

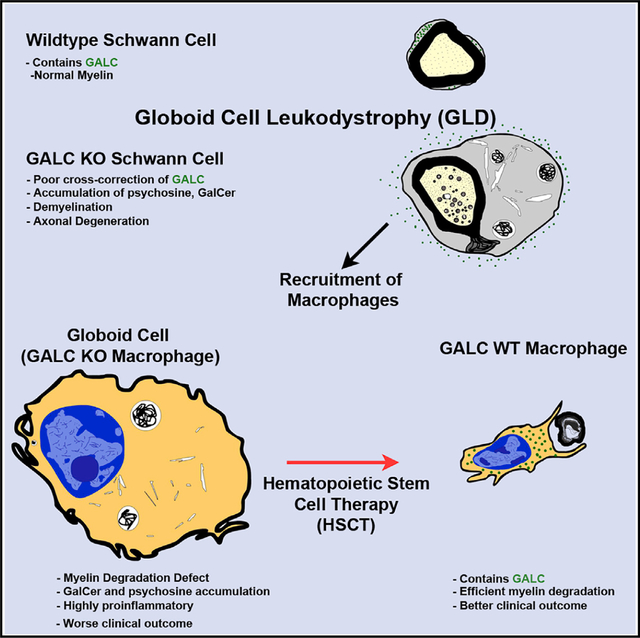
SUMMARY
Many therapies for lysosomal storage disorders rely on cross-correction of lysosomal enzymes. In globoid cell leukodystrophy (GLD), mutations in GALC cause psychosine accumulation, inducing demyelination, a neuroinflammatory “globoid” reaction and neurodegeneration. The efficiency of GALC cross-correction in vivo, the role of the GALC substrate galactosylceramide, and the origin of psychosine are poorly understood. Using a novel GLD model, we show that cross-correction does not occur efficiently in vivo and that Galc-deficient Schwann cells autonomously produce psychosine. Furthermore, macrophages require GALC to degrade myelin, as Galc-deficient macrophages are transformed into globoid cells by exposure to galactosylceramide and produce a more severe GLD phenotype. Finally, hematopoietic stem cell transplantation in patients reduces globoid cells in nerves, suggesting that the phagocytic response of healthy macrophages, rather than cross-correction, contributes to the therapeutic effect. Thus, GLD may be caused by at least two mechanisms: psychosine-induced demyelination and secondary neuroinflammation from galactosylceramide storage in macrophages.
Graphical Abstract
In Brief
Weinstock et al. show that lysosomal GALC is required autonomously by Schwann cells to prevent psychosine formation, demyelination, and subsequent axonal degeneration. Macrophages independently require GALC to serve in phagocytosis and myelin turnover. HSCT likely exerts its therapeutic benefit by restoring phagocyte function rather than cross-correcting myelin cells of GALC.
INTRODUCTION
Globoid cell leukodystrophy (GLD, also Krabbe disease) is a lysosomal storage disorder (LSD) that causes rapid neurological decline and death (Hagberg et al., 1969; Krabbe, 1916). GLD pathogenesis includes intertwined mechanisms of demyelination, neurodegeneration, and inflammation, characterized by the presence of pathognomonic macrophages, globoid cells (Collier and Greenfield, 1924). GLD is caused by mutations in the gene encoding the lysosomal hydrolase galactosylceramidase (GALC) (Malone, 1970; Suzuki and Suzuki, 1970), which catabolizes the myelin lipid galactosylceramide (GalCer) and galactosylsphingosine (psychosine) (Li et al., 2019; Miyatake and Suzuki, 1972). Psychosine accumulates in GLD tissues (Svennerholm et al., 1980) and is thought to initiate and drive the majority of GLD pathology (Suzuki, 1998).
The twitcher mouse is a spontaneous Galc mutant that mirrors severe GLD (Duchen et al., 1980). Hematopoietic stem cell transplantation (HSCT) extends lifespan in twitcher mice (Yeager et al., 1984) and in pre-symptomatic GLD patients (Escolar et al., 2005). HSCT is thought to work by cross-correction, in which donor-derived cells transfer missing enzyme to GALC-deficient cells (reviewed in Sands and Davidson, 2006). This principle is the basis for lysosomal enzyme replacement therapy (ERT) and enhances the “bystander” effect of gene therapy in LSDs. Therefore, cross-correction forms the basis for nearly all approved and prospective clinical therapies among the LSDs.
While many pre-clinical studies have explored delivering Galc to GLD models (reviewed in Mikulka and Sands, 2016), cross-correction of GALC in vivo is controversial (Kondo et al., 2005; Matthes et al., 2015). Furthermore, although GALC is expressed ubiquitously, its specific role and autonomous function among different cell types is unknown. HSCT may also have immunomodulatory benefits (Hoogerbrugge et al., 1988; Reddy et al., 2011), suggesting an intrinsic role for GALC in leukocytes. We propose that any cell type could, in theory, be directly affected by GALC loss of function and may intrinsically contribute to overall GLD progression.
Here, we developed a conditional Galc floxed mouse to address these questions using the peripheral nervous system (PNS) as a model system. The PNS is an ideal site to ask questions about cellular autonomy and cross-correction due to its simplicity and anatomical isolation. We found that Schwann cells, the myelinating glia of the PNS, require GALC to maintain myelin and axonal integrity, synthesizing psychosine in its absence. Interestingly, Galc-deficient Schwann cells are unable to receive GALC from their surrounding environment due, in part, to ineffective uptake. Furthermore, we discovered that, contrary to expectations, psychosine was not sufficient to drive the entirety of disease burden in GLD. Conversely, macrophages deficient for Galc contribute to neurodegeneration by developing a proinflammatory globoid reaction. Thus, we define a novel essential role of GALC in macrophages recruited to sites of demyelination. Based on these data and the analysis of GLD post-mortem human tissues, we propose that the mechanism of HSCT in GLD, and possibly other LSDs, is the restoration of intrinsic degradative functions of macrophages as opposed to cross-correction of neighboring cells.
RESULTS
Galc-Deficient Schwann Cells Are Not Cross-Corrected and Produce Psychosine
We generated a loxP-flanked (floxed) Galc mouse, to ablate Galc in a Cre-dependent fashion. Galc-null mice phenocopied twitcher mice (GalcW339X/W339X) by all tested parameters including survival, weight (Weinstock et al., 2020), and PNS pathology (Figure S1A). Galc KO mice express no Galc mRNA (Figure 1A), enzymatic activity (Figure 1B), and protein in the sciatic nerve (Figure 1C). We ablated Galc in Schwann cells of the PNS (Galc SC cKO, Figure S1B) by crossing Galc floxed mice to the well-characterized Mpz-Cre transgenic line (Feltri et al., 1999). Galc SC cKO sciatic nerves had approximately 15% of wild-type (WT) Galc mRNA levels (Figure 1D). We suspected that this residual Galc mRNA was produced by PNS cells other than Schwann cells. Indeed, primary Schwann cell culture from Galc SC cKO produced virtually no Galc mRNA (Figure 1E). Thus, Mpz-Cre efficiently removed Galc expression in Schwann cells.
Figure 1. Galc Is Efficiently Ablated in Schwann Cells of Conditional Knockout Mice.
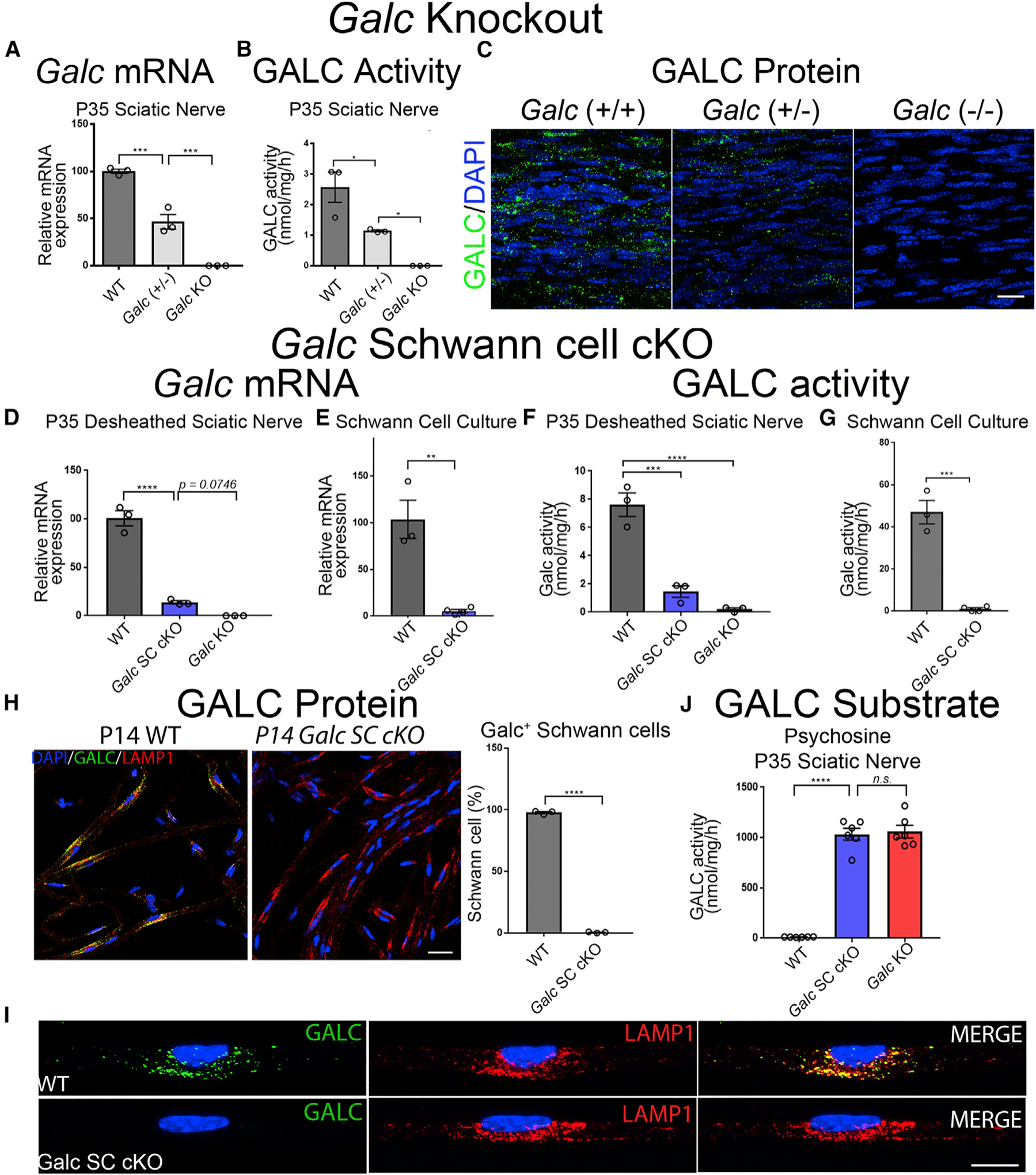
(A–C) Dose-dependent reduction of GALC in P35 sciatic nerves from Galc(+/+), Galc(+/−), and Galc(−/− ) mice.
(A) Galc mRNA expression, normalized to β-Actin, and reported relative to average WT expression.
(B) GALC enzymatic activity, measured as nmol of fluorogenic substrate MUGAL, normalized to protein and time.
(C) GALC immunofluorescence in longitudinal sections of P35 sciatic nerves. Polyclonal anti-GALC antibody (green); DAPI (blue).
(D–I) Efficient Galc ablation in Schwann cells of conditional knockout mice.
(D and E) Galc mRNA expression of desheathed P35 sciatic nerves (D) and primary Schwann cells (E).
(F and G) GALC activity of desheathed P35 sciatic nerves (F) and primary Schwann cells (G).
(H) GALC immunofluorescence and quantification of teased fibers from P14 sciatic nerves. GALC (green); LAMP1 (red); DAPI (blue). (I) Higher magnification of (H).
(J) Psychosine measured by HPLC-MS from P35 sciatic nerves.
Scale bars, 25 μm (C), 30 μm (H), and 14 μm (I). Error bars represent mean ± SEM, n = 3 biological replicates and 3 technical replicates per experiment (n = 6 for J). Statistical significance was calculated by one-way ANOVA (A, B, D, F, and J) or Student’s t test (E, G, and H). In all figures, asterisks represent statistical significance (*p < 0.05, **p < 0.01; ***p < 0.005, ****p < 0.001). n.s., not significant.
GALC activity mirrored mRNA levels (Figure 1F), with 80% reductions in Galc SC cKO sciatic nerves. Since GALC should be secreted and transferred between cells, we expected that this residual GALC would cross-correct Schwann cells of Galc SC cKO. Interestingly, GALC enzymatic activity was nearly absent in Galc SC cKO Schwann cell cultures (Figure 1G), and GALC protein was undetectable by immunofluorescence in Schwann cells of nerve teased fibers (Figures 1H and 1I). Furthermore, the toxic GALC substrate psychosine, was elevated to comparable levels in Galc SC cKO and Galc KO sciatic nerves (Figure 1J; Figure S1C). The cellular identity of the producer of psychosine was previously unknown, and our data first show that Schwann cells are the major producers of psychosine in the PNS. Together, these data illustrate the high efficiency of GALC ablation in Schwann cells of the Galc SC cKO mice and show that cross-correction of endogenous GALC to Schwann cells is minimal.
Cross-Correction of GALC Is Perturbed in Galc-Deficient Cells
To investigate why cross-correction to Schwann cells was not observed, we performed in vitro cross-correction experiments using recombinant human GALC (rhGALC). Primary Schwann cells were cultured from WT and Galc KO mice and incubated with His-HA-tagged rhGALC (modified from Shin et al., 2016). Similar to previous reports (Nagano et al., 1998; Rafi et al., 1996), WT Schwann cells were capable of receiving purified rhGALC from media. In contrast, Galc KO Schwann cells were five times less efficient at receiving rhGALC (Figures 2A and 2B).
Figure 2. Cross-Correction Is Impaired in Galc Mutants.
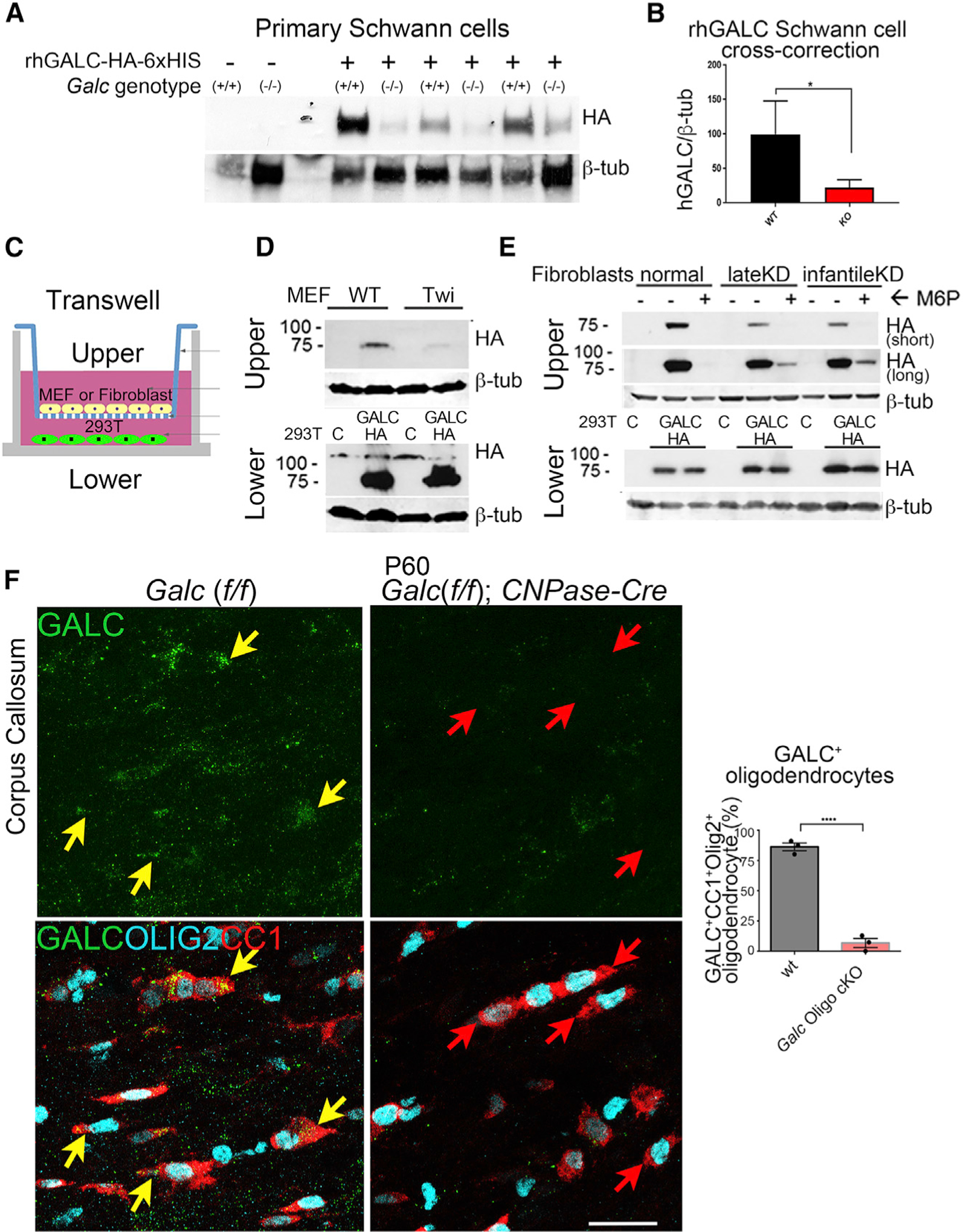
(A) Western blot of primary Schwann cells incubated with recombinant human GALC-HA-6xHis. Each lane represents a separate Schwann cell-culture preparation from a distinct animal. hGALC-HA bands were detected by western blot at the expected size of 80 kDa using an anti HA-HRP antibody.
(B) Quantification of rhGALC uptake from western blot in (A). Bands were quantified by densitometry for rhGALC and normalized to β-tubulin.
(C) Schematic of the transwell GALC uptake experiment for (D) and (E).
(D) Western blot of mouse embryonic fibroblasts in the top chambers or 293T cells in the lower chamber, after the transwell GALC-uptake assay.
(E) Control and GLD fibroblasts using the transwell GALC-uptake assay, as above. Infantile KD carried the homozygous 30 kb GALC deletion and late-onset KD had compound G286D/30 kb-del GALC mutations. 6 mM M6P was added where indicated.
(F) Immunofluorescence of corpus callosum white matter in WT and oligodendrocyte Galc cKO brains. The oligodendrocyte markers CC1 (red) and OLIG2 (cyan) co-localized with GALC (green) in WT brains, depicted by yellow arrows. Instead, KO brains had no GALC in CC1/OLIG2 double-positive oligodendrocytes (red arrows). Scale bar, 30 μm. Quantifications on right-hand side.
Error bars represent mean ± SEM, n = 3 biological replicates. For (B) and (F), a Student’s t test was used to calculate statistical significance.
To determine whether this deficiency was true for all Galc-deficient cells, similar experiments were performed on mouse embryonic fibroblasts (MEFs) from WT and twitcher mice. MEFs were plated in a Boyden chamber in direct contact with medium from HEK cells transfected with hGALC-HA (Figure 2C) (Shin et al., 2016). Again, cross-correction of hGALC to WT MEFs was more efficient than to twitcher MEFs (Figure 2D). Similarly, human fibroblasts isolated from late-onset and early-infantile GLD patients (Shin et al., 2016) received less GALC than controls (Figure 2E). Notably, the mechanism of GALC cross-correction was also perturbed. Control cells received GALC via mannose-6-phosphate (M6P)-dependent uptake, as it was competitively inhibited by co-incubation with M6P (Figure 2E). Instead, patient fibroblasts were still able to uptake some GALC, after co-incubation with M6P (Figure 2E). This suggests that GLD fibroblasts may have reduced GALC uptake via the canonical M6P pathway and may have alternative and inefficient compensatory uptake via other mechanisms.
Finally, to determine whether poor in vivo cross-correction also occurs in myelinating glia of the CNS, oligodendrocyte Galc cKO (Galc Oligo cKO) animals were generated by crossing Galc floxed mice to CNPase-Cre mice (Lappe-Siefke et al., 2003). WT oligodendrocytes of the corpus callosum and cervical spinal cord both express GALC (yellow arrows, Figure 2F; Figure S2). In contrast, most oligodendrocytes in Galc Oligo cKO brains and spinal cords did not contain GALC (Figure 2F, red arrows, and Figure S2), indicating that they also were not able to receive extracellular GALC.
GALC Protects Schwann Cells from Demyelination
The PNS is severely affected in GLD and may underlie the refractory morbidity and mortality of HSCT-treated patients (Wright et al., 2017). Despite this, the extent of disease contribution attributable to Schwann cells or the PNS as a whole is unknown. We thus compared peripheral nerve function in Galc SC cKO and Galc KO mice to define the degree of neuropathy directly attributable to Schwann cell dysfunction.
Galc SC cKO mice develop a clenching phenotype, suggestive of peripheral neuropathy (Figure 3A) and exhibit poor rotarod performance (Figure 3B). Galc SC cKO mice survived normally to at least 1 year of age (data not shown) and had only minor reductions in weight (Figure 3C), suggesting PNS dysfunction does not explain the clinical demise and death seen in KD patients.
Figure 3. Ablation of Schwann Cell Galc Causes a Progressive Peripheral Neuropathy.
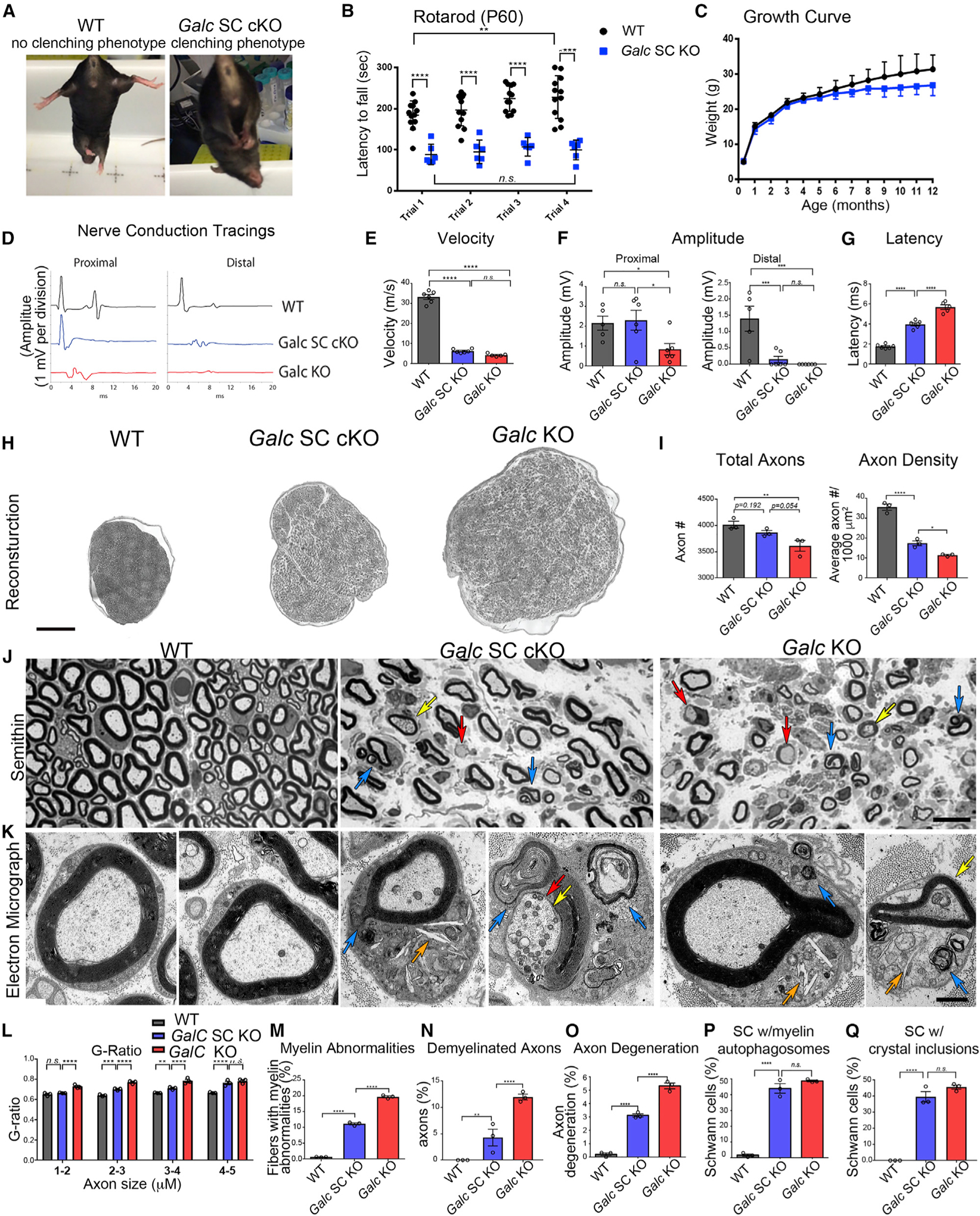
(A) Clenching phenotype of P60 Galc SC cKO mice.
(B) Rotarod analysis of P60 mice. n = 6–12.
(C) Growth curve of mice. n = 6–12.
(D–G) Electrophysiology of sciatic nerves from P35 WT animals.
(D) Representative tracing from nerve conduction at proximal and distal sites.
(E) Nerve conduction velocities from P35 sciatic nerves.
(F) Proximal and distal amplitude from P35 sciatic nerves.
(G) Latency measurements from P35 sciatic nerves.
(H) Representative sciatic nerve reconstructions from P35 mice.
(I) Total axon count and myelinated axon density of whole P35 sciatic nerves (n = 3).
(J) Representative semithin sections of P35 sciatic nerves. Blue arrows indicate myelin abnormalities (outfoldings, infoldings, and degeneration), red arrows indicate demyelinated axons, and yellow arrows indicate axon degeneration.
(K) Representative electron micrographs of Schwann cells from P35 sciatic nerves. Blue arrows indicate degenerated myelin, orange arrows indicate crystals, yellow arrows indicate axon degeneration, and red arrows indicate demyelinated axons.
(L) G-ratio, as a function of axon size, from P35 sciatic nerves. n = 3 per genotype.
(M) Quantification of myelin abnormalities in P35 sciatic nerves.
(N) Quantification of demyelinated axons in P35 sciatic nerves.
(O) Quantification of degenerated axons in P35 sciatic nerves.
(P) Quantification of Schwann cells with accumulated myelin autophagosomes.
(Q) Quantification of Schwann cells with accumulated crystal inclusions.
Scale bars, 150 μm (H), 10 μm (J), and 1 μm (K). Error bars represent mean ± SEM, n = 3 biological replicates. Statistical significance was calculated by one-way ANOVA (E–G, I, and M–Q) or two-way ANOVA (B, C, and L).
By electrophysiological studies at P35, ubiquitous Galc KO mice exhibited severe reductions in nerve conduction velocity, indicative of myelin defects (Figures 3D and 3E); proximal and distal amplitude, indicative of axonal defects (Figure 3F); and increased latency (Figure 3G). Galc SC cKO mice exhibited similar reductions in nerve conduction velocity, suggesting comparable peripheral demyelination but had improved amplitude and latency measurements, implying GALC expression in other cells (i.e., neurons, macrophages) is important for axonal function. Therefore, Galc SC cKO animals have a clear neuropathic phenotype, though less severe than full Galc KO animals.
Reconstructions of P35 sciatic nerves (Figure 3H) semithin sections show that Galc SC cKO nerves were edematous and larger than WT nerves, though not to the degree of full Galc KO nerves, reflected in reduction of total axon count and myelinated axon density (Figure 3I). Galc KO and Galc SC cKO fibers had thin myelin, but Galc SC cKO myelin was thicker than Galc KO (Figures 3J and 3L). Likewise, Galc SC cKO fibers had other GLD pathologic findings, including myelin abnormalities (Figure 3J, blue arrows, Figure 3M), demyelinated fibers (Figure 3J, red arrows, Figure 3N), and degenerating axons (Figure 3J, yellow arrows, Figure 3O). Although qualitatively similar, the occurrence of these features was reduced in Galc SC cKO nerves. The attenuated GLD phenotype is particularly surprising when considering that both genotypes had near-absent GALC activity in Schwann cells and equal levels of the toxic GALC substrate psychosine.
Electron microscopy (EM) of sciatic nerves (Figure 3K) revealed that Schwann cells in both Galc SC cKO and Galc KO nerves accumulate myelin ovoids and autophagosomes (Figure 3K, blue arrows) encircled in double membranes (Figure S3A) and crystalloid inclusions that were proposed to consist of the GALC substrate galactosylceramide (Figure 3K, orange arrows, Figure S3B) (Austin, 1963; Jacobs et al., 1982; Yunis and Lee, 1970). Interestingly, myelin autophagosomes (Figure 3P) and crystal inclusions (Figure 3Q) accumulate to equal levels in Schwann cells of the Galc SC cKO and full Galc KO. In contrast, GalCer levels were reduced in Galc SC cKO nerves (Figure S3C), as previously reported, possibly representing loss of myelin, or that crystallized GalCer is insoluble by conventional purification techniques. Alternatively, inclusions may be cholesterol crystals formed after myelin degradation (Cantuti-Castelvetri et al., 2018). However, cholesterol did not accumulate in Schwann cell cultures of Galc SC cKO (Figure S3D), and cholesterol pathway genes were not uniformly changed to indicate a defect in cholesterol homeostasis (Figure S3E).
Taken together, these data suggest that the absence of GALC in Schwann cells directly accounts for demyelination and the accumulation of psychosine and crystals. However, some other cell type(s) that expresses GALC ameliorate GLD neuropathology in a psychosine-independent manner.
Deletion of GALC in Schwann Cells Causes Progressive Axonal Degeneration
To determine whether ablation of Galc in Schwann cells is sufficient to perturb axonal health, we analyzed older Galc SC cKO mice (Figure 4). The diameter of Galc SC cKO sciatic nerves continued to enlarge (Figure S4). The number of myelinated fibers progressively declined as Galc SC cKO mice aged (Figures 4A and 4B), while demyelinated axons (Figure 4C), degenerated axons (Figure 4D) and onion bulbs (Figures 4A and 4E) progressively increased. The etiology of axonal pathology in GLD has been difficult to elucidate, as GALC may play a cell-specific role in both Schwann cells and neurons, and many studies documented a direct effect of psychosine on the neuronal cytoskeleton and on axonal transport (Cantuti Castelvetri et al., 2013; Castelvetri et al., 2011). The progressive fiber loss in Galc SC cKO mice suggests that primary Schwann cell dysfunction directly contributes to axonal degeneration.
Figure 4. Deletion of Galc in Schwann Cells Causes a Progressive Demyelinating Neuropathy and Axonal Degeneration.
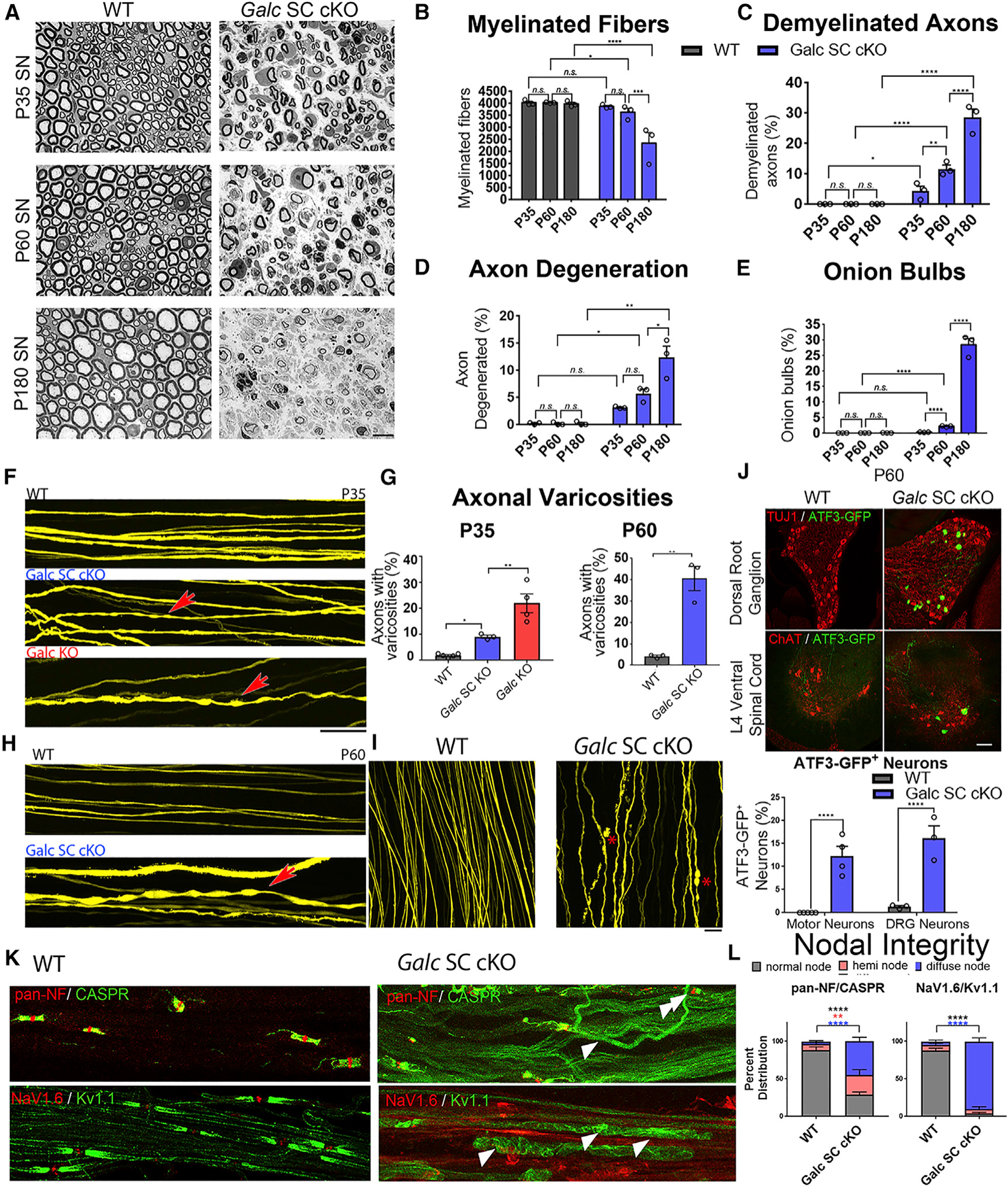
(A) Semithin sections of sciatic nerve from P35, P60, and P180.
(B) Quantification of myelinated fibers in sciatic nerves of WT and Galc SC cKO at different ages.
(C) Percent of demyelinated axons in the sciatic nerves of WT and Galc SC cKO at different ages.
(D) Percent of degenerating axons in the sciatic nerves of WT and Galc SC cKO at different ages.
(E) Percent of fibers with onion bulbs in the sciatic nerves of WT and Galc SC cKO at different ages. (F) Thy1-YFP labelled axons of P35 sciatic nerve for the genotype indicated. Red arrow shows varicosities indicative of axonopathy.
(G) Quantification of varicosities (from F, left and H, right).
(H) Thy1-YFP labelled axons of P60 sciatic nerve in WT and Galc SC cKO nerves.
(I) P60 Thy1-YFP labelled axons showing examples of axon degeneration in Galc SC cKO, indicated by asterisk (red).
(J) ATF3-GFP expression and quantification in P60 DRG neurons or motor neurons.
(K) Evaluation of nodal integrity in sciatic nerve teased fibers stained with antibodies to the nodal and paranodal proteins Neurofascins (pan-NF, red); the paranodal protein Caspr (green), the nodal voltage-gated sodium channel NaV1.6 (red), and the juxtaparanodal potassium channel KV1.1 (green). Arrowheads point to fibers with abnormal diffuse localization of proteins, and double arrowheads point to heminodes.
(L) Quantification of (K).
Scale bars, 10 μm, 100 μm(F and H), and 40 μm(I). Error bars represent mean ± SEM, n = 3 biological replicates and 3 technical replicates per experiment (n = 4 for J). Statistical significance was calculated by two-way ANOVA (B–E), one-way ANOVA (P35, G), or Student’s t test (P60 G, J, and L).
To more sensitively evaluate axonal integrity, we crossed our mutants to Thy1-YFP mice (Ey et al., 2007). Both Galc KO and Galc SC cKO nerves had axonal varicosities at P35 (Figure 4F, arrows), though more severe in Galc KO (Figure 4G). As mice aged, Galc SC cKO developed progressive axonopathy (Figures 4G and 4H) and overt axon degeneration (Figure 4I). To assess neuronal health, we used Atf3-GFP transgenic mice that report ATF3 expression in stressed neurons (Seijffers et al., 2006) (Y.-C.C. and C. Woolf, unpublished data). Consistent with the axonopathy data, P60 Galc SC cKO mice had more ATF3-GFP+ motor and dorsal root ganglia (DRG) neurons than WT (Figure 4J). Finally, we assessed the localization of nodal proteins. In Galc SC cKO axons the structural proteins neurofascin and Caspr, and the functional Na+ and K+ channels (Figures 4K and 4L) were diffused and not well clustered at nodes and paranodes. The abundance of heminodes (Figure 4K, double arrowhead) suggests that the underlying mechanism is segmental demyelination. Taken together, these data suggest that axonal degeneration and neuronal dysfunction can be, at least in part, directly caused by loss of function of Schwann cell GALC. However, GALC absence in neurons or other cell types of the Galc KO mice likely accelerates the axonopathy (Castelvetri et al., 2011).
Macrophages Recruited by Schwann Cells Express GALC and Assist in Myelin Turnover
We hypothesized that Galc SC cKO mice had attenuated pathology due to the presence of GALC in macrophages. Semithin sections of P35 sciatic nerves showed that macrophages in the Galc KO, but not those in Galc SC cKO nerves, had a hypertrophic globoid phenotype and many organelles throughout their distended cytoplasm (Figure 5A). Staining for the macrophage marker F4/80 and western blot for macrophage transcription factor PU.1 confirmed that macrophages were more abundant in Galc KO sciatic nerves (Figures 5B–5D).
Figure 5. Macrophages Recruited to Sites of Demyelination Require GALC for Myelin Turnover.
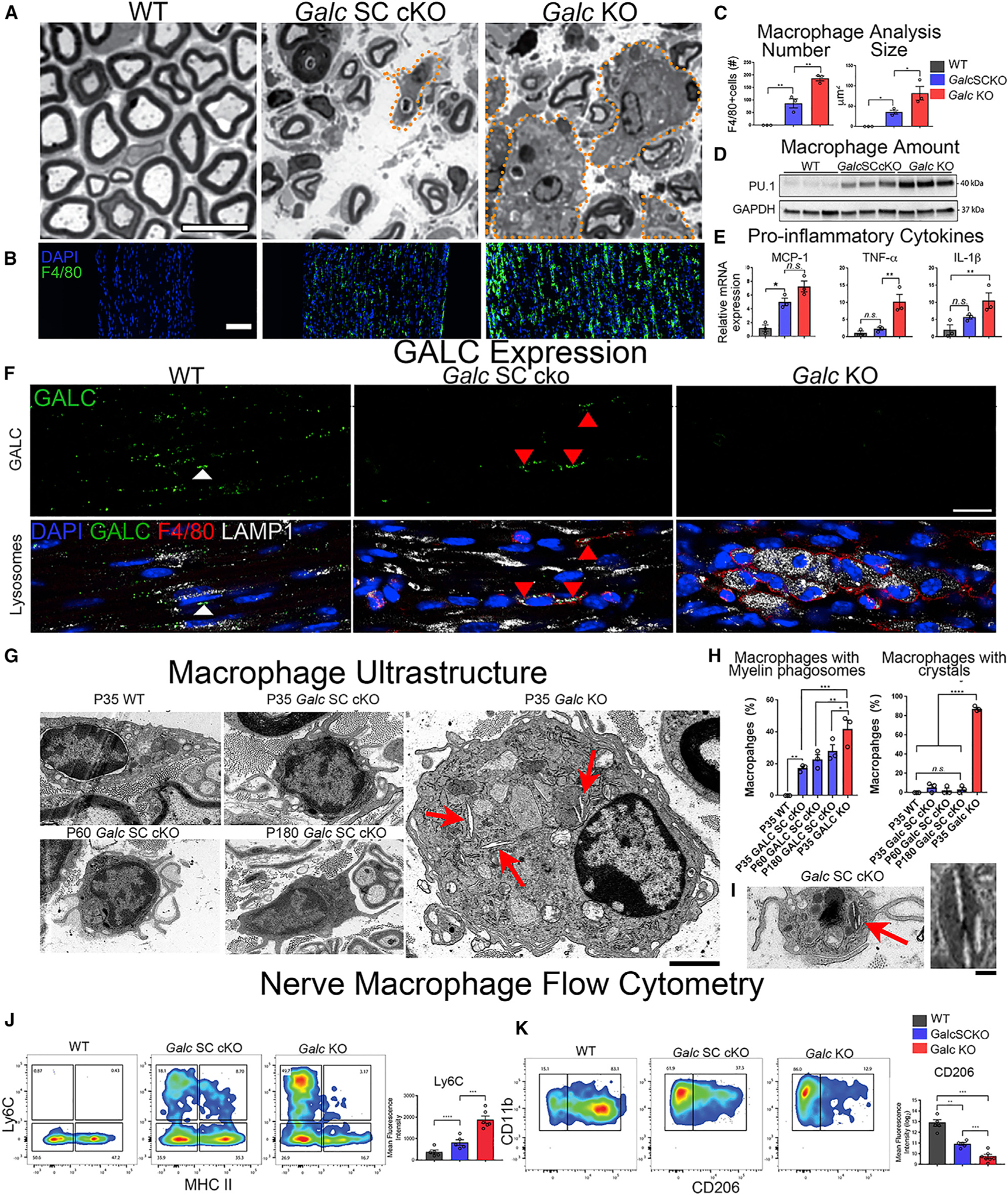
(A) Transverse semithin sections of P35 sciatic nerve (SN). Orange dotted lines outline macrophages.
(B) Immunofluorescence of macrophages in P35 sciatic nerve. Green, anti-F4/80; blue, DAPI.
(C) Quantification of macrophage number and size from (B).
(D) Western blot of macrophage marker PU.1 in P35 sciatic nerve.
(E) Quantitative RT-PCR of Mcp-1, Tnf-α, and Il-1β mRNA isolated from the sciatic nerve and normalized to β-Actin.
(F) Immunofluorescence of GALC expression in P35 nerves. Green, anti-GALC; red, anti-F4/80; blue, DAPI; white, anti-LAMP1. Scale bar, 30 μm.
(G) Electron microscopy (EM) of macrophages from sciatic nerves. Red arrows denote crystal inclusions. Scale bar, 2 μm.
(H) Quantifications of myelin phagosomes and crystals in macrophages from nerve.
(I) Galc SC cKO macrophage with crystal in EM. Inset is high magnification (scale bar, 200 nm).
(J) Representative plots of flow cytometry analysis of CD11b+/F4/80+ macrophages expressing MHC II and Ly6C from nerves (left) with quantification (right).
(K) Representative plots of flow cytometry analysis of CD11b+/F4/80+ macrophages expressing CD11b and CD206 from KO nerves (left) with quantification (right).
Scale bar, 10 μm (A), 50 μm (B), 30 μm (F), and 2 μm (G). Error bars represent mean ± SEM, n = 3–6 biological replicates and 3 technical replicates per experiment (n = 4 for J). One-way ANOVA was used to calculate statistical significance.
Macrophages are recruited to injured nerves upon fibroblast production of the chemoattractant protein MCP-1 in response to Schwann cell demyelination (reviewed in Martini and Willison, 2016). Interestingly, Galc SC cKO and Galc KO sciatic nerves had similar levels of MCP-1 (Figure 5E), suggesting that the macrophage recruiting signal is similar in both genotypes. However, the pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-10β (IL-1β) were increased in Galc KO nerves compared to Galc SC cKO and WT nerves, respectively (Figure 5E). Thus, macrophages are more abundant and larger and may have a greater inflammatory response in Galc KO than in Galc SC cKO nerves.
GALC staining from P35 sciatic nerves show that nerve macrophages express GALC in Galc SC cKO nerves (Figure 5F, arrowheads). Of note, while globoid cell macrophages from Galc KO nerves had abundant LAMP1+ lysosomes, indicative of lysosome storage, Galc-expressing macrophages in the Galc SC cKO nerve did not (Figure 5F). By ultrastructural EM, we found that Galc KO macrophages accumulate myelin phagosomes and crystalloid inclusions (Figures 5G and 5H), similar to Galc-ablated Schwann cells. Galc SC cKO macrophages, on the other hand, rarely accumulate crystals, even as mice aged to 6 months (Figures 5G and 5H).
The differences among these macrophages led us to explore their phenotypic signature by flow cytometry. We first analyzed Cd11b+ myeloid cells from the spleen (Figure S5A), a primary lymphoid organ that serves as a reservoir for circulating monocytes and resident macrophages (Swirski et al., 2009). Consistent with previous reports (Galbiati et al., 2007), Galc KO spleens were smaller than WT littermates (Figure S5B), previously attributed to autonomic dysfunction. Interestingly, Galc SC cKO spleens were instead larger, suggesting a CNS-dependent atrophic mechanism in GLD. Monocytes in Galc KO spleens were reduced, dendritic cells were increased, while macrophages remained unchanged (Figure S5C) and expressed similar amounts of the activation markers CD86 and major histocompatibility complex (MHC) II (Figure S5D) and the monocyte marker Ly6C (Figure S5E). This suggests that the phenotypic signature of circulating macrophages of the Galc KO mouse remains naive and is comparable to WT and Galc SC cKO macrophages.
In nerves, the majority of CD11b+ myeloid cells were F4/80+ macrophages (Figures S5F and S5G). Galc SC cKO and Galc KO nerve macrophages expressed similar M1 and M2 markers (Figure S5I). Interestingly, Galc KO macrophages expressed high levels of Ly6C (Figure 5J) and low levels of CD206 (Figure 5K). Ly6Chi monocytes are recruited to inflammatory environments and differentiate to Ly6Chi macrophages, where they degrade scar tissue, phagocytose inflammatory debris, and subsequently transition to a more restorative Ly6Clow subtype (Ramachandran et al., 2012). Conversely, CD206 renders macrophages phagocytosis competent (Schulz et al., 2019) and is downregulated in globoid cells from twitcher CNS (Kondo et al., 2011). Therefore, Ly6Chi/Cd206low Galc KO macrophages likely represent dysfunctional macrophages with impaired phagocytosis. Overall, these data suggest that macrophages expressing GALC are recruited to nerves to assist in myelin degradation. When these macrophages do not express GALC (i.e., in the Galc KO), they accumulate lysosomes and myelin phagosomes and develop a globoid appearance and an inflammatory and less phagocytic Ly6Chi/Cd206low profile.
Concurrent GALC Ablation in Schwann Cells and Macrophages Recapitulates Peripheral GLD
To directly probe GALC necessity in macrophages, we generated macrophage knockout mice (Galc Mac cKO) using LysM-Cre. LysM-Cre mice crossed to tdTomato reporter mice showed that 80% of spleen macrophages and 90% of sciatic nerve macrophages (Figures S6A and S6B) expressed tdTomato, indicative of high recombination efficiency in resident macrophages of the sciatic nerve. Instead, LysM Cre recombination in microglia was specific but less efficient at approximately 35% (Figures S6C and S6D). Galc mRNA was not reduced in desheathed Galc Mac cKO nerves, likely due to the low number of resident macrophages in non-pathologic nerves (Figure S6E). However, GALC expression by immunofluorescence was reduced in macrophages of the Galc Mac cKO (Figures 6A and 6B). Galc Mac cKO nerves had normal morphology in EM sections (Figure 6E). These data indicate that GALC is not required in macrophages for myelination nor for the myelin remodeling associated with development and early maintenance.
Figure 6. Concurrent Galc Ablation in Schwann Cells and Macrophages Recapitulates Peripheral GLD.
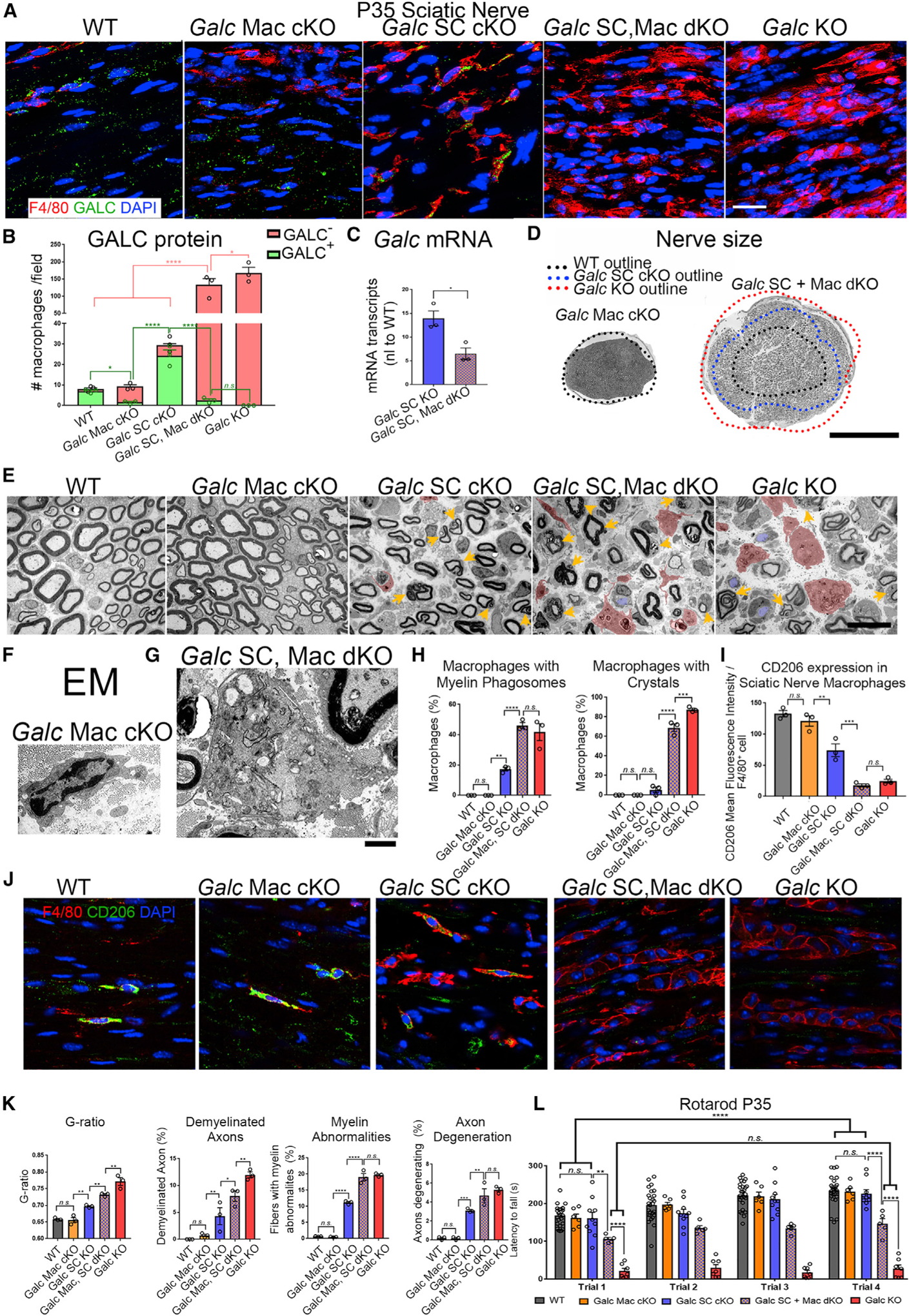
(A) Immunofluorescence of macrophages (anti-F4/80; red) and GALC (green) in sciatic nerve longitudinal frozen sections.
(B) Quantification of GALC+ and GALC– macrophages in P35 SN (from A). Pink and green bars represent the total number of GALC– and GALC+ macrophages, respectively.
(C) Quantitative RT-PCR of Galc mRNA from P35 sciatic nerves. Galc normalized to β-Actin and reported relative to average WT expression.
(D) Reconstructions of P35 sciatic nerves. Black dotted line represents the outline of a WT sciatic nerve and matches the circumference of the Galc Mac cKO nerve diameter. Blue dotted line represents the outline of a Galc SC cKO sciatic nerve. Red dotted line represents the outline of the full Galc KO nerve.
(E) EM of P35 SN shows macrophages pseudo-colored in red and demyelinated axons in blue. Yellow arrows reflect myelin abnormalities including degenerating myelin.
(F) EM of macrophage from P35 Galc Mac cKO sciatic nerves.
(G) EM of macrophage from P35 Galc SC, Mac dKO sciatic nerves.
(H) Quantification of macrophages with myelin phagosomes and crystals.
(I) Quantification of CD206 mean fluorescence intensity per F4/80 macrophage.
(J) Immunofluorescence of macrophages in P35 sciatic nerves with anti-CD206 (green), anti-F4/80 (red), and DAPI (blue).
(K) Quantification of average G-ratio and abnormalities shown in (E).
(L) Rotarod of P35 animals.
Scale bar, 25 μm (A), 300 μm (D), 10 μm (E), and 2 μm (F). Error bars represent mean ± SEM, n = 3 biological replicates and 3 technical replicates per experiment (n ≥ 6 for L). Statistical significance was calculated by one-way ANOVA (B, H, I, and K), Student’s t test (C) or two-way ANOVA (L).
To ask whether GALC was required in macrophages during demyelination in the context of GLD, we generated Galc SC and macrophage double knockouts (Galc SC, Mac dKO). Globoid-like macrophages from Galc KO sciatic nerves expressed tdTomato when crossed to LysM-Cre;tdTomato(tg) mice (Figure S6F). Galc mRNA and protein levels were significantly reduced in the Galc SC, Mac dKO, confirming that a large part of GALC expression in the SC cKO is derived from infiltrating macrophages (Figures 6A–6C). The sciatic nerve diameter of P35 Galc SC, Mac dKO nerves was larger than P35 Galc SC cKO nerves (Figure 6D), suggesting additional pathology. Indeed, macrophages of the Galc SC, Mac dKO were larger and more abundant than those of the Galc SC cKO (Figures 6A, 6B, and 6E), closely resembling the globoid macrophages of the Galc KO by EM (Figure 6G). Galc SC, Mac dKO macrophages accumulated myelin phagosomes and crystalloid inclusions, while macrophages of the Galc Mac cKO did not (Figures 6F–6H). Furthermore, macrophages of the Galc SC, Mac dKO were Cd206low, like Galc KO macrophages, while Galc Mac cKO macrophages were Cd206hi, like WT macrophages (Figures 6I and 6J). These data suggest that globoid cell formation is driven by concomitant macrophage GALC deficiency and macrophage exposure to myelin containing GALC substrates from demyelinating cells.
Interestingly, the total nerve pathology of Galc SC, Mac dKO was worse when compared to the single Galc SC cKO nerve and even approached the pathology of full Galc KO. Specifically, the G-ratio, number of demyelinated axons and number of myelin abnormalities all were worse in the Galc SC, Mac dKO compared to the Galc SC cKO alone (Figures 6E and 6K). Similarly, the amount of axonal degeneration was increased in Galc SC, Mac dKO mice (Figures 6E and 6K), and they performed worse on the rotarod (Figure 6L), though LysM-Cre expression in microglia could contribute to this particular test. To assess the long-term functional consequence of GALC ablation in Schwann cells and macrophages, we analyzed 6-month-old mice by nerve conduction studies (Figure S7). Analogous to P35 data, 6-month-old Galc SC cKO nerves had robust NCV defects but sparred proximal amplitude function (Figure S7). Instead, Galc SC, Mac dKO nerves had impairments in proximal amplitude and delays in latency compared to Galc SC cKO nerves. This suggests that the recruitment of GALC-expressing macrophages to sites of demyelination is beneficial to the health of Schwann cells, myelin turnover, and ultimately neuronal andaxonal health and function, while the recruitment of GALC-deficient macrophages further aggravates pathology.
Galc KO Macrophages Have Defects in Myelin and GalCer Degradation
Galc SC, Mac dKO mice highlight the importance of GALC in macrophages recruited to GLD nerves. However, it remains unclear why macrophages need GALC to reduce neuropathology and as to whether the globoid cell reaction occurs in response to the GALC substrate GalCer or psychosine.
To elucidate the cellular mechanism of GALC deficiency in macrophages, we turned to in vitro bone-marrow-derived macrophages (BMDMs) cultured from WT or Galc KO mice. To test the ability of Galc KO macrophages to digest myelin, BMDMs were incubated with IgG-opsonized myelin for 3 h and analyzed thereafter. Western blots of the myelin proteins PMP2 and MPZ showed that Galc KO BMDMs accumulated more non-degraded myelin compared to WT BMDMs (Figure 7A). To confirm that this myelin degradation defect was due to the degrading capacity of lysosomes, we repeated the assay in the presence of the proteasome inhibitor bortezomib and found that the myelin degrading capacity of Galc KO BMDMs became 5-fold worse than WT BMDMs (Figure 7B). Conversely, when we tested proteasome function of BMDMs by repeating the assay with the lysosome inhibitor chloroquine, Galc KO BMDMs degraded myelin proteins at approximately the same rate as WT BMDMs (Figure 7B). Therefore, GALC is required by macrophages for efficient degradation of myelin via the lysosome.
Figure 7. Galc-Deficient Macrophages Have a Defect in Myelin Degradation.
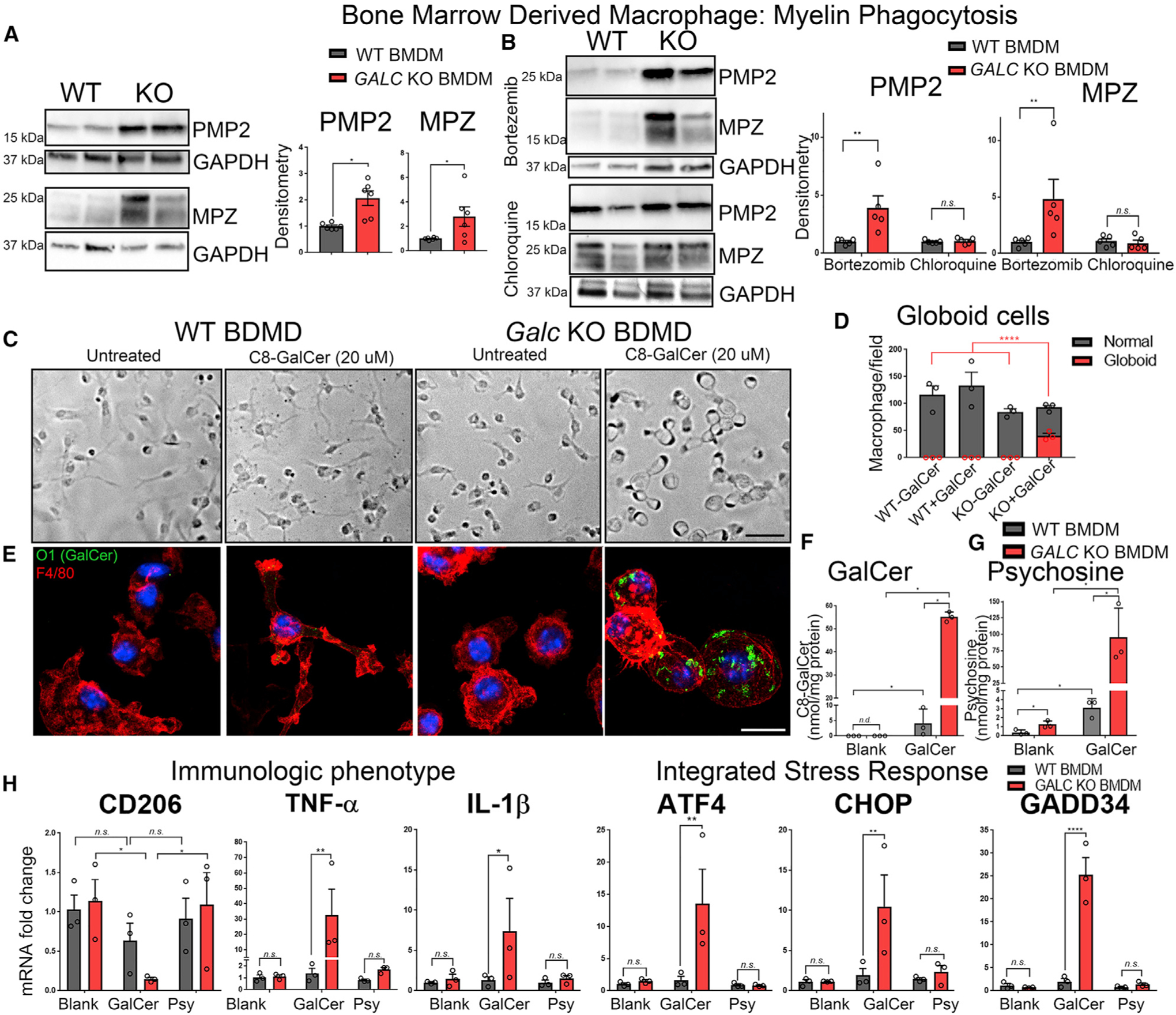
(A) Western blot and quantification of Peripheral Myelin Protein 2 (PMP2) and Myelin Protein Zero (MPZ) after myelin phagocytosis assay.
(B) Western blot and quantification of PMP2 and MPZ when macrophages were co-incubated with myelin and either (i) the proteasome inhibitor bortezomib or (ii) the lysosome inhibitor chloroquine.
(C and D) Bright-field images (C) and quantification (D) of bone-marrow-derived macrophages in culture treated with either blank or C8-galactosylceramide for 24 h.
(E) Immunofluorescence of macrophages (anti-F4/80; red) cultured with C8-GalCer and stained for O1 (anti-GalCer; green) and DAPI (Blue).
(F and G) HPLC-MS measurement of C8-GalCer (F) and psychosine (G) from macrophages treated with 20 μM C8-GalCer.
(H) qPCR of BMDMs incubated with 20 μM DMSO (blank), 20 μM C8-GalCer, or 5 μM psychosine for markers related to the immunological phenotype and integrated stress response, normalized to β-Actin, and reported relative to average WT expression.
Scale bars, 60 μm (C) and 15 μm (E). Error bars represent mean ± SEM, n = 3 biological replicates and 3 technical replicates per experiment (n ≥ 5 for A and B). Statistical significance was calculated by Student’s t test (A), one-way ANOVA (D), or two-way ANOVA (B, F, G, and H).
To determine whether GalCer causes globoid-like changes in macrophages, BMDMs were cultured with 20 μM C8-galactosylceramide (GalCer) for 24 h. WT BMDMs cultured with GalCer exhibited an activated morphology after 24 h (Figure 7C). Strikingly, Galc KO BMDMs incubated with GalCer developed a distended globoid-like morphology (Figures 7C and 7D). Immunofluorescence for GalCer and quantification using mass spectrometry showed that GALC KO BMDMs, but not WT BMDMs, accumulated GalCer (Figures 7E and 7F). Psychosine levels in GalCer-treated BMDMs were also increased in Galc KO cells, likely due to direct conversion of GalCer to psychosine by acid ceramidase (Li et al., 2019). Psychosine was equally toxic in WT and KO macrophages, while GalCer was not (Figures S8A and S8B). Interestingly, GALC KO BMDMs treated with C8-GalCer, but not psychosine, recapitulated a globoid cell profile including reduced CD206 and increased TNF-α and IL-1β (Figure 7H). Similar findings were seen for markers of the integrated stress response (Figure 7H). Taken together, these data indicate that Galc loss of function in macrophages causes a lysosomal defect in degrading myelin, which in turn results in GalCer accumulation and psychosine production, ultimately causing cellular stress and a pro-inflammatory globoid reaction.
HSCT Reduces Globoid Cells in Early Infantile Krabbe Disease
HSCT is beneficial to pre-symptomatic patients with early infantile Krabbe Disease (EIKD). Semithin sections from nerves of transplanted EIKD patients harboring the homozygous 30 kb deletion (Table S1) show improvements in axonal integrity and myelin thickness (Figures 8A and 8A′). The mechanism of action of HSCT is thought to be via secretion of GALC by monocytes or macrophages and uptake by myelinating glia (cross-correction). Our data suggest that GALC may not be able to efficiently transfer from macrophages to pathologic myelinating glia. To confirm this, we performed HSCT on twitcher mice (Figures S9A–S9Z) and asked whether Schwann cells were incorporating GALC. Only a minority of GALC+ macrophages were found in sciatic nerves (Figures S9K and S9L), probably reflecting the low engraftment using this method (Marshall et al., 2018). GALC+ macrophages had fewer lysosomes and expressed CD206 (Figures S9R–S9Z′). Only occasional Schwann cells showed scant GALC reactivity, suggesting that cross-correction may occur but to inefficient levels (Figures S9M–S9Q′). We hypothesized, instead, that GALC+ HSCT-derived macrophages are able to better degrade myelin and benefit GLD by decreasing the GalCer and psychosine load and by immunomodulation. To ask whether this hypothesis is consistent with findings in humans, nerves were stained with the CD68 macrophage marker. Untreated EIKD nerves had widespread macrophage infiltration with globoid appearance (Figure 8B). Strikingly, the EIKD subject who received HSCT had drastically reduced macrophages, suggesting reduced neuroinflammation (Figure 8B). These phenotypes occurred independently of patient lifespan. Furthermore, EM showed that Schwann cells of both untreated and treated EIKD patients had crystal inclusions (Figure 8C), while only macrophages of the untreated subject were globoid with crystalloid inclusions (Figure 8D). Overall, these data suggest that patients with EIKD who receive HSCT have healthy macrophages and few globoid cells. Finally, analysis of nerves from late-onset KD (LOKD) patients show that nerve macrophages are reduced and not globoid in appearance (Figure S9A′). This would suggest that a minimal amount of GALC activity (typically estimated to be between 1% and 5% of control levels) may be sufficient to prevent a globoid reaction. Accordingly, one might suspect that the storage-induced inflammatory reaction might be a major differentiator between EIKD and LOKD.
Figure 8. Macrophages Are Reduced and Have Normal Morphology in Nerves of EIKD Patients Who Received HSCT.
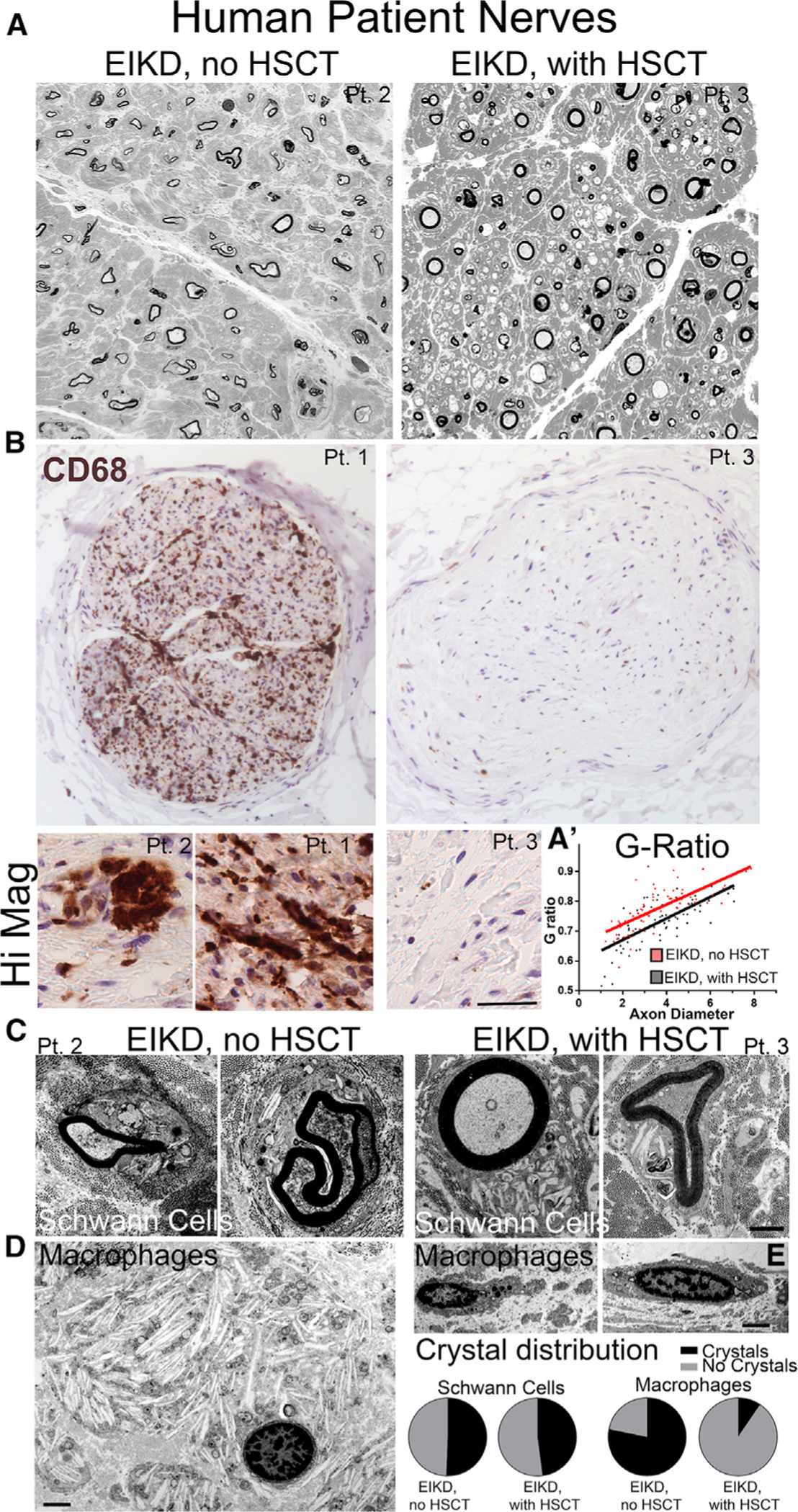
(A) Semithin sections of brachial plexus nerves from EIKD patients who either did or did not receive HSCT show loss of fibers and thin myelin but is worse without HSCT treatment.
(B) CD68 IHC staining of femoral nerves from EIKD patients show abundant macrophages only in non-HSCT-treated patients
(C–E) Nerve EM show accumulation of crystals in Schwann cells from both patients (C) and in globoid cells from non-treated patients (D). Crystal deposits are significantly reduced and morphology is normal in macrophages from HSCT-treated patients (E).
Scale bar, 100 μm (B), 35 μm in high-magnification panels, and 2 μm (C–E).
DISCUSSION
Cellular Cascade of GLD
Demyelination, neurodegeneration, and inflammation are the hallmarks of GLD. Due to the ubiquitous nature of GALC, previous studies were unable to discern whether these events follow in a linear and sequential cascade or instead reflect intertwined mechanisms, each requiring cell autonomous GALC. By using a conditional mutagenic approach in the PNS, we developed a simplified model system to study cellular autonomy of GALC in myelinating glia and the innate immune system. Our initial characterization revealed that GALC is essential to prevent demyelination in Schwann cells and is independently required for proper functioning of injury-recruited macrophages.
Autonomous Galc ablation in Schwann cells generates the majority of nerve psychosine in GLD, which is accompanied by widespread demyelination. Degenerating myelin and crystals also accumulated in Schwann cells. This confirms that myelinating glia require GALC for long-term myelin stability. Furthermore, Schwann cell GALC is also required for protection of axonal and neuronal health, at least in part independent of GALC expression in neurons.
Despite near-total ablation of Schwann cell Galc, resulting in identical levels of psychosine to the global Galc KO nerves, the overall degree of pathology was incomplete in mice with Schwann cell-restricted Galc deletion. Many neuropathic features, including demyelination and axonal degeneration, were reduced. On the contrary, pathologic features relating to macrophage function, including storage-laden globoid cells and inflammation, were pointedly absent when Galc remained expressed in macrophages. This led us to demonstrate that macrophages require GALC for autonomous functions during demyelination but not during normal nerve development.
During demyelination, as in Galc SC cKO nerves, macrophages that express GALC are recruited to assist in myelin degradation. These macrophages are beneficial to overall nerve health. Instead, when injury-recruited macrophages lack GALC, they have a molecular profile associated with poor phagocytosis and are unable to efficiently degrade myelin. This leads to lysosomal dysfunction and the accumulation of galactosylceramide, some of which is converted to psychosine. The ultimate consequence of this process is a proinflammatory globoid cell reaction with further myelin and axonal pathology. These findings are consistent with recent data suggesting that GALC haploinsuffiency perturbs remyelination and is a risk factor in multiple sclerosis (Sawcer et al., 2011; Scott-Hewitt et al., 2017).
Psychosine and GalCer Toxicity
Since the advent of the psychosine hypothesis (Miyatake and Suzuki, 1972), the Krabbe field has explored numerous mechanisms and pathways by which psychosine causes toxicity, including a direct effect on neuronal health (reviewed in Spassieva and Bieberich, 2016). We thus expected that psychosine would be toxic to surrounding cells. We were surprised, therefore, when pathology did not directly correlate with psychosine levels. This suggests that a non-psychosine-based GLD mechanism exists to explain the full pathology. Because psychosine levels in global KO and in Schwann cell-specific cKO nerves were similar, psychosine, once generated, may not be readily degraded by other GALC-expressing cells in the endoneurium. Alternatively, psychosine may not be freely diffusible between cells, even if the fact that psychosine is found in circulating blood may argue otherwise.
The pathogenic role of the major GALC substrate GalCer is unclear. Presumed GalCer inclusions are found in lysosomes of GLD globoid cells, oligodendrocytes, and Schwann cells (Suzuki and Grover, 1970; Yunis and Lee, 1970). Furthermore, direct implantation of GalCer, but not psychosine, in WT rat brains produced a “globoid reaction” (Andrews and Menkes, 1970). Ultimately though, GalCer does not accumulate at a whole tissue level (Hers, 1965) in brain or nerve (Suzuki, 1998) and has not been shown to be toxic in culture. Our work argues that GalCer nonetheless plays an important part in GLD toxicity. We saw that GalCer was sufficient to induce a globoid reaction in GALC-deficient BMDMs. Induced globoid cells expressed more proinflammatory cytokines and effectors of the integrated stress response. Remarkably, GALC-deficient macrophages in vivo have decreased expression of CD206, a surface receptor that enables macrophage phagocytosis (Schulz et al., 2019) and short-term GalCer induction in Galc-deficient BMDMs was sufficient to decrease CD206 expression, suggesting a direct role of GalCer in impairing macrophage function. Crystal accumulation in cells and particularly innate immune cells can be toxic due to the formation of the NLRP3 inflammasome (Duewell et al., 2010; Rajamäki et al., 2010) or lysosome membrane permeabilization (Ono et al., 2018), such as in gout (Martinon et al., 2006), asbestos, silica lung disease (Dostert et al., 2008), and atherosclerosis (Duewell et al., 2010). Similarly, myelin-derived crystals, thought to be composed of cholesterol, induce pathology in a demyelinating model (Cantuti-Castelvetri et al., 2018). Therefore, a number of GalCer-based toxicities could occur in GLD.
GALC Cross-Correction
A major finding of our study is that endogenous GALC was unable to efficiently cross-correct Galc-deficient cells in vivo. Furthermore, we found that Galc deficiency impaired the uptake of GALC in Schwann cells, oligodendrocytes, MEFs, and human fibroblasts. These data are surprising, especially because cross-correction is considered the basis for a number of therapies of LSDs (reviewed in Coutinho et al., 2012; Sands and Davidson, 2006). The majority of previous cross-correction studies have been analyzed in vitro and have, with a few notable exceptions, rarely compared the uptake ability of WT cells to their disease mutant counterparts (Kondo et al., 2005; Luddi et al., 2001; Nagano et al., 1998; Rafi et al., 1996). These data may also explain why Fabry disease females with one mutant copy, once thought to be “carriers” may develop clinical findings despite significant αGAL protein present (MacDermot et al., 2001; Wilcox et al., 2008). In addition, a recent study in postmortem brains from HSCT-treated metachromatic leukodystrophy patients also showed no evidence of cross-correction (Wolf et al., 2020). Whether this phenomenon extends to other LSDs remains an important unanswered question and may explain why certain corrective therapies have had only modest clinical improvements despite overall increased enzymatic activity.
HSCT for GLD
HSCT is the mainstay of treatment for pre-symptomatic KD. The mechanism of action for HSCT is poorly understood and is generally attributed to cross-correction, though immunomodulatory benefits of HSCT have also been proposed (Hoogerbrugge et al., 1988; Reddy et al., 2011). Our mouse data argue against the notion that donor-derived cells efficiently cross-correct neuronal and peripheral glial cells. Instead, we propose that allogenic transplantation restores the beneficial function of GALC-expressing macrophages and microglia in GLD, thereby reducing substrate accumulation and disease burden. In line with these findings, analysis of nerves from early infantile KD patients showed that HSCT normalized the number and globoid morphology of peripheral macrophages. This correlated with slightly decreased neuropathic findings in nerves from HSCT-treated patients, that were strikingly similar to the differences found between Galc SC cKO and Galc SC, Mac dKO nerves.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, M. Laura Feltri (mlfetri@buffalo.edu).
Material Availability
There are no restrictions on any data or materials presented in this paper. All unique/stable regents generated in this study, including plasmids generated, are available from the Lead Contact with a completed Materials Transfer Agreement. Floxed Galc mice are freely available from the Lead Contact and D. Shin (daesungs@buffalo.edu).
Data and Code Availability
No dataset/code is associated with this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were maintained in C57BL/6 background (Charles River Laboratories International, Inc.). Both male and female mice were used in all experiments. Animal housing and experimentation strictly adhered to Department of Laboratory and Animal Resource (DLAR) core facility at Roswell Park Cancer Institute (RPCI), and both RPCI and UB Institutional Animal Care and Use Committee protocols (IACUC approval UB# 1188 and 1254M). DNA was extracted from toe clippings or ear punching. Galc floxed mice were designed by L.W. and D.S. and generated by D.S. (Weinstock et al., 2020). Galc twitcher mice (JAX#000845) (Duchen et al., 1980) were used in comparison to validate Galc knockout (KO) mice. To produce Galc KO mice resembling twitcher mouse, Galc floxed mice were crossed with CMV-Cre (JAX# 006054) (Schwenk et al., 1995). Recombination of the floxed allele was passed in the germ-line of these mice, separated from Cre expression, and defined as a null allele (denoted by “ − “). P0-Cre (JAX#017927) mice were used to produce recombination in Schwann cells (Feltri et al., 1999). LysM-Cre (JAX#004781) mice were used to produce recombination in macrophages (and granulocytes) (Clausen et al., 1999). CNPase-Cre mice were used to produce recombination in oligodendrocytes (Lappe-Siefke et al., 2003). Td-Tomato Mice (JAX#007905) were used to test Cre specificity by expression of tdTomato fluorescence (Madisen et al., 2010). For a detailed characterization of neuronal/axonal morphology, the reporter Thy1.1-YFP mouse (JAX# 003709), which expresses YFP in subsets of neurons, allowed the fluorescent labeling of their projecting axons (Porrero et al., 2010). ATF3-GFP Mice (Y.-C.C. and C. Woolf, unpublished data) express GFP under control of the neuronal stress marker ATF3.
Human Tissues
Parents of Krabbe disease patients consented to autopsy and tissue donation to the University of Pittsburgh Pediatric Neurodegenerative Brain Bank. All banking procedures were approved by the Committee for Oversight of Research and Clinical Training Involving Decedents (CORID) at the University of Pittsburgh. The University of Buffalo granted an IRB exemption for the analysis of post-mortem human samples. Subject information of patient tissues used in this study can be found in Table S1.
Cell Culture
Primary Schwann cell culture were prepared from sciatic nerves dissected from P10-P15 mice (or from other ages if specified) under aseptic conditions. Nerves were placed in Leibovitz L-15 media supplemented with penicillin/streptomycin. Nerves were desheathed of epineurium and then transferred to high glucose DMEM supplemented with in 0.25% dispase II and 0.05% collagenase. Nerves were physically dissociated and incubated at 37C, 5%CO2 for 24 hours. Media and tissue was pipetted for further dissociation and transferred to plates pre-coated with poly-L-lysine and Laminin 211. Cells were filtered through a 70 μm cell strainer, plated in concentrated bubbles, and incubated for 24 hours. The following day, media and debris were removed and cells were grown in DMEM media supplemented with N2, penicillin/streptomycin, FBS and L-glutamine for 3–4 days in culture.
For bone marrow derived macrophages, isolation was performed using a previously published protocol (Weischenfeldt and Porse, 2008). 5-week-old mice were sacrificed by cervical dislocation. Femurs and tibias are collected without breaking bones, using sterile technique. Bone marrow was flushed using ice cold PBS, centrifuged, and resuspended in BMDM-complete media (RPMI 1640, 10% FBS, 1% glutamine, 1% Penicillin/Streptomycin, and 200 ng/mL rm-CSF). Bone marrow collected cells were plated on bacterial grade cell culture plates and grown at 37°C in 5% CO2 for 5 days. BMDMs were then scraped and replated on cell culture grade plastic dishes in BMDM-complete media. Cells were ready for downstream applications after 24 hours.
Mouse embryonic fibroblasts (MEFs) were collected according to previously published protocol (Durkin et al. Bio-protocol 2013). Human GLD and control fibroblasts were purchased from Telethon biobank and cultured as previously published (Shin et al., 2016). HEK293T cells, MEFs and human fibroblasts were grown in DMEM supplemented with 10% FBS, 1% glutamine, 1% penicillin/streptomycin (Shin et al., 2016). Nerve-derived macrophages were isolated as described in the Flow Cytometry section.
METHOD DETAILS
Isolation of Genomic DNA
For routine genotyping, toes were digested in 75 μL 25mM NaOH / 0.2 mM EDTA at 95°C for 45 minutes, and neutralized with 75 μL of 40 mM Tris HCl (pH 5.5). Samples were centrifuged and used for PCR reactions. For recombination PCRs, tissues were dissected and digested at 50°C overnight in 500 μL SNET buffer (20mM tris-Cl pH 8.0, 5mM EDTA pH 8.0, 400 mM NaCl, 1% w/v SDS) and 10 μL of proteinase K (PK, 20mg/mL in H2O). 500 μL of Phenol/Chloroform (1:1) was then added, vortexed and centrifuged. Supernatants were collected and 2x volume of 100% ethanol was added and centrifuged. Pellets were washed with 70% ethanol, dried, and resuspended in autoclaved double distilled H20.
RNA Isolation
Total RNA was isolated from mouse sciatic nerves using TRIzol reagent following the manufacturers’ instruction. For frozen nerves or tissues, samples were frozen in liquid N2 following dissection and stored at −80°C. A cooled pestle was used to pulverize tissues to powder. Powder was resuspended in TRIzol reagent. For cells, TRIzol was added directly to PBS-washed cell culture plate. After incubation for 5 minutes at room temperature, chloroform was added, shaken vigorously and centrifuged. The upper aqueous phase containing RNA was collected and 1 μL glycogen was added (20mg/mL). RNA was precipitated with isopropanol, pelleted, and washed twice with 75% ethanol. Pellets were resuspend in 10 μL DEPC-dH2O. RNA was quantified (OD at 260 nm) using a spectrophotometer (Thermo Fisher scientific; nanodrop 2000C) and analyzed for purity by 260/280 ratio.
cDNA Preparation and RT PCR
Invitrogen Kit (Superscript III) was used to convert RNA to cDNA using oligo dT and random hexamers provided. Following RT reaction, provided RNase H was added to each sample. cDNA was collected and stored at −80°C. For RT-PCR, SYBR Green and Taqman systems were used for different primers. For both systems the RT-PCR reaction was 50°C for 3 minutes, 95°C for 10 minutes, 95°C for 15 s, 60°C for 1 minutes (40 cycles). The amount of cDNA used in RT-PCR reactions were determined by standard curves in accordance with Applied Biosystems protocols. Each cDNA sample was tested in triplicate for the presence of each gene of interest and for the standard (18S rRNA or β-actin) on the same plates. Target and reference gene PCR amplification was performed. Assays on Demands (Applied Biosystems Instruments): 18S (Hs99999901_s1), Ddit3/Chop (Mm00492097_m1), XBP1s (Mm03464496_m1), Hspa5/BIP (Hs99999174_m1), CD206 (Mm01329362_m1). SYBR primers used can be found in Table S2. Mouse Galc mRNA primers were designed spanning exons 9 (forward primer) and 10 (reverse primer). All samples were analyzed in triplicate and the relative expression of the target RNAs was calculated using the ΔΔCt of the gene of interest compared to the housekeeper gene.
GALC enzymatic activity
GALC activity from, tissue or cell culture, was determined by a previously described method (Martino et al., 2009). Tissues and cells were homogenized in 10 mmol/L sodium phosphate buffer pH 6.0 with 0.1% Nonidet NP40 and sonicated.15 μg of protein extract (50 μL) was added to 100 μL of 1.5 mmol/L MUGAL substrate (fluorogenic substrate 4-methylumbelliferyl derivative 4-MU-β-d-galactoside) resuspended in 0.1/0.2 mol/L citrate/phosphate buffer, pH 4.0. 55 μM AgNO3 was also added to the mixture. Reactions were incubated for 30 min at 37°C. Reactions were stopped by adding 50 μL of 0.2 mol/L glycine/NaOH, pH 10.6. Fluorescence was measured using a spectrofluorometer (ex = 360 nm, em = 446 nm; Biotek Cytation5 imaging reader).
Trans-cardiac Perfusion
Brain immunohistochemistry was performed on animals perfused with 4% paraformaldehyde. Mice were anesthetized with avertin and monitored until sedated. Once unconscious, the thoracic wall was removed and the right atria was punctured. 20 mL of PBS was perfused into the left ventricle, followed immediately by 20 mL of 4% PFA. Tissues were post fixed in 4% PFA for 24 hours at 4 C, followed by sucrose and OCT embedding. Tissues were frozen in OCT and stored at −80C.
Immunofluorescence
Nerves were fixed for 30 minutes in 4% paraformaldehyde, washed 2x in PBS for 5 minutes, left overnight in 20% sucrose at 4°C and then frozen in OCT. Longitudinal sections (10 μm) were cut on a cryostat (Leica CM 1950), collected on a slide and circled with raw pap-pen boundary. Sections were rehydrated in PBS for 10 min at room temperature, blocked for 1hr at room temperature in blocking buffer (3% BSA in PBS; 2% normal goat serum, 0.5% Triton X-100). Primary antibodies were either purchased, affinity purified as described previously (GALC (Lee et al., 2010), CASPR (Peles et al., 1997)) or prodced from hybridoma cells (anti-GalCer O1 (Sommer and Schachner, 1981)). Primary antibodies were diluted in blocking buffer and applied to sections for 1 hour, at room temperature, in a humidified chamber. After washing 3x with PBS, secondary antibodies were diluted in blocking buffer and applied to sections for 1 hour at room temperature. DAPI was added for 8 minutes at room temperature. Sections were then washed 3x with PBS and mounted in Vectashield mounting media. Staining for lysosomes (including GALC) was similar to general protocol, but block included 0.05% saponin. Unless otherwise specified, all imaging was processed using Leica confocal microscope (Leica TCS SP5 II, Leica DMI 5000 CS).
Cholesterol Staining, Filipin
Primary Schwann cells were cultured from WT and Galc SC cKO nerves from P60 animals as described above. On day 3 of culture Schwann cells grown on coverslips were processed with cholesterol assay kit (abcam), according to manufacturer’s directions. Filipin III stock solution was prepared in 100% ethanol. Cell culture medium was removed from wells and cells were fixed with cell-based assay fixative solution for 10 minutes. Cells were washed with cholesterol detection wash buffer, three times, for five minutes each. Filipin stock solution was diluted in cholesterol detection assay buffer and applied to each well for 60 minutes. Cells were washed with wash buffer, twice, for five minutes. Coverslips were mounted on microscope slides with Vectashield and immediately processed on Apotome microscope (Zeiss Observer.Z1 AX10).
Flow cytometry
Spleens of P35 euthanized mice were removed, homogenized between frosted glass slides, flushed through a 70 μm filter, then subjected to ACK lysis in order to remove anucleated cells. Peripheral nerves of P35 mice, including sciatic nerves, trigeminal nerves and brachial plexus nerves, were dissected and pooled together. Pooled nerves were placed in Leibovitz L-15 media supplemented with penicillin/streptomycin. Nerves (with epineurium) were transferred to high glucose DMEM supplemented with in 0.25% dispase II and 0.05% collagenase. Nerves were manually dissociated using forceps and incubated at 37°C, shaking, for 45 minutes. Media and tissue was pipetted for further dissociation and filtered through two sequential 250 μm cell strainers. Both splenic and peripheral nerve cells were blocked for 15 minutes with Fc blocking reagent (anti-CD16/32, clone 93, Biolegend) in BSA-containing solution to reduce non-specific binding. Cell surface markers were stained with the appropriate combination of antibodies for 20 minutes, washed, and kept on ice. For staining of intracellular antigens, cells were fixed with 2% formalin for 20 minutes at room temperature, permeabilized with Perm/Wash reagent for 15 minutes (BD Cytofix/Cytoperm, BD Biosciences), then stained with appropriate antibodies in Perm/Wash reagent for 30 minutes. All samples were analyzed by BD LSR II flow cytometer. Data was analyzed with FlowJo software against isotype and single color controls. Myeloid cells were identified by positive staining for CD11b. Further subsets were defined as in Figure S8.
Panel 1: CD11b-BV711 (M1/70), Ly6G-PE (1A8), F4/80-APC (BM8), Ly6C-PE/Cy7 (HK1.4), CD11c-FITC (HL-3, BD PharMingen), CD86-APC/Cy7 (GL-1), MHC II-PerCP (M5/114.15.2)
Panel 2: CD11b-BV510 (M1/70), F4/80-APC/Cy7 (BM8), Nos2-APC (CXNFT), Arg1-FITC (IC5868F, R&D Systems), CD206-PE/Cy7 (C068C2), Egr2-PE (erongr2, Invitrogen), TNFa-BV711 (MP6-XT22), IL10-BV421 (JES5–16E3), IL12-PerCP/Cy5.5 (C15.6)
Teased Fiber Preparation
Slides were prepared using vectabond (Tespa). Nerves were fixed in 4% PFA for 30 minutes and then washed 3x with PBS. Nerves were desheathed with forceps and a 27-gauge needle. Bundles of fibers were separated in PBS using 27-gauge needles and then gently teased apart to single fibers on tespa coated slides. Slides were allowed to dry for at least one hour before staining. Staining protocol replicated as above.
Cloning and Purification of rhGALC
hGALC cDNA was previously ligated between the BamHI and XhoI sites of the pCMVTag4a vector (Shin et al., 2016) and included an HA tag. This plasmid construct was used for transwell fibroblast experiments. The plasmid was further modified to introduce an in-frame HA-6xHis-STOP at a SalI site by using primers listed in Table S2. The plasmid was sequenced after cloning. HEK293T cells were cultured and transfected with rhGALC-HA-His using lipofectamine 3000 reagent. HEK cells were cultured in serum-free media for 72 hours. Media was collected every 24 hours and stored at 4°C. After 72 hours, media was pooled, filtered and added to columns packed with His-Pur resin. Resin was washed (PBS, 10 mM imadizole) then eluted (PBS, 150 mM imadizole) in 8 mL. Eluant was concentrated using Amicon ultra15 10K columns, 4,000 x g, 4°C, 30 min. rhGALC-HA-His was quantified for protein using BCA method. Primary Schwann cells were cultured as described previously. After 4 days in culture, Schwann cells were treated with 5 μg/mL rhGALC-HA-His in serum-free media for 24 hours. Following incubation with enzyme, media was removed and cells were washed. Cells were then collected for western blot analysis. D-mannose 6-phosphate (M6P) (Sigma M6876) was added at a final concentration of 6 mM as described in (Sukehisa Nagano et al., Clinica Chimica Acta 276 (1998) 53–61) to inhibit CI-MPR mediated uptake.
Western Blotting
Tissue or cells were lysed in RIPA buffer supplemented with phosphatase and protease inhibitors. For sciatic nerves, the samples were flash frozen in liquid nitrogen and pulverized with a pestle. The crushed powder was resuspended in lysis buffer. For cells, wells were washed with PBS 1x and RIPA buffer was added to each well on ice. Cells were scrapped off in RIPA buffer and collected. Lysates were disrupted on a cell homogenizer for 10 minutes at 4°C and left on ice for 20 minutes. Lysates were centrifuged and the supernatant was removed. Protein concentration analysis was determined with a BCA protein assay kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Samples were prepared with 6x Laemmli buffer and lysis buffer. 5 μg to 20 μg of protein were loaded per lane and resolved using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions with a mini-Protean II gel electrophoresis apparatus and included a Precision Plus Standard Protein Dual color from (Biorad) to enable band size identification. Separated proteins were transferred to a PVDF blotting membrane, in a mini gel transfer tank. Non-specific binding sites on the membrane were blocked for 1 hour at room temperature using 5% non-fat free milk powder in TBS/0.05% Tween 20. Primary antibodies in 3% BSA in TBS/0.05% Tween 20 were incubated overnight at 4°C. Membranes were washed in TBST (0.05%) 3x for 5 minutes and incubated for 1 hour at room temperature with secondary antibody. Secondary antibodies were either horseradish peroxidase-conjugated (HRP) secondary antibody diluted in 5% milk TBST or Li-Cor secondary fluorescent antibody diluted in Li-Cor odyssey buffer. Blots incubated with HRP- conjugated secondary antibody were developed with ECL chemiluminescent reagent with film on a Chemi-doc apparatus (BIO RAD), while Li-Cor antibodies were developed using the Odyssey CLx apparatus. (Li-Cor).
Semithin Morphology and Electron Microscopy
Nerves were dissected and fixed in 2% glutaraldehyde. Nerves were washed in phosphate buffer 0.12M pH 7.4 at RT and incubated in osmium tetroxide 1% (phosphate buffer 0.12M pH 7.4) for two hours in room temperature in the dark. Nerves were then washed twice (phosphate buffer 0.12M pH 7.4) and dehydrated in increasing ethanol concentrations (50%, 70%, 90% and 100%) followed by incubation in propylene oxide. Propylene oxide was then evaporated and nerves were embedded in Epon 100%. Resin was polymerized at 60°C overnight. Nerves were then cut to semithin sections at 1 μm thickness or ultrathin sections at 80–85 nm thickness. Semithin sections were stained with toluidine blue 2% (phosphate buffer 0.12M pH 7.4). Ultrathin sections with saturated uranyl acetate (in dH2O) and lead citrate.
For quantification of axon density, 10 fields per nerve were imaged and analyzed. For quantification of myelin abnormalities, all axons in cross section of reconstructed sciatic nerves were counted and normalized to total myelinated axons. G-ratio analyses were performed on EM images, in which 75–100 axons were quantified per nerve. For axon degeneration and demyelination, 300 axons were analyzed per nerve, on EM images, and normalized as a percentage of total axons. For myelin ovoid and crystal analysis, Schwann cells and macrophages were assessed on EM micrographs. Schwann cells were identified by presence of basal lamina while macrophages were identified by characteristic morphologic features including microvilli.
Nerve Conduction Studies
Electrophysiologic studies were performed as previously described (Della-Flora Nunes et al., 2017). Mice (35 days or 6 months) were anesthetized with 2,2,2 tribromoethanol (avertin; Sigma-Aldrich) 10mg/mL in H2O and placed under a heating lamp. The sciatic nerve conduction velocity was obtained with steel monopolar needle electrodes. One pair of stimulating electrodes was inserted subcutaneously near the nerve at the ankle, a second pair of electrodes was placed at the sciatic notch, and a third pair over the dorsum of the spine. The compound motor action potential (CMAP) was recorded with an active electrode inserted in muscles in the middle of the paw and a reference needle in the skin between the first and second digits.
Rotarod Analysis
Rotarod analyses were performed as previously described (Della-Flora Nunes et al., 2017). All mice were tested in two sessions of three trials each per day (6-hour rest between the two daily sessions) for two consecutive days. Analyses measuring 35 day old animals occurred at P34 and P35; those measuring P60 animals occurred at P59 and P60. Only naive mice were used for Rotarod tests (no training occurred prior to trial 1). Rotarod conditions were set to acceleration of 5 rotations per minute2, starting at a minimum velocity of 4 rotations per minute and accelerating to a maximum velocity of 40 rotations per minute. Each trial consisted of one acclimating run that was not scored. The next three runs were recorded and averaged. Each run was stopped when the mouse fell or passed completely underneath the rod (180 degrees of rotation).
Pig Myelin Purification
Pig myelin was purified for use in myelin phagocytosis assay. Frozen pig nerves were stored at −80°C until used. Myelin was purified according to (Larocca and Norton, 2006). Pig nerves were pulverized using a mortar and pestle on dry ice. Nerve homogenate was further homogenized in a dounce homogenizer in 0.27 M sucrose solution, containing protease inhibitors. Connective tissue was eliminating by filtering homogenate through a cheese cloth. Equal volumes of nerve/0.27M sucrose solution was overlaid over 0.83M sucrose in ultracentrifuge tubes. The solution was centrifuged for 45 min at 82,000 x g, at 4°C. Using a Pasteur pipet, the crude myelin fraction was isolated at the 0.27/0.83M sucrose interface. Osmotic shock was then carried out by resuspending the combined myelin layers in Tris Cl buffer. The myelin/Tris buffer was then ultracentrifuged for 15 min at 82,000 x g, at 4°C. The pellet was resuspended in Tris buffer, and the protein concentration was estimated by the BCA method.
BMDM Myelin phagocytosis assays
Experiments were modified from previously described methods (Mosley and Cuzner, 1996; Trotter et al., 1986). Bone marrow derived macrophages (BMDMs) were plated in complete BMDM media. Pig myelin was pre-incubated with rabbit anti-PMP2 antibody (proteintech) and rabbit anti-MPZ antibody (abcam) at a concentration of 0.1 μL/μg of myelin protein, for two hours at 37°C. Opsonized myelin was added to serum free BMDM media at a concentration of 200 μg/mL, and delivered to macrophages for 3 hours. Pharmacologic inhibitors were added to opsonized myelin when indicated. The concentration of bortezomib was 20 nM and the concentration of chloroquine was 100 μM. After assay, cells were washed in PBS and collected for protein analysis.
BMDM GalCer and psychosine treatment
Bone marrow derived macrophages (BMDMs) were plated in complete BMDM media. C8-galacotsylceramide or psychosine was purchased from Avanti polar lipids and resuspended at 10 mM in DMSO. Lipids were aliquoted and stored at −20°C until further use. At use, lipids were warmed in the incubator for 10–15 min and added to cells in serum-free media for 24 hours. GalCer was delivered at a concentration of 20 μM. Psychosine was delivered at a concentration of 20 μM for cell viability experiments or 5 μM for qPCR experiments. Cells were washed in PBS prior to downstream applications.
Cell Viability Assays
Cells were assessed for cell viability using the cell counting kit 8 (Dojindo) in 96 well plates. After treating BMDMs with lipids (C8-GalCer and psychosine), 10 μL of WST-8 reagent was added to each well. Cells were incubated at 37°C, for 4 hours. Plates were read at 450 nm absorbance on a plate reader and normalized according to manufacturer’s instructions.
Bone Marrow Transplantation
WT and twitcher mice were transplanted with bone marrow, as previously described (Marshall et al., 2018). Briefly, P2 mice were injected intravenous treatment of bone marrow stem cells harvested from 6–8 week old syngeneic WT mice (30 million cells each treatment). Mice were sacrificed at P35 and nerves were 4% PFA post-fixed and sucrose embedded as described previously.
Psychosine Measurement
Nerves were snap frozen in liquid nitrogen and stored at −80°C until ready to perform experiments. Nerves were pulverized and homogenized in PBS. A fraction of PBS-homogenate was refrozen and shipped to for analysis by collaborators X.H. and M.G. or intact frozen nerves were sent to D.N. and E.R.B. For the latter, psychosine was quantified according to (Galbiati et al., 2007).
For the former, the other fraction of PBS-homogenate was mixed with 10X RIPA lysis buffer to make 1X RIPA buffer. Samples were then sonicated and analyzed for protein quantification by BCA. To 5 μL of tissue/PBS homogenate, 250 μL of 1 nM d5-psychosine (Avanti Polar Lipids) in methanol was added. Psychosine was extracted at 37°C for 2 h with orbital shaking (250 rpm). After extraction, the residual was centrifuged at 13,000 x g for 5 min. The supernatant was loaded onto an Oasis MCX column (1 cc, 30 mg, Waters Corp., #186000252), which was preconditioned with 1 mL of methanol, following by 1 mL of water. After sample loading, the cartridge was washed with 1 mL of water with 2% formic acid, 1 mL of methanol, and then 1 mL of 80:20 methanol:water (v:v) with 5% NH4OH. The column was washed with 0.8 mL of methanol with 5% NH4OH, which was collected and solvent evaporated using a SpeedVac vacuum concentrator. The residue was stored at −20°C until analyzed. The residue was reconstituted with 100 μL mobile phase prior to UPLC-MS/MS analysis.
For UPLC-MSMS analysis, an ACQUITY UPLC I-Class system from Waters was used for the separation of glucosyl- and galactosyl-sphingosine (psychosine). A 2.1 × 50 mm BEH Amide column (Waters Corp., #186004800) with a guard column (Waters Corp., #186004799) was used. The column was held at 40°C. Mobile phase A was water with 2 mM ammonium acetate and 0.1% NH4OH, mobile phase B was 95:5 acetonitrile:water (v:v) with 2 mM ammonium acetate and 0.1% NH4OH. The flow rate was 0.3 mL/min. The weak needle wash was 95:5 acetonitrile:water (v:v), and the strong needle wash was 50:50 acetonitrile: water. Ten μL of sample was injected onto the column, which was developed with the following gradient: elution started with 99.5% mobile phase B, decreased to 95% mobile phase B over 4.5 min (linear gradient), decreased to 60% mobile phase B at 5.5 min (linear gradient) and was held at 60% mobile phase B for cleaning until 6.0 min. At 6.01 min the solvent was switched back to initial conditions (99.5% mobile phase B) and was held for 9 min for re-equilibration.
The UPLC system was coupled to a Xevo TQ-S (Waters) tandem mass spectrometer, which was operated in the multiple reaction monitoring (MRM) mode. ESI source parameters are as follows: capillary 3.5kV, source temperature 150°C, desolvation temperature 600°C, cone gas flow 50 L/Hr, desolvation gas flow 1000 L/Hr and collision gas flow ON (mL/Min). MRM transitions of psychosine and d5-psychsoine had SRM transition (m/z) of 462.30 > 282.20, and 467.35 > 287.35 respectively. Cone voltage was 20V and collision energy was 21V.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data collection and analyses were performed blind to genotypes and conditions of experiments. Quantification of images and mean fluorescent intensity was done using ImageJ (Schneider et al., 2012). Statistical details of experiments can be found in figure legends, including statistical tests used, value of n and dispersion measures. Additional details may be found in methods and main text.
Statistical tests were performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, San Diego California USA, https://www.graphpad.com/, and included Student t test, One way ANOVA and Two way ANOVA as desribed through text. Statistical significance was defined as p < 0.05 and were denoted by asterisks. * = p < 0.05, ** = p < 0.01; *** = p < 0.005; **** = p < 0.001. Data throughout text are presented as mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CASPR | A gift from Elior Peles, Weizmann Institute. (Peles et al., 1997) | #A6061 RRID:AB_231422 0 |
| Mouse anti-APC (CC-1) | Millipore | Millipore Cat# OP80, RRID:AB_2057371 |
| Mouse anti-CD-68 | Dako (Agilent) | Agilent Cat# M0814, RRID:AB_2314148 |
| Rabbit anti-CD206 | Proteintech | Cat# 18704–1-AP RRID:AB_105972 32 |
| Goat anti-Choline Acetyltransferase (ChAT) | Millipore | Millipore Cat# AB144P, RRID:AB_2079751 |
| Rat anti-mouse F4/80 | Bio-Rad | Bio-Rad Cat# MCA497GA, RRID:AB_323806 |
| Chicken anti-GALC | A gift from Chris Lee and Christopher Eckman. (Lee et al., 2010) | CL1021AP |
| Rabbit anti-GAPDH | Sigma-Aldrich | Sigma-Aldrich Cat# G9545, RRID:AB_796208 |
| Rat anti-HA High Affinity | Roche | Roche Cat# 11867423001, RRID:AB_390918 |
| Anti-HA-Peroxidase, High Affinity | Roche | Roche Cat# 12013819001, RRID:AB_390917 |
| Rabbit anti-IBA1 (for immunocytochemistry) | Wako Chemicals | Cat# 019–19741 RRID:AB_839504 |
| Rabbit anti-Kv1.1 (KCNA1) | Alomone Labs | Cat# APC-009, RRID:AB_204014 4 |
| Rabbit anti-LAMP-1 | Abeam | Abcam Cat# ab24170, RRID:AB_775978 |
| Chicken anti-MPZ | Aves | Aves Labs Cat# PZO, RRID:AB_2313561 |
| Rabbit anti-MPZ | Abeam | Abcam Cat# ab31851, RRID:AB_2144668 |
| Mouse anti-Nav1.6 clone K87A/10 | UC Davis / NeuroMab | RRID:MMRRC_06 5966-UCD |
| Mouse anti-Pan-Neurofascin L11A/41 | Millipore | Millipore Cat# MABN621 RRID: AB_10672370 |
| Anti-galactocerebroside (O1) | A gift from Pablo Paez, SUNY Buffalo. (Sommer and Schachner, 1981) | O1 Hybridoma |
| Rabbit anti-human OLIG2 | Proteintech | Proteintech Cat# 13999–1-AP, RRID:AB_2157541 |
| Rabbit anti-PMP2 | Proteintech | Proteintech Cat# 12717–1-AP, RRID:AB_2166978 |
| Rabbit anti-PU.1 (Spi-1) (T-21) | Santa Cruz | Santa Cruz Biotechnology Cat# sc-352, RRID:AB_632289 |
| Mouse anti-Tubulin β3 (Tuj1) | Biolegend | Covance Cat# MMS-435P, RRID:AB_2313773 |
| Rabbit anti-β-Tubulin | Novus | Novus Cat# NB600–936, RRID:AB_1000065 6 |
| Brilliant Violet 711 anti-mouse/human CD11b antibody (M1/70) | BioLegend | Cat# 101241 RRID:AB_112187 91 |
| PE anti-mouse Ly-6G antibody (1A8) | BioLegend | Cat# 127607 RRID:AB_118610 4 |
| APC anti-mouse F4/80 antibody (BM8) | BioLegend | Cat# 123115 RRID:AB_ 893493 |
| PE/Cy7 anti-mouse Ly-6C antibody (HK1.4) | BioLegend | Cat# 128017 RRID:AB_ 1732093 |
| FITC Hamster Anti-Mouse CD11C (HL3) | BD PharMingen | Cat# 553801 RRID:AB_395060 |
| APC/Cy7 anti-mouse CD86 antibody (GL-1) | BioLegend | Cat# 105029 RRID: AB_2074993 |
| PerCP anti-mouse I-A/I-E antibody (M5/114.15.2) | BioLegend | Cat# 107623 RRID: AB_893586 |
| Brilliant Violet 510 anti-mouse/human CD11b antibody (M1/70) | BioLegend | Cat# 101245 RRID:AB_256139 0 |
| APC/Cyanine7 anti-mouse F4/80 antibody (BM8) | BioLegend | Cat# 123117 RRID: 893489 |
| iNOS Monoclonal antibody (CXNFT), APC, eBioscience | Thermo Fisher Scientific | Cat#17-5920-82 RRID:AB_257324 4 |
| Human/Mouse Arginase 1/ARG1 Fluorescein- conjugated antibody | R&D Systems | Cat#IC5868F RRID:AB_107181 18 |
| PE/Cy7 anti-mouse CD206 (MMR) antibody (C068C2) | BioLegend | Cat# 141719 RRID:AB_256224 7 |
| EGR2 Monoclonal Antibody (erongr2), PE, eBioscience | Thermo Fisher Scientific | Cat# 12-6691-82 RRID:AB_107178 04 |
| Brilliant Violet 711 anti-mouse TNF-α antibody (MP6-XT22) | BioLegend | Cat# 506349 RRID:AB_262980 0 |
| Brilliant Violet 421 anti-human IL-10 antibody (JES3–9D7) | BioLegend | Cat# 501421 RRID:AB_108969 47 |
| PerCP/Cy5.5 anti-mouse IL-12/IL-23 p40 (monomer, dimer, heterodimer) antibody (C15.6) | BioLegend | Cat# 505211 RRID:AB_256622 4 |
| TruStain FcX (anti-mouse CD16/32) antibody (93) | BioLegend | Cat#101319 RRID:AB_157497 3 |
| Alexa Fluor 488-AffiniPure F(ab’)2 Fragment Donkey anti-Chicken IgY | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 703-546-155, RRID:AB_2340376 |
| Rhodamine Red-X-AffiniPure Donkey Anti-Rabbit IgG | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 711-295-152, RRID:AB_2340613 |
| Alexa Fluor 647 AffiniPure F(ab’)2 Fragment Donkey Anti-Rabbit IgG | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 711-606-152, RRID:AB_2340625 |
| Goat anti-Mouse lgG2b Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A- 21145, RRID:AB_2535781 |
| Alexa Fluor 594-AffiniPure Goat Anti-Mouse IgG, Fc subclass 2a Specific antibody | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 115-585-206, RRID:AB_2338886 |
| Cy3-AffiniPure Donkey Anti-Goat IgG (H+L) antibody | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 705-165-003, RRID:AB_2340411 |
| Alexa Fluor 488-AffiniPure Donkey Anti-Rat IgG antibody | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 712-545-153, RRID:AB_2340684 |
| Rhodamine (TRITC)-AffiniPure Donkey Anti-Rat IgG antibody | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 712-025-153, RRID:AB_2340636 |
| Donkey Anti-Rabbit Rabbit IgG (H+L) Polyclonal, HRP-Conjugated antibody | Novus | Novus Cat# NB 7185, RRID:AB_524677 |
| Peroxidase-AffiniPure Donkey Anti-Chicken IgY (IgG) antibody | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 703-035-155, RRID:AB_1001528 3 |
| Peroxidase-AffiniPure Goat Anti-Rat IgG, Light Chain Specific | Jackson ImmunoResearch Labs | Jackson ImmunoResearch Labs Cat# 112-035-175, RRID:AB_2338140 |
| Goat anti-Mouse IgM, mu chain, FITC conjugate antibody, Millipore | Millipore | Millipore Cat# AP128F, RRID:AB_1121579 1 |
| Bacterial and Virus Strains | ||
| DH5a | Thermo Fisher Scientific | Catalog number: 1825801 2 |
| Biological Samples | ||
| Mouse Nerve RNA | This paper | N/A |
| Mouse Schwann cell RNA | This paper | N/A |
| Mouse BMDM RNA | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2,2,2-tribromoethanol (avertin) | Millipore Sigma | Cat#T48402 |
| 4-methylumbelliferone β-D-galactopyranoside | Sigma-Aldrich | Cat#M1633 |
| 4-methylumbelliferone sodium salt | Sigma-Aldrich | Cat#M1508 |
| Amicon Ultra-15 | Fisher Scientific | Cat#UFC901024 |
| C8 galactosyl ceramide | Avanti Polar Lipids | Cat#860538P |
| Diethyl pyrocarbonate (DEPC) | Fisher Scientific | Cat#FERR0601 |
| D-Mannose 6-phosphate | Sigma-Aldrich | Cat#M6876 |
| DNA 100bp ladder | Goldbiotechnology | Cat#D001–2500 |
| Pierce ECL Western Blotting | Thermo Fisher Scientific | Cat#32106 |
| Pierce ECL Plus Western Blotting | Thermo Fisher Scientific | Cat#32132 |
| GE Healthcare Amersham | Fisher Scientific | Cat#45-001-507PM |
| HisPur Cobalt Resin | Thermo Fisher Scientific | Cat#89964 |
| Imidazole | Millipore Sigma | Cat#T6330 |
| β-Mercaptoethanol | Sigma-Aldrich | Cat#M6250 |
| Phosphatase Cocktail 2 | Sigma-Aldrich | Cat#P5726 |
| Phosphatase Cocktail 3 | Sigma-Aldrich | Cat#P0044 |
| PMSF | Sigma-Aldrich | Cat#45-P7626 |
| Precision Plus Protein Dual Color Standards | Bio-Rad | Cat#161–0394 |
| Psychosine (Galactosyl Sphingosine, d18:1) | Avanti Polar Lipids | Cat#860537P |
| Deuterated Psychosine Quantitative Mass Spec Standard | Avanti Polar Lipids | Cat#330714W |
| Saponin | Sigma-Aldrich | Cat#47036 |
| Thermo Scientific Pierce Tissue Strainers (250 microns) | Fisher Scientific | Cat#87791 |
| Vectashield Antifade Mounting Medium | Vector Labs | Cat#H-1000 |
| Vetabond reagent for tissue section (TESPA) | Vector Labs | Cat#SP-1800 |
| B27 Supplement | Thermo Fisher Scientific | Cat#17504–044 |
| Collagen | Corning | Cat#354236 |
| Collagenase type I | Fischer Scientific | Cat#NC9633623 |
| Dispase II | Sigma-Aldrich | Cat#D4693 |
| Forskolin | EMD Millipore | Cat#344270 |
| Human Recombinant Laminin 211 | Biolamina | Cat#LN211–03 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000008 |
| Recombinant Human Nrg1-β1 | R&D Systems | Cat#396-HB-050 |
| Recombinant Mouse M-CSF | Biolegend | Cat#576404 |
| RPMI-1640 medium | Thermo Fisher Scientific | Cat#21870076 |
| Nerve Growth Factor NGF | Envigo | Cat#BT.5017 |
| Opti-MEM | Thermo Fisher Scientific | Cat#11058–021 |
| Critical Commercial Assays | ||
| Cell counting kit-8 | Dojindo | Cat#CK04–05 |
| Cholesterol Assay Kit (aka Filipin) | Abeam | Cat#Ab133116 |
| Fixation/Permeabilization Solution Kit | BD Cytofix/Cytoperm | Cat#554714 |
| Reverse Transcriptase (superscript III Kit) | Thermo Fisher Scientific | Cat#18080–051 |
| Taqman Gene Expression Assay: 18S (Hs99999901_s1) | Thermo Fisher Scientific | Cat#4331182 |
| Taqman Gene Expression Assay: Ddit3/CHOP (Mm00492097_m1) | Thermo Fisher Scientific | Cat#4331182 |
| Taqman Gene Expression Assay: XBP1 (Mm03464496_m1) | Thermo Fisher Scientific | Cat#4331182 |
| Taqman Gene Expression Assay: HSPa5/BiP (Hs99999174_m1) | Thermo Fisher Scientific | Cat#4331182 |
| Taqman Gene Expression Assay: MRC1/CD206 (Mm01329362_m1) | Thermo Fisher Scientific | Cat#4331182 |
| Experimental Models: Cell Lines | ||
| Human: Dermal Fibroblasts | Telethon biobank (Shin et al., 2016) | N/A |
| Mouse: Primary Schwann cells | This paper | N/A |
| Mouse: Primary bone marrow derived macrophages | This paper | N/A |
| Mouse: Primary embryonic fibroblasts | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.C-Tg(GALCfl/fl) | This paper. (Weinstock et al., 2020) | N/A |
| Mouse: B6.CE-Galctwi/J | The Jackson Laboratory | Cat#000845 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | The Jackson Laboratory | Cat#006054 |
| Mouse: B6N.FVB-Tg(Mpz-cre)26Mes/J (aka PO Cre) | The Jackson Laboratory | Cat#017927 |
| Mouse: B6.129P2-Lyz2tm1(cre)Ifo/J (aka LysM Cre) | The Jackson Laboratory | Cat#004781 |
| Mouse: B6;129S6-Gt(ROSA)26Sortm14(CAG-dTomato)Hze/J (aka tdTomato) | The Jackson Laboratory | Cat#007908 |
| Mouse: B6.Cg-Tg(Thy1-YFP)HJrs/J | The Jackson Laboratory | Cat# 003782 |
| Mouse: ATF3-GFP | Y.-C.C. and C. Woolf unpublished data | N/A |
| Oligonucleotides | ||
| Primers for qPCR, See Table S2 | This paper | N/A |
| Primers for cloning hGALC-6xHis-HA, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| hGALC-HA | (Shin et al., 2016)) | N/A |
| hGALC-6xHis-HA | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.net/Welcome |
| Graph Pad Prism 7 | GraphPad | Graphpad.com |
Highlights.
Schwann cell Galc prevents psychosine-induced demyelination and neurodegeneration
GALC transfer (cross-correction) is minimal in globoid cell leukodystrophy (GLD) cells
Storage-laden macrophages (globoid cells) are direct mediators of GLD pathogenesis
HSCT for GLD provides functional macrophages that are competent of phagocytosis
ACKNOWLEDGMENTS
This work was funded by grants NIH R01 NS111715 to M.L.F. and L.W., NIH R01 NS112327 to D.S., NIH F30 NS090835 to N.I.W., NIH R01NS065808 to E.R.B., the Empire State Development Corporation Krabbe Disease Grants U446 and W753 to L.W., and the Hunter’s Hope Foundation. We thank E. Hurley and G.G. Shackleford for expert technical assistance and K. Mincorczyk, A. Lauko, K. Diaz, and I. Yu for their contribution; C. Eckman, Mayo Clinic for anti-GALC antibody; E. Peles, Weizmann Institute for anti-Caspr antibodies; C. Woolf, Harvard Medical School for ATF3-GFP mice; Klaus Nave MPI Gottingen for CNPase Cre mice. We are most grateful to the families who have donated their loved one’s tissues, made possible by Partners for Krabbe Research and the NDRD Brain and Tissue Bank, UPMC Pittsburgh PA.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.03.031.
DECLARATION OF INTERESTS
E.R.B. is consultant for Lysosomal Therapeutics and for E-escape-Bio. M.L.E. consults for Passagebio and is PI in the development of a treatment for Krabbe disease using HSCT and gene therapy. Neither have had any decision making in this current study.
REFERENCES
- Andrews JM, and Menkes JH (1970). Ultrastructure of experimentally produced globoid cells in the rat. Exp. Neurol 29, 483–493. [DOI] [PubMed] [Google Scholar]
- Austin JH (1963). Studies in globoid (KRABBE) Leukodystrophy. II. Controlled thin-layer Chromatographic studies of globoid body fractions in seven patients. J. Neurochem 10, 921–930. [DOI] [PubMed] [Google Scholar]
- Cantuti Castelvetri L, Givogri MI, Hebert A, Smith B, Song Y, Kaminska A, Lopez-Rosas A, Morfini G, Pigino G, Sands M, et al. (2013). The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3b and deregulation of molecular motors. J. Neurosci 33, 10048–10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil M-T, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lütjohann D, et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. [DOI] [PubMed] [Google Scholar]
- Castelvetri LC, Givogri MI, Zhu H, Smith B, Lopez-Rosas A, Qiu X, van Breemen R, and Bongarzone ER (2011). Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol 122, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Förster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Collier J, and Greenfield J (1924). The encephalitis periaxialis of Schilder. A clinical and pathological study, with an account of two cases, one of which was diagnosed during life. Brain 47, 489–519. [Google Scholar]
- Coutinho MF, Prata MJ, and Alves S (2012). Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol. Genet. Metab 105, 542–550. [DOI] [PubMed] [Google Scholar]
- Della-Flora Nunes G, Mueller L, Silvestri N, Patel MS, Wrabetz L, Feltri ML, and Poitelon Y (2017). Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia 65, 1626–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, and Tschopp J (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen LW, Eicher EM, Jacobs JM, Scaravilli F, and Teixeira F (1980). Hereditary leucodystrophy in the mouse: the new mutant twitcher. Brain 103, 695–710. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med 352, 2069–2081. [DOI] [PubMed] [Google Scholar]
- Ey B, Kobsar I, Blazyca H, Kroner A, and Martini R (2007). Visualization of degenerating axons in a dysmyelinating mouse mutant with axonal loss. Mol. Cell. Neurosci 35, 153–160. [DOI] [PubMed] [Google Scholar]
- Feltri ML, D’antonio M, Previtali S, Fasolini M, Messing A, and Wrabetz L (1999). P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann. N Y Acad. Sci 883, 116–123. [PubMed] [Google Scholar]
- Galbiati F, Basso V, Cantuti L, Givogri MI, Lopez-Rosas A, Perez N, Vasu C, Cao H, van Breemen R, Mondino A, and Bongarzone ER (2007). Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J. Neurosci 27, 13730–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg B, Kollberg H, Sourander P, andÅkesson HO (1969). Infantile globoid cell leucodystrophy (Krabbe’s disease). A clinical and genetic study of 32 Swedish cases 1953–1967. Neuropadiatrie 1, 74–88. [DOI] [PubMed] [Google Scholar]
- Hers HG (1965). Inborn lysosomal diseases. Gastroenterology 48, 625–633. [PubMed] [Google Scholar]
- Hoogerbrugge PM, Suzuki K, Suzuki K, Poorthuis BJ, Kobayashi T, Wagemaker G, and van Bekkum DW (1988). Donor-derived cells in the central nervous system of twitcher mice after bone marrow transplantation. Science 239, 1035–1038. [DOI] [PubMed] [Google Scholar]
- Jacobs JM, Scaravilli F, and De Aranda FT (1982). The pathogenesis of globoid cell leucodystrophy in peripheral nerve of the mouse mutant twitcher. J. Neurol. Sci 55, 285–304. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Wenger DA, Gallo V, and Duncan ID (2005). Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc. Natl. Acad. Sci. USA 102, 18670–18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Adams JM, Vanier MT, and Duncan ID (2011). Macrophages counteract demyelination in a mouse model of globoid cell leukodystrophy. J. Neurosci 31, 3610–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe K (1916). A new familial, infantile form of diffuse brain-sclerosis. Brain 39, 74–114. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave K-A (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet 33, 366–374. [DOI] [PubMed] [Google Scholar]
- Larocca JN, and Norton WT (2006). Isolation of myelin. Curr. Protoc. Cell Biol Published online January 2007. 10.1002/0471143030.cb0325s33. [DOI] [PubMed] [Google Scholar]
- Lee WC, Kang D, Causevic E, Herdt AR, Eckman EA, and Eckman CB (2010). Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J. Neurosci 30, 5489–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu Y, Benitez BA, Nagree MS, Dearborn JT, Jiang X, Guzman MA, Woloszynek JC, Giaramita A, and Yip BK (2019). Genetic ablation of acid ceramidase in Krabbe disease confirms the psychosine hypothesis and identifies a new therapeutic target. Proc. Natl. Acad. Sci. USA 116, 20097–20103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luddi A, Volterrani M, Strazza M, Smorlesi A, Rafi MA, Datto J, Wenger DA, and Costantino-Ceccarini E (2001). Retrovirus-mediated gene transfer and galactocerebrosidase uptake into twitcher glial cells results in appropriate localization and phenotype correction. Neurobiol. Dis 8, 600–610. [DOI] [PubMed] [Google Scholar]
- MacDermot KD, Holmes A, and Miners AH (2001). Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet 38, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone M (1970). Deficiency in a degradative enzyme system in globoid leukodystrophy. Trans. Am. Soc. Neurochem 1, 56. [Google Scholar]
- Marshall MS, Issa Y, Jakubauskas B, Stoskute M, Elackattu V, Marshall JN, Bogue W, Nguyen D, Hauck Z, Rue E, et al. (2018). Long-term improvement of neurological signs and metabolic dysfunction in a mouse model of Krabbe’s disease after global gene therapy. Mol. Ther 26, 874–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, and Willison H (2016). Neuroinflammation in the peripheral nerve: Cause, modulator, or bystander in peripheral neuropathies? Glia 64, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino S, Tiribuzi R, Tortori A, Conti D, Visigalli I, Lattanzi A, Biffi A, Gritti A, and Orlacchio A (2009). Specific determination of β-galactocerebro sidase activity via competitive inhibition of β-galactosidase. Clin. Chem 55, 541–548. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- Matthes F, Andersson C, Stein A, Eistrup C, Fogh J, Gieselmann V, Wenger DA, and Matzner U (2015). Enzyme replacement therapy of a novel humanized mouse model of globoid cell leukodystrophy. Exp. Neurol 271, 36–45. [DOI] [PubMed] [Google Scholar]
- Mikulka CR, and Sands MS (2016). Treatment for Krabbe’s disease: Finding the combination. J. Neurosci. Res 94, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake T, and Suzuki K (1972). Globoid cell leukodystrophy: additional deficiency of psychosine galactosidase. Biochem. Biophys. Res. Commun 48, 539–543. [DOI] [PubMed] [Google Scholar]
- Mosley K, and Cuzner ML (1996). Receptor-mediated phagocytosis of myelin by macrophages and microglia: effect of opsonization and receptor blocking agents. Neurochem. Res 21, 481–487. [DOI] [PubMed] [Google Scholar]
- Nagano S, Yamada T, Shinnoh N, Furuya H, Taniwaki T, and Kira J (1998). Expression and processing of recombinant human galactosylceramidase. Clin. Chim. Acta 276, 53–61. [DOI] [PubMed] [Google Scholar]
- Ono H, Ohta R, Kawasaki Y, Niwa A, Takada H, Nakahata T, Ohga S, and Saito MK (2018). Lysosomal membrane permeabilization causes secretion of IL-1β in human vascular smooth muscle cells. Inflamm. Res 67, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, and Schlessinger J (1997). Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 16, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrero C, Rubio-Garrido P, Avendaño C, and Clascá F (2010). Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res 1345, 59–72. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Fugaro J, Amini S, Luzi P, de Gala G, Victoria T, Dubell C, Shahinfar M, and Wenger DA (1996). Retroviral vector-mediated transfer of the galactocerebrosidase (GALC) cDNA leads to overexpression and transfer of GALC activity to neighboring cells. Biochem. Mol. Med 58, 142–150. [DOI] [PubMed] [Google Scholar]
- Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, and Eklund KK (2010). Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE 5, e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, et al. (2012). Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 109, E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AS, Kim JH, Hawkins-Salsbury JA, Macauley SL, Tracy ET, Vogler CA, Han X, Song S-K, Wozniak DF, Fowler SC, et al. (2011). Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J. Neurosci 31, 9945–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands MS, and Davidson BL (2006). Gene therapy for lysosomal storage diseases. Mol. Ther 13, 839–849. [DOI] [PubMed] [Google Scholar]
- Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, et al. ; International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2 (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Severin Y, Zanotelli VRT, and Bodenmiller B (2019). In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci. Rep 9, 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, and Rajewsky K (1995). A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23, 5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt NJ, Folts CJ, Hogestyn JM, Piester G, Mayer-Pröschel M, and Noble MD (2017). Heterozygote galactocerebrosidase (GALC) mutants have reduced remyelination and impaired myelin debris clearance following demyelinating injury. Hum. Mol. Genet 26, 2825–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Allchorne AJ, and Woolf CJ (2006). The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell. Neurosci 32, 143–154. [DOI] [PubMed] [Google Scholar]
- Shin D, Feltri ML, and Wrabetz L (2016). Altered trafficking and processing of GALC mutants correlates with globoid cell leukodystrophy severity. J. Neurosci 36, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer I, and Schachner M (1981). Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev. Biol 83, 311–327. [DOI] [PubMed] [Google Scholar]
- Spassieva S, and Bieberich E (2016). Lysosphingolipids and sphingolipidoses: Psychosine in Krabbe’s disease. J. Neurosci. Res 94, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K (1998). Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem. Res 23, 251–259. [DOI] [PubMed] [Google Scholar]
- Suzuki K, and Grover WD (1970). Krabbe’s leukocystrophy (globoid cell leukodystrophy). An ultrastructural study. Am. J. Obstet. Gynecol 106, 385–396. [PubMed] [Google Scholar]
- Suzuki K, and Suzuki Y (1970). Globoid cell leucodystrophy (Krabbe’s disease): deficiency of galactocerebroside β-galactosidase. Proc. Natl. Acad. Sci. USA 66, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svennerholm L, Vanier MT, and Månsson JE (1980). Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J. Lipid Res 21, 53–64. [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo J-L, Kohler RH, Chudnovskiy A, Waterman P, et al. (2009). Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter J, DeJong LJ, and Smith ME (1986). Opsonization with antimyelin antibody increases the uptake and intracellular metabolism of myelin in inflammatory macrophages. J. Neurochem 47, 779–789. [DOI] [PubMed] [Google Scholar]
- Weinstock NI, Kreher C, Favret J, Bongarzone E, Wrabetz L, Feltri ML, and Shin D (2020). Brainstem development requires galactosylceramidase and is critical for the pathogenesis of Krabbe disease. bioRxiv 10.1101/2020.03.25.007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, and Porse B (2008). Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc Published online December 1, 2008. 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, et al. ; Fabry Registry (2008). Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol. Genet. Metab 93, 112–128. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Breur M, Plug B, Beerepoot S, Westerveld ASR, van Rappard DF, de Vries SI, Kole MHP, Vanderver A, van der Knaap MS, et al. (2020). Metachromatic leukodystrophy and transplantation: remyelination, no cross-correction. Ann. Clin. Transl. Neurol 7, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MD, Poe MD, DeRenzo A, Haldal S, and Escolar ML (2017). Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 89, 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager AM, Brennan S, Tiffany C, Moser HW, and Santos GW (1984). Prolonged survival and remyelination after hematopoietic cell transplantation in the twitcher mouse. Science 225, 1052–1054. [DOI] [PubMed] [Google Scholar]
- Yunis EJ, and Lee RE (1970). Tubules of globoid leukodystrophy: a right-handed helix. Science 169, 64–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No dataset/code is associated with this paper.