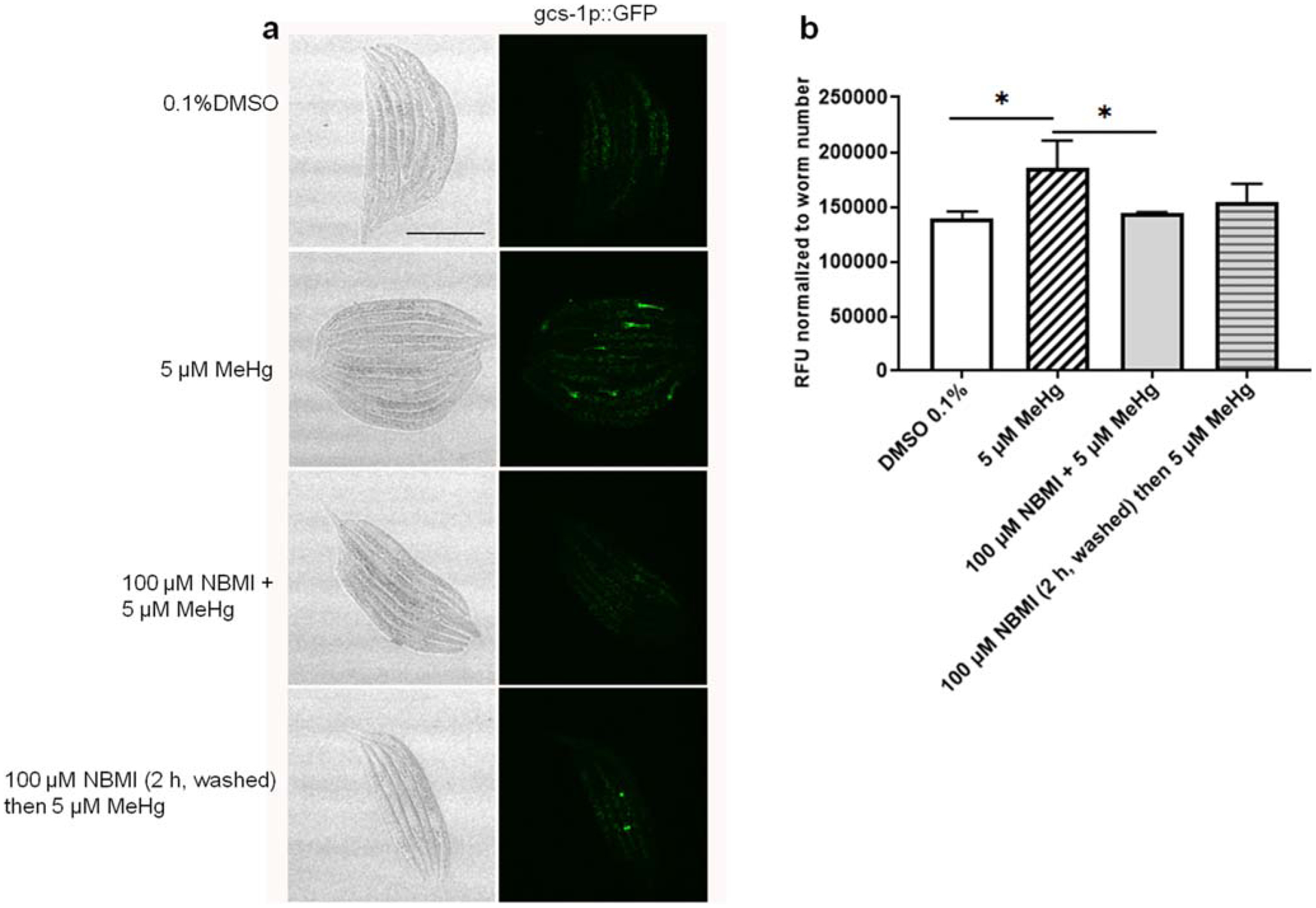
NBMI treatment inhibits MeHg- induced upregulation of gsc-1. a Bright field and fluorescence images of worms carrying gcs-1:: GFP. b Mean fluorescence level of worms treated with MeHg or NBMI. For each group, data were derived from at least 30 worms. Comparisons were made with one-way ANOVA and Tukey’s multiple comparisons test. The horizontal bars represent a statistical significant difference between the groups. *P < 0.05, **P < 0.01, and ***P < 0.001
Previously, we found that MeHg treatment is associated with a decreased fluorescence level of the mitochondria specific probe, mitoTracker Red (Ke and Aschner 2019). MeHg treatment (5 μM) for 24 h significantly decreased the fluorescence (p = 0.029, Fig. 7). Co-treatment with 100 μM NBMI significantly increased the fluorescence to a level statistically indistinguishable from the untreated group (p = 0.066, Fig. 7), and significantly higher than in the MeHg alone treated group (Fig. 7). Consistent with the gene reporter data (Fig. 5 and Fig. 6), pre-treatment with NBMI for 2 h had no statistically significant effect on the fluorescence level compared to the MeHg alone treated group (p = 0.397, Fig. 7).
Fig. 7.
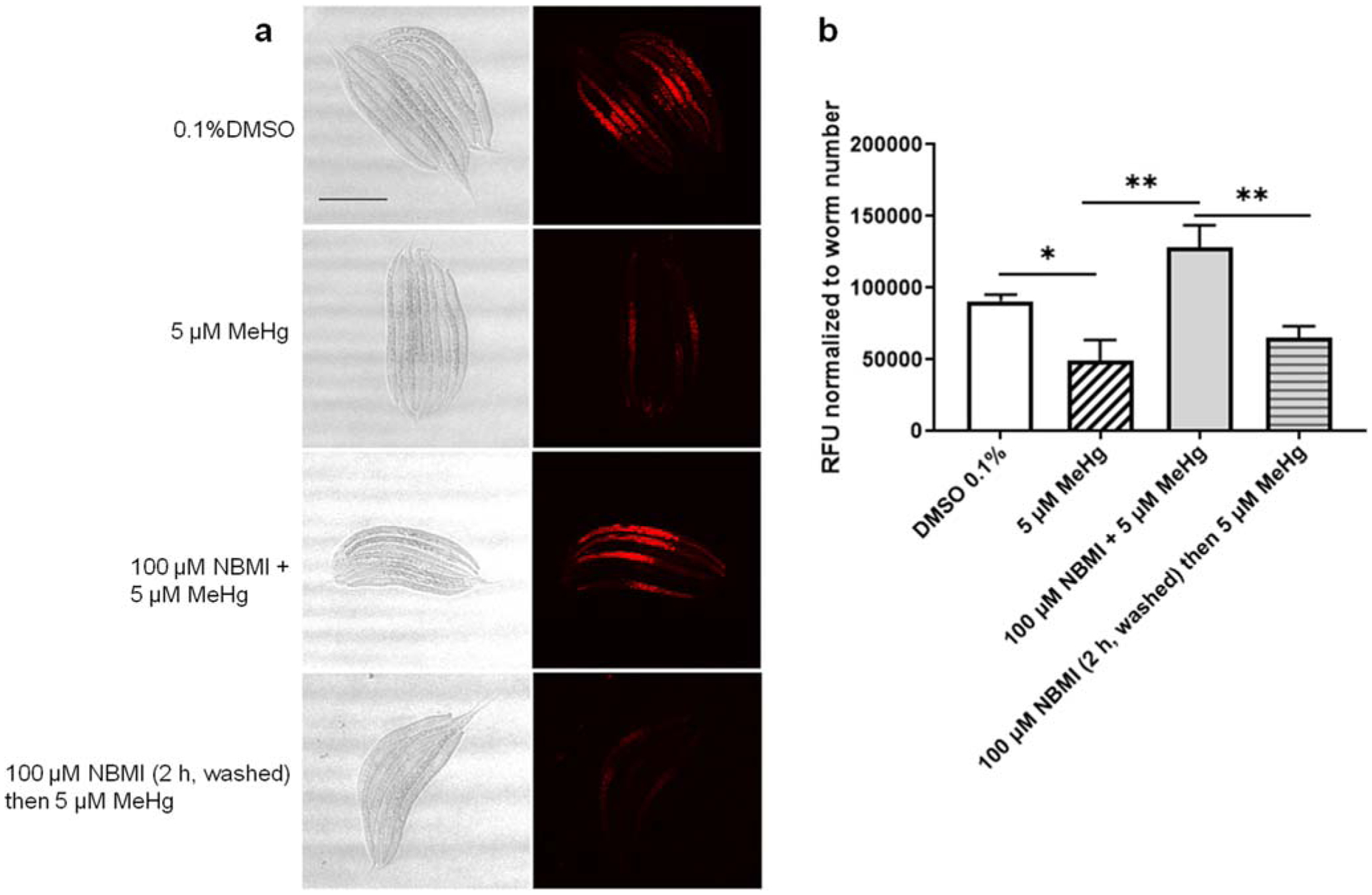
NBMI treatment inhibits MeHg- induced sequestration of thiols in mitochondria. a Red fluorescence images of worms probed with mitochondria specific marker- mitotracker red. b Mean fluorescence level of worms treated with MeHg or NBMI. For each group, data were derived from at least 30 worms. Comparisons were made with one-way ANOVA and Tukey’s multiple comparisons test. The horizontal bars represent a statistical significant difference between the groups. *P < 0.05, **P < 0.01, and ***P < 0.001
Discussion
The complexing agent NBMI has been previously shown to afford therapeutic benefits against inorganic Hg toxicity in a rat model (Clarke et al. 2012b); however, an evaluation of its efficacy against organomercury (MeHg) toxicity in vivo has yet to be reported. The present study showed that upon acute co-treatment of NBMI significantly mitigated MeHg-induced death (Fig. 1a). However, post-treatment with NBMI had no effect on the acute toxicity of MeHg (Fig. 1a). Pre-treatment with NBMI protected against the acute effects of MeHg, yet increasing pre-treatment time had no additional protection (Fig. 1b). These results suggest that co-treatment of NBMI with MeHg affords maximal protection.
One of the toxic mechanisms of MeHg is protein function inhibition or alteration by complexing with native membrane and intracellular proteins that possess structural and functional thiol groups (Tiernan et al. 2015). Post-treatment with NBMI suggests that the latter is unable to reverse the MeHg-induced damage. Empirically, MeHg has a higher binding affinity for selenol-containing proteins than thiol-containing molecules; however, the in vivo affinity and toxicity is also dictated by the concentration and accessibility of the target groups. The affinity of MeHg for the thiol groups (log K, where K is the association constant) is on the order of 1015–23, significantly higher than its affinity constants for other organic ligands (Aschner and Aschner 1990). It is also important to note that the Hg-S bond is liable, and -Hg- moves very rapidly between free thiol groups, preferentially being attacked by the thiol groups with a higher nucleophilicity (Nogara et al. 2019). Hence, membrane channels and mitochondrial respiratory enzymes that possess sulfhydryl groups with high affinity for Hg electrophilic forms can be oxidized by MeHg (Abd-Elfattah and Shamoo 1981; Cambier et al. 2009; Glaser et al. 2014; Glaser et al. 2013). Pre-treatment with NBMI partially mitigated MeHg toxicity (Fig. 1c). The lesser efficacy of pre-treatment (Fig. 1c) versus co-treatment (Fig. 1a) raises several possibilities. First, upon pre-treatment, there is no (or smaller likelihood) in vitro interaction between NBMI and MeHg. However, for the co-treatment, the bioactive level of MeHg in the treatment buffer might be significantly reduced by NBMI due to in vitro interaction. Second, the complexation of NBMI with MeHg is presumably in a 1:2 M ratio (Zaman et al. 2007), hence the NBMI-uncomplexed MeHg concentration is around 105 μM in the 125 μM MeHg/10 μM NBMI treatment group, which is a lethal dose (Fig. 1c). Accordingly, the complete inhibition of death rate in the 125 μM MeHg group by co-treatment with 10 μM NBMI (Fig. 1a), suggests that the in vitro reaction cannot fully account for the protective effect of NBMI. It should be noted that the above inferences are based on the premise that in acute toxicity, MeHg-NBMI complex is significantly less toxic than MeHgCl. However, it remains to be investigated whether the MeHg-NBMI is indeed less toxic than other organic complexes of MeHg. Due to the insolubility of MeHg-NBMI complex in the water, experiments comparing the lethality by injecting the MeHg-NBMI complex could dissect out these possibilities.
In patients with MD, the severity of illness in MeHg poisoning is related to the threshold level of remaining cerebellar granule cells (Eto 1997). However, in C. elegans, it remains to be determined if MeHg-induced death is caused by the death of specific group of cells. C. elegans hermaphrodite has only 302 neurons, nearly all of which are dispensable for viability except CAN (Forrester and Garriga 1997) and M4 neurons (Avery and Horvitz 1989). MeHg-induced death in C. elegans may result from the death of essential neurons (CAN and M4) or a group of susceptible cell populations. Characterization of the cellular basis of MeHg-induced death in C. elegans can facilitate testing antidotes for targeted therapy. In a previous animal study testing efficacy of NBMI, it was found that, in rats injected with Hg, after a short time window, given 10 times dose of NBMI, it could significantly reduce the death rate (Clarke et al. 2012b). The treatment paradigm mimics the clinical use of a chelator. However, these evaluations were on inorganic Hg2+, not MeHg (Clarke et al. 2012b). Overall, further research on the cellular basis of MeHg-induced death in C. elegans may help to clarify the effects of NBMI on the acute toxicity of MeHg.
Another possible effect of NBMI is that the transport of MeHg could be altered. In the current study, the mean Hg level in NBMI treated worms is 20% less than worms treated with MeHg alone (Fig. 2d), while the difference does not reach a significant level (Fig. 2d), consistent with a pilot study on NBMI in human subjects exposed to Hg vapor (Schutzmeier et al. 2018) and with the rats study with Hg2+ (Clarke et al. 2012a). Previous studies have shown that the genes homologous to human lat-1 possibly involve in MeHg transport in worms (Caito et al. 2013). However, we failed to find analogous effects of MeHg or NBMI on these transporters (Fig. 2). The discrepancy between the current and Caito’s study (Caito et al. 2013) can be attributed to different experimental conditions (treatment buffers and MeHg doses).
To further enquiry the mechanism of NBMI, the genes skn-1 and sod-1 were analyzed. In C. elegans, skn-1 is a homolog of the mammalian transcriptional factor nrf-2 (Martinez-Finley et al. 2013), which plays key roles in detoxifying MeHg (Unoki et al. 2018). It has been reported that C. elegans with reduced expression of skn-1 upon RNAi are vulnerable to the toxic effects of MeHg (Vanduyn et al. 2010), whereas sod-1 mutant mice are susceptible to its toxicity (Johnson et al. 2011). In the current study, we found that the MeHg-induced increase in skn-1 or sod-1 expression was unaltered by pre-treatment with NBMI (Fig. 3). However, the lethality data showed that pre-treatment with NBMI afforded some protection, with ~ 50% death rate in the 125 μM MeHg group (Fig. 1c). Multiple mechanisms were advanced for the up-regulation of SKN-1 upon MeHg exposure, among which a direct binding of MeHg with cysteine residues of SKN-1 negative regulator was believed to be an important switch for SKN-1 activation (Ke et al. 2019). As one of the major defensive mechanisms, skn-1 gene expression level might not be closely associated with the death pathways in response to MeHg. Future research on the mechanisms of MeHg-induced transcriptional regulation of skn-1 could fill the gap in the understanding of skn-1 in MeHg toxicity.
Motor activities have been shown to be compromised in patients of MD (Eto 1997). Studies in Iraq also showed that ataxia is the earliest clinical finding in MeHg poisoning (Bakir et al. 1973). Moreover, low level of MeHg (0.5 ppm) affect DAergic neuron-mediated motor functions in rats (Gimenez-Llort et al. 2001). In an early investigation, we noted that chronic MeHg treatment induced mCherry aggregation in dendrites of CEP neurons, showing bright puncta morphology (Ke et al. 2020). In order to evaluate the efficacy of NBMI against chronic toxicity of MeHg, the dendritic morphology of CEP DAergic neurons was assessed in a 10 days MeHg exposure model (Fig. 4). The bright, sphere-like shape puncta are only observed in MeHg-treated worms (Fig. 4e). The increased number of DAergic puncta reveals that C. elegans DAergic neuronal damage is a vulnerable target for MeHg, which is attenuated by NBMI (Fig. 4c). As NBMI treatment followed MeHg exposure, the puncta in the NBMI/MeHg group could be induced by the active form of MeHg during the six-day exposure (Fig. 4a). Thus, NBMI treatment probably prevents further neuronal damage from the active form of MeHg. The irreversible and degenerated neuronal damage marks the clinic feature of severe MeHg poisoning, which cannot be attenuated by antidotes for reducing body burden of MeHg (Nierenberg et al. 1998). Nevertheless, the significant effect of NBMI on MeHg-induced DAergic neuronal damage deserves further evaluation of its effects in a clinic context.
Further evidence from the antioxidant response reporter strains (Fig. 5 and Fig. 6) supports the notion that co-treatment with NBMI mitigated the chronic effects of MeHg, while pre-treatment NBMI had no effect. Previously, we found that MeHg could dose-dependently decrease the fluorescence of the mitochondria probe mitoTracker red (Ke and Aschner 2019). The evidence derived in the current study corroborates that the fluorescence level emitted from mitoTracker red is significantly decreased by MeHg treatment in live C. elegans, and co-treatment of NBMI rather than pre-treatment of NBMI restored the fluorescence to the level comparable to the control (Fig. 7). Though it is currently unclear, the decreased signal of mitoTracker red could be related to a decreased mitochondrial potential (Yin et al. 2007), a loss of mitochondrial free thiols, or both following MeHg treatment (Kholmukhamedov et al. 2013). Thus whether NBMI treatment can restore the damaged mitochondrial potential deserve further investigation. Furthermore, the mild effect of pre-treatment with NBMI is partly caused by a low absorption rate of NBMI in worms. The absorption rate of NBMI in rats is 13~15% by gavage (Clarke et al. 2012b). In C. elegans, the absorption rate of NBMI is unknown. Future research, using different treatment methods, such as a mixture of NBMI with bacteria food could facilitate drug delivery in C. elegans and maximize the effect of pre-treatment regime.
In summary, the current study found that NBMI has some protective effects in acute as well as chronic MeHg toxicity paradigms. The co-treatment of NBMI had maximal protection against MeHg toxicity, as shown by reduced worms’ death rate and structural damage of DAergic neurons, as well as mitigated antioxidant response in worms treated with NBMI. However, NBMI had no effect on the transport of MeHg. Further experiments to explore the cellular and molecular basis of NBMI’s efficacy are warranted.
Funding Information
This work was supported by the National Institutes of Health to MA (NIEHS R01ES007331). The authors thank the Analytical Imaging Facility (AIF) at Albert Einstein College of Medicine, which is sponsored by NCI cancer center support grant P30CA013330 and Shared Instrumentation Grant (SIG) 1S10OD023591-01. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). NBMI was kindly provided by Mr. Ragnar Klingberg.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- Abd-Elfattah AS, Shamoo AE (1981) Regeneration of a functionally active rat brain muscarinic receptor by D-penicillamine after inhibition with methylmercury and mercuric chloride. Mol Pharmacol 20: 492–497 [PubMed] [Google Scholar]
- Aschner M (2012) Considerations on methylmercury (MeHg) treatments in in vitro studies. Neurotoxicology 33:512–513. 10.1016/j.neuro.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Aschner JL (1990) Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neurosci Biobehav Rev 14:169–176. 10.1016/s0149-7634(05)80217-9 [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR (1989) Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron 3: 473–485. 10.1016/0896-6273(89)90206-7 [DOI] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A et al. (1973) Methylmercury poisoning in Iraq. Science 181:230–241. 10.1126/science.181.4096.230 [DOI] [PubMed] [Google Scholar]
- Bakir F, Al-Khalidi A, Clarkson TW, Greenwood R (1976) Clinical observations on treatment of alkylmercury poisoning in hospital patients. Bull World Health Organ 53(Suppl):87–92 [PMC free article] [PubMed] [Google Scholar]
- Bridges CC, Joshee L, Zalups RK (2009) Effect of DMPS and DMSA on the placental and fetal disposition of methylmercury. Placenta 30: 800–805. 10.1016/j.placenta.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caito SW, Zhang Y, Aschner M (2013) Involvement of AAT transporters in methylmercury toxicity in Caenorhabditis elegans. Biochem Biophys Res Commun 435:546–550. 10.1016/j.bbrc.2013.04.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier S, Benard G, Mesmer-Dudons N, Gonzalez P, Rossignol R et al. (2009) At environmental doses, dietary methylmercury inhibits mitochondrial energy metabolism in skeletal muscles of the zebra fish (Danio rerio). Int J Biochem cell B 41:791–799. 10.1016/j.biocel.2008.08.008 [DOI] [PubMed] [Google Scholar]
- Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari EM et al. (2007) Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology 239:195–203. 10.1016/j.tox.2007.07.009 [DOI] [PubMed] [Google Scholar]
- Clarke D, Buchanan R, Gupta N, Haley B (2012a) Amelioration of Acute Mercury Toxicity by a Novel, Non-Toxic Lipid Soluble Chelator N, N’bis-(2-mercaptoethyl)isophthalamide: Effect on Animal Survival, Health, Mercury Excretion and Organ Accumulation. Toxicol Environ Chem 94:616–640. 10.1080/02772248.2012.657199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke D, Buchanan R, Gupta N, Haley B (2012b) Amelioration of acute mercury toxicity by a novel, non-toxic lipid soluble chelator N,N ‘ bis-(2-mercaptoethyl)isophthalamide: effect on animal survival, health, mercury excretion, and organ accumulation. Toxicological and Environmental Chemistry 94:616–640. 10.1080/02772248.2012.657199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW (2002) The three modern faces of mercury environ health. Perspect 110(Suppl 1):11–23. 10.1289/ehp.02110s111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Cox C, Greenwood MR, Amin-Zaki L et al. (1981) Tests of efficacy of antidotes for removal of methylmercury in human poisoning during the Iraq outbreak. J Pharmacol Exp Ther 218:74–83 [PubMed] [Google Scholar]
- Davies FC (1991) Minamata disease: A 1989 update on the mercury poisoning epidemic in Japan. Environ Geochem Health 13:35–38. 10.1007/BF01783494 [DOI] [PubMed] [Google Scholar]
- Davis LE, Kornfeld M, Mooney HS, Fiedler KJ, Haaland KY et al. (1994) Methylmercury poisoning: long-term clinical, radiological, toxicological, and pathological studies of an affected family. Ann Neurol 35:680–688. 10.1002/ana.410350608 [DOI] [PubMed] [Google Scholar]
- Ekino S, Susa M, Ninomiya T, Imamura K, Kitamura T (2007) Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J Neurol Sci 262:131–144. 10.1016/j.jns.2007.06.036 [DOI] [PubMed] [Google Scholar]
- Eto K (1997) Pathology of Minamata disease. Toxicol Pathol 25:614–623. 10.1177/019262339702500612 [DOI] [PubMed] [Google Scholar]
- Forrester WC, Garriga G (1997) Genes necessary for C. elegans cell and growth cone migrations. Development 124:1831–1843 [DOI] [PubMed] [Google Scholar]
- Gimenez-Llort L, Ahlbom E, Dare E, Vahter M, Ogren S et al. (2001) Prenatal exposure to methylmercury changes dopamine-modulated motor activity during early ontogeny: age and gender-dependent effects. Environ Toxicol Pharmacol 9:61–70 [DOI] [PubMed] [Google Scholar]
- Glaser V, Moritz B, Schmitz A, Dafre AL, Nazari EM et al. (2013) Protective effects of diphenyl diselenide in a mouse model of brain toxicity. Chem Biol Interact 206:18–26. 10.1016/j.cbi.2013.08.002 [DOI] [PubMed] [Google Scholar]
- Glaser V, Martins Rde P, Vieira AJ, Oliveira Ede M, Straliotto MR et al. (2014) Diphenyl diselenide administration enhances cortical mitochondrial number and activity by increasing hemeoxygenase type 1 content in a methylmercury-induced neurotoxicity mouse model. Mol Cell Biochem 390:1–8. 10.1007/s11010-013-1870-9 [DOI] [PubMed] [Google Scholar]
- Johnson FO, Yuan Y, Hajela RK, Chitrakar A, Parsell DM, Atchison WD (2011) Exposure to an environmental neurotoxicant hastens the onset of amyotrophic lateral sclerosis-like phenotype in human Cu2+/Zn2+ superoxide dismutase 1 G93A mice: glutamate-mediated excitotoxicity. J Pharmacol Exp Ther 338:518–527. 10.1124/jpet.110.174466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke T, Aschner M (2019) Bacteria affect Caenorhabditis elegans responses to MeHg toxicity. Neurotoxicology 75:129–135. 10.1016/j.neuro.2019.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke T, Goncalves FM, Goncalves CL, dos Santos AA, JBT R et al. (2019) Post-translational modifications in MeHg-induced neurotoxicity Bba-Mol Basis Dis 1865:2068–2081. 10.1016/j.bbadis.2018.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke T, Tsatsakis A, Santamaria A, Antunes Soare FA, Tinkov AA et al. (2020) Chronic exposure to methylmercury induces puncta formation in cephalic dopaminergic neurons in Caenorhabditis elegans Neurotoxicology. 10.1016/j.neuro.2020.01.003 [DOI] [PMC free article] [PubMed]
- Kholmukhamedov A, Schwartz JM, Lemasters JJ (2013) Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats: mitotracker probes and mitochondrial membrane potential. Shock 39:543. 10.1097/SHK.0b013e318292300d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Martinez-Finley EJ, Caito S, Slaughter JC, Aschner M (2013) The role of skn-1 in methylmercury-induced latent dopaminergic neurodegeneration. Neurochem Res 38:2650–2660. 10.1007/s11064-013-1183-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers GJ, Davidson PW, Cox C, Shamlaye C, Cernichiari E et al. (2000) Twenty-seven years studying the human neurotoxicity of methylmercury exposure. Environ Res 83:275–285. 10.1006/enrs.2000.4065 [DOI] [PubMed] [Google Scholar]
- Nierenberg DW, Nordgren RE, Chang MB, Siegler RW, Blayney MB et al. (1998) Delayed cerebellar disease and death after accidental exposure to dimethylmercury. N Engl J Med 338:1672–1676. 10.1056/NEJM199806043382305 [DOI] [PubMed] [Google Scholar]
- Nogara PA, Oliveira CS, Schmitz GL, Piquini PC, Farina M, Aschner M, Rocha JBT (2019) Methylmercury’s chemistry: From the environment to the mammalian brain. Biochim Biophys Acta Gen Subj 1863:129284. 10.1016/j.bbagen.2019.01.006 [DOI] [PubMed] [Google Scholar]
- Schutzmeier P, Focil Baquerizo A, Castillo-Tandazo W, Focil N, Bose-O’Reilly S (2018) Efficacy of N,N’bis-(2-mercaptoethyl) isophthalamide on mercury intoxication: a randomized controlled trial. Environ Health 17:15. 10.1186/s12940-018-0358-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secor JD, Kotha SR, Gurney TO, Patel RB, Kefauver NR et al. (2011) Novel lipid-soluble thiol-redox antioxidant and heavy metal chelator, N,N’-bis(2-mercaptoethyl)isophthalamide (NBMI) and phospholipase D-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI) attenuate mercury-induced lipid signaling leading to protection against cytotoxicity in aortic endothelial cells. Int J Toxicol 30:619–638. 10.1177/1091581811422413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbernagel SM, Carpenter DO, Gilbert SG, Gochfeld M, Groth E 3rd et al. (2011) Recognizing and preventing overexposure to methylmercury from fish and seafood consumption: information for physicians. J Toxicol 2011:983072. 10.1155/2011/983072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiernan CT, Edwin EA, Hawong HY, Rios-Cabanillas M, Goudreau JL et al. (2015) Methylmercury impairs canonical dopamine metabolism in rat undifferentiated pheochromocytoma (PC12) cells by indirect inhibition of aldehyde dehydrogenase. Toxicol Sci 144:347–356. 10.1093/toxsci/kfv001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki T, Akiyama M, Kumagai Y, Goncalves FM, Farina M et al. (2018) Molecular pathways associated with Methylmercury-induced Nrf2 modulation front. Genet 9:373. 10.3389/fgene.2018.00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanduyn N, Settivari R, Wong G, Nass R (2010) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci 118:613–624. 10.1093/toxsci/kfq285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Pears C, Woollard A (2017) An enhanced C. elegans based platform for toxicity assessment. Sci Rep 7:9839. 10.1038/s41598-017-10454-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JB et al. (2007) Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 1131:1–10. 10.1016/j.brainres.2006.10.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman KM, Blue LY, Huggins FE, Atwood DA (2007) Cd, hg, and Pb compounds of Benzene-1,3-diamidoethanethiol (BDETH(2)). Inorg Chem 46:1975–1980. 10.1021/ic0607639 [DOI] [PubMed] [Google Scholar]