

Abstract
In genome-wide association studies, signals associated with rare variants and interactions between genes are hard to detect even when the sample size is in tens of thousands. To overcome these problems, we examine the concept of supervariant. Like the classic concept of the gene, a supervariant is a combination of alleles in multiple loci, but the contributing loci can be anywhere in the genome. We hypothesize that supervariants are easy to detect and the aggregated signals are more stable in their associations with the disease than that from a single nucleoid polymorphism. Using the UK Biobank databases, we develop a ranking and aggregation method for identifying supervariants. Specifically, we examine 9,377 breast cancer cases with 46,861 controls matched by sex and age. In our simulations, the use of supervariants outperforms single-nucleotide polymorphism-based association method in detecting rare variants and signals with interactive structure. In real data analysis, we identify supervariants on Chromosomes 1, 2, 3, 5, 6, 7, 8, 9, 10, 11, 16, and 22 which cover previously reported loci that have associations with breast or other cancers, and several novel loci on Chromosomes 2, 5, 9, and 12. These findings demonstrate the validity of supervariants and its potential of discovering replicable and novel results for complex disease.
Keywords: depth importance, gene-gene interaction, GWAS, random forest
1 |. INTRODUCTION
Genome-wide association studies (GWAS) have been popular in detecting association between single-nucleotide polymorphisms (SNPs) and disease, where the association is usually determined via the test of the marginal effect of an SNP. This has led to successful findings for many complex diseases including breast cancer and type 2 diabetes (Ferreira et al., 2019; Xue et al., 2018). However, the risk attributed to genetic factors is still far from satisfactory; for instance, the common genetic variation only explains 18% of the twofold familial relative risk for breast cancer (Ferreira et al., 2019). One of the major impediments is the focus on the additive effects of individual SNPs. Although there have been growing efforts in identifying gene-gene interactions for complex diseases (Banerjee, Vats, Kushwah, & Srivastava, 2019; Chen, Liu, Zhang, & Zhang, 2007), the success has been limited by the lack of effective approaches of identifying potential interactions.
To overcome the aforementioned bottleneck, we consider the concept of supervariant. On one hand, like the classic concept of the gene, a supervariant consists of a combination of alleles in multiple loci. On the other hand, the loci contributing to a supervariant can be anywhere in the genome while a gene refers to a physically connected region of the chromosome. The combination of alleles for a supervariant is expected to reflect both the individual and interactive effects of contributing alleles. We hypothesize that the supervariants are easy to detect and the aggregated signals are more stable in their associations with the disease than that from a single SNP.
Specifically, we introduce a ranking and aggregation approach to identifying supervariants. It is based on the rank but generally weak association between an individual locus and a disease of interest. SNPs are first divided into sets by their physical positions that may or may not be adjacent to each other, and then ranked within the sets by their importance in terms of disease discrimination ability. To account for integrated effects of multiple SNPs, the importance of an individual SNP is measured by the so-called depth-importance in a tree and forest based framework (Chen et al., 2007; Zhang & Singer, 2010). A supervariant is formed by a certain number of top SNPs within the set. The number of top SNPs is selected by considering all possible numbers of top SNPs and selecting the one with the strongest association between the resulting supervariant and disease.
With access to the UK Biobank databases (Sudlow et al., 2015), we apply our method to identify supervariants for breast cancer and empirically demonstrate the validity and power of using supervariants. We consider breast cancer because it is not only a common and complex disease but also has been extensively studied in the genetic literature. The UK Biobank databases, including genetic data on ~500,000 individuals, have become valuable sources for genetic studies of complex diseases (Anderson et al., 2018; Ferreira et al., 2019).
2 |. MATERIALS AND METHODS
2.1 |. Methodology details
We consider the following generalization of the logistic model:
where F is a function not limited to be linear, and Yi indicates the disease status, vi includes all genotyped variants, and zi denotes all confounding covariates to be adjusted. We consider the tree-based method to fit the unknown function F which allows potential nonlinear relations and any possible interactions.
To facilitate the identification of supervariants, we adopt the transformation in Song and Zhang (2014) which relies on an ordering of the estimated effect sizes of all variants. To be consistent with the nonparametric spirit, we use the depth importance score (Chen et al., 2007) as a proxy to the effect size of each variant, and to provide the ordering. The idea of depth importance score is to measure the importance of the variable by the depth at which a variable is used as a splitting node in a tree classifier. The assumption behind such measure is that an important variable tends to be used in the early stage of the construction of a tree. To provide a more stable measure, we also consider the ensemble approach to estimating the importance. Specifically, for a given set g of SNPs, we construct forest f consisting of a total number of |f| trees. Each tree in the forest is built without pruning based on a randomly selected subset of variants in g. Then, the depth importance score of variant vjg, the jth variant in set g, in tree T is defined as
where Lt is the depth of node t and Gt is the χ2 independence test statistics of node t. Then, the overall depth importance score is given by
over all |f| trees in the forest f. Once we obtain the ordering of variants within the set g, let djg be the index of the variant with the jth largest depth importance score. Define
where Jg is the total number of variants considered in set g, 1 ≤i ≤n and n is the total number of subjects. In other words, for each subject, the transformation returns the rank of first variant with minor allele within the depth-importance-ordered variants list of set g. This transformation considers the dominant mode of transmission. Similarly, we can define a transformation based on the recessive mode of transmission. Define
where Jg is the total number of variants considered in set g, 1 ≤i ≤n and n is the total number of subjects. In this way, the transformation returns the rank of the first variant with two copies of minor allele within the ordered list of set g.
Finally, to obtain a supervariant, we inspect all possible thresholds for variable Xg with observations x1g,…,xng. For each threshold, the variable is turned into binary; that is, for a threshold c, Sg = I(Xg < c), where I is the indicator function, and c∈{x1g,…,xng}. A univariate logistic regression is carried out to investigate its effect, and the final threshold is the one that gives the smallest p value among all possible thresholds. This leads to the supervariant constructed with top variants in the depth-importance-ordered variants list and the total number of variants used to form the supervariant is the same as the final threshold. Here, the threshold is selected to enhance the association between the disease and the supervariant to be formed. After supervariants are identified, univariate, and/or multivariable regression can be performed in both discovery and verification datasets. Hence, the tree and forest methods are used to select putative supervariants for further evaluation, and not to determine nor to fit the final model of analysis.
2.2 |. Data processing
We apply our proposed method at 41,502,298 genetic variants of UK Biobank imputed SNP datasets after genotyping quality controls. Specifically, we consider the biallelic variants coded based on the number of copies of the minor allele. We remove variants with low call rate (missing probability ≥ 0.1) and disrupted Hardy–Weinberg equilibrium (p < 1 × 10−7). We divide the whole SNP dataset into 2,734 nonoverlapping local sets by the physical position so that each set consists of SNPs within a segment of physical length 1 Mb; for instance, SNPs on Chromosome 1 with base-pair position value falling in 1 to 999,999 are in SNP set 1, and those with base-pair position value between 1,000,000 to 1,999,999 are in SNP set 2. This is similar to commonly used sliding window, except no overlapping so that the same SNP will not appear in multiple supervariants. Our method still works without this constraint but we find it reasonable not to allow the same SNP to appear repeatedly. Otherwise, there would be many more possible supervariants with many of them overlapping each other and highly correlated. Information of linkage disequilibrium (LD) is not used in the identification of the supervariants.
To reduce the confound of population structure, we only keep individuals considered to have recent British ancestry using the quality control information provided by UK Biobank. We select individuals with self-reported breast cancer as cases and those without any self-reported cancer diseases as control candidates. We use the difference between the reported date of breast cancer and the date of birth as the age of cases. We use the difference between the date of the last survey on cancer and the date of birth as the age of controls. Individuals without age information are excluded. We also exclude individuals whose genetic and self-reported genders are inconsistent. The construction and selection of supervariants do not involve further covariates for simplicity.
We randomly select 60% of the cases and create a nominally unrelated subset (without relatives closer than third cousins) following existing procedures (Bycroft et al., 2018). This gives the cases of the discovery set. We match each case subject with five control subjects with same age and gender to complete the discovery set. In addition, we avoid picking controls who are relatives of any selected individuals. We use the remaining 40% cases and the same 1:5 matching rule for controls to generate the validation set. In the end, after sample quality control, we obtain a discovery set with 5,653 cases and 28,241 controls, and a validation set with 3,724 cases and 18,620 controls. There are 150 and 95 male cases included in discovery and validation set, respectively, and in total 1,470 male samples in combined datasets.
2.3 |. Simulation setup
In the simulation, we use the SNP data on Chromosome 17 from UK Biobank breast cancer dataset because there is no significant signal identified on this chromosome from this dataset. We randomly sample 10,000 subjects from the controls of discovery dataset. We then randomly sample 30 SNP sets and 500 random SNPs (with minor allele frequency (MAF) > 0.01) from each set to form the whole synthetic genetic dataset. The physical order of selected 15,000 (=500 × 30) SNPs is kept. The disease status is generated according to the following model: Yi ~ Bernoulli(pi), and
where xiBj_k is the kth SNP in SNP set j for subject i, 1 ≤ i ≤ 10, 000, 1 ≤ j ≤ 30, and 1 ≤ k ≤ 500. Therefore, have 12 true signals in total from six different SNP sets with three different structures, individual signal, interactive signal with group sizes 2 and 3, respectively. After the disease status generation, the whole dataset is divided into two sets, one for discovery and one for validation to mimic the procedure we adopt in the real data application. Each set consists of 5,000 samples and the case-control ratio is kept close. We apply the traditional single SNP-based association method and the proposed method with transformation based on the dominant mode of transmission to detect associations. The whole process is repeated 100 times.
2.4 |. Analysis of UK Biobank breast cancer dataset
To calculate the depth importance score of SNPs in each SNP set in the discovery dataset, we construct a forest with 3,000 trees, given the average size of one SNP set is about 15,000 SNPs, and each tree is constructed with randomly selected one sixtieth of total SNPs in the given set with RTEE (Zhang & Singer, 2010). Transformations based on both dominant and recessive modes of transmission are considered. We use 3.66 × 10−5 (i.e., 0.1/2,734) as the threshold for supervariant-level association as we consider 2,734 SNP sets. We use 0.1/2,734 instead of 0.05/2,734 to limit false-positive errors while being more inclusive of potentially important SNPs. The association between any of the discovered supervariants is assessed by a univariate logistic regression with age and gender properly controlled.
To demonstrate the potential of supervariant, we also conduct the traditional single SNP-based association analysis as a benchmark for comparison. We consider the same discovery, verification, and combined sets for fair comparison. Age and gender are included as control variables.
3 |. RESULTS
3.1 |. Simulation results
We first provide a summary of the simulation setup. The average case-control ratio of 100 repetitions is about 0.225 (close to 1-to-5 ratio which is used in breast cancer data analysis). On average, 3.44 out of 12 signal SNPs have its MAF less than 0.05 in the simulation repetitions.
For the use of the traditional single SNP-based association method, an SNP is considered to be significant on discovery set if its p value is less than 3.33 × 10−6 (i.e., 0.05/15,000), and a supervariant is significant if its p value on discovery set is less than .0017 (i.e., 0.05/30 as there are in total 30 supervariants, one for each set).
We first consider the number of SNP sets that each method selects. In total, there are six sets with true signals and 24 sets without signal. On average, on the discovery dataset, the traditional single SNP-based association method covers 5.40 (out of six) sets with true signals, while the proposed method identified 5.94 supervariants from six sets with true signals. At the same time, on the discovery set, there are on average 9.78 identified supervariants coming from 24 sets without signal, and only 0.07 such sets are selected by the traditional single SNP-based association method. However, if we also require p value of a supervariant to be less than .01 on the validation set, then on average the number of verified supervariants coming from sets without signal is less than 1, and there are still 5.82 supervariants covering six sets with true signals.
Because some SNPs are in LD with others, for both methods, if any SNP in the neighborhood of the true signal SNP is selected, we consider this signal SNP as being identified. In the simulation, we take 60 nearby SNPs, 30 SNPs on the left and 30 on the right, as the neighborhood. The frequency of true signal identification for two methods is shown in Table 1. Supervariant clearly outperforms the traditional single SNP-based association method.
TABLE 1.
Frequency of true signal identification in simulation
| SNP | B1_1 | B2_1 | B3_1 | B3_301 | B4_1 | B4_301 | B5_1 | B5_201 | B5_401 | B6_1 | B6_201 | B6_401 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GWAS | 0.95 | 0.92 | 0.67 | 0.6 | 0.62 | 0.69 | 0.31 | 0.42 | 0.28 | 0.46 | 0.5 | 0.55 |
| Supervariant | 0.99 | 0.92 | 0.75 | 0.74 | 0.75 | 0.74 | 0.6 | 0.62 | 0.58 | 0.64 | 0.65 | 0.7 |
Abbreviations: GWAS, Genome-wide association studies; SNP, single-nucleotide polymorphism.
The concept of supervariant is proposed to enhance the association study of rare variants and interactions. The power of identifying rare variants and their interactions is given in Tables 2 and 3, respectively. Here, we consider an interaction being identified if all true signal SNPs within the same set, such as the two B3_1 and B3_301 on SNP set 3, are identified at the same time. From Table 2, the proposed method is much more powerful than the traditional single SNP-based association method, even when a rare variant is inside a group signal. Similarly, in terms of interaction, Table 3 shows that the proposed method outperforms the traditional single SNP-based association method by a large margin.
TABLE 2.
Frequency of rare variant signal identification in simulation
| Single SNP signal | Within group signal | Within group of two | Within group of three | |
|---|---|---|---|---|
| GWAS | 0.729 | 0.199 | 0.228 | 0.181 |
| Supervariant | 0.833 | 0.284 | 0.376 | 0.225 |
Abbreviations: GWAS, Genome-wide association studies; SNP, single-nucleotide polymorphism.
TABLE 3.
Frequency of interaction identification in simulation
| Overall | Group of two | Group of three | |
|---|---|---|---|
| Genome-wide association studies | 0.22 | 0.36 | 0.08 |
| Supervariant | 0.38 | 0.53 | 0.24 |
3.2 |. Breast cancer dataset analysis results
3.2.1 |. Traditional single SNP-based association results
The Manhattan plot based on the discovery dataset is shown in Figure 1, and details of verified top SNPs in each LD block are given in Table 4. Here, an SNP is verified if its p value is less than 5 × 10−8 on discovery set and is less than .01 on validation set. Regional plots of verified regions with multiple SNPs are shown in Figure 2, and the plots are based on the combined dataset.
FIGURE 1.
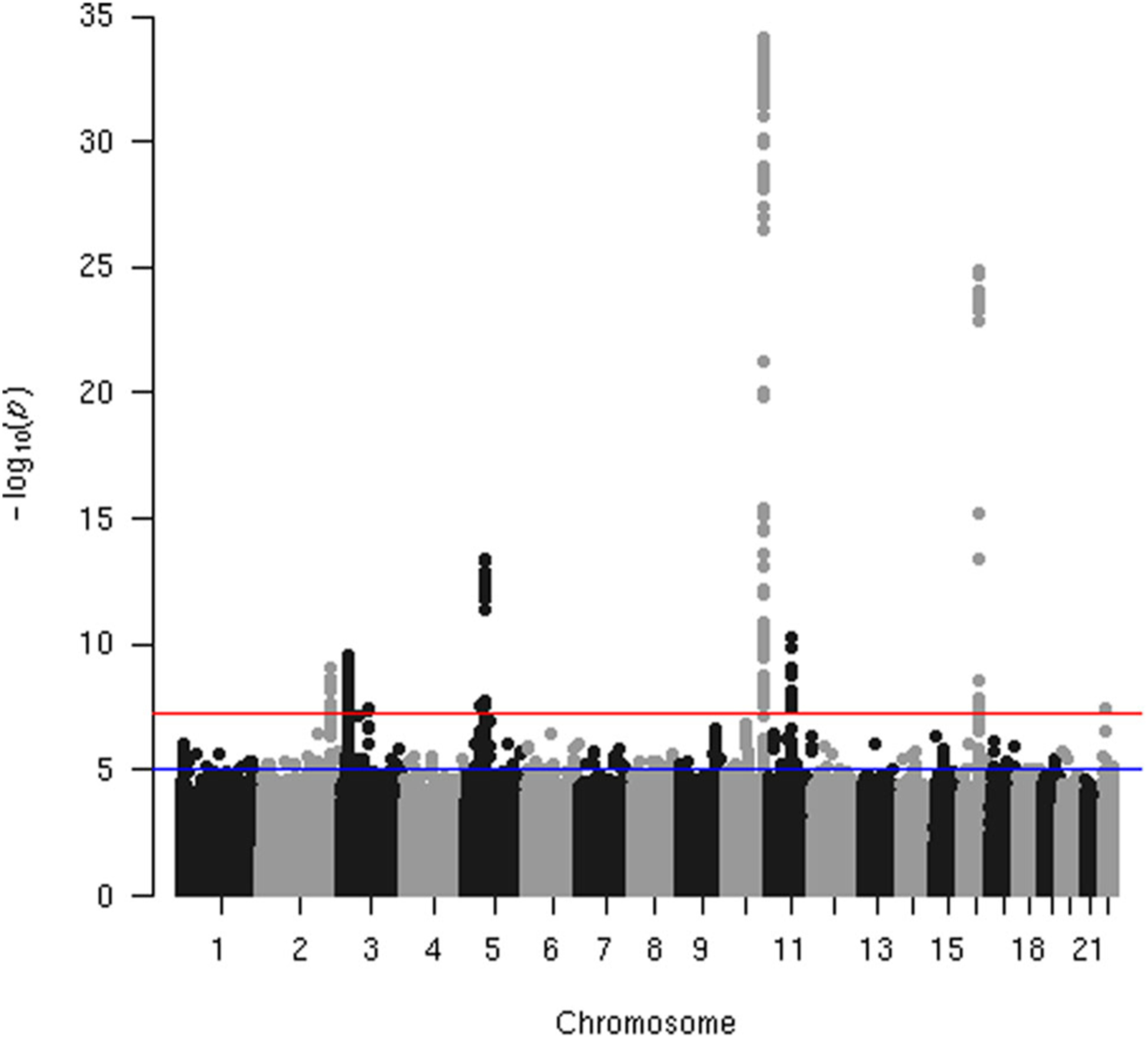
Manhattan plot of single SNP-based association analysis on discovery dataset. SNP, single-nucleotide polymorphism
TABLE 4.
Verified top SNPs in each LD block from GWAS
| Chr | SNP name | Position | Minor allele | Major allele | MAF | OR | p |
|---|---|---|---|---|---|---|---|
| 2 | rs4442975 | 217920769 | G | T | 0.491 | 1.15 | 4.56 × 10−18 |
| 3 | rs11715126 | 27401247 | A | G | 0.414 | 0.89 | 1.55 × 10−12 |
| 5 | rs10043344 | 44926518 | A | T | 0.39 | 1.11 | 4.61 × 10−10 |
| 5 | rs7714232 | 56011357 | T | A | 0.165 | 1.22 | 5.56 × 10−22 |
| 10 | rs2981575 | 123346116 | G | A | 0.4 | 1.29 | 2.41 × 10−54 |
| 11 | rs661204 | 69330983 | A | G | 0.123 | 1.25 | 9.14 × 10−23 |
| 16 | rs4784227 | 52599188 | T | C | 0.244 | 1.27 | 6.54 × 10−39 |
| 22 | rs62237617 | 28761148 | T | C | 0.002 | 2.86 | 8.39 × 10−16 |
Abbreviations: GWAS, Genome-wide association studies; LD, linkage disequilibrium; MAF, minor allele frequency; OR, odds ratio; SNP, single-nucleotide polymorphism.
FIGURE 2.
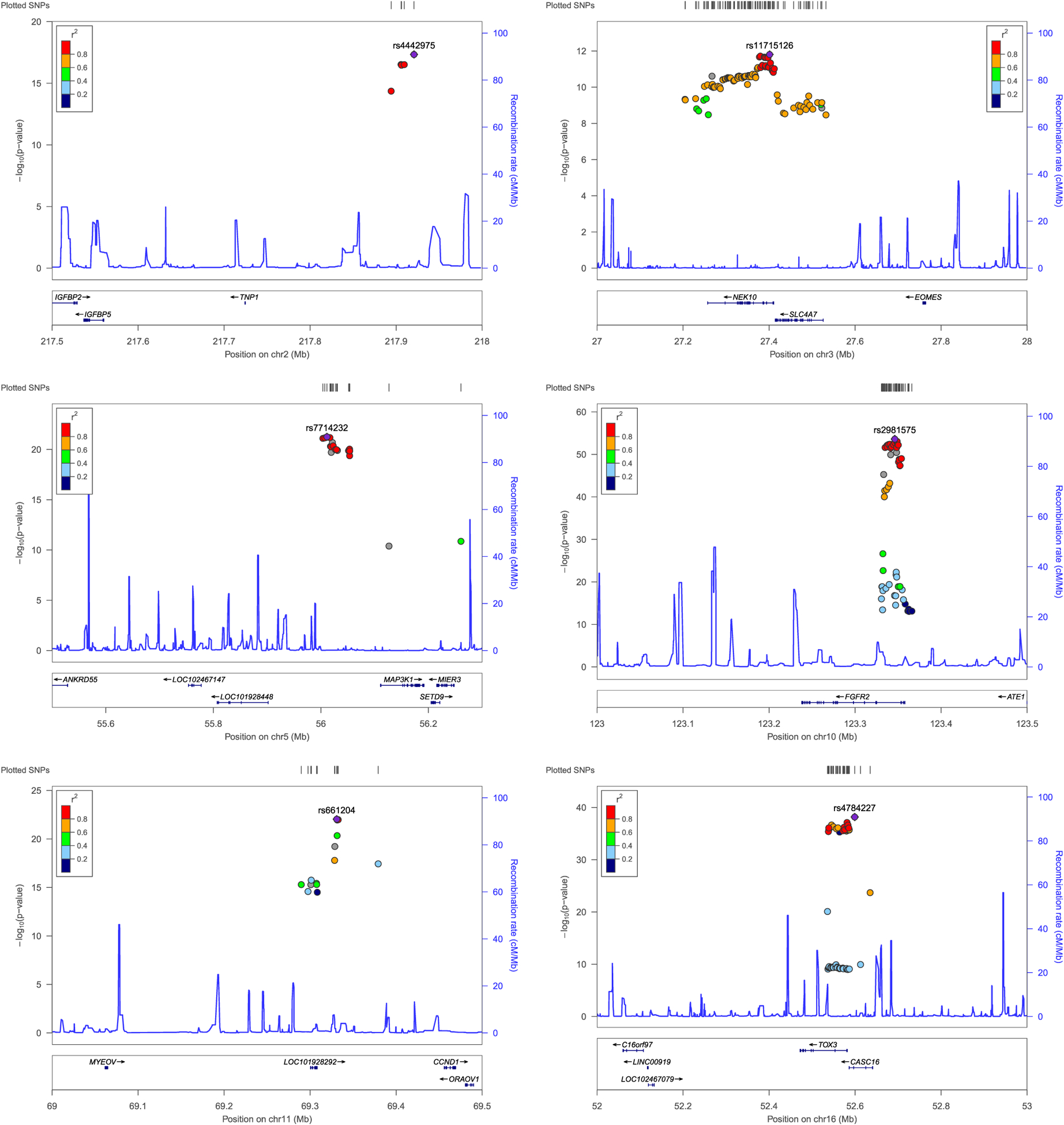
Regional plots of verified GWAS regions with multiple SNPs. GWAS, genome-wide association studies; SNPs, single-nucleotide polymorphism
3.2.2 |. Significant supervariants associated with breast cancer
We find 510 supervariants with p values below 3.66 × 10−5 (96 from recessive model and 414 from dominant model), and 24 of them has p value less than .01 in the validation dataset (7 from recessive model and 17 from dominant model). We also assess the association of these 24 supervariants in the combined dataset (discovery plus validation datasets), and all of them have p values in the range of 10−7 (see Table 5). The specific formation of the 24 verified supervariants are given in Table 6 where the odds ratio and corresponding p values are calculated on the combined dataset. The LD heat maps of the SNPs in the verified supervariants involving multiple SNPs are displayed in Figure 3.
TABLE 5.
Marginal effects of 24 verified supervariants on discovery, validation, and combined datasets
| Supervariant | Discovery dataset | Validation dataset | Combined dataset | |||
|---|---|---|---|---|---|---|
| Recessive | Odds ratio (OR) | p | OR | p | OR | p |
| Chr1_122 | 0.853 | 2.43 × 10−5 | 0.89 | 8.74 × 10−3 | 0.875 | 8.13 × 10−7 |
| Chr2_218 | 1.189 | 2.39 × 10−9 | 1.31 | 1.55 × 10−11 | 1.25 | 6.37 × 10−19 |
| Chr3_28 | 0.832 | 1.10 × 10−5 | 0.864 | 2.56 × 10−3 | 0.85 | 1.07 × 10−7 |
| Chr6_152 | 1.186 | 1.70 × 10−5 | 1.3 | 3.84 × 10−6 | 1.25 | 1.24 × 10−9 |
| Chr9_84 | 1.378 | 2.55 × 10−5 | 1.32 | 2.80 × 10−3 | 1.35 | 2.55 × 10−7 |
| Chr10_124 | 1.391 | 6.55 × 10−19 | 1.37 | 4.54 × 10−12 | 1.388 | 8.65 × 10−30 |
| Chr16_53 | 1.529 | 4.69 × 10−13 | 1.47 | 1.66 × 10−7 | 1.5 | 4.60 × 10−19 |
| Dominant | OR | p | OR | p | OR | p |
| chr2_203 | 1.136 | 1.24 × 10−5 | 1.117 | 2.11 × 10−3 | 1.128 | 9.80 × 10−8 |
| chr2_214 | 1.159 | 9.91 × 10−6 | 1.118 | 6.20 × 10−3 | 1.142 | 2.52 × 10−7 |
| chr3_28 | 0.842 | 1.14 × 10−8 | 0.907 | 9.26 × 10−3 | 0.867 | 1.26 × 10−9 |
| chr5_13 | 0.618 | 6.49 × 10−6 | 0.712 | 8.46 × 10−3 | 0.654 | 2.27 × 10−7 |
| chr5_45 | 1.171 | 2.32 × 10−7 | 1.182 | 8.33 × 10−6 | 1.176 | 8.88 × 10−12 |
| chr5_56 | 1.233 | 2.89 × 10−12 | 1.236 | 1.05 × 10−8 | 1.235 | 1.75 × 10−19 |
| chr5_57 | 1.25 | 5.21 × 10−13 | 1.250 | 4.23 × 10−9 | 1.25 | 1.31 × 10−20 |
| chr5_174 | 1.201 | 2.55 × 10−7 | 1.130 | 4.75 × 10−3 | 1.172 | 7.32 × 10−9 |
| chr7_156 | 1.378 | 1.40 × 10−5 | 1.279 | 5.24 × 10−3 | 1.337 | 2.88 × 10−7 |
| chr8_129 | 1.162 | 1.59 × 10−6 | 1.152 | 2.22 × 10−4 | 1.158 | 1.41 × 10−9 |
| chr9_111 | 0.857 | 2.25 × 10−7 | 0.895 | 2.46 × 10−3 | 0.872 | 3.03 × 10−9 |
| chr10_65 | 0.83 | 4.86 × 10−8 | 0.879 | 1.96 × 10−3 | 0.849 | 6.42 × 10−10 |
| chr10_124 | 1.418 | 1.69 × 10−27 | 1.387 | 9.95 × 10−17 | 1.406 | 1.54 × 10−42 |
| chr11_70 | 1.222 | 5.57 × 10−11 | 1.265 | 3.95 × 10−10 | 1.239 | 1.68 × 10−19 |
| chr12_97 | 0.879 | 1.12 × 10−5 | 0.886 | 8.56 × 10−4 | 0.882 | 3.66 × 10−8 |
| chr16_53 | 1.323 | 9.50 × 10−22 | 1.289 | 1.93 × 10−12 | 1.309 | 1.57 × 10−32 |
| chr22_29 | 2.536 | 2.07 × 10−8 | 3.700 | 2.87 × 10−9 | 2.897 | 7.41 × 10−16 |
TABLE 6.
SNPs corresponding to 24 verified supervariants
| Recessive | Chr | SNP name | Position | Minor allele | Major allele | MAF | OR | p |
|---|---|---|---|---|---|---|---|---|
| Chr1_122 | 1 | rs12026807 | 121274278 | G | A | 0.48 | 0.875 | 8.13 × 10−7 |
| Chr2_218 | 2 | rs6721996 | 217909463 | G | A | 0.491 | 1.247 | 5.67 × 10−18 |
| rs4442975 | 217920769 | G | T | 0.491 | 1.253 | 1.88 × 10−18 | ||
| Chr3_28 | 3 | rs73055736 | 27392625 | A | T | 0.416 | 0.855 | 4.75 × 10−7 |
| rs73055746 | 27399466 | C | T | 0.417 | 0.857 | 6.57 × 10−7 | ||
| rs12637322 | 27389740 | C | T | 0.416 | 0.854 | 4.52 × 10−7 | ||
| rs2370959 | 27397148 | A | G | 0.414 | 0.852 | 3.28 × 10−7 | ||
| rs55676236 | 27391598 | C | A | 0.419 | 0.859 | 9.24 × 10−7 | ||
| rs112238765 | 27388820 | A | C | 0.416 | 0.855 | 5.29 × 10−7 | ||
| rs73055753 | 27403304 | C | T | 0.416 | 0.854 | 4.17 × 10−7 | ||
| Chr6_152 | 6 | rs6913578 | 151949806 | C | A | 0.325 | 1.222 | 1.14 × 10−8 |
| rs7740686 | 151948173 | T | A | 0.333 | 1.216 | 1.47 × 10−8 | ||
| rs6557160 | 151949582 | C | A | 0.333 | 1.214 | 1.88 × 10−8 | ||
| rs58164038 | 151956201 | G | A | 0.329 | 1.237 | 1.46 × 10−9 | ||
| rs3757322 | 151942194 | G | T | 0.336 | 1.205 | 5.39 × 10−8 | ||
| Chr9_84 | 9 | rs12551463 | 83471165 | G | T | 0.162 | 1.35 | 2.55 × 10−7 |
| Chr10_124 | 10 | rs2981584 | 123350216 | A | C | 0.4 | 1.388 | 8.65 × 10−30 |
| Chr16_53 | 16 | rs112149573 | 52581245 | T | G | 0.241 | 1.453 | 1.65 × 10−17 |
| rs3095606 | 52584173 | G | A | 0.262 | 1.443 | 2.13 × 10−19 | ||
| rs1362548 | 52563951 | C | G | 0.26 | 1.427 | 4.71 × 10−18 | ||
| rs3112578 | 52585440 | C | T | 0.245 | 1.433 | 9.50 × 10−17 | ||
| rs1345388 | 52556293 | C | T | 0.259 | 1.433 | 2.74 × 10−18 | ||
| rs3095607 | 52584295 | G | T | 0.262 | 1.444 | 1.80 × 10−19 | ||
| rs17271951 | 52538040 | C | T | 0.254 | 1.45 | 9.33 × 10−19 | ||
| rs4784226 | 52583143 | T | C | 0.242 | 1.442 | 6.27 × 10−17 | ||
| rs4784227 | 52599188 | T | C | 0.244 | 1.455 | 5.63 × 10−18 | ||
| rs3803661 | 52586477 | A | G | 0.262 | 1.44 | 2.97 × 10−19 | ||
| rs12930156 | 52581424 | T | C | 0.262 | 1.441 | 3.28 × 10−19 | ||
| rs3803662 | 52586341 | A | G | 0.262 | 1.44 | 3.27 × 10−19 | ||
| rs7500427 | 52545277 | A | G | 0.258 | 1.443 | 6.64 × 10−19 | ||
| rs12600239 | 52538900 | T | C | 0.257 | 1.444 | 9.56 × 10−19 | ||
| rs35668161 | 52538825 | A | C | 0.254 | 1.452 | 5.97 × 10−19 | ||
| rs9936081 | 52549646 | A | G | 0.258 | 1.437 | 1.46 × 10−18 | ||
| rs8045285 | 52579986 | G | A | 0.246 | 1.452 | 5.26 × 10−18 | ||
| Dominant | Chr | SNP name | Position | Minor allele | Major allele | MAF | OR | p |
| Chr2_203 | 2 | rs3769823 | 202122995 | A | G | 0.28 | 1.128 | 9.80 × 10−8 |
| Chr2_214 | 2 | rs7580977 | 213473493 | T | G | 0.21 | 1.102 | 3.12 × 10−5 |
| rs190740846 | 213176997 | C | T | 0.009 | 1.201 | 2.55 × 10−2 | ||
| rs61521361 | 213496258 | A | T | 0.344 | 1.095 | 9.06 × 10−5 | ||
| rs2054613 | 213484354 | G | C | 0.354 | 1.097 | 7.25 × 10−5 | ||
| rs10174150 | 213487269 | C | T | 0.354 | 1.095 | 8.93 × 10−5 | ||
| rs11679805 | 213494947 | T | A | 0.344 | 1.095 | 8.37 × 10−5 | ||
| rs5838347 | 213547305 | AT | A | 0.406 | 1.132 | 2.86 × 10−7 | ||
| rs6736536 | 213429063 | T | C | 0.341 | 1.086 | 3.39 × 10−4 | ||
| rs13410624 | 213499043 | G | A | 0.344 | 1.096 | 7.43 × 10−5 | ||
| rs2054615 | 213484752 | T | C | 0.354 | 1.097 | 7.08 × 10−5 | ||
| rs7601545 | 213483894 | A | G | 0.354 | 1.097 | 7.08 × 10−5 | ||
| rs66535530 | 213499028 | T | C | 0.299 | 1.099 | 3.44 × 10−5 | ||
| rs6435714 | 213442905 | T | G | 0.342 | 1.085 | 3.56 × 10−4 | ||
| rs67447343 | 213490466 | C | G | 0.347 | 1.093 | 1.31 × 10−4 | ||
| rs6749009 | 213482684 | A | G | 0.354 | 1.097 | 7.26 × 10−5 | ||
| rs67535717 | 213492898 | A | G | 0.354 | 1.095 | 1.00 × 10−4 | ||
| rs62186335 | 213475881 | T | C | 0.21 | 1.102 | 3.30 × 10−5 | ||
| rs6712252 | 213488021 | T | C | 0.354 | 1.096 | 8.24 × 10−5 | ||
| rs2068404 | 213484997 | C | T | 0.354 | 1.097 | 7.08 × 10−5 | ||
| rs931216 | 213493825 | T | C | 0.3 | 1.098 | 3.90 × 10−5 | ||
| rs7606156 | 213485596 | A | G | 0.356 | 1.095 | 8.88 × 10−5 | ||
| rs13020448 | 213438055 | T | C | 0.342 | 1.084 | 4.33 × 10−4 | ||
| rs931218 | 213496624 | A | C | 0.299 | 1.099 | 3.70 × 10−5 | ||
| rs6708862 | 213487661 | T | C | 0.354 | 1.096 | 8.30 × 10−5 | ||
| rs6435717 | 213535690 | A | G | 0.384 | 1.121 | 1.40 × 10−6 | ||
| rs10221625 | 213541714 | T | C | 0.362 | 1.123 | 6.87 × 10−7 | ||
| rs7606703 | 213473115 | C | A | 0.21 | 1.102 | 2.99 × 10−5 | ||
| rs4673675 | 213539145 | C | T | 0.363 | 1.122 | 8.61 × 10−7 | ||
| rs72935050 | 213609001 | A | T | 0.073 | 1.06 | 7.35 × 10−2 | ||
| rs17330768 | 213494330 | G | T | 0.3 | 1.099 | 3.75 × 10−5 | ||
| rs11676391 | 213485772 | C | T | 0.354 | 1.097 | 7.08 × 10−5 | ||
| rs6736377 | 213428760 | A | G | 0.341 | 1.086 | 3.45 × 10−4 | ||
| rs140149813 | 213243507 | G | C | 0.004 | 1.259 | 6.02 × 10−2 | ||
| rs6749157 | 213482781 | C | G | 0.354 | 1.097 | 7.30 × 10−5 | ||
| rs6711917 | 213487687 | T | C | 0.356 | 1.095 | 1.00 × 10−4 | ||
| rs2128324 | 213490883 | A | G | 0.356 | 1.094 | 1.13 × 10−4 | ||
| rs4673676 | 213539645 | T | C | 0.363 | 1.118 | 1.65 × 10−6 | ||
| 2:213537990_TACCC_T | 213537990 | T | TACCC | 0.365 | 1.119 | 1.75 × 10−6 | ||
| rs142052382 | 213493752 | GA | G | 0.334 | 1.091 | 2.16 × 10−4 | ||
| rs34741122 | 213498134 | G | A | 0.299 | 1.099 | 3.53 × 10−5 | ||
| rs10177496 | 213436989 | A | G | 0.221 | 1.086 | 3.30 × 10−4 | ||
| rs57917865 | 213495985 | T | G | 0.347 | 1.092 | 1.39 × 10−4 | ||
| rs10221626 | 213541719 | A | G | 0.362 | 1.123 | 6.87 × 10−7 | ||
| rs199875963 | 213536531 | G | GT | 0.389 | 1.116 | 4.06 × 10−6 | ||
| rs4673669 | 213477128 | A | C | 0.355 | 1.094 | 1.16 × 10−4 | ||
| rs13394068 | 213533178 | C | T | 0.358 | 1.106 | 1.56 × 10−5 | ||
| Chr3_28 | 3 | rs12637322 | 27389740 | C | T | 0.416 | 0.862 | 3.11 × 10−10 |
| rs11714071 | 27377648 | G | A | 0.415 | 0.863 | 5.21 × 10−10 | ||
| Chr5_13 | 5 | rs560776922 | 12563406 | G | A | 0.006 | 0.655 | 7.28 × 10−4 |
| rs560646329 | 12573113 | C | T | 0.006 | 0.687 | 2.13 × 10−3 | ||
| rs141667948 | 12970825 | C | T | 0.007 | 0.637 | 4.51 × 10−5 | ||
| Chr5_45 | 5 | rs10941679 | 44706498 | G | A | 0.255 | 1.185 | 9.68 × 10−14 |
| rs10043344 | 44926518 | A | T | 0.39 | 1.167 | 1.91 × 10−10 | ||
| Chr5_56 | 5 | rs79299334 | 55991699 | A | G | 0.075 | 1.21 | 7.71 × 10−10 |
| rs16886128 | 55998085 | C | A | 0.103 | 1.165 | 3.23 × 10−8 | ||
| rs74773564 | 55995869 | T | C | 0.075 | 1.211 | 5.79 × 10−10 | ||
| rs77367410 | 55992290 | G | A | 0.075 | 1.212 | 4.91 × 10−10 | ||
| rs16886113 | 55995035 | G | T | 0.079 | 1.216 | 1.19 × 10−10 | ||
| rs74626386 | 55990052 | T | G | 0.089 | 1.21 | 5.83 × 10−11 | ||
| Chr5_57 | 5 | rs61489170 | 56021469 | GA | G | 0.165 | 1.25 | 1.44 × 10−20 |
| rs7714232 | 56011357 | T | A | 0.165 | 1.254 | 4.45 × 10−21 | ||
| rs12653202 | 56016918 | C | A | 0.162 | 1.254 | 5.96 × 10−21 | ||
| rs66893416 | 56051596 | A | G | 0.164 | 1.245 | 8.40 × 10−20 | ||
| Chr5_174 | 5 | rs187944270 | 173955315 | A | G | 0.01 | 1.303 | 4.21 × 10−4 |
| rs2909714 | 173814304 | T | C | 0.031 | 1.145 | 3.16 × 10−3 | ||
| 5:173903211_AT_A | 173903211 | A | AT | 0.005 | 0.898 | 3.71 × 10−1 | ||
| rs555707527 | 173219789 | G | A | 0.003 | 1.424 | 9.70 × 10−3 | ||
| rs962985 | 173767987 | C | A | 0.233 | 1.098 | 5.70 × 10−5 | ||
| rs141509709 | 173926457 | T | C | 0.018 | 1.158 | 1.24 × 10−2 | ||
| rs2913479 | 173779289 | C | T | 0.23 | 1.09 | 1.88 × 10−4 | ||
| rs2913491 | 173810101 | A | T | 0.401 | 1.097 | 1.51 × 10−4 | ||
| Chr7_156 | 7 | rs9718441 | 155094172 | T | C | 0.269 | 1.096 | 5.35 × 10−5 |
| rs56181358 | 155867553 | A | G | 0.09 | 1.072 | 1.83 × 10−2 | ||
| rs73174921 | 155844449 | A | G | 0.103 | 1.075 | 1.00 × 10−2 | ||
| rs10245853 | 155097216 | G | A | 0.273 | 1.099 | 2.98 × 10−5 | ||
| 7:155202635_CTG_C | 155202635 | C | CTG | 0.253 | 1.022 | 3.50 × 10−1 | ||
| rs10264491 | 155259716 | G | C | 0.346 | 1.01 | 6.65 × 10−1 | ||
| rs141589981 | 155119687 | C | T | 0.008 | 1.204 | 3.16 × 10−2 | ||
| rs143721983 | 155248281 | T | C | 0.01 | 0.819 | 2.38 × 10−2 | ||
| rs28637616 | 155845220 | T | C | 0.102 | 1.073 | 1.24 × 10−2 | ||
| rs6459701 | 155296003 | G | A | 0.108 | 1.06 | 3.89 × 10−2 | ||
| rs73174937 | 155863201 | G | A | 0.09 | 1.081 | 8.54 × 10−3 | ||
| rs28705370 | 155843988 | A | G | 0.104 | 1.074 | 1.06 × 10−2 | ||
| rs6605564 | 155149763 | A | G | 0.262 | 1.087 | 2.50 × 10−4 | ||
| rs12534832 | 155242699 | C | A | 0.388 | 1.051 | 3.56 × 10−2 | ||
| rs73174920 | 155843148 | T | C | 0.103 | 1.075 | 1.06 × 10−2 | ||
| Chr8_129 | 8 | rs12550713 | 128370949 | G | C | 0.405 | 1.152 | 4.37 × 10−9 |
| rs10110330 | 128370755 | A | G | 0.405 | 1.153 | 3.32 × 10−9 | ||
| rs594868 | 128344602 | A | G | 0.397 | 1.147 | 1.17 × 10−8 | ||
| Chr9_111 | 9 | rs659713 | 110893949 | T | G | 0.376 | 0.875 | 9.04 × 10−9 |
| rs522463 | 110886254 | G | T | 0.381 | 0.878 | 1.95 × 10−8 | ||
| rs519679 | 110885947 | C | G | 0.381 | 0.878 | 1.73 × 10−8 | ||
| 9:110893720_GTATT_G | 110893720 | GTATT | G | 0.373 | 0.871 | 3.28 × 10−9 | ||
| rs497006 | 110885781 | C | T | 0.381 | 0.878 | 1.67 × 10−8 | ||
| rs628931 | 110893030 | A | G | 0.376 | 0.877 | 1.33 × 10−8 | ||
| rs631475 | 110885650 | C | A | 0.382 | 0.876 | 1.09 × 10−8 | ||
| rs676256 | 110895353 | C | T | 0.379 | 0.874 | 6.27 × 10−9 | ||
| rs520613 | 110886052 | C | T | 0.381 | 0.878 | 1.89 × 10−8 | ||
| rs648354 | 110887106 | G | A | 0.38 | 0.879 | 2.46 × 10−8 | ||
| rs525142 | 110886534 | G | A | 0.381 | 0.878 | 1.72 × 10−8 | ||
| rs662694 | 110887996 | C | G | 0.38 | 0.879 | 2.30 × 10−8 | ||
| rs7862747 | 110892899 | C | A | 0.378 | 0.875 | 9.66 × 10−9 | ||
| rs630965 | 110885479 | C | T | 0.382 | 0.876 | 1.14 × 10−8 | ||
| rs3119744 | 110895863 | A | C | 0.377 | 0.876 | 9.71 × 10−9 | ||
| rs527071 | 110886745 | C | A | 0.38 | 0.879 | 2.59 × 10−8 | ||
| rs857609 | 110888677 | G | C | 0.38 | 0.88 | 2.92 × 10−8 | ||
| Chr10_65 | 10 | rs34511355 | 64276964 | C | A | 0.142 | 0.849 | 6.42 × 10−10 |
| Chr10_124 | 10 | rs2981584 | 123350216 | A | C | 0.4 | 1.391 | 3.14 × 10−41 |
| rs2981575 | 123346116 | G | A | 0.4 | 1.394 | 7.12 × 10−42 | ||
| rs1219651 | 123344501 | A | G | 0.398 | 1.392 | 1.50 × 10−41 | ||
| rs2912774 | 123348662 | T | G | 0.4 | 1.395 | 8.39 × 10−42 | ||
| rs4752571 | 123342567 | C | T | 0.398 | 1.39 | 4.02 × 10−41 | ||
| rs2981583 | 123350523 | A | G | 0.399 | 1.384 | 2.71 × 10−39 | ||
| 10:123340431_GC_G | 123340431 | GC | G | 0.409 | 1.398 | 2.62 × 10−41 | ||
| Chr11_70 | 11 | rs625668 | 69308369 | A | G | 0.122 | 1.227 | 5.02 × 10−15 |
| rs657686 | 69332670 | G | A | 0.123 | 1.294 | 2.05 × 10−23 | ||
| rs554219 | 69331642 | G | C | 0.123 | 1.292 | 2.95 × 10−23 | ||
| Chr12_97 | 12 | rs11836367 | 96027467 | T | C | 0.35 | 0.88 | 3.66 × 10−8 |
| Chr16_53 | 16 | rs4784227 | 52599188 | T | C | 0.244 | 1.31 | 1.57 × 10−32 |
| Chr22_29 | 22 | rs62237617 | 28761148 | T | C | 0.002 | 2.90 | 7.41 × 10−16 |
Note: Statistics are calculated with corresponding recessive or dominant mode of transmission on the combined dataset.
Abbreviations: MAF, minor allele frequency; OR, odds ratio; SNP, single-nucleotide polymorphism.
FIGURE 3.
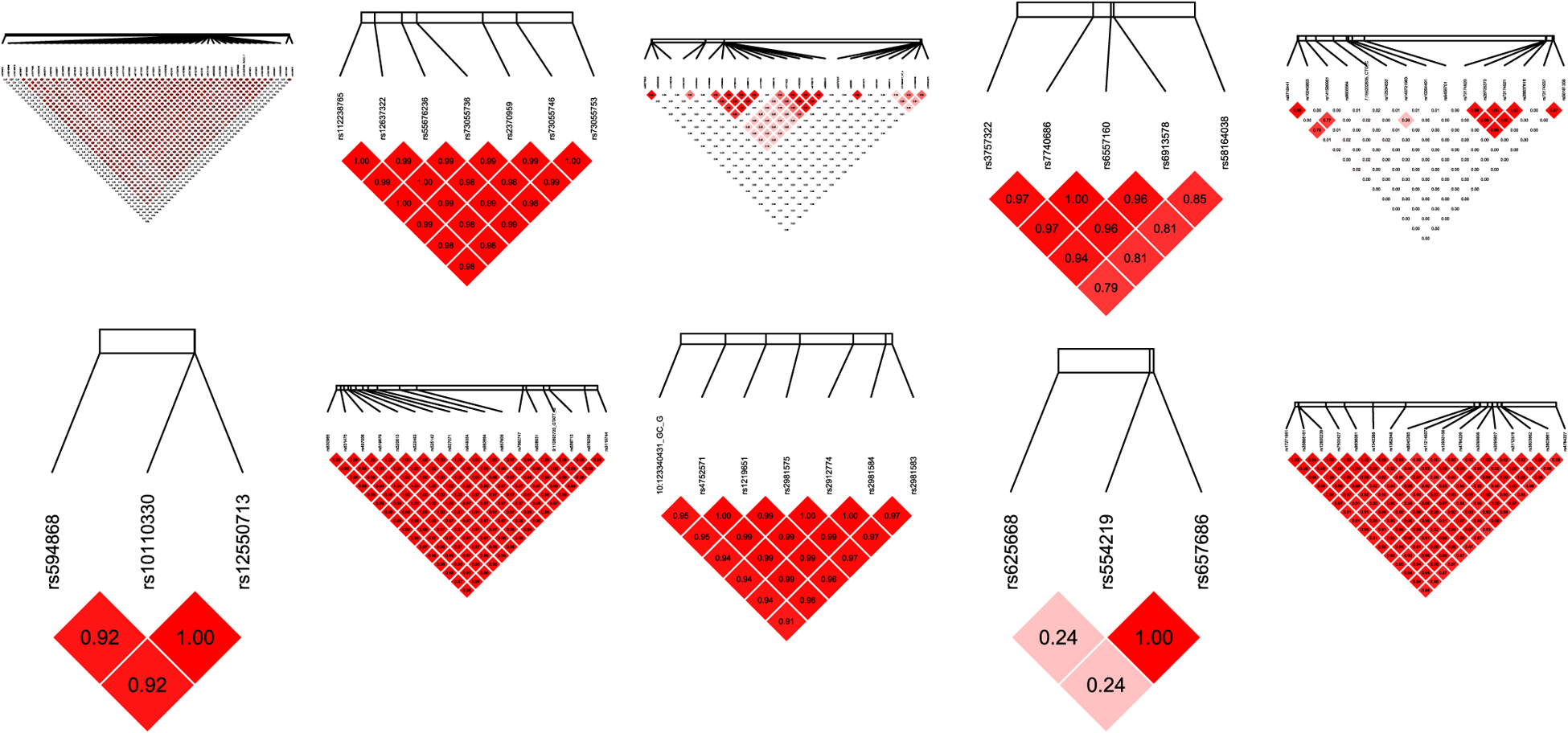
The linkage disequilibrium heat map of SNPs in the verified supervariants. The top row from left to right are for SNPs selected on Chromosomes 2, 3, 5, 6, and 7, respectively. The bottom row from left to right are for SNPs selected on Chromosomes 8, 9, 10, 11, and 16, respectively. SNP, single-nucleotide polymorphism
3.2.3 |. Comparison with single SNP-based association results
We find that our verified supervariants cover SNP signals in all LD blocks identified by the traditional single SNP-based association method. Moreover, we identify 13 additional supervariants that are verified. In addition, we find that there exist specific studies that provide support for associations between some SNPs in the supervariants we identified and the breast cancer or cancer in general (see Table 7). The confirmation of the known genes demonstrates the validity of the supervariants and its capability of unifying existing results. Moreover, we find several novel loci in gene LOC107985979 on Chromosome 2, LOC105374655, LOC105377739, LINC01411, and LINC01485 on Chromosome 5, and LOC107987084 on Chromosome 9, and a novel SNP rs11836367 on Chromosome 12 and have not yet been previously associated to breast cancer.
TABLE 7.
Support from existing literature
Related to other types of cancer.
rs6557160 is considered to join the regulation of ESR1 (Dunning et al., 2016).
Gene expression analysis.
4 |. DISCUSSION
In this study, we introduce the concept of supervariant, similar to but different from the classic concept of the gene, to group any number of loci together as the basis of genetic risk factor. Supervariant is designed to enhance the risk detection, and its associations with the disease are expected to be more stable than that of single SNP as there are usually (but not necessarily) a collection of SNPs involved in a supervariant. We propose a ranking and aggregation method to facilitate the search of disease-associated supervariants. We demonstrate in simulations that supervariant can be more powerful than the traditional single SNP-based association method in detecting rare variants and signals with interactive structure. We apply our method to a UK Biobank breast cancer dataset, and are able to replicate several previously reported breast cancer-associated genes documented in the literature as well as to identify several novel genes for further investigation.
Our results show that supervariants usually manifest stronger association signals than individual SNPs. As can be seen from Tables 5 and 6, SNPs on Chromosome 7 have p values at most at the level of 10−5 individually on the combined dataset while the supervariant Chr7_156 has a p value of level 10−7. The same is true for Chr2_218, Chr5_13, Chr5_56, and Chr5_174 where the supervariants have p values smaller than that of any contributing SNPs. This means that if a given significance level is used to detect association, a supervariant is more likely to be retained than the contributing SNPs. Of note, the number of candidate supervariants is much smaller than the number of SNPs, and therefore, in terms of the control of false-discovery, the use of supervariants is advantageous over the use of SNPs.
Furthermore, we demonstrate that supervariants are able to group SNPs in multiple genetic regions together. For instance, on Chromosome 6, SNPs located in CCDC170 and those that may participate in the regulation of ESR1 are grouped together. The same is true for Chr7_156 where SNPs in INSIG1, CNPY1, and BLACE are components of the same supervariant, and it also happens for Chr16_53 where SNPs in TOX3 and CASC16 are components of the same supervariant. Thus, not only can several risk loci be detected at the same time, but it may also indicate the existence of interactive effects between those SNPs. Such interactions may have implications to the underlying mechanisms involving multiple genes. It is also interesting to observe that supervariant captures the association between breast cancer and gene INSIG1 and CNPY1 on Chromosome 7, which is previously discovered via gene expression analysis (Jiang et al., 2017; Sadanandam et al., 2011).
All supervariants identified by our method pass the tests with Bonferroni correction on a combined dataset including several novel SNPs in gene LOC107985979 on Chromosome 2, in gene LOC105374655, LOC105377739, LINC01411, and LINC01485 on Chromosome 5, and gene LOC107987084 on Chromosome 9, and a novel SNP rs11836367 on Chromosome 12. Although there is limited knowledge on these SNPs and genes, it points to a direction for further investigation.
Certainly, there are genes in the literature that we are not able to identify with the proposed method by using this particular dataset. In a recent large breast cancer GWAS analysis, 122,977 number of cases and 105,974 number of controls (Ferreira et al., 2019) were used. It would be useful to apply the proposed method to this large sample of breast cancer dataset.
SNPs in a supervariant may be in high LD if the initial SNP sets are selected by their physical distance. However, whether SNPs are in high LD or not, their associations are not assessed individually, even though they are ranked by their individual effects.
It is worth noting that there is a critical difference between supervariant and haplotype. For haplotype analysis, we need to first infer the haplotypes through possible origins of the transmitted alleles. Supervariants are simply a combination of genotypes in multiple loci and do not depend on the origins of the transmission.
While highly promising, the use and identification of supervariants warrant further investigation. One aspect could be considered is how to best segment genotypes on all chromosomes to form SNP sets. In this study, we set the initial SNP sets to be physically close for convenience. However, LD, gene or pathway information may be considered to form more informative SNP sets for identification. Another aspect to consider is how to best rank the SNPs within the sets. In general, ranking variables is a challenging task in its own right.
The concept of supervariant and the ranking and aggregation method are proposed to identify sets of SNPs or mutations that can be used in the future for causal inference beyond association analysis or prediction. The ideas in the related literature such as polygenic risk scores, which involves two datasets but is largely prediction oriented, may be useful to improve our method. This is another possible direction for future work.
ACKNOWLEDGMENTS
The work of Heping Zhang is supported in part by the U.S. National Institutes of Health (R01HG010171 and R01MH116527) and the National Science Foundation (Grant no. DMS1722544). This study has been conducted using the UK Biobank Resource under Application Number 42009. The authors would like to thank the Yale Center for Research Computing for the guidance and use of the research computing infrastructure.
Funding information
National Science Foundation, Grant/Award Number: DMS1722544; National Human Genome Research Institute, Grant/Award Number: R01HG010171; National Institute of Mental Health, Grant/Award Number: R01MH116527
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
DATA AVAILABILITY STATEMENT
The genetic datasets used in the study are the genotypes with imputation from the UK Biobank databases (Field ID: 22801–22822). The imputation was performed by a collaborative group headed by the Wellcome Trust Centre for Human Genetics. Restrictions apply to the availability of these data, which were used under license for this study. More detailed information about the datasets can be found at http://www.ukbiobank.ac.uk/wp-content/uploads/2014/04/imputation_documentation_May2015-1.pdf.
REFERENCES
- Anderson JJ, Darwis ND, Mackay DF, Celis-Morales CA, Lyall DM, Sattar N, … Pell JP (2018). Red and processed meat consumption and breast cancer: UK Biobank cohort study and meta-analysis. European Journal of Cancer, 90, 73–82. [DOI] [PubMed] [Google Scholar]
- Banerjee M, Vats P, Kushwah AS, & Srivastava N (2019). Interaction of antioxidant gene variants and susceptibility to type 2 diabetes mellitus. British Journal of Biomedical Science, 76(4), 166–171. [DOI] [PubMed] [Google Scholar]
- Betts JA, Marjaneh MM, Al-Ejeh F, Lim YC, Shi W, Sivakumaran H, … Wiegmans AP (2017). Long noncoding RNAs CUPID1 and CUPID2 mediate breast cancer risk at 11q13 by modulating the response to DNA damage. The American Journal of Human Genetics, 101(2), 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, … Cortes A (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature, 562(7726), 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Wen W, Qu S, Li G, Egan KM, Chen K, … Blot WJ (2011). Replication and functional genomic analyses of the breast cancer susceptibility locus at 6q25.1 generalize its importance in women of Chinese, Japanese, and European ancestry. Cancer Research, 71(4), 1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu CT, Zhang M, & Zhang H (2007). A forest-based approach to identifying gene and gene–gene interactions. Proceedings of the National Academy of Sciences of the United States of America, 104(49), 19199–19203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FJ, Kuchenbaecker KB, Michailidou K, Mendoza-Fandino GA, Nord S, Lilyquist J, … Aittomäki K (2016). Identification of four novel susceptibility loci for estrogen receptor negative breast cancer. Nature Communications, 7(1), 11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Gao M, Yin Z, Yan L, & Cui L (2018). Association between lncRNA CASC8 polymorphisms and the risk of cancer: A meta-analysis. Cancer Management and Research, 10, 3141–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J, … Dicks E (2016). Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nature Genetics, 48(4), 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, … Wareham N (2007). Genome-wide association study identifies novel breast cancer susceptibility loci. Nature, 447(7148), 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejerman L, Chen GK, Eng C, Huntsman S, Hu D, Williams A, … Ingles S (2012). Admixture mapping identifies a locus on 6q25 associated with breast cancer risk in US Latinas. Human Molecular Genetics, 21(8), 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, Gamazon ER, Al-Ejeh F, Aittomäki K, Andrulis IL, Anton Culver H, … Asseryanis E (2019). Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nature Communications, 10(1), 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoussaini M, Edwards SL, Michailidou K, Nord S, Cowper-Sal R, Desai K, … Beesley J (2014). Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nature Communications, 5, 4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi Y, Soucy P, Adoue V, Michailidou K, Canisius S, Lemaçon A, … Baynes C (2016). Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget, 7(49), 80140–80163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Liu P, & Li X (2017). G9A performs important roles in the progression of breast cancer through upregulating its targets. Oncology Letters, 13(6), 4127–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HC, Lee JY, Sung H, Choi JY, Park SK, Lee KM, … Park M (2012). A genome-wide association study identifies a breast cancer risk variant in ERBB4 at 2q34: Results from the Seoul Breast Cancer Study. Breast Cancer Research, 14(2), R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström S, Ablorh A, Chapman B, Gusev A, Chen G, Turman C, … Hofmann O (2016). Deep targeted sequencing of 12 breast cancer susceptibility regions in 4611 women across four different ethnicities. Breast Cancer Research, 18(1), 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, … Wang Q (2013). Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature Genetics, 45(4), 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, … Bolla MK (2017). Association analysis identifies 65 new breast cancer risk loci. Nature, 551(7678), 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odefrey F, Stone J, Gurrin LC, Byrnes GB, Apicella C, Dite GS, … Hopper JL (2010). Common genetic variants associated with breast cancer and mammographic density measures that predict disease. Cancer Research, 70(4), 1449–1458. [DOI] [PubMed] [Google Scholar]
- Pan Z, Bao Y, Zheng X, Cao W, Cheng W, & Xu X (2016). Association of polymorphisms in intron 2 of FGFR2 and breast cancer risk in Chinese women. Cytology and Genetics, 50(5), 312–317. [PubMed] [Google Scholar]
- Sadanandam A, Futakuchi M, Lyssiotis CA, Gibb WJ, & Singh RK (2011). A cross-species analysis of a mouse model of breast cancer-specific osteolysis and human bone metastases using gene expression profiling. BMC Cancer, 11(1), 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, & Zhang H (2014). TARV: Tree-based analysis of rare variants identifying risk modifying variants in CTNNA2 and CNTNAP2 for alcohol addiction. Genetic Epidemiology, 38(6), 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, … Liu B (2015). UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Medicine, 12(3), 1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udler MS, Ahmed S, Healey CS, Meyer K, Struewing J, Maranian M, … Platte R (2010). Fine scale mapping of the breast cancer 16q12 locus. Human Molecular Genetics, 19(12), 2507–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialle-Castellano A, Laduron S, De Plaen E, Jost E, Dupont S, Ameye G, … van Baren N (2004). A gene expressed exclusively in acute B lymphoblastic leukemias. Genomics, 83(1), 85–94. [DOI] [PubMed] [Google Scholar]
- Wang Y, He Y, Qin Z, Jiang Y, Jin G, Ma H, … Shen H (2014). Evaluation of functional genetic variants at 6q25.1 and risk of breast cancer in a Chinese population. Breast Cancer Research, 16(4), 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu Z, Tong H, Peng H, Xian Z, Li L, … Xie S (2019). Linc01194 acts as an oncogene in colorectal carcinoma and is associated with poor survival outcome. Cancer Management and Research, 11, 2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderle M, Olmes G, Nabieva N, Häberle L, Jud SM, Hein A, … Hoyer J (2018). Risk, prediction and prevention of hereditary breast cancer–large-scale genomic studies in times of big and smart data. Geburtshilfe und Frauenheilkunde, 78(5), 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue A, Wu Y, Zhu Z, Zhang F, Kemper KE, Zheng Z, … McRae AF (2018). Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nature Communications, 9(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, & Singer BH (2010). Recursive partitioning and applications. Berlin, Germany: Springer Science & Business Media. [Google Scholar]
- Zheng Y, Nie P, & Xu S (2020). Long noncoding RNA CASC21 exerts an oncogenic role in colorectal cancer through regulating miR-7–5p/YAP1 axis. Biomedicine & Pharmacotherapy, 121, 109628. [DOI] [PubMed] [Google Scholar]