

Abstract
Purpose of review
Virtually all viruses have evolved molecular instruments to circumvent cell mechanisms that may hamper their replication, dissemination, or persistence. Among these is p53, a key gatekeeper for cell division and survival that also regulates innate immune responses. This review summarizes the strategies used by different viruses and discusses the mechanisms deployed by SARS-CoV to target p53 activities.
Recent findings
We propose a typology for the strategies used by different viruses to address p53 functions: hit and run (e.g. IAV, ZIKV), hide and seek (e.g. HIV1), kidnap and exploit (e.g. EBV, HSV1), dominate and suppress (e.g. HR HPV). We discuss the mechanisms by which SARS nsp3 protein targets p53 for degradation and we speculate on the significance for Covid-19 pathogenesis and risk of cancer.
Summary
p53 may operate as an intracellular antiviral defense mechanism. To circumvent it, SARS viruses adopt a kidnap and exploit strategy also shared by several viruses with transforming potential. This raises the question of whether SARS infections may make cells permissive to oncogenic DNA damage.
Keywords: coronavirus, nsp3, p53, replication, RING finger and CHY zinc finger domain-containing protein 1, severe acute respiratory syndrome coronavirus 2, virus
INTRODUCTION
As for 1 October 2020, the current outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been confirmed in 39 023 292 people and has caused 1 099 586 reported deaths worldwide (source: WHO website, accessed 14 October 2020). Coronavirus disease-2019 (Covid-19), the main clinical manifestation of this infection, is a complex syndrome with common symptoms encompassing fever, cough, fatigue, shortness of breath, breathing difficulties, and loss of smell and taste. About 5% of patients with Covid-19 develop severe symptoms including pulmonary inflammation, pneumonia, and acute respiratory distress syndrome. There is also evidence that patients who have developed Covid-19 may experience long-lasting health effects, such as breathing difficulties, persistent fatigue, and neurological symptoms [1].
SARS-CoV-2 is a novel beta-coronavirus belonging to the sarbecovirus subgenus of Coronaviridae family. Its genomic sequence is closely related to those of two bat SARS-like viruses, SL-CoVZXC21 and SL-CoVZC45 (88%). It also shows similarities with human SARS-CoV-1, the virus responsible for the SARS outbreak in 2002–2004 (79%), and MERS-CoV, responsible for an outbreak in the middle east in 2012 (50%) [2]. The SARS-CoV-2 uses the same cell entry receptor, ACE2, to infect human cells [3], consistent with the observed clinical similarities between the two viruses. The viral genome is 29.9 kb long and encodes for up to 29 distinct peptides. Our knowledge of the structure and functions of these proteins is still limited and part of current concepts on their roles are based on their similarities with other coronaviruses, notably SARS-CoV-1. Upon entry into host cells, the viral RNA genome is translated into proteins that compose the structure of the viral particle, accessory proteins that facilitate several aspects of viral replication and packaging and nonstructural proteins involved in the processing of other viral proteins, in RNA replication and in producing factors that interfere with the host cell machineries to create a cellular ecosystem that supports virus replication and expansion [4,5].
In the process of infecting their hosts, virtually all viruses have developed strategies and molecular instruments to deactivate and control cell regulatory mechanisms that may hamper their replication, dissemination, or persistence. To this end, viruses have evolved specific proteins that target the cell decision centers that regulate cell proliferation and survival, as well innate immune response mechanisms used by cells to fight off viral infections, such as interferon-gamma-mediated antiviral responses. Among these decision centers and mechanisms, the p53 tumor suppressor protein plays a major role. This stress-inducible factor can, directly and indirectly, control multiple pathways, controlling DNA replication and repair, cell proliferation, programmed cell death, metabolism, and innate immune responses. This broad spectrum of suppressive effects makes p53 a most legitimate target for inactivation by mutation, deletion, or other mechanisms in many different cancers [6]. The p53 protein was initially discovered as a cellular target of the Large-T antigen (LT) of the oncogenic Simian Virus 40 (SV40). Over the years, multiple studies have identified an amazing diversity of molecular devices deployed by every known virus family to hijack, control, or impair p53 functions. In the case of persistent oncogenic viruses, such as high-risk human papillomaviruses (HPV 16, 18, 31, 45), these devices are so specific and powerful that they permanently inactivate p53 functions, causing an oncogenic effect similar to TP53 mutation [7]. Nononcogenic viruses also produce proteins interacting with p53 or p53 regulators, expanding the concept that controlling p53 is an essential mechanism to support efficient virus replication, propagation, and, in some instances, persistence [8]. In this review, we briefly summarize current concepts on the mechanisms by which viruses interfere with the p53 pathway and their consequences. Next, we discuss the evidence to date on how SARS-CoV-2 targets the p53 pathways and we elaborate on the possible functional effects of this interference for the replication, pathogenesis, and potential long-term effects of infection.
Box 1.

no caption available
VIRAL STRATEGIES TO CIRCUMVENT p53
From a virus perspective, the presence of an active p53 in the host cell represents a threat that must be neutralized or circumvented to allow the virus to establish its replication and propagation program. The most immediate challenges posed by p53 are stress-induced programmed death of the host cell and induction of antiviral innate or adaptive immune responses. Programmed cell death, mainly through apoptosis, is a radical cell-protective response that effectively destroys the virus host cell. It can be triggered by the extent of unprogrammed nucleic acid and protein neosynthesis caused by viral replication, which is seen by the cell as a form of intrinsic DNA and protein damage. The p53 protein operates as a stress-activated switch button for many pro-apoptotic pathways and targeting this switch ensures that the virus can control the survival and lifespan of its host cell. On the other hand, several p53 target genes have been identified within pathways involved in inflammation responses, pathogen sensing, cytokine/chemokine production and immune checkpoint regulation [9–11]. These transcriptional targets include key components of the cell antiviral response, including interferon (IFN) regulatory factor 5 (IRF5), IRF9, protein kinase RNA-activated (PKR), Toll-like receptor 3 (TLR3), IFN-stimulated gene 15 (ISG15), and monocyte chemoattractant protein 1 (MCP-1; also known as CCL2, CC-chemokine ligand 2). Moreover, the expression of the TP53 gene is enhanced in response to signalling through IFN a-b receptor signalling, suggesting that p53 is part of a positive feedback loop that enhances antiviral IFN responses. Studies in p53-deficient cells and in experimental mouse models lacking p53 (trp53-/-) have revealed that they are often more permissive to infections than their wild-type p53 counterparts [12,13]. This effect was observed even in the presence of caspase inhibitors that block p53 apoptosis, highlighting the role of p53-mediated antiviral IFN responses in the control of these infections.
Table 1 lists the molecular mechanisms evolved by selected viruses infecting humans to target the p53 pathway. These mechanisms are amazingly diverse in their molecular detail. The particular mechanisms selected by each virus is dependent upon cell tropism, replication--propagation cycle, and modalities of latency or persistence. In the next section, we detail four broad strategies used by viruses to target the p53 pathway: hit and run; hide and seek; kidnap and exploit; and dominate and suppress. Each strategy is a reflection of how the virus exploits its host cell(s) for its own replicative and propagation purposes, revealing the exquisite adaptation of the virus to a specific ecological niche.
Table 1.
Selected examples of viral strategies to target and circumvent the p53 pathway
| Virus | Viral proteins involved | Interaction with p53 | References |
| Influenza A virus (IAV) | NP | NP enhances p53 stabilization and apoptosis, enhancing virus release from infected cells | [12,14–15] |
| Zika virus (ZIKV) | ZCP | ZIKV triggers p53-induced cell death in neural cells and progenitors | [18,19] |
| HIV type 1 (HIV-1) | Nef Tat Vif | Nef may inhibit p53 function during early steps of viral replication; Vif may increase p53 function and contribute to cytopathic effects. Inhibition of SIRT1 by tat may modulate p53 acetylation and activation | [20–22] |
| Human herpes simplex virus 1 (HSV-1) | ICP0 ICP22 ICP27 | ICP27 is a transcriptional target of p53. ICP0 targets HAUSP and enhances p53 degradation. p53 regulates ICP0 degradation. ICP22 prevents the negative regulation of ICP0 by p53 | [23,24,25] |
| Epstein--Barr virus (EBV) | BZLF1 EBNA1 EBNA3C LMP1 | BZLF1 induces Mdm2-independent p53 degradation. EBNA1 prevents p53 and Mdm2 degradation, modulating p53-dependent repair and apoptosis. EBNA3C repress DNA-binding and transcriptional activity. LMP1 promotes p53 accumulation and impair cell-cycle arrest and apoptosis | [26–28,29] |
| High -risk papilloma viruses (HPV 16, 18, 31, 45) | E6, E7 | E6 binds E6AP and induces p53 degradation. Disruption of pRb by E7 abrogate p53 downregulation of DREAM | [30–31,33] |
| Hepatitis B virus (HBV) | HBx | HBx re-directs and re-wire p53 transcriptional activity. Mutant HBx form an oncogenic complex with mutant TP53 p.R249S | [35–36] |
| SARS-CoV viruses | Nsp2, nsp3 | Nsp2 interacts with prohibitin (PHB) 1 and 2, involved mitochondrial biogenesis, causing ROS release and inducing p53 through DNA damage Nsp3 is a multidomain protein that binds and activates RCHY1, inducing Mdm2-independent p53 degradation | [4,13,46] |
Hit and run
This strategy is exemplified by Influenza Virus (IAV), a member of the Orthomyxovirdae family of RNA viruses and one of the most common pathogens to cause human respiratory infections. Speed is a key issue for IAV: the time course of its infection--replication--propagation sequence cycle is very short. This cytolytic virus induces host cell apoptosis as a mechanism for enhancing virion release from infected cells. It, therefore, uses an unusual strategy of hitting onto p53 in order to activate its capacity to induce programmed cell death. To achieve this, the viral nucleoprotein NP interferes with the binding to p53 of Mdm2, the main E3-ubiquitin ligase responsible for maintaining p53 at low levels in normal conditions by targeting it for proteasome-mediated degradation [14]. Simultaneously, NP inhibits another E3-ligase, RNF43, further contributing to stabilize p53 [15]. Whereas these effects concur to the strategy of the virus to use apoptosis for its cytolytic propagation, it raises questions on how the virus escapes IFN-mediated antiviral responses expected to be upregulated by p53. Transcriptome analysis in p53-competent versus deficient lung cells infected by IAV has identified that p53 could inhibit the expression of IFITM1, IFITM2, and IFITM3 [16], three members of the interferon-induced membrane protein family, which have been shown to restrict infectivity of diverse pathogens, including IAV, dengue, rabies, or Ebola viruses. IFITMs appear to restrict infectivity by interfering with virus--endosome fusion, therefore, preventing the release of infectious materials within the cell. Furthermore, upon infection by IAV, p53 appears to upregulate its target gene endoplasmic reticulum endopeptidase 1 (ERAP1), a regulator of the expression of Major Histocompatibility Complex 1 (MHC1), thus supporting a role for p53 in enhancing the cytolytic T-cell response [17]. Taken together, NP-mediated p53 activation may enable IAV to exploit p53 function to increase apoptosis and T-cell-mediated cytolysis, while controlling specific aspects of the antiviral IFN response. It follows that p53 activity is probably essential to ensure a clean course of IAV infection, with efficient replication, cytolysis, and propagation. Indeed, p53-deficient mice show delayed cytokine and antiviral gene responses in lung and bone marrow, decreased dendritic cell activation, and reduced IAV-specific CD8+ T-cell immunity, resulting in a more severe IAV-induced disease compared with their wild-type counterparts [12].
Another illustration of the hit and run strategy is given by the Zika virus (ZIKV), a member of the Flaviviridae family of single-stranded, positive sense RNA viruses primarily transmitted from monkeys to humans by Aedes mosquitoes. ZIKV causes mainly asymptomatic to mild flu-like symptoms but, when transmitted to pregnant women, it causes increased risk of microcephaly in fetuses and of neurocognitive disorders in infants. ZIKV infects several cell types, including myeloid and epithelial cells as well as neuronal progenitors but it is not known whether it uses similar p53-targeting strategies in all infected cell types. In neuronal progenitors, ZIKV increases total p53 levels and nuclear accumulation and activates p53-dependent activation of pro-apoptotic pathways [18]. Reminiscent of the NP protein of IAV, this activation is mediated by interference of ZIKV main capsid protein ZCP with Mdm2-mediated p53 degradation [19]. ZIKV-induced p53 activation is associated with patterns of oxidative and genotoxic stress similar to those observed in severe forms of genetic microcephaly. Taken together, these observations imply that p53 activation is a critical step in neuronal damage causing microcephaly in unborn infants from infected mothers.
Hide and seek
This viral strategy for targeting p53 consists into two successive phases: first attenuation of p53 functions (hide) and second positive mobilization of p53 (seek) to assist the virus in the different phases of its life cycle. A typical example of such a hide-and-seek strategy is given by HIV-1, a lentivirus (member of the Retroviridea family), which latently infects CD4 lymphocyte, provoking a loss in T-helper functions that causes the AIDS. HIV-1 has evolved distinct proteins that target p53 either during the early or late phases of the virus life cycle. First, the early protein Nef, one of the first proteins expressed after virus entry into host cells, directly interacts with p53 and destabilizes it, impairing p53 function [20]. This effect is thought to attenuate the capacity of p53 to activate stress-induced cell death as well as DNA-damage responses that may interfere with the integration of the provirus into the host cell genome. Second, the Tat protein, the main transactivator of HIV-1 gene expression, neutralizes the p53 deacetylase SIRT1, and thus contributes to the regulation of p53 transcriptional capacity [21]. Third, the Vif protein, expressed during the latent phase, neutralizes Mdm2-mediated degradation of p53, causing increased expression of negative regulators of host cell proliferation and survival, such as p21/CDKN1A and Bax [22]. Taken together, these contrasted effects ensure that the virus readily establishes itself and integrates its genome into host cells by down-regulating p53, then uses p53-dependent suppressive pathways to attenuates the activity of CD4+ cells, thus precluding the development of an antiviral adaptive immune response.
Kidnap and exploit
This strategy is a somewhat more sophisticated form of hide and seek, in which the virus manipulates p53 using a whole range of molecular devices, not only to attenuate p53-mediated antiviral effects but also to exploit p53 as a factor that controls and facilitate viral replication. Examples of this strategy include members of the Herpesviridae family, such as herpes simplex virus 1 (HSV-1) and Epstein--Barr virus (EBV). The genome of these viruses is constituted of double-stranded linear DNA containing 74–85 distinct genes. Infection by HSV-1 is frequently asymptomatic. Its main manifestation is benign herpetic cutaneous and mucosal lesions but the virus can also cause latent, recurrent infections because of its persistence in neurons. There is evidence that p53 entertains complex interactions with the products of immediate-early viral genes, namely ICP0, ICP27, and ICP22, causing p53 to act as host cell regulator of HSV-1 infection [23,24]. First, p53 binds to DNA sequences in the promoter of ICP27 and activates the expression of this essential factor for viral replication during the early phases of infection. This positive role of p53 is tightly regulated by ICP0 and ICP22. ICP0 binds and neutralizes herpes-associated ubiquitin-specific protease (USF7/HAUSP), the main ubiquitin protease involved in deubiquitinating p53 [25]. This interaction leads to p53 destabilization, preventing its stress-induced accumulation. Of note, p53 can bind to and degrade ICP0 in an autoregulatory feedback loop. This effect is prevented by ICP22, which binds to p53 and impairs its function, thus inactivating its negative effects on ICP0 [23].
Similarly, complex interactions are observed in the case of EBV, which infects both B cells and epithelial cells. This common virus is best known as the main cause of infectious mononucleosis. It also causes lymphoproliferative disorders, among which Burkitt lymphoma, and is involved in several other forms of cancers as well as in a broad range of autoimmune or neurological disorders. The defining feature of its life cycle is its aptitude to switch on latency programs inducing persistence and ultimately transformation in B cells and/or in epithelial cells. EBV targets and regulates p53 in both early (replication) and latent phases. During initial infection and replication, the early viral protein BZLF1 impairs p53 function by promoting its degradation independently of Mdm2 through activation of the elongin BC–cullin 5–SOCS box ubiquitin–protein ligase complex [26]. This effect neutralizes p53 protective functions during active viral replication. During the latent phase, p53 is taken up in a cocoon of viral regulatory proteins that positively and negatively control its activity and functions. These proteins include EBNA1, EBNA3C, and LMP1. Similar to HSV1's ICP0, EBNA1 binds and inhibits USF7/HAUSP, promoting p53 degradation by the Mdm2 pathway [27]. EBNA3C down-regulates p53 through induction of Aurora kinase B [28]. LMP1 is essential for EBV latency and is considered as having a pro-oncogenic effect. Surprisingly, LMP1 promotes aberrant ubiquitination states of p53 that enables it to escape Mdm2-dependent degradation. In turn, p53 concurs to stimulate LMP1 expression by transactivating interferon response factor (IFR) 5, which itself upregulates the LIMP1 promoter [29,30]. Adding to this complex regulatory network, EBNA3C binds to p53 C-terminus and regulates its DNA-binding and transcriptional activity.
Taken together, these observations suggest that both EBV and HSV1 have developed elaborate mechanisms in which different viral proteins cooperate to hijack p53 function, shielding it from its usual cell regulators and capturing it in a web of positive and negative control loops in order to serve viral replication and/or latency purposes, while attenuating p53-mediated proapoptotic or antiviral effects. Of note, the capture of p53 function by viral proteins during the latent phase of EBV represents a particularly vicious form of hijacking and exploitation, in which a presumed antiviral p53-dependent pathway involving IRF5 is mobilized to promote the expression of LMP1, the main and essential factor for EBV persistence.
Dominate and suppress
This strategy consists in radical elimination of p53 function by a dominant viral antigen, thus acting as an oncogene. The paradigmatic example of this strategy is given by high-risk human papillomaviruses (HPV). These nonenveloped viruses with double-stranded circular DNA genomes belong to the Papillomaviridea family. Human HPV constitute about 200 genotypes, which infect epithelial cells, including mucosal (alpha) HPV subtypes and or cutaneous (beta) HPV subtypes. HPV infections are benign and generally asymptomatic but in some instances, their persistence may cause warts or precancerous lesions. A group of alphaHPV is dubbed ‘high risk’ and causes anogenital and oral cancers (HR HPV, mainly HPV 16, 18, 31, and 45). During the early stages of infection, two viral proteins, E1 and E2, play essential roles in the transcription and replication of the viral genome. Replication is strictly controlled, leading to low levels of HPV expression, presumably to limit the extent of adaptive immune responses. The p53 protein binds to E2 and represses HPV replication [31]. Concomitantly, HR HPV expresses two other early proteins, E6 and E7 that interfere with the cell proliferation and survival machinery, thus enabling the expansion and survival of the pool of infected epithelial cells. Although E7 binds pRb and impair its capacity to inhibit cell cycle entry into S phase, E6 binds and activates the cellular E3 ubiquitin ligase E6AP, which ubiquitinates p53 and directly targets it to proteasome-dependent degradation, causing levels of p53 to remain extremely low in infected cells [32]. Importantly, E6 proteins of non-HR HPV appear to be less effective in inducing p53 degradation, indicating that this event is critical for cell transformation induced by HR-HPV [33]. To compound this effect, E7 also contributes to impair p53-mediated suppressive effects by disrupting DREAM, a transcriptional activation complex containing pRb, which is regulated by p53 and controls the expression of over 200 genes involved in cell-cycle regulation and survival [34].
The examples above underpin the complexity of the strategies used by viruses to address the challenges posed to viral propagation by the suppressive pathways controlled by p53. Further, they show that viruses have evolved very specific mechanisms to make use of the p53 pathway to fulfill their own needs. The mechanisms used by viruses not only outline their rich adaptivity but also identify convergent control points. For example, many viruses have developed molecular weaponry that targets p53 protein stability, either by modulating Mdm2-dependent p53 degradation, by interfering with deubiquitinases, such as USF7/HAUSP, or my mobilizing alternative, Mdm2-independent E3 ligases. This functional convergence in the modus operandi of most viruses highlights the crucial role of rapid p53 accumulation in mediating appropriate homeostatic responses to multiple forms of stress, including the one imposed by viral infection, replication, propagation, and persistence.
When comparing these strategies, fundamental differences appear between viruses that cause primary acute infections and those that have a latent, persistent phase. Whereas the former often use hit and run strategies, persistent viruses have co-opted stable molecular devices capable of dominantly repressing p53 functions, thus acting as potential oncogenes. Perhaps, the most sophisticated strategy for interacting with p53 is the one developed by Hepatitis B Virus (HBV), a double-stranded DNA member of the Hepadnaviridae family, which may persist in mature hepatocytes, inducing chronic hepatitis, fibrosis, and ultimately hepatocellular carcinoma. The viral protein Hbx is a multifunctional molecular ‘swiss knife’ that interacts with multiple homeostatic mechanisms of the host cell, including p53 [35]. By interacting with p53 and altering its sequence-specific binding to the response elements, HBx appears to modulate the transcription of genes, p53 target genes, involved in cell proliferation and death, suggesting a role of HBx in cell fate determination [36]. However, the two proteins achieve their most accomplished cooperation during the evolution of chronic hepatitis, which entails the integration of Hbx into the host genome and its expression as a mutant protein. This event is frequent in patients who are chronically infected by HBV in regions of the world where HBV is endemic. In these areas, hepatocarcinogenesis often occurs as a result of a synergistic effect between chronic HBV and TP53 mutagenesis induced by aflatoxin, a carcinogen that contaminates staple diets in many hot-humid, low-resource countries. In the tumors that develop in such an epidemiological context, aflatoxin-induced mutant p53 (p.R249S) and mutant Hbx form a complex and cooperate to act as a dominant oncogene [37], a strategy that could be defined as partnership in crime.
HOW DO SEVERE ACUTE RESPIRATORY SYNDROME CORONAVIRUSES INTERFERE WITH THE p53 PATHWAY?
Information on how SARS-CoV-2 targets and manipulates the p53 pathway are still scarce and limited. However, more detailed information is available for SARS-CoV-1. As the two viruses share about 89% overall genome homology and show many common features in their mechanisms of infection and pathogenesis, it is reasonable to speculate that both viruses may use similar molecular mechanisms to target and circumvent p53.
Coronaviruses have long (∼30 kb) single-stranded RNA genome and their replication is a highly orchestrated process involving a set of nonstructural replicase proteins, which control the transcription and replication of the viral genome (Fig. 1, panel a) and also interfere with the antiviral response mechanisms of the host cells. Because of their nature, length, and cytoplasmic localization, the viral genomes are potentially extremely sensitive to the host cell's antiviral innate response. SARS-CoV-1 and 2 genomes are structured in different reading frames. The replicase unit encompasses 16 nonstructural proteins (nsp 1--16), encoded by two open reading frames (ORF1a and ORF1b) covering the 5′-terminal two-third segment of the viral genome (Fig. 1, panel b). The production of these replicases is initiated by the translation of ORF1a and b through a ribosomal frameshift mechanism. This process supports the synthesis of two large precursor polyproteins, PP1a and PP1b, which are further processed into individual polypeptides by two cysteine proteases domains encoded within PP1a itself, the papain-like protease, PLpro and a 3C-like protease (3CLpro). The cleavage of PP1a and PP1b at multiple sites by these proteases enables the release and maturation of the 16 nsp proteins, collectively involved in the assembly of a membrane-associated replicase complex directly responsible for the transcription and replication of the viral genome. To achieve this, several nsp proteins (nsp3, nsp4, and nsp6) harbor transmembrane domains that mediate the recruitment of the complex to a network of vesicles originating from the endoplasmic reticulum (ER) [38]. This structural organization provides an environment that contributes to shield the viral genomes from the host cell's innate immune surveillance. The multidomain protein nsp3 is the largest replicase protein (1922 residues for SARS-CoV-1, 1945 residues for SARS-CoV-2, 292 kDa) [4]. Overall, nsp3 plays essential roles in viral replication as a scaffold for the assembly of other nsp into the replicase complex and for its anchorage to membranes, as well as for the downregulation of innate immune response. It contains multiple catalytic activities in addition to the cysteine protease activity responsible for the cleavage and release of nsp 1, 2, 3, and 4. Specifically, the PLpro domain also carries deubiquitinating (DUB) and deISGylating (DISG) activities. These activities hydrolyze lysine-bond ubiquitin or the ubiquitin-like protein interferon-induced gene 15 (ISG15), two signaling mechanisms mobilized in the host cell's innate immune response [39,40]. Through these mechanisms, PLpro impairs the production of important cytokines, such as CCL5 and CXCL10, interferes with transcription factors NFkappaB and IRF3, and down-regulates type-I interferon production. It is, therefore, of interest that genome alignments across CoVs from different species identify nsp3 as a hotspot for adaptive mutations. Interestingly, several of the missense mutations identified so far in SARS-CoV-2 appear to fall in regions encoding nsp2 and nsp3 [41].
FIGURE 1.
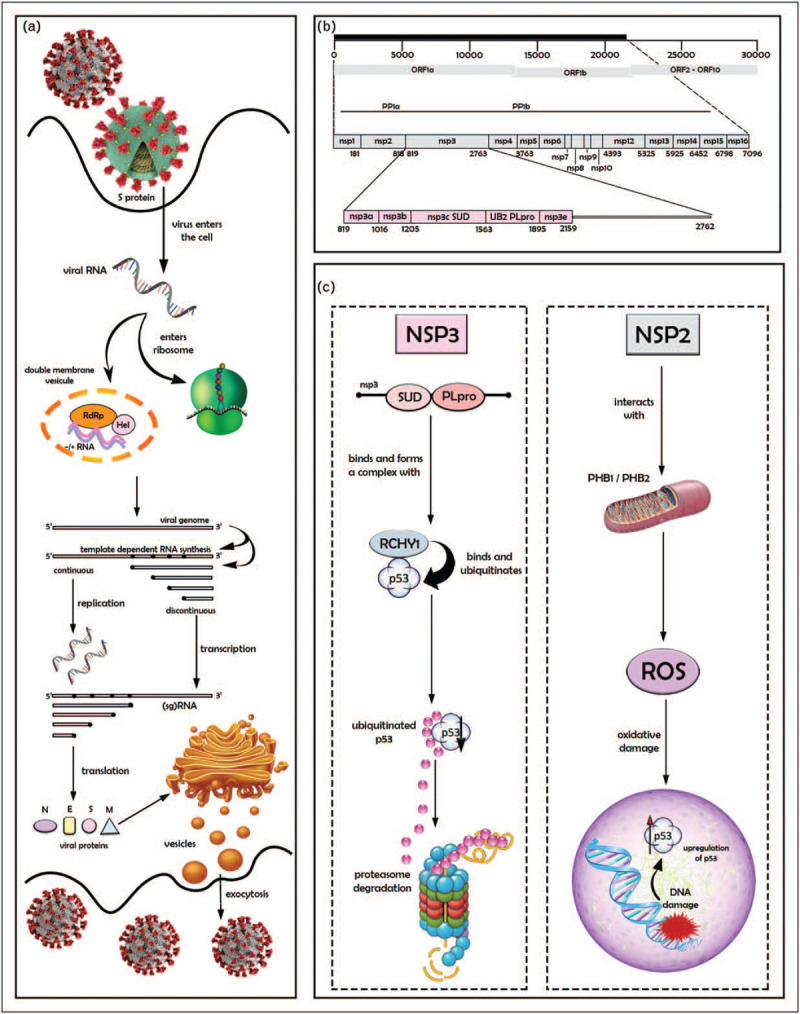
Severe acute respiratory syndrome coronaviruses’ lifecycle and possible mechanisms of interference with p53 and its regulatory pathways. Panel a: severe acute respiratory syndrome-related coronavirus (SARS-CoV and SARS-CoV-2) lifecycle, from entry into cells to release of newly synthetized virions. Viral particles recognize host receptors via spike glycoprotein (S protein), enter host cells by membrane fusion, releasing the RNA genome into the cytosol, where it is translated into the replicase proteins (see panel b). Replication occurs in virus-induced double-membrane vesicles (DMVs) derived from the endoplasmic reticulum (ER), in which incoming positive-strand genome serves as a template for full-length negative-strand RNA and sub genomic (sg)RNA, the translation of which results generates structural proteins and accessory proteins (N, S, M, and E). Maturation into the ER–Golgi complex leads to virion assembly and release from the plasma membrane. N, S, M, E: nucleocapsid, spike, membrane and envelope viral proteins, respectively. Panel b: viral genome and open reading frames, highlighting ORF1a encoding the viral polyprotein PP1a and PP1b (top) supporting the production of 16 nonstructural proteins (middle), including the multidomain protein nsp3 (bottom). Panel c: two antagonist mechanisms of interference with p53. Left, interaction of SUD-PLpro domains of nsp3 with RCHY1, inducing p53 degradation by the ubiquitin-proteasome pathway. Right, interaction of nsp2 with prohibitins (PHB) 1 and 2, disrupting mitochondrial metabolism and causing the release of reactive oxygen species (ROS), which in turn may activate p53 through a DNA-damage-dependent pathway.
Figure 1, panel C highlights two distinct molecular mechanisms by which products of the viral PLpro precursor peptide may interfere with the p53 pathway. The most direct pathway involves the viral nsp3 protein. Evidence from experimental studies in vitro and in-cell model systems has identified that nsp3 proteins from SARS-CoV-1, MERV-CoV, and NL63-CoV interfere with the RING finger and CHY zinc finger domain-containing protein 1 (RCHY1), an E3 ubiquitin ligase also known as Pirh-2 that promotes p53 degradation independently of Mdm2 [13]. Given the near-complete conservation of structures and activities between the SARS viruses, it is highly plausible that such an activity is also shared by SARS-CoV-2 nsp3. The interacting domain in nsp3 overlaps with both PLpro and the adjacent SARS unique domain (SUD), a structural domain not present in other coronaviruses. SUD itself is located upstream of PLpro and has a two-domain architecture, constituting a N-terminal subdomain (SUD-N) and a so-called middle subdomain (SUD-M). SUD, as well as each of its subdomains separately, specifically interacts with stretches of G nucleotides forming G-quadruplexes in both RNA and DNA [42]. SUD may, therefore, directly regulate viral and/or cellular RNA that contain such structures. With respect to interaction between RCHY1 and SUD, the critical protein regions involved encompass residues 95–144 of RCHY1 and 389–652 of the SUD-NM subdomain. Association with SUD appears to increase the stability of RCHY1, enhancing RCHY1-mediated ubiquitination, and consequently, degradation of p53. Similar to SUD, the PLpro domain physically interacts with and stabilizes RCHY1, thus triggering the degradation of endogenous p53. Both domains appear to act synergistically in binding RCHY1 and in enhancing p53 degradation [13]. Studies comparing SARS-CoV-1 replication and growth in syngeneic p53-competent and deficient cells have shown that virus titers were up to 1000-fold higher in the absence of functional p53 [13]. Thus, degradation of p53 caused by the interaction between nsp3 and RCHY1 may represent an efficient mechanism to remove a natural barrier to viral replication and propagation. Interestingly, the difference in virus production between p53+/+ and p53−/− cells does not appear to be because of increased virus-induced apoptosis in the former. The main mechanism appears to be the downregulation of p53-dependent antiviral responses. Overall, these observations suggest that SARS viruses have developed at least two complementary nsp3-dependent pathways to circumvent innate immunity, one involving the deubiquitinase and deISGylase activities of PLpro, and the other the RCHY1-targeting activity of PLpro-SUD.
Another, separate mechanism by which SARS viruses may interfere with the p53 pathway involves the nsp2 protein. A search for nsp2 interactors using multidimensional proteomics technology has identified prohibitin 1 and 2 (PHB1, PHB2), two proteins with primarily mitochondrial localization, which plays pleiotropic signaling in signaling pathways controlling cell survival, metabolism, and inflammation [43]. Within the mitochondria, PHB1 and PHB2 interact with each other to form ring-like structures of 20–25 nm diameter that provide a scaffold for the spatial organization of mitochondrial enzymatic activities. As such, they play an essential role in the maintenance of mitochondrial activity [44]. Their depletion increases mitochondrial permeability and leakage of reactive oxygen species, causing intracellular oxidative damage. PHB1 and PHB2 can also translocate in other cell compartments. In the nucleus, they associate as cofactors with various epigenetic regulators and transcription factors, including p53, for which they act both as an activator and a chaperone. Consequently, prohibitin depletion impairs p53 transcriptional activities [45,46]. Whereas the precise functional impact of nsp2 on PHB1 and PHB2 remains to be evaluated, the hypothesis is that this interaction disrupts the capacity of prohibitins to exert their scaffolding roles in the mitochondria and their chaperone/cofactor roles in the nucleus. The consequences of such effects for the p53 pathway would be both direct and indirect. First, PHB disruption may directly impair the transactivation of p53-dependent genes. Second, and perhaps more significant, mitochondrial dysfunction, resulting in oxidative stress may generate strong DNA damage signals causing acute p53 accumulation and activation.
The contribution of nsp2 to the replication of SARS viruses remains elusive. Indeed, deletion of nsp2 from the SARS-CoV-1 genome results in only a modest reduction in viral titers, and nsp2 is currently thought to be dispensable for replication [47]. However, this mechanism may contribute to other phases of the pathogenic process induced by SARS viruses, notably the acute tissue damage that develops in severely affected patients. In this respect, it is interesting to note that transcriptomic analysis of bronchial lavage fluids and peripheral mononuclear cells reveals signatures of SARS-CoV-2-induced activation of apoptosis and P53 signaling pathway [48].
CONCLUSION
The observations summarized above underscore the fact that, like most other virus families, SARS-CoV viruses have developed and evolved adapted molecular tools targeting p53 and its pathway. The strategy adopted by these viruses shows similarities with the kidnap and exploit strategy highlighted above for EBV and HSV1. The hallmark of this strategy is the hijacking of p53 by viral antigens that induce alternative pathways for p53 degradation, thus not only impairing p53 suppressor functions but also shielding it from Mdm2, its usual regulator under stress conditions. This mechanism, in effect, displaces p53 from its normal cell response pathway towards a virus response pathway, enabling the virus to circumvent or even possibly harness for its own benefit components of the pathways controlled by p53. The impact of this mechanism on the pathogenesis of SARS-CoV infections, and specifically on the course of Covid-19, remains highly speculative. Two scenarios may be considered:
p53 may serve as an antiviral factor that constraints the replication and propagation of SARS-CoV viruses. The speed and level at which the virus replicates may, therefore, depend upon the efficiency of nsp3-induced degradation of p53 mediated by RCHY1.
In parallel to impairing p53, SARS-CoV viruses also unroll molecular programs that lead to oxidative cell and DNA damage, with the potential of activating p53 to high levels resulting in rapid and massive apoptosis. This mechanism may contribute to the severity of pulmonary inflammation and respiratory distress that occurs in severe forms of Covid-19. Here too, p53 may operate as a rheostat, with the extent of cell and tissue damage depending upon the intensity of p53-mediated responses.
Placing these two scenarios one after the other, it is possible to speculate that, upon infection, nsp3 rapidly switches off p53 function, shielding the virus from innate immune responses and enabling replication. When replication and virus production declines, p53 becomes again available for activation by DNA damage and other cell stress response pathways. At this time the host cell has accumulated considerable oxidative damage, causing p53 to switch from impaired to a hyperactive status. This, in turn, may contribute to launching a sequence of events that fuel severe inflammation and tissue damage. Whether balancing and normalizing p53 activity may represent an accessible and affordable target for Covid-19 therapy remains to be evaluated.
With respect to cancer risk, it should be noted the molecular mechanisms used by SARS viruses to impair p53 bear similarities with those leveraged by several DNA or RNA viruses with transforming potential. Such mechanisms, consisting in the targeting of various p53 degradation mechanisms by viral proteins, are utilized by members of the Herpes virus family, by EBV as well as by HR HPVs. In the latter case, long-term inhibition of p53 via persistent E6-mediated degradation has similar functional consequences as TP53 inactivating mutation. Whereas the p53 impairment mediated by SARS-CoV nsp3 is supposed to be short-lived, it implies that during a certain amount of time, cells may be deprived from the capacity of p53 to act as a guardian of the genome against oncogenic DNA damage, despite being exposed to a high level of potentially oncogenic stress. Of note, the duration of SARS-CoV-2 infection is still not definitively evaluated. Several studies have evoked the possibility that SARS-CoV-2 may become persistent into some tissues, with diverse consequences regarding long-term effects of infection [49–50]. Regarding SARS-CoV-1, proximal tubular epithelial cells of the kidney have been suggested to be a site of productive and persistent viral replication favoring the emergence of viral variants [51]. However, in the case of SARS-CoV-2, there is currently no experimental evidence of long-term persistence and this question needs to be further assessed. Considering the specificity and power of the weapons deployed by SARS viruses to target p53, this hypothesis cannot be just brushed aside. The public health implications would be major and would require that infected patients would be cautiously monitored over a long period of time.
Acknowledgements
The authors thank Dr Klas Wiman, Karolinska Institute Stockholm, for critical reading and comments on the manuscript.
Financial support and sponsorship
Research on p53 and infections at IAB Grenoble is supported by ANR grant ‘Toxop53’.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Wiersinga WJ, Rhodes A, Cheng AC, et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 2020; 324:782–793. [DOI] [PubMed] [Google Scholar]
- 2.Lu R, Zhao F, Li J, et al. Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020; 395:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA 2020; 117:11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Báez-Santos YM, St. John SE, Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antivir Res 2015; 115:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies JP, Almasy KM, McDonald EF, et al. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. bioRxiv 2020; 14:2020.07.13.201517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med 2016; 6:a026179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tornesello ML, Annunziata C, Tornesello AL, et al. Human oncoviruses and p53 tumor suppressor pathway deregulation at the origin of human cancers. Cancers 2018; 10:213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aloni-Grinstein R, Charni-Natan M, Solomon H, et al. p53 and the viral connection: back into the future. Cancers 2018; 10:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine AJ. P53 and the immune response: 40 years of exploration-a plan for the future. Int J Mol Sci 2020; 21:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowe J, Shatz M, Resnick M, et al. Modulation of immune responses by the tumor suppressor p53. Biodiscovery 2013; 8:2. [DOI] [PubMed] [Google Scholar]
- 11.Muñoz-Fontela C, Mandinova A, Aaronson SA, et al. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol 2016; 16:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muñoz-Fontela C, Pazos M, Delgado I, et al. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J Immunol 2011; 187:6428–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma-Lauer Y, Carbajo-Lozoya J, Hein MY, et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PL pro via E3 ubiquitin ligase RCHY1. Proc Natl Acad Sci USA 2016; 113:E5192–E5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Deng F, Yan W, et al. Stabilization of p53 in influenza A virus-infected cells is associated with compromised MDM2-mediated ubiquitination of p53. J Biol Chem 2012; 287:18366–18375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nailwal H, Sharma S, Mayank AK, Lal SK. The nucleoprotein of influenza A virus induces p53 signaling and apoptosis via attenuation of host ubiquitin ligase RNF43. Cell Death Dis 2015; 6:e1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muñoz-Fontela C, Macip S, Martinez-Sobrido L, et al. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med 2008; 205:1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Niu D, Lai L, Ren EC. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat Commun 2013; 4:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghouzzi VE, Bianchi FT, Molineris I, et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. Cell Death Dis 2016; 8:e2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teng Y, Liu S, Guo X, et al. An integrative analysis reveals a central role of P53 activation via MDM2 in Zika virus infection induced cell death. Front Cell Infect Microbiol 2017; 7:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenway AL, McPhee DA, Allen K, et al. Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis. J Virol 2002; 76:2692–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thakur BK, Chandra A, Dittrich T, et al. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem Biophys Res Commun 2012; 424:245–250. [DOI] [PubMed] [Google Scholar]
- 22.Izumi T, Io T, Matsui M, et al. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc Natl Acad Sci 2010; 107:20798–20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maruzuru Y, Fujii H, Oyama M, et al. Roles of p53 in herpes simplex virus 1 replication. J Virol 2013; 87:9323–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruzuru Y, Koyanagi M, Takemura N, et al. p53 is a host cell regulator during herpes simplex encephalitis. J Virol 2016; 90:6738–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everett RD, Meredith M, Orr A, et al. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J 1997; 16:1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato Y, Shirata N, Murata T, et al. Transient increases in p53-responsible gene expression at early stages of Epstein-Barr virus product. Cell Cycle 2010; 9:807–814. [DOI] [PubMed] [Google Scholar]
- 27.Saridakis V, Sheng Y, Sharkari F, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1. Mol Cell 2005; 18:25–36. [DOI] [PubMed] [Google Scholar]
- 28.Jha HC, Yang K, El-Naccache DW, et al. EBNA3C regulates p53 through induction of Aurora kinase B. Oncotarget 2015; 20:5788–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Li W, Xiao L, et al. Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53. Cell Cycle 2012; 11:2327–2336. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Lingel A, Geiser V. Tumor suppressor p53 stimulates the expression of Epstein-Barr virus latent membrane protein 1. J Virol 2017; 91:e00312–e00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massimi P, Pim D, Bertoli C, et al. Interaction between the HPV-16 E2 transcriptional activator and p53. Oncogene 1999; 18:7748–7754. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Zapien D, Ruiz FX, Poirson J, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016; 529:541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White EA, Kramer RE, Tan MJA, et al. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J Virol 2012; 86:13174–13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischer M, Uxa S, Stanko C, et al. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci Rep 2017; 7:2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng SA, Lee C. Hepatitis B Virus X gene and hepatocarcinogenesis. J Gastroenterol 2011; 46:974–990. [DOI] [PubMed] [Google Scholar]
- 36.Chan C, Thurnherr T, Wang J, et al. Global re-wiring of p53 transcription regulation by the hepatitis B virus X protein. Mol Oncol 2016; 10:1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouas DA, Shi H, Hautefeuille AH, et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: interaction with hepatitis B virus X protein. Carcinogenesis 2010; 31:1475–1482. [DOI] [PubMed] [Google Scholar]
- 38.Knoops K, Kikkert M, Van der Worm SHE, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 2008; 6:e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindner HA, Lytvyn V, Qi H, et al. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch Biochem Biophys 2007; 466:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ratia K, Kilianski A, Baez-Santos YM, et al. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog 2014; 10:e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chouljenko VN, Lin XQ, Storz J, et al. Comparison of genomic and predicted amino acid sequences of respiratory and enteric bovine coronaviruses isolated from the same animal with fatal shipping pneumonia. Gen Virol 2001; 82 (Pt 1):2927–2933. [DOI] [PubMed] [Google Scholar]
- 42.Tan J, Vonrhein C, Smart OS, et al. The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog 2009; 5:e1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Signorile A, Sgaramella G, Bellomo F, De Rasmo D. Prohibitins: a critical role in mitochondrial functions and implication in diseases. Cells 2019; 8:71–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshinaka T, Kosako H, Yoshizumi T, et al. Structural basis of mitochondrial scaffolds by prohibitin complexes: insight into a role of the coiled-coil region. iScience 2019; 19:1065–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fusaro G, Dasgupta P, Rastogi S, et al. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem 2003; 278:47853–47861. [DOI] [PubMed] [Google Scholar]
- 46.Chander H, Halpern M, Resnick-Silverman L, et al. Skp2B attenuates p53 function by inhibiting prohibitin. EMBO Rep 2010; 11:220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham RL, Sims AC, Brockway SM, et al. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. JVI 2005; 79:13399–13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. 11 Emerg Microbes Infect 2020; 9:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alpalhão M, Ferreira JA, Filipe P. Persistent SARS-CoV-2 infection and the risk for cancer. Med Hypotheses 2020; 143:109882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh KA, Jordan K, Clyne B, et al. SARS-CoV-2 detection, viral load and infectivity over the course of an infection. J Infect 2020; 81:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pacciarini F, Ghezzi S, Canducci F, et al. Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. J Virol 2008; 8:5137–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]