

Adhesins are critical for the formation and maturation of bacterial biofilms. We identify a second adhesin in P. fluorescens, called MapA, which appears to play a role in biofilm maturation and whose regulation is distinct from the previously reported LapA adhesin, which is critical for biofilm initiation. Analysis of bacterial adhesins shows that LapA-like and MapA-like adhesins are found broadly in pseudomonads and related organisms, indicating that the utilization of different suites of adhesins may be broadly important in the Gammaproteobacteria.
KEYWORDS: Pseudomonas fluorescens, RTX, adhesin, biofilm, cyclic di-GMP
ABSTRACT
Mechanisms by which cells attach to a surface and form a biofilm are diverse and differ greatly among organisms. The Gram-negative gammaproteobacterium Pseudomonas fluorescens attaches to a surface through the localization of the large type 1-secreted RTX adhesin LapA to the outer surface of the cell. LapA localization to the cell surface is controlled by the activities of a periplasmic protease, LapG, and an inner membrane-spanning cyclic di-GMP-responsive effector protein, LapD. A previous study identified a second, LapA-like protein encoded in the P. fluorescens Pf0-1 genome: Pfl01_1463. Here, we identified specific growth conditions under which Pfl01_1463, here called MapA (medium adhesion protein A) is a functional adhesin contributing to biofilm formation. This adhesin, like LapA, appears to be secreted through a Lap-related type 1 secretion machinery, and its localization is controlled by LapD and LapG. However, differing roles of LapA and MapA in biofilm formation are achieved, at least in part, through the differences in the sequences of the two adhesins and different distributions of the expression of the lapA and mapA genes within a biofilm. LapA-like proteins are broadly distributed throughout the Proteobacteria, and furthermore, LapA and MapA are well conserved among other Pseudomonas species. Together, our data indicate that the mechanisms by which a cell forms a biofilm and the components of a biofilm matrix can differ depending on growth conditions and the matrix protein(s) expressed.
IMPORTANCE Adhesins are critical for the formation and maturation of bacterial biofilms. We identify a second adhesin in P. fluorescens, called MapA, which appears to play a role in biofilm maturation and whose regulation is distinct from the previously reported LapA adhesin, which is critical for biofilm initiation. Analysis of bacterial adhesins shows that LapA-like and MapA-like adhesins are found broadly in pseudomonads and related organisms, indicating that the utilization of different suites of adhesins may be broadly important in the Gammaproteobacteria.
INTRODUCTION
Biofilm formation is a critical mode of growth for many microorganisms, including bacteria (1), archaea (2), and fungi (3). The mechanisms by which individual cells can attach to abiotic surfaces, and to each other, to form a biofilm are highly varied. However, in all cases, formation of a biofilm involves the production of adhesive molecules by the cells. These molecules are most commonly polysaccharide or protein based, and these factors remain adhered to the substratum while also often remaining tethered to the cells that produced them (4–8).
In bacteria, many different mechanisms of surface attachment by cells have been described. The biofilm matrix is typically composed of a variety of molecules, including a combination of polysaccharides, proteins, and extracellular DNA, used to join cells together into a biofilm (4, 9–12). In general, a combination of multiple molecules makes up the biofilm matrix, but some organisms use different molecules as their major biofilm matrix components. Examples include the production of a polysaccharide called Vibrio polysaccharide (VPS) and the matrix proteins RbmA, RbmC, and Bap1 by Vibrio cholerae (13–15) and the production of amyloid proteins called curli fibers by Escherichia coli (16). For many bacteria, biofilm formation is important for the effective colonization of their natural niches in the soil, on plants, or in the water column (17–19). Biofilm formation has also been implicated as an important virulence factor for many pathogenic bacteria (20–22). It is therefore important to understand both the mechanisms by which bacteria can form a biofilm and the ways in which that process is regulated.
An organism with one of the best-understood mechanisms of surface attachment and regulation of biofilm formation is Pseudomonas fluorescens. In P. fluorescens, surface attachment is achieved predominantly through the activity of a single large adhesion protein, LapA (23, 24). LapA is a noncanonical type 1-secreted, RTX adhesin which is composed of a C-terminal secretion signal as well as a central portion containing numerous repeat domains, multiple calcium binding domains, and a von Willibrand factor A domain that all contribute to adhesion (25–27). LapA also has an N-terminal globular domain that folds while the protein is in the process of being secreted, thus anchoring the protein in its TolC-like outer membrane pore (28, 29). LapA is an ∼520-kDa adhesin (25–27) and was thus named the large adhesion protein A and for lapa, the Spanish word for limpet, a marine mollusk that attaches tightly to rocks and other surfaces (23). The localization of LapA to the cell surface is dependent on the activity of two regulatory proteins, namely, LapG, a periplasmic cysteine protease that can proteolytically process LapA, removing the globular N terminus and releasing LapA from the cell surface, and LapD, an inner membrane-localized GGDEF/EAL-containing, cyclic di-GMP (c-di-GMP)-responsive effector protein that binds LapG in response to cytoplasmic c-di-GMP, preventing LapG’s access to the N terminus of LapA (25, 28, 30–34). Together, LapD and LapG function to transduce cytoplasmic c-di-GMP signals across the inner membrane and determine whether LapA is retained at the cell surface or not (28, 34, 35). While the biofilms formed by some lab strains and environmental isolates of P. fluorescens have been shown to contain appreciable levels of polysaccharides (36, 37), in the best studied lab strain, Pf0-1, P. fluorescens polysaccharides have not previously been shown to play an appreciable role in biofilm formation. Instead, LapA has been shown to be the key determinant of biofilm formation (23, 28, 31). The simplicity of the use of a single adhesin protein to determine surface attachment and biofilm formation has made P. fluorescens an attractive model system for the study of biofilm formation and its regulation by c-di-GMP.
In this study, we demonstrate that biofilm formation in P. fluorescens is not determined solely by the action of LapA but instead by the action of two, related proteins, LapA and the herein described medium adhesion protein A (MapA). We show that while these two proteins share much of the regulatory machinery controlling cell surface localization, the differences in transcriptional regulation, their differential expression within mature biofilms, and their distinct sizes and sequences suggest that these two large adhesins play different roles in the development of a mature biofilm by P. fluorescens. We also demonstrate that LapA- and MapA-like proteins can be found in various combinations in the Gammaproteobacteria.
RESULTS
LapA is not necessary for the formation of a biofilm under all medium conditions.
Previous work characterizing the role of the adhesive protein LapA in biofilm formation had suggested that biofilm formation by P. fluorescens Pf0-1 is dependent on the function of LapA (23, 25, 28, 29). However, recent work by Smith et al. (29) indicated that P. fluorescens Pf0-1 encodes a second putative large adhesin. LapA is approximately 520 kDa, while this second adhesin, encoded by the Pfl01_1463 gene, is predicted to be approximately 300 kDa. We therefore named this new putative adhesin MapA (medium adhesion protein A), since the data shown below support a role for MapA as a biofilm adhesin.
We first assessed biofilm formation by mutants lacking one or both of the lapA and mapA genes in K10-T medium, a high-phosphate medium containing glycerol and tryptone as carbon and nitrogen sources that is commonly used in studies of P. fluorescens Pf0-1 biofilm formation. After 6 h of growth in this medium, the ΔmapA mutant formed the same amount of biofilm as that of wild-type (WT) Pf0-1, while the ΔlapA mutant formed no biofilm, similarly to the ΔlapA ΔmapA double mutant (Fig. 1A). This pattern was also observed after 16 h of growth in K10-T medium (Fig. 1B and E).
FIG 1.

Biofilm formation by adhesin mutants. (A to D) Quantification of biofilm formed by WT P. fluorescens Pf0-1, ΔlapA and ΔmapA single mutants, and the ΔlapA ΔmapA double mutant after 6 h of growth in K10-T medium (A), 16 h of growth in K10-T medium (B), 6 h of growth in KA medium (C), and 16 h of growth in KA medium (D). For statistical tests, hypothesis testing was conducted in GraphPad Prism 8 using two-tailed t tests. P values are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; nonsignificant (ns), P > 0.05. (E) Representative image of biofilm formed by WT P. fluorescens Pf0-1 and ΔlapA, ΔmapA, and ΔlapA ΔmapA mutants after 16 h of growth in the indicated medium and stained with 0.1% (wt/vol) crystal violet.
In order to find a role for MapA in biofilm formation, we sought to assess the biofilm formed by a ΔlapA or ΔmapA single mutant or a ΔlapA ΔmapA double mutant under a growth condition that fosters robust biofilms. Arginine has been shown to strongly promote biofilm formation by Pseudomonas species, likely through stimulation of the production of c-di-GMP (38–41). In addition, arginine is commonly found in root exudates and is therefore likely encountered by P. fluorescens when growing in the soil (42–44). After trying several medium formulations (data not shown), we found that the condition under which a ΔlapA mutant is most capable of biofilm formation is a modification of K10-T medium in which no glycerol or tryptone is added but rather it is supplemented with 0.4% (wt/vol) l-arginine HCl (here referred to as KA medium), as described in Materials and Methods. Of note, we have shown previously that growth on arginine increases the level of cellular c-di-GMP and stimulates biofilm formation by Pseudomonas (39, 41), perhaps accounting for our ability to observe an impact of MapA on biofilm formation in this medium formulation.
When grown in KA medium, WT P. fluorescens Pf0-1 can form a biofilm by 6 h, although less well than it can in K10-T medium (Fig. 1C). After 6 h of growth, the ΔlapA mutant has a significantly higher level of biomass attached to the well than the ΔlapA ΔmapA mutant, although the ΔmapA single mutant shows no reduction in biofilm compared to the WT. When cells were allowed to grow for 16 h in KA medium, the relative deficit in biofilm formation by the ΔlapA mutant was reduced, while the significant deficit of the ΔlapA ΔmapA mutant was still observed (Fig. 1D and E). In addition, the biofilm formed by the ΔmapA mutant in KA medium after 16 h of growth was slightly (but not significantly) lower than that of the WT (P = 0.09, unpaired two-tailed t test). Interestingly, when the ΔlapA ΔmapA mutant was grown in KA medium for 16 h, there was still modest diffuse staining observed in the well (Fig. 1E), perhaps indicating an additional factor contributing to biofilm formation under these conditions. These data indicate that MapA is capable of significantly contributing to biofilm under some medium conditions and is able to do so in the absence of LapA when grown in KA medium.
Genetic studies indicate that MapA localization is controlled by the regulatory proteins LapD and LapG.
Localization of LapA to the cell surface is dependent on the activity of the proteins LapD and LapG. The structure of the N-terminal portion of LapA is essential for its retention in the outer membrane pore LapE and thus for anchoring LapA to the bacterial cell surface (25, 29). In silico analysis of the N-terminal portion of LapA and MapA indicates a high degree of sequence and secondary structure similarity as well as the presence of the functionally important TAAG motif required for LapG processing (35) (Fig. 2A, red box).
FIG 2.

LapD and LapG contribute to MapA-dependent biofilm formation. (A) Alignment of LapA and MapA N-terminal portions generated using MUSCLE (65) and visualized using Jalview (85) with secondary structure prediction using JPred4 (83). Similar residues are indicated by color. Predicted secondary structure is indicated by colored shapes: green arrows, beta sheets; red bars, alpha helix. The LapG processing site TAAG is indicated by a red box. (B) Quantification of biofilm assay assessing biofilm formation of adhesin and regulatory mutants grown for 16 h in KA medium. (C) Quantification of biofilm assay assessing biofilm formation of adhesin and regulatory mutants grown for 16 h in K10-T medium. For statistical tests, hypothesis testing was conducted in GraphPad Prism 8 using two-tailed t tests with Holm-Sidak’s multiple comparison correction. P values are indicated by asterisks as follows: **, P < 0.01; ***, P < 0.001; ns, P > 0.05. In panel C, all asterisks denote P values of t tests between WT and the indicated mutant with Holm-Sidak’s multiple comparison correction.
Previous work demonstrated that the regulatory protease, LapG, is capable of proteolytically processing the N-terminal portion of MapA in vitro (29). To assess the regulatory role of LapD and LapG in the localization of MapA, we examined the level of biofilm formation of mutants lacking lapD or lapG when grown in KA medium (Fig. 2B). We found that while a ΔlapA mutant can form a biofilm in KA medium, the biofilm is significantly reduced in a ΔlapD mutant compared to the ΔlapA mutant, consistent with LapD being required to prevent constitutive processing of LapA and MapA by LapG. We also observed a reduction in the biofilm formed by the ΔlapAG ΔmapA mutant compared to the ΔlapAG mutant, indicating that MapA does contribute to the biofilm formed under these conditions. Interestingly, when cells were grown in KA medium (Fig. 2B), no significant difference in biofilm formation was observed between the WT and the ΔlapG mutant or between the ΔlapA mutant and the ΔlapAG mutant. These data suggest that while LapG is capable of processing both LapA and MapA, cells grown in KA medium have very little LapG activity, so the loss of LapG does not lead to an appreciable increase in biofilm formation, in contrast to cells grown in K10-T medium (Fig. 2C). Finally, when grown in KA medium, the ΔlapD mutant forms less biofilm than the ΔlapA ΔmapA mutant, although this difference is not statistically significant when the P value is adjusted to control the family-wise error rate. This observation may indicate the existence of an additional, unknown, LapD-regulated factor that modestly contributes to biofilm formation under these conditions, an observation consistent with the biofilm data shown in Fig. 1D and E. Taken together, these genetic data are consistent with the previous biochemical data (29), indicating that MapA is a substrate of LapG and its localization is likely regulated by LapD.
MapA is localized to the cell surface.
The sequence similarity between the N termini of LapA and MapA, the role for MapA in biofilm formation, the impact of loss of function in LapD on MapA-dependent biofilm formation, as shown here, and the published observation that the N terminus of MapA is processed by LapG (29) are all consistent with the conclusion that MapA is a cell surface-localized adhesin. To assess the cell surface localization of MapA, we built a hemagglutinin (HA)-tagged variant of MapA at the native locus on the chromosome, and this strain showed no difference in biofilm formation compared to the untagged WT strain when grown in KA medium (Fig. 3A, left); we similarly saw no impact of the HA tagging of MapA on the biofilm formed in the lapA mutant background (Fig. 3A, right).
FIG 3.
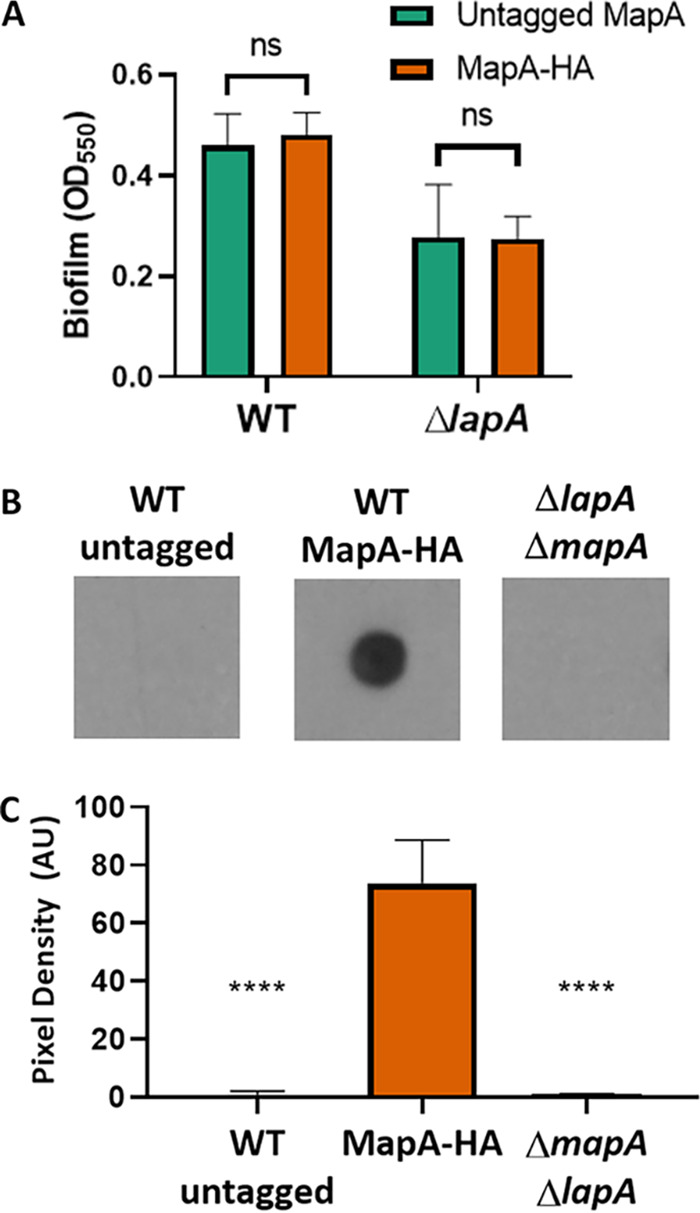
MapA is localized to the cell surface. (A) Quantification of crystal violet staining of wells of a 96-well plate after 16 h of growth in KA. Results for P. fluorescens Pf0-1 in both the untagged MapA and the tagged MapA variant bearing 3 tandem HA tags are shown. The phenotype is shown either in strains carrying a functional LapA or in a strain for which the lapA gene has been deleted. When compared using Student's t test, neither P. fluorescens Pf0-1 (P = 0.25) nor the ΔlapA mutant (P = 0.9) formed a significantly different level of biofilm when bearing an HA-tagged variant of MapA. (B) Representative dot blot of cell surface-localized MapA for the indicated strains. The WT (untagged strains) and a strain deleted for both the mapA and lapA genes serve as negative controls. (C) Three independent dot blot experiments were quantified using ImageJ as follows. The pixel values for the blots were inverted for ease of plotting. The region of interest (ROI) was defined for the control LapA dot blot (not shown), and then the mean gray value was determined for each spot using the predefined ROI. The background was subtracted from each spot; the background was determined by measuring the mean gray value in a section of the blot not containing any samples. The adjusted mean pixel density was plotted here for three biological replicates. There was a significant reduction in signal for both control strains compared to the strain expressing MapA-HA as assessed by t test. ****, P < 0.001; ns, P > 0.05.
We next performed dot blot studies that specifically assess the MapA associated with the bacterial cell surface. In these studies, cells are harvested by centrifugation, washed, spotted on a nitrocellulose filter, and then probed with an antibody to the HA-tagged variant to MapA. As shown in Fig. 3B and C, while no signal is detected for the untagged WT strain (left), a strong signal is detected for the strain carrying the HA-tagged MapA (center), and as an additional negative control, this signal is absent from a strain in which both the lapA and mapA genes are deleted (right). These data are consistent with MapA, like LapA, being cell surface localized.
Genetic evidence that components of the T1SS of LapA may be promiscuous.
Homologs of LapA are commonly encoded adjacent to their cognate type 1 secretion system (T1SS) (23, 45). This is also true for MapA. The mapA gene is adjacent to three genes encoding a putative T1SS (Fig. 4). We have named these genes mapB, mapC, and mapE, encoding a predicted inner membrane-spanning ABC transporter, a membrane fusion protein, and an outer membrane pore protein, respectively. Given that TolC, a homolog of LapE and MapE, is capable of functioning in association with multiple different ABC transporters and membrane fusion proteins (46–50) and the sequence similarity between these proteins (both LapE and MapE share roughly 35% similarity with TolC), we next sought to assess the extent to which LapA and MapA might share the components of their secretion systems. To investigate the ability of LapA and MapA to be secreted through either secretion machinery, we assessed the level of biofilm formed by mutants lacking combinations of adhesin and secretion machinery genes grown for 16 h in KA.
FIG 4.

Schematic of the lap and map loci. The organization of the genes encoding the LapA and MapA adhesins and their secretion machinery is shown. Gene names are indicated above the arrows, and the P. fluorescens Pfl0-1 gene numbers are indicated below the arrows. Arrows indicate the location and direction of transcription of the ORFs. Arrow direction indicates the orientation of the gene in the genome.
The biofilms produced by the ΔlapA, ΔmapA, and ΔmapE mutants were not different from one another but were significantly reduced compared to that produced by the WT (Fig. 5A). The biofilm formed by the ΔlapA ΔmapE mutant was reduced compared to those produced by both the ΔlapA and ΔmapE mutants but was not as low as that produced by the ΔlapA ΔmapA or ΔlapE ΔmapE mutants. This intermediate degree of biofilm formation indicates that when LapA and MapE are absent, MapA is likely able to be secreted, at least in part, via some portion of the LapBCE T1SS and thereby contribute to biofilm formation.
FIG 5.

Genetic evidence that MapA can utilize the Lap secretion system. (A) Biofilm formation by strains in KA medium carrying mutations in adhesin genes and genes encoding the Map secretion system outer membrane pore component. (B) Biofilm formation in KA medium for strains carrying mutations in adhesin genes and genes encoding the Lap secretion system outer membrane pore component. For statistical tests, hypothesis testing was conducted in GraphPad Prism 8 using two-tailed t tests with Holm-Sidak’s multiple comparison correction. P values are indicated by asterisks as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, P > 0.05.
Next, we assessed the ability of LapA to be secreted in the absence of LapE (Fig. 5B). The ΔlapE ΔmapA mutant formed a biofilm that was not different from that formed by the ΔlapE ΔmapE mutant. This observation suggests that while MapA may utilize some portion of the LapBCE T1SS, LapA is likely not secreted through the MapBCE T1SS. Additionally, it was observed that deletion of the lapE gene resulted in a greater decrease in the biofilm formed than the deletion of the lapA gene. This supports a model in which MapA is capable of being secreted through both secretion systems and is therefore able to partially suppress the impact of the loss of LapA on biofilm formation. Alternatively, the differential expression of LapA and MapA may also explain the inability of LapA to use the Map secretion machinery.
LapA and MapA are both required for the typical three-dimensional structure of the biofilm.
We next sought to elucidate the functional significance of encoding two large adhesins in the context of the formation of a mature biofilm using a flow cell system. After 96 h of growth in a modified version of the previously described BMM medium (51) (see Materials and Methods) in the microfluidic chamber, the WT forms a thick biofilm that fills most of the flow cell chamber (Fig. 6A). The ΔlapA mutant is severely deficient in biofilm formation, and only small clumps of cells are observed attached to the surface (Fig. 6B), as reported previously (23). The ΔmapA mutant appears able to attach to the surface of the chamber, but the thickness of the biofilm is reduced compared to that of the WT (Fig. 6C). Finally, the ΔlapA ΔmapA mutant results in a biofilm comprised of sparsely distributed clumps of cells (Fig. 6D). Together, these clear deficiencies in biofilm structure support a role for both adhesins in the formation of the biofilm.
FIG 6.
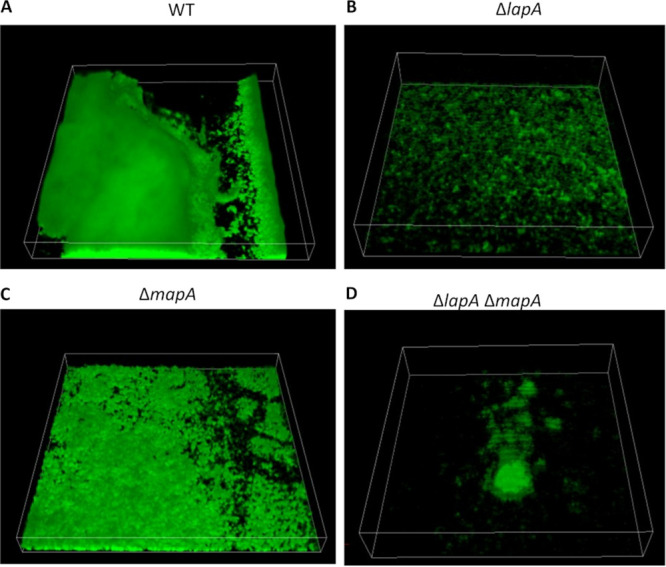
Confocal spinning-disk microscopy images of biofilm formed by adhesin mutants in flow cells. The indicated strains were grown as described in Materials and Methods and incubated at room temperature under constant flow for 96 h before imaging. Shown are representative images taken from one of two biological replicates. (A) The WT forms a thick biofilm that fills most of the flow cell chamber. (B) The ΔlapA mutant is severely deficient in biofilm formation, and only small clumps of cells are observed attached to the surface. (C) The ΔmapA mutant appears able to attach to the surface of the chamber, but the thickness of the biofilm is reduced compared to that of WT. (D) The ΔlapA ΔmapA mutant results in a biofilm comprised of sparsely distributed clumps of cells.
The lapA and mapA genes are differentially expressed in a P. fluorescens Pf0-1 biofilm.
Given the differential impact on biofilm formation for the lapA and mapA mutants, we hypothesized that LapA and MapA may be produced in different regions of a biofilm. In order to test this hypothesis, we constructed a reporter strain in which the promoter of each adhesin is driving the expression of a different fluorescent protein at a neutral site on the chromosome, while the native loci remain unchanged and thus functional. In this strain, the promoter of the lapA gene drives expression of green fluorescent protein (GFP) while the promoter of the mapA gene drives production of mRuby. Thus, this strain produces LapA and MapA from their native loci, while the GFP and mRuby constructs function as reporters of the activity of the lapA and mapA promoters, respectively. Figure 7A depicts a graphical representation of this adhesin reporter construct.
FIG 7.

Differential expression of lapA and mapA genes in a biofilm. (A) Schematic of adhesin reporter construction indicating the promoter and the gene encoding the fluorescent protein gene. PlapA is driving the expression of the green fluorescent protein gene, and PmapA is driving the expression of the mRuby gene. Both fluorescent protein genes have a transcriptional terminator of the rrnB gene from E. coli downstream of the genes to prevent transcriptional readthrough. (B) Confocal image of a biofilm grown in a microfluidic device. The image of a 96-h biofilm was taken with a 40× oil immersion lens objective. Green is the GFP channel excited by a 488-nm laser and indicates lapA-expressing cells. Magenta is the mRuby channel excited by a 560-nm laser and indicates mapA-expressing cells. Shown is a representative image taken from one of two biological replicates.
We grew this adhesin reporter strain in microfluidic devices under constant flow in a variant of the KA medium (see Materials and Methods for details) for 96 h and then imaged the biofilms formed by spinning-disk confocal microscopy to assess expression of the lapA or mapA reporters (Fig. 7B). Figure 7B shows a typical field of view in which a thick region of biofilm can be seen spanning from the bottom to the top of the center of the field of view, while the regions on either side of this central area of dense biofilm are visibly more porous and thinner. We found that only lapA is expressed in the areas where the biofilm is thin or less dense (Fig. 7B, green, right portion of the field of view), whereas robust lapA and mapA expression could both be detected in regions where the biofilm is thicker and denser (Fig. 7B, magenta). The expression of the mapA gene is localized to this dense biofilm region and is not observed in the more porous regions of the biofilm. In addition, mapA expression seemed to be localized to the region closest to the point of attachment to the glass cover slip, as shown by the side projections on the right and the bottom of Fig. 7B (arrow).
The observations that LapA and MapA seem to be capable of supporting biofilm formation under different growth conditions and are both required for proper three-dimensional biofilm structure support a model wherein these two adhesins play different roles during biofilm formation. However, it is not clear from these data whether those different roles are due solely to differences in transcriptional regulation and that LapA and MapA are otherwise functionally redundant or, alternatively, that the proteins have distinct functions/roles in the context of biofilm formation due to their distinct amino acid sequences. To distinguish between these possibilities, we created a strain in which mapA expression is placed under the control of the lapA promoter, referred to as Pswap. The region described here as the lapA promoter is defined as spanning from the base adjacent to the lapE open reading frame (ORF) to the base adjacent to the lapA ORF (Fig. 4). The region described as the mapA promoter region is the equivalent region spanning from the base adjacent to the mapE ORF to the base adjacent to the mapA ORF (Fig. 4). We reasoned that this construct would lead to both mapA and the mapBCE genes encoding its cognate T1SS being transcriptionally regulated in a fashion similar to that of lapA and its cognate T1SS. We hypothesized that LapA and MapA function differently due to their different amino acid sequences and that, therefore, placing mapA under the transcriptional control of the lapA promoter would not rescue the biofilm defect of the ΔlapA mutant.
We tested biofilm formation of the WT and the ΔlapA, Pswap, and ΔlapA Pswap mutants when grown in KA medium for 6 and 16 h (Fig. 8A and B). If MapA were functionally equivalent to LapA, then we would expect the ΔlapA Pswap mutant to behave like a complemented lapA mutant and form approximately WT levels of biofilm. Instead, we observed no increase in biofilm formation between the ΔlapA and ΔlapA Pswap mutants at either time point (Fig. 8A and B).
FIG 8.

Expressing mapA under the control of the lapA promoter does not rescue loss of LapA function. (A and B) Quantification of biofilm formed by WT P. fluorescens Pf0-1 and ΔlapA, Pswap, and ΔlapA Pswap mutants after 6 h of growth in KA medium (A) or 16 h of growth in KA medium (B). (C) Cell surface expression of MapA in the WT and Pswap strains. The cell surface expression of LapA is shown as an additional control. Shown is one representative experiment of three biological replicates. (D) Quantification of the cell surface expression of HA-MapA in the WT and Pswap strains and of HA-LapA in the WT strain, as indicated. Quantification was performed as described in the legend for Fig. 3. For statistical tests, all hypothesis testing was conducted using a two-tailed t test. *, P < 0.05; ns, P > 0.05.
To confirm that MapA was expressed and, more importantly, surface localized in the ΔlapA Pswap strain, we performed a dot blot experiment as described above. We found that the ΔlapA Pswap strain did indeed yield cell surface HA-MapA at a level that was not significantly different from the level of MapA produced by the WT strain (Fig. 8C and D, compare left and center of panel C). As an additional positive control, we also blotted for the cell surface levels of LapA in a strain carrying the HA-LapA-expressing construct (Fig. 8C, right). Given that we observed no change in biofilm formation between the lapA and ΔlapA Pswap mutants (Fig. 8A and B) and that the level of MapA in the Pswap strain was ∼75% that of the WT strain but not significantly different, the simplest explanation for these observations is that MapA cannot functionally substitute for LapA.
Lap-like systems are broadly distributed throughout the Proteobacteria.
The use of multiple adhesins in biofilm formation has been described in other organisms, for example, BapA and SiiE in Salmonella enterica (52–54) and LapA and LapF in Pseudomonas putida (45, 55–57). We were therefore interested in assessing how widespread this phenomenon may be throughout the bacterial domain in the context of LapA-like adhesins. To assess the distribution of these proteins, we modified a previously described approach to identify LapD and LapG homologs in other organisms and then examined those genomes for evidence of RTX adhesins that may be targets of the LapG homolog in the same organism (29). (See Materials and Methods for a description of this approach. All code and the raw data used in this analysis are available at https://github.com/GeiselBiofilm/Collins-MapA.)
Using this approach, we identified 3,740 unique species encoding both LapD and LapG homologs. We next retrieved all available complete, annotated protein coding sequences for organisms of LapD- and LapG-encoding organisms that are available in the NCBI GenBank database. The phylogenetic class classification of these organisms is indicated in Table 1. The vast majority of these organisms whose genome encodes a LapA-like protein are members of the Proteobacteria, with most of those being members of the Gammaproteobacteria. The observation that we identified more organisms that encode putative LapD and LapG homologs in the Gammaproteobacteria than in other Proteobacteria is not necessarily indicative of an enrichment of these proteins in that class, as only 12% of Gammaproteobacteria represented in the GenBank database were found to encode LapD and LapG homologs. Indeed, 53% of the Zetaproteobacteria and 42% of the Hydrogenophilalia were found to encode both LapD and LapG, indicating that this system may be more highly represented in classes other than the Gammaproteobacteria. However, we identified almost no LapD and LapG homologs outside the Proteobacteria (Table 1), indicating that these proteins may be largely restricted to this phylum.
TABLE 1.
Number of genomes found to encode both putative LapD and LapG homologs in each of the given taxa
| Taxon | No. of genomes encoding predicted LapD and LapG homologs/total no. of genomes (%) |
|---|---|
| Gammaproteobacteria | 2,779/23,115 (12) |
| Betaproteobacteria | 435/16,142 (2.7) |
| Alphaproteobacteria | 301/12,355 (2.4) |
| Deltaproteobacteria/Epsilonproteobacteria | 173/11,567 (1.5) |
| Zetaproteobacteria | 23/43 (53) |
| Hydrogenophilalia | 10/24 (42) |
| Deferribacteres | 4/7,688 (0.05) |
| Bacteroidetes/Chlorobi group | 1/499 (0.2) |
| Nitrospira | 1/406 (0.2) |
Having collected a list of LapD- and LapG-encoding organisms, we next retrieved amino acid sequences for all annotated genes in these organisms and searched these sequences for features that identify them as putative LapA-like proteins (see Materials and Methods for details). Briefly, the criteria for this characterization are the following: (i) having a size in excess of 1,000 amino acids, (ii) the presence of putative RTX repeats in the C-terminal portion of the protein, and (iii) the presence of a putative LapG cleavage site in the N-terminal portion of the protein. Through this search, we identified 1,020 organisms encoding predicted LapA-like proteins in addition to LapG and LapD homologs (see File S1 in the supplemental material). Through this approach, we were able to identify LapA-like proteins that have been previously described (55, 58–60), supporting the utility of this approach. Among organisms found to encode LapA-like proteins, approximately one-quarter were found to encode multiple LapA-like adhesins (Table 2). Interestingly, while many LapD- and LapG-encoding organisms encoded proteins that could be identified as LapA-like proteins, 2,720 organisms in our data set were found to encode LapD and LapG homologs but were not found to encode proteins that we could readily identify as LapA-like by use of our criteria, which could be due to the lack of such proteins or the difficulties in annotation of such large adhesins.
TABLE 2.
Number of putative LapA-like adhesins predicted to be encoded in genomes in which putative LapD and LapG homologs were identified
| No. of adhesins | No. of organisms |
|---|---|
| 1 | 352 |
| 2 | 109 |
| 3 | 20 |
| 4 | 3 |
Pseudomonads encode LapA-like proteins with a distinct size distribution.
RTX adhesins are typically large and are often the largest proteins produced by the bacteria in which they are found. Most of the length of these proteins is composed of tandem repeats. These repeats have in some cases been described to function as extender domains which hold the adhesive C-terminal domains far away from the cell surface (61, 62). Thus, while the functions of the various domains of LapA-like proteins are still in most cases unclear, the protein length may be an important characteristic. We therefore sought to assess the distribution of LapA-like protein sizes found within the three most highly represented genera in our data set, Vibrio, Shewanella, and Pseudomonas (Fig. 9). By plotting the distribution of LapA-like proteins, we can assess the sizes of adhesins encoded by organisms containing one or more such proteins.
FIG 9.
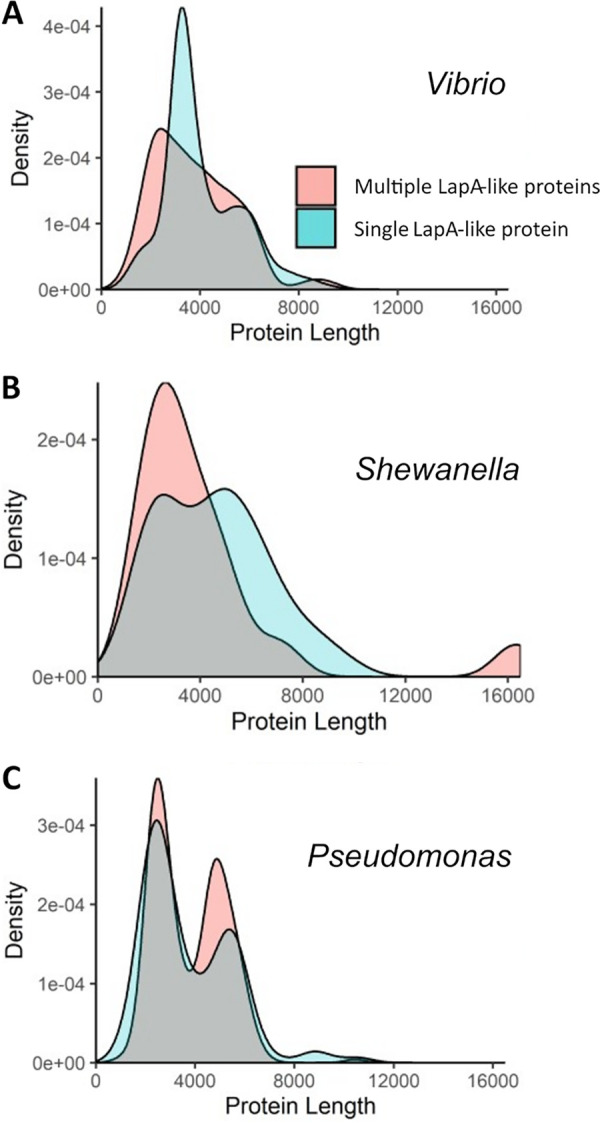
Size distribution of LapA-like proteins encoded by selected genera. All organisms found to encode both LapD and LapG and putative LapA-like proteins were grouped based on whether they are predicted to encode a single or multiple LapA-like proteins. We then assessed the distribution of the protein sizes of adhesins encoded by organisms with a single (blue shading) or multiple adhesins (orange shading). Shown are density plots representing the size distributions of LapA-like proteins found in the following genera: Vibrio (A), Shewanella (B), and Pseudomonas (C).
LapA-like proteins encoded by Vibrio species with just one such protein exhibit a striking peak at a length of around 3,000 amino acids and a shoulder at around 5,000 amino acids (Fig. 9A). The major peak of organisms encoding a single LapA-like protein corresponds to a large number of V. cholerae isolates, including V. cholerae O1 biovar El Tor, that encode an almost identical adhesin, while the shoulder at around 5,000 amino acids corresponds to a variety of different species. Interestingly, there is some diversity in the number and size of LapA-like proteins encoded by V. cholerae isolates, with some encoding two proteins and others just one such protein. In addition, there appear to be several distinct LapA-like variants encoded among isolates of V. cholerae. Some of these variant proteins appear to share very little sequence similarity to one another, while others appear to be mostly identical to another variant, differing mostly due to a large indel. For example, the LapA-like protein encoded by the V. cholerae O1 biovar El Tor is highly similar to a LapA-like protein encoded by V. cholerae TMA 21, but the El Tor protein is ∼880 amino acids shorter. How these putative LapA-like proteins differ functionally from one another is unclear. However, the variation seen within the V. cholerae species may indicate that this protein is important for adaptation to different lifestyles of these organisms.
Within the genus Shewanella, no one species is represented dramatically more than any other in our data set. Accordingly, we do not see a dramatic peak in the protein size distribution corresponding to the adhesin encoded by a single species. Most of the species within this genus encode proteins that differ from those of other species both in size and at the sequence level, leading to a smooth curve when the size distribution is plotted (Fig. 9B). Interestingly, the largest LapA-like protein in our data set is encoded by Shewanella woodyi ATCC 51908 by the gene Swoo_0477. This protein is predicted to be 16,322 amino acids, which is over 5,000 amino acids longer than the next longest protein we identified as a LapA-like protein. While we are not aware of any experimental evidence to support the enormous size of this protein, it is nonetheless to our knowledge the largest predicted LapA-like protein.
The distribution of protein sizes observed within the Pseudomonas genus has a bimodal distribution, with peaks around 3,000 and 5,000 amino acids, the approximate sizes of MapA and LapA, respectively, among both organisms that encode a single and multiple LapA-like proteins (Fig. 9C). There are many isolates in our data set that have not been assigned a species designation and which encode proteins with sizes approximately equal to that of LapA and MapA. These isolates may represent oversampling of a small number of species that could exaggerate the observed distribution. However, there are also a considerable number of Pseudomonas isolates with species designations that exhibit this pattern.
Pseudomonas species encoding multiple LapA-like proteins are mostly P. fluorescens lineage members.
Having identified which Pseudomonas species encode LapD, LapG, and LapA-like proteins, we next sought to assess the phylogenetic distribution of these proteins within the genus. We therefore constructed a phylogenetic tree of the Pseudomonas species that are represented in the GenBank database (Fig. 10). The phylogenetic tree presented here has a topology similar to that of trees generated through the same approach in previous studies (63, 64) (see Fig. 10 legend and Materials and Methods for details).
FIG 10.

Phylogenetic distribution of the Lap proteins among members of the Pseudomonas genus. Shown is a phylogenetic tree of Pseudomonas lineages. The innermost colored ring around the tree indicates whether LapD and LapG are encoded by the indicated species (as determined by finding the intersection of the species lists retrieved from the NCBI Conserved Domain Database for pfam06035 and pfam16448 domains) and how many LapA-like proteins were identified in that species using the analysis described in Materials and Methods. The outermost three rings indicate the percent similarity between the LapA-like proteins identified in each species and either LapA or MapA. Amino acid sequences of putative LapA-like proteins were aligned with LapA and MapA in pairwise alignments using MUSCLE (65) (https://github.com/GeiselBiofilm/Collins-MapA/tree/master/Supplemental_File_Alignments), and the percent sequence similarity was determined by counting the positions identified as similar by the MUSCLE program. The percent similarity of whichever was most similar (either LapA or MapA) is indicated, with darker color indicating higher similarity. The order of the three rings indicating similarity with LapA and MapA is organized by the relative size of the LapA-like proteins encoded by each organism. The innermost of the three rings is the largest LapA-like protein encoded by that organism. The second and third rings represent alignments of the second- and third-largest LapA-like proteins, respectively, identified in organisms that are predicted to encode multiple LapA-like proteins. The multilocus sequence analysis (MLSA) phylogenetic tree here shows the clustering of representatives Pseudomonas species found in the GenBank database based on the analysis of concatenated alignments of 16S rRNA, gyrB, rpoB, and rpoD genes. Distance was calculated using the Jukes-Cantor model, and the tree was constructed using neighbor joining. The P. aeruginosa, P. fluorescens, and P. pertucinogena lineages described by Mulet et al. (64) are highlighted with the indicated colors. The namesake species of the groups described by García-Valdés and Lalucat (86) are in bold font. The P. aeruginosa and P. fluorescens lineages described by Mulet et al. (64) and the P. pertucinogena lineage described by Peix et al. (63) are also apparent in our tree and are indicated using the indicated shading (colored according to the lineages described by Peix et al. [63]). The most notable difference between the tree presented here and previously reported trees is the presence of a distinct P. luteola and P. duriflava clade, while Peix et al. (63) presented them as part of the monophyletic P. aeruginosa lineage. However, the placement of P. luteola within the P. aeruginosa lineage is not strongly supported. The study by Mulet et al. (64) placed P. luteola outside the P. aeruginosa lineage, and the bootstrap support for Peix et al. (63) positioning the P. luteola and P. duriflava clade within the P. aeruginosa lineage is less than 50%. Three other species were not present in previously published phylogenies of Pseudomonas and do not fit neatly into the three lineages described previously: P. pohangensis, P. hussainii, and P. acidophila. Both P. pohangensis and P. hussainii are positioned between the P. pertucinogena lineage and the other two lineages. P. acidophila is positioned outside the rest of the genus, consistent with a recent study that argued that P. acidophila should be reclassified as Paraburkholderia acidophila (87). While the P. pertucinogena lineage described by Peix et al. (63) is well supported by the tree we present here, the species P. pertucinogena is absent from our tree. This is because we included only species present in the GenBank database in our tree. At the time of writing, there is an available genome sequence record of P. pertucinogena (BioProject accession no. PRJNA235123), but the absence of an assembled genome from GenBank means that the organism was not included in our analysis of the distribution of LapA-like proteins. Nevertheless, the tree topology is similar overall to the previously reported trees outlined above.
Through our analysis, we identified LapD and LapG homologs throughout the P. aeruginosa and P. fluorescens lineages of the Pseudomonas genus (Fig. 10, innermost ring). However, LapD and LapG homologs were identified in only 2 of the 17 (11.7%) representatives of the Pseudomonas pertucinogena lineage, and only one of those was found to encode a LapA-like protein. Within the P. aeruginosa and P. fluorescens lineages, roughly the same proportions of species were found to encode LapD and LapG (56% and 59%, respectively). In addition, the proportions found to encode both LapD and LapG but no detectable LapA-like protein were also similar between the two lineages, with 9 of 50 species (18%) in the P. aeruginosa lineage and 26 of 147 species (17.7%) in the P. fluorescens lineage. Interestingly, these two lineages differ dramatically in number of adhesins detected in species that were found to encode LapD, LapG, and a LapA-like protein. Within the P. aeruginosa lineage, the number of species encoding a single adhesin is 15 (30%) and the number encoding more than one is 4 (8%). Within the P. fluorescens lineage, the number encoding a single adhesin is 32 (21%) and the number encoding multiple adhesins is 29 (20%). It is therefore clear that species encoding multiple LapA-like proteins are mostly found within the P. fluorescens lineage.
While species encoding LapD, LapG, and LapA-like proteins are found across both the P. aeruginosa and the P. fluorescens lineages, the distribution of these proteins within those lineages is not uniform. There are multiple clades within each lineage that seem to lack identifiable homologs for any of the three proteins. For example, within the P. aeruginosa lineage, the clade containing P. luteola and P. duriflava and the clade containing P. oryzihabitans are predominated by species that do not encode identifiable Lap proteins. Within the P. fluorescens lineage, the clade containing P. syringae is also predominated by species lacking identifiable Lap proteins. Interestingly, while species in the clade containing P. syringae mostly lack Lap proteins, P. syringae encodes both LapD and LapG as well as two identifiable LapA-like proteins.
LapA and MapA share high levels of sequence similarity with LapA-like proteins encoded by other P. fluorescens lineage members.
Many members of the P. fluorescens lineage encode multiple LapA-like proteins, and the distribution of sizes of these proteins is similar to the sizes of LapA and MapA. Therefore, we hypothesized that the LapA-like proteins encoded by Pseudomonas species in our data set are similar to LapA and MapA, while those encoded by organisms from other genera are less similar to LapA and MapA. To assess this idea, we conducted pairwise alignments of the amino acid sequences of every LapA-like protein that we identified in our analysis with both LapA and MapA using MUSCLE v3.8 (65) (see File S2 in the supplemental material and https://github.com/GeiselBiofilm/Collins-MapA/tree/master/Supplemental_File_Alignments).
We found that within the Pseudomonas genus, many organisms encode adhesins that are highly similar to LapA and MapA (Fig. 10, rings of green and magenta squares). For example, P. syringae encodes two LapA-like proteins: the larger of the two is 87% identical and 90% similar to LapA, while the smaller of the two is 97% identical and 99% similar to MapA (Fig. 10; File S2). Interestingly, most of the organisms within the P. fluorescens lineage that encode multiple adhesins were found to encode proteins with very high similarity to LapA and MapA. However, LapA-like proteins encoded by members of the P. aeruginosa lineage have relatively lower similarity to LapA and MapA proteins.
Outside the Pseudomonas genus, the occurrence of proteins with very high similarity to LapA or MapA is lower. However, many organisms do encode proteins with moderate similarity (over 50% across the entire length of the protein) to LapA or MapA. In some cases, the similarity between these proteins is mostly localized to the N- and C-terminal portions of the proteins that contain the anchor domain and RTX repeats, respectively, which are characteristic of LapA-like proteins (66, 67). In other cases, it appears that similarity between adhesins is localized to the central portion of the protein. For example, the LapA-like protein encoded by Aeromonas salmonicida has 38% amino acid identity (68% similarity) to MapA across the whole length of the protein. However, there is a highly similar region of these proteins located in the first half of their central portion (54% identity, 85% similarity between positions 393 to 1620), while the N- and C-terminal portions of the proteins have relatively low sequence identity (https://github.com/GeiselBiofilm/Collins-MapA/tree/master/Supplemental_File_Alignments). This pattern is in contrast to the typically high conservation of the N and C termini and the low conservation of the central portion of LapA-like proteins (66, 67).
DISCUSSION
In this paper, we present MapA, a second type 1-secreted, large RTX adhesin that contributes to biofilm formation by P. fluorescens. We demonstrate that MapA is surface localized and can contribute to biofilm formation in the absence of LapA, at least under one growth condition we have identified to date. We also present genetic data showing that MapA surface localization is likely controlled by the previously described c-di-GMP effector protein, LapD, and the periplasmic protease, LapG (see model in Fig. 11A).
FIG 11.

Model of the mechanisms by which LapA and MapA contribute to biofilm formation. (A) Model for c-di-GMP regulation of cell surface localization of LapA and MapA. Representations of the LapA (left) and MapA (right) adhesins anchored in the outer membrane via LapE and MapE, respectively, are shown. Also shown is the periplasmic protease LapG and the inner membrane, c-di-GMP receptor LapD, which regulates LapG activity. (B) Representation of the expression of lapA and mapA in a biofilm. Indicated are possible roles for nutrient and/or oxygen limitation in controlling the expression of the mapA gene.
Our bioinformatic analysis indicates the distribution of similar multiple-adhesin systems throughout the Proteobacteria. Through our analysis, we identified many LapD and LapG homolog-encoding organisms whose genomes code for differing numbers of putative LapA-like proteins. However, we also identified many organisms that seem to encode LapD and LapG homologs but no identifiable LapA. Even within the Pseudomonas genus, we identified multiple species that seemed to lack any identifiable LapA- or MapA-like proteins (Fig. 10). It is possible that LapA-like proteins are present in these organisms and that our approach was not able to identify them or that the protein sequence database that we used as the source of our data is incomplete due to misannotation of these large, often repeat-rich proteins. In addition, the search parameters we used to identify LapA-like proteins are based on the characterization of a small number of proteins characterized to date (67), so it is possible that we have designed our search in such a way that some authentic LapA-like proteins were excluded. However, in the case of P. aeruginosa, a LapD and a LapG homolog are present but no LapA-like protein exists. Instead, LapG is able to proteolytically process a noncanonical target CdrA and regulate its localization to the cell surface (68). CdrA is secreted through a type Vb secretion process and is seemingly evolutionarily unrelated to LapA. Therefore, we hypothesize that some of the organisms we have identified that lack LapA-like proteins may encode proteins that have convergently evolved to be regulated by LapG. Thus, our analysis may provide a good basis on which to identify organisms that may encode other novel LapG-regulated proteins.
Interestingly, while both the P. aeruginosa and P. fluorescens lineages contain many organisms encoding LapD, LapG, and LapA-like proteins, the P. pertucinogena lineage contains only two such species. The presence of LapD and LapG homologs in organisms outside the Pseudomonas genus suggest that these proteins were lost in the P. pertucinogena lineage rather than gained in the other Pseudomonas lineages. P. pertucinogena lineage members are thought to be more niche specialized than other Pseudomonas species and have reduced genome size and metabolic versatility (69–72). Thus, it is possible that the genome reduction that is thought to have occurred in the P. pertucinogena lineage is responsible for the apparent absence of LapD, LapG, and LapA-like proteins in these organisms.
Through our analysis of the sequence similarity between LapA-like proteins, we have found that many organisms both within the Pseudomonas genus and in other genera encode LapA-like proteins with high sequence similarity to LapA or MapA. Within the Pseudomonas genus, these LapA-like proteins often have high sequence similarity to LapA and MapA across the whole length of the protein, particularly within the P. fluorescens lineage. Organisms from other genera were found to encode LapA-like proteins with lower sequence similarity with LapA and MapA over the whole length of the protein. However, in some cases, portions of the central region of these proteins appear to be highly identical. The precise function of the diverse repeated sequences found in LapA-like proteins is poorly understood (67). In some cases, an extender-like function has been described which positions functional or substrate-binding domains of the protein away from the cell surface (61, 62). However, in the case of LapA, the repeats are thought to directly bind to hydrophobic surfaces (26). It is therefore possible that LapA-like proteins with high identity over parts of their central portion contain conserved substrate-binding domains or bind to surfaces with similar physical characteristics.
How MapA contributes to biofilm formation is still not entirely clear, and complicating our analysis is the fact that our genetic studies indicate that MapA may utilize the Lap secretion system for surface localization. Loss of MapA function results in a defect in biofilm formation in static 96-well dish assays, at least under a select medium condition. Similarly, MapA was required for robust biofilm formation in a microfluidic device. The phenotype of the mapA mutants is also clearly distinct from that of strains lacking LapA. What is the basis for this difference in phenotype? One obvious explanation is that the LapA protein is ∼200 kDa larger than MapA, and there are clear differences in the sequence of the central repeat regions. Differential expression of the proteins might also impact their differential contributions to biofilm formation. For example, we observed differential spatial expression patterns of the mapA and lapA genes in biofilms grown in microfluidic chambers under flow. However, our studies using the Pswap construct, which allows mapA to be expressed via the lapA promoter, argue against this hypothesis. We note that mapA expression is seen only in large, thicker regions of biofilms, which presumably would be oxygen or nutrient limited. Thus, we postulate that oxygen, nutrients, or energy status of the cells may contribute to the regulation of mapA gene expression (Fig. 11B). This observation is consistent with the fact that loss of MapA shows a phenotype in static assays when only arginine is present but not when the medium contains a complex mixture of peptides and glycerol. Furthermore, given that arginine has been shown previously to stimulate the c-di-GMP level in pseudomonads (39, 41), the involvement of MapA in biofilm formation may also be a c-di-GMP-regulated process. Studies are ongoing to dissect the role of transcriptional regulation in the different roles of LapA and MapA in P. fluorescens biofilm formation.
Our findings here are reminiscent of previous studies with P. putida, which showed that this organism utilizes two adhesins during biofilm formation. The P. putida adhesins, LapA and LapF, are required for biofilm formation and play distinct roles in building the three-dimensional structure of the biofilm (6, 55–57). P. putida LapA is involved in initial attachment to the substratum, while LapF is required for the three-dimensional structure of the biofilm via contributing cellular cohesion (6, 45, 56, 57). It is possible that the LapA and MapA of P. fluorescens play similar roles. The lapA mutant of P. fluorescens has a reported defect in early biofilm formation (23, 25), and the data we present here show that MapA is likely produced only in the heart of a mature biofilm. Furthermore, the mapA mutant strain appears to initiate a biofilm, but that biofilm does not fully mature, a phenotype similar to that observed for the lapF mutant of P. putida (56). Interestingly, a key difference between the two strains is that while the P. putida LapA protein is thought to be processed by LapG, LapF was not identified by our search for LapA-like proteins as it does not contain key features required for LapG processing. Thus, LapF may be part of a distinct class of LapA-like adhesins that are secreted and permanently anchored to the cell surface, as LapF appears to not be processed by LapG. Interestingly, we showed previously that the LapA-like PA1874 protein of P. aeruginosa, which shares features of the N-terminal retention module with the LapA protein of P. fluorescens, is also not processed by LapG and lacks the canonical TAAG processing site for LapG (29). Thus, Lap-like proteins which are cell surface localized but lacking LapG processing sites might be another variation on the theme of Lap-like adhesion systems. Finally, in P. putida KT2440, the lapA and lapF genes are differentially regulated by FleQ, RpoS, GacS/GacA, and Fis (56, 73–76). Our observation that MapA appears to be produced at high levels only in the center of the biofilm, which is typically associated with a region of the biofilm lacking nutrients, and in medium that promotes high levels of c-di-GMP production suggests that the mapA gene may also be under the control of RpoS and FleQ, respectively. Future studies should explore the role of these transcription factors in controlling expression of the lapA and mapA genes in P. fluorescens.
Our analysis here suggests that Lap-like systems are conserved across the pseudomonads and in other Proteobacteria. The observation that some pseudomonads and other Proteobacteria can express two discrete LapA-like proteins adds to the complex strategies that these bacteria can utilize to establish their extracellular matrix during biofilm formation. In the case of P. fluorescens and P. putida, their two respective LapA-like proteins appear to play distinct roles in biofilm initiation versus biofilm formation, likely driven in part by their distinct protein sequences and their differential regulation. While exopolysaccharides and extracellular DNA have received the majority of attention as matrix components, the observation that organisms like P. fluorescens, P. putida, V. cholerae, and S. enterica all use multiple adhesins to form a biofilm indicates the importance of such extracellular proteins as matrix components.
MATERIALS AND METHODS
Strains and media.
The strains used in this study can be found in Table 3. P. fluorescens Pf0-1 and E. coli S17 (77, 78) were used throughout this study. P. fluorescens and E. coli were grown at 30°C when containing plasmids; E. coli strains were otherwise grown at 37°C. Media used in this study include lysogeny broth (LB), K10T-1 (77), and two new modifications of previously used media. The first, KA medium, is a modification of K10-T in which the changes are the removal of glycerol and tryptone and the addition of 0.4% (wt/vol) l-arginine HCl. The second medium was used for growing P. fluorescens Pf0-1 in microfluidic devices and is a modification of BMM medium that was previously described (51). This BMM medium was modified by reducing the concentration of glycerol and (NH4)2SO4 by 16-fold and adding l-arginine. The final concentrations of each in this medium were therefore 0.009375% (vol/vol) glycerol, 472.5 μM (NH4)2SO4, and 0.025% (wt/vol) l-arginine mono-HCl (Sigma). When E. coli was grown harboring the allelic exchange plasmid pMQ30 or the expression plasmid pMQ72, the growth medium was supplemented with 10 μg/μl gentamicin. When E. coli was grown harboring the plasmid pMQ56 or the helper plasmid pUX-BF13, the growth medium was supplemented with 50 μg/μl carbenicillin. When P. fluorescens was grown harboring pMQ72, the growth medium was supplemented with 30 μg/μl gentamicin. Allelic exchange was performed as previously reported (79).
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| Saccharomyces cerevisiae InvSc1 | MATa/MAT leu2/leu2 trp1-289/trp1-289 ura3-52/ura3-52 his3D1/his3D1 | Invitrogen |
| Escherichia coli S17-1 (λpir) | Tpr Smr recA thi pro hsdR-M+RP4::2-Tc::Mu::Km Tn7 λpir | 78 |
| Pseudomonas fluorescens Pf0-1 | ||
| SMC 1358 | WT Pf0-1 (both LapA and MapA untagged) | |
| SMC 4798 | LapA-HA; HA tag inserted after residue 4093 | 88 |
| SMC 8581 | MapA-HA | This study |
| SMC 7905 | LapA-HA MapA-HA | This study |
| SMC 8586 | ΔlapA MapA-HA | This study |
| SMC 7977 | ΔmapA LapA-HA | This study |
| SMC 8570 | ΔlapA ΔmapA | This study |
| SMC 8582 | ΔlapD LapA-HA MapA-HA | This study |
| SMC 8584 | ΔlapG LapA-HA MapA-HA | This study |
| SMC 8594 | ΔlapAG MapA-HA | This study |
| SMC 8569 | ΔlapAG ΔmapA | This study |
| SMC 8588 | ΔmapE LapA-HA MapA-HA | This study |
| SMC 8589 | ΔlapA ΔmapE MapA-HA | This study |
| SMC 8591 | ΔlapE ΔmapE LapA-HA | This study |
| SMC 7954 | ΔlapE LapA-HA | 29 |
| SMC 8636 | ΔlapE ΔmapA LapA-HA | This study |
| SMC 8638 | PmapA::PlapA (also known as Pswap) LapA-HA MapA-HA | This study |
| SMC 8639 | ΔlapA PmapA::PlapA MapA-HA | This study |
| SMC 8642 | mTn7 PlapA-GFP PmapA-mCherry LapA-HA MapA-HA (also known as Pfluor) | This study |
| Plasmids | ||
| SMC 7221 | mTn7/pMQ56 | 79 |
| SMC 1852 | pUX-BF13 tnsA tnsC tnsD tnsE mob ori R6K bla MCS | 89 |
| SMC 2765 | pMQ30 | 79 |
| SMC 7975 | pMQ30 ΔmapA | This study |
| SMC 7907 | pMQ30 MapA-HA | This study |
| SMC 8703 | pMQ30 ΔmapE | This study |
| SMC 8637 | pMQ30 PmapA::PmapA | This study |
| SMC 8635 | pMQ56 mTn7 plapA-GFP, pmapA-mCherry | This study |
Static biofilm assay.
Pseudomonas fluorescens strains were struck out on LB plates overnight. Single colonies were picked and grown overnight in liquid LB medium with shaking. A 1.5-μl volume of overnight culture was added to 100 μl of growth medium in a 96-well U-bottom polystyrene plate (Costar). Plates were covered and placed in a humidified chamber at 30°C for either 6 or 16 h, as indicated, at which time the liquid in the wells was discarded and the wells were stained with 125 μl of 0.1% (wt/vol) crystal violet (CV) at room temperature for 20 min and then rinsed two times with water. Wells were allowed to dry, and then the stain was dissolved using 150 μl of a solution of water, methanol, and acetic acid (45:45:10 ratio by volume) for 10 min at room temperature. A 100-μl volume of the solubilized CV solution was transferred to a flat-bottom 96-well plate, and the optical density at 550 nm (OD550) was recorded.
Construction of HA-tagged MapA.
The 3×HA tag was amplified by PCR from P. fluorescens Pf0-1 in which LapA had been HA tagged. This tag was inserted into the genomic copy of the Pfl01_1463 (mapA) gene between the codon encoding H2601 and G2602 by allelic exchange, as previously described (79).
Assessment of surface localization of adhesins by dot blot.
WT P. fluorescens Pf0-1 and mutants were grown with agitation in LB medium at 30°C for ∼16 h, and then 100 μl of this inoculum was added to 25 ml of KA medium. This subculture was incubated with agitation at 30°C for 24 h. After incubation, the 25-ml cultures were centrifuged at 5,000 rpm for 5 min in a microcentrifuge, and cell pellets were resuspended in 1 ml of fresh medium. All cultures were washed once with fresh medium and normalized such that the OD600 of each culture was the same. Five microliters of washed, normalized culture was spotted onto a nitrocellulose membrane and allowed to dry. The membrane was then incubated in blocking buffer for 1 h at 25°C (Tris-buffered saline [TBS] with Tween 20 [TBST] with 3% bovine serum albumin [BSA]; Sigma) before being probed for LapA or MapA containing an HA epitope tag with an anti-HA epitope tag antibody (BioLegend) at a 1:2,000 dilution in TBST with 3% BSA and 0.02% sodium azide overnight at 4°C. Excess antibody was washed away with 3× TBST rinses before the membrane was incubated with anti-mouse secondary antibody (Bio-Rad) conjugated to horseradish peroxidase at a 1:15,000 dilution in TBST at 25°C for 1 h. The membrane was then washed three times in TBST and once in TBS (Bio-Rad) before being exposed to 2 ml of Western Lightning Plus-ECL enhanced chemiluminescence substrate (PerkinElmer). The membrane was then exposed to BioMax XAR film (Carestream), and the film was processed on a Kodak X-Omat 2000 film processor. The processed films were scanned in grayscale, and the dots were quantified in ImageJ (NIH) by reversing the pixel values, defining a region of interest (ROI), and then using that defined object to measure the mean gray value of each spot and the background of the film, which was defined as an area of the film where no spot was present. The background measurement was subtracted from the mean pixel density of each spot.
Microfluidic devices.
Microfluidic devices were constructed as previously described (80). Specifically, the devices were constructed by bonding polydimethylsiloxane (PDMS) castings to size #1.5, 36- by 60-mm cover glass (ThermoFisher, Waltham MA) (81, 82). Bacterial strains were grown in 5 ml of LB medium with 15 μg/μl gentamicin overnight at 30°C with agitation. A 300-μl volume of overnight culture was pelleted, and the supernatant was removed before being resuspended in 1 ml of the modified BMM medium described above. Strains were inoculated into channels of the microfluidic devices and allowed to colonize for 1 h at room temperature, 21 to 24°C, before a constant flow of 0.5 μl/min of the modified BMM (described above) was provided. Medium flow was achieved using syringe pumps (Pico Plus Elite; Harvard Apparatus) and 5-ml syringes (27-gauge needle) fitted with number 30 Cole-Palmer polytetrafluoroethylene (PTFE) tubing (inside diameter, 0.3 mm). Tubing was inserted into holes bored in the PDMS with a catheter punch driver.
Imaging.
Biofilms were imaged using an Andor W1 spinning-disk confocal microscope, an Andor Zyla camera, and a Nikon Ti microscope. A 488-nm laser was used to excite GFP, while a 560-nm laser line was used to excite mRuby. Images were captured using Nikon NIS software.
LapD and LapG homolog prediction and LapA-like protein identification.
The approach to identifying LapD- and LapG-encoding organisms and LapA-like proteins was modified from a previously described approach (29). The code is posted at https://github.com/GeiselBiofilm/Collins-MapA. Briefly, LapG and LapD homologs were defined as ORF coding proteins with the pfam06035 and pfam16448 domains, respectively. The NCBI Conserved Domains Database (CDD) was utilized to generate a list of LapG- and LapD-encoding bacteria, and the programming language R (83) was used to determine the intersecting LapD- and LapG-encoding bacteria by text matching their species name identifiers. The protein annotations of these genomes were downloaded from the NCBI genome database, and each annotated locus with a size greater than 1,000 amino acids was interrogated for the presence of a LapG cleavage site within amino acids 70 to 150 [(T/A/P)AA(G/V)] and at least one RTX motif [DX(L/I)X4GXDX(L/I)XGGX3D].
Phylogenetic tree construction.
Representatives of each Pseudomonas species in the GenBank database were selected based on genome quality (i.e., fully assembled genome, or highest N50 if genome in contigs or scaffolds). In the case of species for which multiple strains with high-quality genomes were available, common lab strain representatives were chosen. A list of the strains used in the construction of this tree is given in File S3 in the supplemental material. The genome assembly files were retrieved from the GenBank ftp server and used to construct a phylogenetic tree according to a previously described method, as follows (63, 64). Orthologs of the 16S rRNA, gyrB, rpoB, and rpoD genes were identified by BLASTn version 2.6.0 (using the setting −gapopen 2) using the sequences of P. fluorescens Pf0-1 genes as queries (Pfl01_R12, 16S rRNA; Pfl01_0004, gyrB; Pfl01_5086, rpoB; Pfl01_5149, rpoD). Any species for which no BLAST matches could be found for any of the query genes were removed. Multiple sequence alignment was conducted using MUSCLE version 3.8.31 (65) for each gene, and alignments were concatenated. Concatenated aligned sequences were analyzed using PAUP*4.0a (build 167) (84). Distance was calculated using a Jukes-Cantor model, and a tree was constructed using neighbor-joining.
Statistics.
Statistical analysis was conducted in GraphPad Prism 8, and the statistical test performed is indicated in each figure legend or in the test. Unless otherwise indicated, each experiment was performed in at least triplicate, with three or more technical replicates per experiment.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carey Nadell and his laboratory for assistance with microfluidic chambers and imaging.
The work was supported by NIH grant no. R01 GM123609 to G.A.O. We also acknowledge the Molecular Biology Core, which receives funding from a Norris Cotton Cancer Center Core grant from the NIH (no. P30-CA023108).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Flemming H, Wuertz S. 2019. Bacteria and archaea on Earth and their abundance in biofilms. Nat Rev Microbiol 17:247–260. doi: 10.1038/s41579-019-0158-9. [DOI] [PubMed] [Google Scholar]
- 2.van Wolferen M, Orell A, Albers S. 2018. Archaeal biofilm formation. Nat Rev Microbiol 16:699–713. doi: 10.1038/s41579-018-0058-4. [DOI] [PubMed] [Google Scholar]
- 3.Fanning S, Mitchel A. 2012. Fungal biofilms. PLoS Pathog 8:e1002585. doi: 10.1371/journal.ppat.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mann EE, Wozniak DJ. 2012. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36:893–916. doi: 10.1111/j.1574-6976.2011.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Houdt R, Michiels CW. 2005. Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res Microbiol 156:626–633. doi: 10.1016/j.resmic.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Gil M, Quesada JM, Ramos-González MI, Soriano MI, de Cristóbal RE, Espinosa-Urgel M. 2013. Interplay between extracellular matrix components of Pseudomonas putida biofilms. Res Microbiol 164:382–389. doi: 10.1016/j.resmic.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 7.Berk V, Fong JCN, Dempsey GT, Develioglu ON, Zhuang X, Liphardt J, Yildiz FH, Chu S. 2012. Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337:236–239. doi: 10.1126/science.1222981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Götz F. 2002. Staphylococcus and biofilms. Mol Microbiol 43:1367–1378. doi: 10.1046/j.1365-2958.2002.02827.x. [DOI] [PubMed] [Google Scholar]
- 9.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487–1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 10.Cue D, Lei MG, Lee CY. 2012. Genetic regulation of the intercellular adhesion locus in staphylococci. Front Cell Infect Microbiol 2:38. doi: 10.3389/fcimb.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster TJ, Geoghegan JA, Ganesh VK, Höök M. 2014. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12:49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobley L, Harkins C, MacPhee CE, Stanley-Wall NR. 2015. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol Rev 39:649–669. doi: 10.1093/femsre/fuv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong JCN, Syed KA, Klose KE, Yildiz FH. 2010. Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiology 156:2757–2769. doi: 10.1099/mic.0.040196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yildiz FH, Schoolnik GK. 1999. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci U S A 96:4028–4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong JNC, Yildiz FH. 2015. Biofilm matrix proteins. Microbiol Spectr 3:3. doi: 10.1128/microbiolspec.MB-0004-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsén A, Jonsson A, Normark S. 1989. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 338:652–655. doi: 10.1038/338652a0. [DOI] [PubMed] [Google Scholar]
- 17.Vogeleer P, Tremblay YDN, Mafu AA, Jacques M, Harel J. 2014. Life on the outside: role of biofilms in environmental persistence of Shiga-toxin producing Escherichia coli. Front Microbiol 5:317. doi: 10.3389/fmicb.2014.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramey BE, Koutsoudis M, von Bodman SB, Fuqua C. 2004. Biofilm formation in plant-microbe associations. Curr Opin Microbiol 7:602–609. doi: 10.1016/j.mib.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Zampini M, Pruzzo C, Bondre VP, Tarsi R, Cosmo M, Bacciaglia A, Chhabra A, Srivastava R, Srivastava BS. 2005. Vibrio cholerae persistence in aquatic environments and colonization of intestinal cells: involvement of a common adhesion mechanism. FEMS Microbiol Lett 244:267–273. doi: 10.1016/j.femsle.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 20.Donlan RM. 2001. Biofilm formation: a clinically relevant microbiological process. Clin Infect Dis 33:1387–1392. doi: 10.1086/322972. [DOI] [PubMed] [Google Scholar]
- 21.Tamayo R, Patimalla B, Camilli A. 2010. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect Immun 78:3560–3569. doi: 10.1128/IAI.00048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Høiby N. 2014. A personal history of research on microbial biofilms and biofilm infections. Pathog Dis 70:205–211. doi: 10.1111/2049-632X.12165. [DOI] [PubMed] [Google Scholar]
- 23.Hinsa SM, Espinosa-Urgel M, Ramos JL, O'Toole GA. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol Microbiol 49:905–918. doi: 10.1046/j.1365-2958.2003.03615.x. [DOI] [PubMed] [Google Scholar]
- 24.Ivanov IE, Boyd CD, Newell PD, Schwartz ME, Turnbull L, Johnson MS, Whitchurch CB, O'Toole GA, Camesano TA. 2012. Atomic force and super-resolution microscopy support a role for LapA as a cell-surface biofilm adhesin of Pseudomonas fluorescens. Res Microbiol 163:685–691. doi: 10.1016/j.resmic.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyd CD, Smith TJ, El-Kirat-Chatel S, Newell PD, Dufrêne YF, O'Toole GA. 2014. Structural features of the Pseudomonas fluorescens biofilm adhesin LapA required for LapG-dependent cleavage, biofilm formation, and cell surface localization. J Bacteriol 196:2775–2788. doi: 10.1128/JB.01629-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Kirat-Chatel S, Beaussart A, Boyd CD, O'Toole GA, Dufrêne YF. 2014. Single-cell and single-molecule analysis deciphers the localization, adhesion, and mechanics of the biofilm adhesin LapA. ACS Chem Biol 9:485–494. doi: 10.1021/cb400794e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Kirat-Chatel S, Boyd CD, O'Toole GA, Dufrêne YF. 2014. Single-molecule analysis of Pseudomonas fluorescens footprints. ACS Nano 8:1690–1698. doi: 10.1021/nn4060489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newell PD, Boyd CD, Sondermann H, O'Toole GA. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol 9:e1000587. doi: 10.1371/journal.pbio.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith TJ, Font ME, Kelly CM, Sondermann H, O'Toole GA. 2018. An N-terminal retention module anchors the giant adhesin LapA of Pseudomonas fluorescens at the cell surface: a novel subfamily of type I secretion systems. J Bacteriol 200:e00734-17. doi: 10.1128/JB.00734-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinsa SM, O'Toole GA. 2006. Biofilm formation by Pseudomonas fluorescens WCS365: a role for LapD. Microbiology 152:1375–1383. doi: 10.1099/mic.0.28696-0. [DOI] [PubMed] [Google Scholar]
- 31.Newell PD, Monds RD, O'Toole GA. 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci U S A 106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyd CD, Chatterjee D, Sondermann H, O'Toole GA. 2012. LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0-1, is a calcium-dependent protease. J Bacteriol 194:4406–4414. doi: 10.1128/JB.00642-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatterjee D, Boyd CD, O'Toole GA, Sondermann H. 2012. Structural characterization of a conserved, calcium-dependent periplasmic protease from Legionella pneumophila. J Bacteriol 194:4415–4425. doi: 10.1128/JB.00640-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooley RB, O’Donnell JP, Sondermann H. 2016. Coincidence detection and bi-directional transmembrane signaling control a bacterial second messenger receptor. Elife 5:e21848. doi: 10.7554/eLife.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Navarro MVAS, Newell PD, Krasteva PV, Chatterjee D, Madden DR, O'Toole GA, Sondermann H. 2011. Structural basis for c-di-GMP-mediated inside-out signaling controlling periplasmic proteolysis. PLoS Biol 9:e1000588. doi: 10.1371/journal.pbio.1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ude S, Arnold DL, Moon CD, Timms-Wilson T, Spiers AJ. 2006. Biofilm formation and cellulose expression among diverse environmental Pseudomonas isolates. Environ Microbiol 8:1997–2011. doi: 10.1111/j.1462-2920.2006.01080.x. [DOI] [PubMed] [Google Scholar]
- 37.Spiers AJ, Bohannon J, Gehrig SM, Rainey PB. 2003. Biofilm formation at the air-liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol Microbiol 50:15–27. doi: 10.1046/j.1365-2958.2003.03670.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Beskrovnaya P, Melnyk RA, Hossain SS, Khorasani S, O’Sullivan LR, Wiesmann CL, Bush J, Richard JD, Haney CH. 2018. A genome-wide screen identifies genes in rhizosphere-associated Pseudomonas required to evade plant defenses. mBio 9:e00433-18. doi: 10.1128/mBio.00433-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caiazza NC, O'Toole GA. 2004. SadB is required for the transition from reversible to irreversible attachment during biofilm formation by Pseudomonas aeruginosa PA14. J Bacteriol 186:4476–4485. doi: 10.1128/JB.186.14.4476-4485.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramos-González MI, Travieso ML, Soriano MI, Matilla MA, Huertas-Rosales Ó, Barrientos-Moreno L, Tagua VG, Espinosa-Urgel M. 2016. Genetic dissection of the regulatory network associated with high c-di-GMP levels in Pseudomonas putida KT2440. Front Microbiol 7:1093. doi: 10.3389/fmicb.2016.01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernier SP, Ha D-G, Khan W, Merritt JH, O'Toole GA. 2011. Modulation of Pseudomonas aeruginosa surface-associated group behaviors by individual amino acids through c-di-GMP signaling. Res Microbiol 162:680–688. doi: 10.1016/j.resmic.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sulochana M. 1962. Amino acids in root exudates of cotton. Plant Soil 16:312–326. doi: 10.1007/BF01381342. [DOI] [Google Scholar]
- 43.Gitte RR, Rai PV, Patil RB. 1978. Chemotaxis of Rhizobium sp. towards root exudate of Cicer arietinum L. Plant Soil 50:553–566. doi: 10.1007/BF02107208. [DOI] [Google Scholar]
- 44.Collins JC, Reilly EJ. 1968. Chemical composition of the exudate from excised maize roots. Planta 83:218–222. doi: 10.1007/BF00385025. [DOI] [PubMed] [Google Scholar]
- 45.Fuqua C. 2010. Passing the baton between laps: adhesion and cohesion in Pseudomonas putida biofilms. Mol Microbiol 77:533–536. doi: 10.1111/j.1365-2958.2010.07250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Touzé T, Eswaran J, Bokma E, Koronakis E, Hughes C, Koronakis V. 2004. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol Microbiol 53:697–706. doi: 10.1111/j.1365-2958.2004.04158.x. [DOI] [PubMed] [Google Scholar]
- 47.Létoffé S, Delepelaire P, Wandersman C. 1996. Protein secretion in gram-negative bacteria: assembly of the three components of ABC protein-mediated exporters is ordered and promoted by substrate binding. EMBO J 15:5804–5811. doi: 10.1002/j.1460-2075.1996.tb00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi N, Nishino K, Yamaguchi A. 2001. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol 183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tikhonova EB, Dastidar V, Rybenkov VV, Zgurskaya HI. 2009. Kinetic control of TolC recruitment by multidrug efflux complexes. Proc Natl Acad Sci U S A 106:16416–16421. doi: 10.1073/pnas.0906601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wandersman C, Delepelaire P. 1990. TolC, an Escherichia coli outer membrane protein required for hemolysin secretion. Proc Natl Acad Sci U S A 87:4776–4780. doi: 10.1073/pnas.87.12.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahlstrom KM, Collins AJ, Doing G, Taroni JN, Gauvin TJ, Greene CS, Hogan DA, O'Toole GA. 2018. A multimodal strategy used by a large c-di-GMP network. J Bacteriol 200:e00703-17. doi: 10.1128/JB.00703-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Velden AW, Bäumler AJ, Tsolis RM, Heffron F. 1998. Multiple fimbrial adhesins are required for full virulence of Salmonella typhimurium in mice. Infect Immun 66:2803–2808. doi: 10.1128/IAI.66.6.2803-2808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Latasa C, Roux A, Toledo-Arana A, Ghigo J-M, Gamazo C, Penadés JR, Lasa I. 2005. BapA, a large secreted protein required for biofilm formation and host colonization of Salmonella enterica serovar Enteritidis. Mol Microbiol 58:1322–1339. doi: 10.1111/j.1365-2958.2005.04907.x. [DOI] [PubMed] [Google Scholar]
- 54.Morgan E, Bowen AJ, Carnell SC, Wallis TS, Stevens MP. 2007. SiiE is secreted by the Salmonella enterica serovar Typhimurium pathogenicity island 4-encoded secretion system and contributes to intestinal colonization in cattle. Infect Immun 75:1524–1533. doi: 10.1128/IAI.01438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Espinosa-Urgel M, Salido A, Ramos J-L. 2000. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J Bacteriol 182:2363–2369. doi: 10.1128/jb.182.9.2363-2369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez-Gil M, Yousef‐Coronado F, Espinosa‐Urgel M. 2010. LapF, the second largest Pseudomonas putida protein, contributes to plant root colonization and determines biofilm architecture. Mol Microbiol 77:549–561. doi: 10.1111/j.1365-2958.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- 57.Martínez-Gil M, Romero D, Kolter R, Espinosa-Urgel M. 2012. Calcium causes multimerization of the large adhesin LapF and modulates biofilm formation by Pseudomonas putida. J Bacteriol 194:6782–6789. doi: 10.1128/JB.01094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kitts G, Giglio KM, Zamorano-Sánchez D, Park JH, Townsley L, Cooley RB, Wucher BR, Klose KE, Nadell CD, Yildiz FH, Sondermann H. 2019. A conserved regulatory circuit controls large adhesins in Vibrio cholerae. mBio 10:e02822-19. doi: 10.1128/mBio.02822-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ambrosis N, Boyd CD, ÓToole GA, Fernández J, Sisti F. 2016. Homologs of the LapD-LapG c-di-GMP effector system control biofilm formation by Bordetella bronchiseptica. PLoS One 11:e0158752. doi: 10.1371/journal.pone.0158752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pérez-Mendoza D, Coulthurst SJ, Humphris S, Campbell E, Welch M, Toth IK, Salmond G. 2011. A multi-repeat adhesin of the phytopathogen, Pectobacterium atrosepticum, is secreted by a type I pathway and is subject to complex regulation involving a non-canonical diguanylate cyclase. Mol Microbiol 82:719–733. doi: 10.1111/j.1365-2958.2011.07849.x. [DOI] [PubMed] [Google Scholar]
- 61.Guo S, Stevens CA, Vance TDR, Olijve LLC, Graham LA, Campbell RL, Yazdi SR, Escobedo C, Bar-Dolev M, Yashunsky V, Braslavsky I, Langelaan DN, Smith SP, Allingham JS, Voets IK, Davies PL. 2017. Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci Adv 3:e1701440. doi: 10.1126/sciadv.1701440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vance TDR, Graham LA, Davies PL. 2018. An ice-binding and tandem beta-sandwich domain-containing protein in Shewanella frigidimarina is a potential new type of ice adhesin. FEBS J 285:1511–1527. doi: 10.1111/febs.14424. [DOI] [PubMed] [Google Scholar]
- 63.Peix A, Ramírez-Bahena M-H, Velázquez E. 2018. The current status on the taxonomy of Pseudomonas revisited: an update. Infect Genet Evol 57:106–116. doi: 10.1016/j.meegid.2017.10.026. [DOI] [PubMed] [Google Scholar]
- 64.Mulet M, Lalucat J, García-Valdés E. 2010. DNA sequence-based analysis of the Pseudomonas species. Environ Microbiol 12:1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x. [DOI] [PubMed] [Google Scholar]
- 65.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith TJ, Sondermann H, O'Toole GA. 2018. Type 1 does the two-step: type 1 secretion substrates with a functional periplasmic intermediate. J Bacteriol 200:e00168-18. doi: 10.1128/JB.00168-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Collins AJ, Smith TJ, Sondermann H, O’Toole GA. From input to output: the Lap/c-di-GMP biofilm regulatory circuit. Annu Rev Microbiol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cooley RB, Smith TJ, Leung W, Tierney V, Borlee BR, O'Toole GA, Sondermann H. 2016. Cyclic di-GMP-regulated periplasmic proteolysis of a Pseudomonas aeruginosa type Vb secretion system substrate. J Bacteriol 198:66–76. doi: 10.1128/JB.00369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bollinger A, Thies S, Katzke N, Jaeger K-E. 2020. The biotechnological potential of marine bacteria in the novel lineage of Pseudomonas pertucinogena. Microb Biotechnol 13:19–31. doi: 10.1111/1751-7915.13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kawai Y, Yabuuchi E. 1975. Pseudomonas pertucinogena sp. nov., an organism previously misidentified as Bordetella pertussis. Int J Syst Evol Microbiol 25:317–323. doi: 10.1099/00207713-25-4-317. [DOI] [Google Scholar]
- 71.Sánchez D, Mulet M, Rodríguez AC, David Z, Lalucat J, García-Valdés E. 2014. Pseudomonas aestusnigri sp. nov., isolated from crude oil-contaminated intertidal sand samples after the Prestige oil spill. Syst Appl Microbiol 37:89–94. doi: 10.1016/j.syapm.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 72.Mulet M, Sánchez D, Rodríguez AC, Nogales B, Bosch R, Busquets A, Gomila M, Lalucat J, García-Valdés E. 2018. Pseudomonas gallaeciensis sp. nov., isolated from crude-oil-contaminated intertidal sand samples after the Prestige oil spill. Syst Appl Microbiol 41:340–347. doi: 10.1016/j.syapm.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Moor H, Teppo A, Lahesaare A, Kivisaar M, Teras R. 2014. Fis overexpression enhances Pseudomonas putida biofilm formation by regulating the ratio of LapA and LapF. Microbiology 160:2681–2693. doi: 10.1099/mic.0.082503-0. [DOI] [PubMed] [Google Scholar]
- 74.Ainelo H, Lahesaare A, Teppo A, Kivisaar M, Teras R. 2017. The promoter region of lapA and its transcriptional regulation by Fis in Pseudomonas putida. PLoS One 12:e0185482. doi: 10.1371/journal.pone.0185482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blanco-Romero E, Redondo-Nieto M, Martínez-Granero F, Garrido-Sanz D, Ramos-González MI, Martín M, Rivilla R. 2018. Genome-wide analysis of the FleQ direct regulon in Pseudomonas fluorescens F113 and Pseudomonas putida KT2440. Sci Rep 8:13145. doi: 10.1038/s41598-018-31371-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martínez-Gil M, Ramos-González MI, Espinosa-Urgel M. 2014. Roles of cyclic di-GMP and the Gac system in transcriptional control of the genes coding for the Pseudomonas putida adhesins LapA and LapF. J Bacteriol 196:1484–1495. doi: 10.1128/JB.01287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monds RD, Newell PD, Schwartzman JA, O'Toole GA. 2006. Conservation of the Pho regulon in Pseudomonas fluorescens Pf0-1. Appl Environ Microbiol 72:1910–1924. doi: 10.1128/AEM.72.3.1910-1924.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 79.Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Simmons M, Bond M, Drescher K, Bucci V, Nadell CD. 2019. Evolutionary dynamics of phage resistance in bacterial biofilms. bioRxiv doi: 10.1101/552265. [DOI]
- 81.Weibel DB, Diluzio WR, Whitesides GM. 2007. Microfabrication meets microbiology. Nat Rev Microbiol 5:209–218. doi: 10.1038/nrmicro1616. [DOI] [PubMed] [Google Scholar]
- 82.Sia SK, Whitesides GM. 2003. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis 24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 83.R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 84.Swofford DL. 2003. PAUP*: phylogenetic analysis using parsimony (and other methods), version 4. Sinauer Associates, Sunderland, MA. [Google Scholar]
- 85.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.García-Valdés E, Lalucat J. 2016. Pseudomonas: molecular phylogeny and current taxonomy, p 1–23. In Pseudomonas: molecular and applied biology. Springer, New York, NY. [Google Scholar]
- 87.Horsman ME, Marous DR, Li R, Oliver RA, Byun B, Emrich SJ, Boggess B, Townsend CA, Mobashery S. 2017. Whole-genome shotgun sequencing of two β-proteobacterial species in search of the bulgecin biosynthetic cluster. ACS Chem Biol 12:2552–2557. doi: 10.1021/acschembio.7b00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monds RD, Newell PD, Gross RH, O'Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol Microbiol 63:656–679. doi: 10.1111/j.1365-2958.2006.05539.x. [DOI] [PubMed] [Google Scholar]
- 89.Bao Y, Lies DP, Fu H, Roberts GP. 1991. An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 109:167–168. doi: 10.1016/0378-1119(91)90604-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.