

Vibrio cholerae, the bacterial pathogen that is responsible for the disease cholera, uses biofilms to aid in survival, dissemination, and persistence. VpsR, which directly senses the second messenger c-di-GMP, is a major regulator of this process. Together with c-di-GMP, VpsR directly activates transcription by RNA polymerase containing σ70 from the vpsL biofilm biogenesis promoter. Using biochemical methods, we demonstrate for the first time that VpsR/c-di-GMP directly activates σ70-RNA polymerase at the first genes of the vps and ribomatrix operons. In this regard, it functions as either a class I or class II activator. Our results broaden the mechanism of c-di-GMP-dependent transcription activation and the specific role of VpsR in biofilm formation.
KEYWORDS: transcription, biofilm, VpsR, c-di-GMP, RNA polymerase, rbmA, vpsU, rbmF, vpsL, gene regulation, Vibrio cholerae
ABSTRACT
Vibrio cholerae biofilm biogenesis, which is important for survival, dissemination, and persistence, requires multiple genes in the Vibrio polysaccharides (vps) operons I and II as well as the cluster of ribomatrix (rbm) genes. Transcriptional control of these genes is a complex process that requires several activators/repressors and the ubiquitous signaling molecule, cyclic di-GMP (c-di-GMP). Previously, we demonstrated that VpsR directly activates RNA polymerase containing σ70 (σ70-RNAP) at the vpsL promoter (PvpsL), which precedes the vps-II operon, in a c-di-GMP-dependent manner by stimulating formation of the transcriptionally active, open complex. Using in vitro transcription, electrophoretic mobility shift assays, and DNase I footprinting, we show here that VpsR also directly activates σ70-RNAP transcription from other promoters within the biofilm formation cluster, including PvpsU, at the beginning of the vps-I operon, PrbmA, at the start of the rbm cluster, and PrbmF, which lies upstream of the divergent rbmF and rbmE genes. In this capacity, we find that VpsR is able to behave both as a class II activator, which functions immediately adjacent/overlapping the core promoter sequence (PvpsL and PvpsU), and as a class I activator, which functions farther upstream (PrbmA and PrbmF). Because these promoters vary in VpsR-DNA binding affinity in the absence and presence of c-di-GMP, we speculate that VpsR’s mechanism of activation is dependent on both the concentration of VpsR and the level of c-di-GMP to increase transcription, resulting in finely tuned regulation.
IMPORTANCE Vibrio cholerae, the bacterial pathogen that is responsible for the disease cholera, uses biofilms to aid in survival, dissemination, and persistence. VpsR, which directly senses the second messenger c-di-GMP, is a major regulator of this process. Together with c-di-GMP, VpsR directly activates transcription by RNA polymerase containing σ70 from the vpsL biofilm biogenesis promoter. Using biochemical methods, we demonstrate for the first time that VpsR/c-di-GMP directly activates σ70-RNA polymerase at the first genes of the vps and ribomatrix operons. In this regard, it functions as either a class I or class II activator. Our results broaden the mechanism of c-di-GMP-dependent transcription activation and the specific role of VpsR in biofilm formation.
INTRODUCTION
Vibrio cholerae, the causative agent of the enteric disease cholera, is responsible for 3 to 5 million infections as well as 100,000 to 120,000 deaths per year (1, 2). During its life cycle, V. cholerae lives in both the planktonic and biofilm state. In the human host and the aquatic reservoir, V. cholerae forms biofilms to aid in survival, transmission, and persistence (3–7). Not only do biofilms shield the bacteria from harsh environmental conditions and stresses, but they also protect the bacteria from protozoa and bacteriophage predation as well as nutrient limitation (3, 4, 8–10). Biofilms are comprised of matrix proteins, extracellular DNA, and Vibrio polysaccharides (VPS). Over 50% of the Vibrio biofilm matrix mass is comprised of VPS (11–16), which is excreted after initial attachment to surfaces (17). The biofilm structure modulator A protein, RbmA, and the biofilm matrix protein, Bap1, are also secreted from the cell surface, promoting cell-cell adhesion and cell-VPS adherence (14, 15, 17).
Genes involved in VPS synthesis are located on V. cholerae’s larger chromosome and organized into two operons, vps-I and vps-II (12, 13). vps-I contains 12 genes, including vpsU and vpsA to vpsK, while vps-II contains six genes, vpsL to vpsQ (12, 13). These two operons are separated by 8.3 kbp that contain six ribomatrix genes, rbmA, rbmC, and rbmBDEF, which also contribute to biofilm formation (Fig. 1A) (12, 14, 15).
FIG 1.

Vibrio cholerae genomic organization of biofilm biogenesis operons and sequences of their promoters. (A) VPS synthesis genes are comprised of two vps operons, vps-I (white arrows) and vps-II (black arrows). The ribomatrix protein production gene cluster is comprised of the rbm genes (gray arrows) and bap1 (not shown). Direction of transcription is indicated by the arrows. Smaller black arrows indicate the positions of the promoters upstream of vpsU, rbmA, rbmF, and vpsL. Image is approximately to scale. (B) Sequences of promoters PrbmA, PrbmF, PvpsU, and PvpsL. Determined VpsR protection sites observed with DNase I footprinting on the template strand are italicized in red with the match to the determined consensus sequence in bold; the predominant +1 TSSs from both V. cholerae RNA and in vitro transcription RNA are enlarged and in bold with a black arrow above. (Note that the match to the consensus for the VpsR binding site at vpsU is in the opposite orientation relative to the promoter.) Other 5′ ends for rbmA and rbmF RNA observed in vivo are underlined and in bold, and the other prominent 5′ end for vpsU RNA seen in vivo is green, in bold, and underlined. Assigned −10 elements are boxed with solid lines, and the −35 region located 17 bp upstream is shown in a dotted box. A potential extended −10 promoter for vpsU transcription from the alternate 5′ end is indicated by the green bar. Information for PvpsL was determined previously and is included here for comparison (31).
Since biofilm formation is a complex process requiring the assembly of multiple gene products, it is not surprising that this process is highly regulated with numerous signaling pathways, transcriptional repressors and activators, and regulatory sRNAs (18). Among these effectors is the ubiquitous signaling molecule, cyclic di-GMP (c-di-GMP), a keystone regulator of biofilm formation in many bacterial species (19). c-di-GMP is formed from two GTP molecules by diguanylate cyclase enzymes that contain a GGDEF domain and then degraded into pGpG or two GMP molecules by phosphodiesterase enzymes containing either an EAL or HD-GYP domain, respectively. Generally, high levels of c-di-GMP increase biofilm formation, while low levels of c-di-GMP promote motility (20).
A major mechanism by which c-di-GMP controls biofilm formation and motility is through regulation of transcription (21). Bacterial RNA polymerase (RNAP), which is responsible for transcription, is comprised of five subunits (β, β′, two αs, and ω) and a specificity factor, σ (22). While σ70 is the housekeeping σ factor that is needed for exponential growth, alternate σs are used under different conditions or times of stress (22). During transcription initiation, σ70-RNAP first forms an unstable short-lived complex called the closed complex (RPc) (23, 24). Kinetically favorable isomerizations then transition RPc to the stable open complex (RPo) (25, 26). Within the RPo, major conformational changes occur within RNAP, the DNA bends approximately 90°, and the transcription bubble forms from −12/−11 to approximately +5 relative to the +1 transcriptional start site (TSS) (27). This bubble allows the single-stranded template DNA to descend into the active site for the start of transcription (27). Although RNAP alone can transcribe from some promoters, yielding a basal level of expression, many promoters require activators to work by stimulating RNAP directly or by acting as antirepressors of regulators that inhibit RNAP. Among the activators that directly stimulate σ70-RNAP, many function by a class I or a class II mechanism or a combination of the two (22, 28). In class I transcription activation, activators bind to regions upstream of the core promoter, while in class II transcription activation, activators interact directly upstream or overlapping the −35 element (22, 28). Furthermore, class I activators typically help recruit RNAP, while class II activators both recruit and isomerize RNAP (22).
In V. cholerae, the transcriptional activators VpsR and VpsT are required to activate transcription from the biofilm formation operons (21, 29–36). Though both regulators bind c-di-GMP with a dissociation constant (Kd) of 1.6 μM and 3.2 μM for VpsR and VpsT, respectively (32, 35), the binding of c-di-GMP to VpsT increases its binding to DNA (30, 34, 35, 37), while the binding of c-di-GMP to VpsR has no effect on its affinity for the vpsL promoter (PvpsL) (18, 31). This allows VpsT to work as an antirepressor of the histone-like repressor H-NS, which represses the vps-I and vps-II operons (38, 39). In contrast, we have previously shown that VpsR is a direct activator of PvpsL (30–32). It functions as a class II activator by interacting with a region overlapping the −35 element to subsequently initiate transcription with c-di-GMP and σ70-RNAP (31). c-di-GMP, together with σ70-RNAP and VpsR, is required to generate the active transcription complex at PvspL as demonstrated in both potassium permanganate footprinting and DNase I footprinting (31). This was a surprising finding given that based on amino acid homology, VpsR is classified as an enhancer-binding protein (EBP), which typically uses the alternate σ, σ54, and ATP hydrolysis to activate transcription. However, VpsR is an atypical EBP. Not only does VpsR lack the GAFTGA motif responsible for interacting with σ54, but VpsR is also missing the conserved residues within the Walker B domain involved in ATP hydrolysis. In vitro transcriptions demonstrate that VpsR does not require ATP hydrolysis to initiate transcription with σ70-RNAP and c-di-GMP (31).
In addition to genes directly involved in biofilm formation in V. cholerae, VpsR, together with c-di-GMP, activates the expression of many other genes that are thought to be adaptive in a biofilm lifestyle. These genes include the transcriptional regulators vpsT, aphA, and tfoY as well as epsC, the first gene of the type II secretion system operon (32, 36, 40, 41). Although VpsR directly binds to the DNA upstream of these genes, the mechanism by which it activates their expression has not been elucidated.
In this study, we report that VpsR also directly activates PrbmA, PrbmF, and PvpsU, making it a primary, direct regulator of promoters throughout the V. cholerae biofilm biogenesis operons. As we found with PvpsL, this transcription requires σ70-RNAP and c-di-GMP. Analogous to PvpsL, VpsR binding at PvpsU overlaps the −35 element, while the VpsR binding sites at PrbmA and PrbmF are located farther upstream from the core promoter. Though binding affinities vary in the presence and absence of c-di-GMP at each promoter, specific DNA-protein interactions do not change in the presence of the signaling molecule. Our findings demonstrate that VpsR is an adaptable, direct regulator, functioning as both a class I and class II transcription activator, to induce multiple promoters of genes essential for biofilm formation in V. cholerae.
RESULTS
VpsR binds the promoter regions of rbmA, rbmF, and vpsU with or without c-di-GMP.
Previously, VpsR has been shown to bind upstream of the aphA, epsC, vpsL, and vpsT genes in electrophoretic mobility shift assays (EMSAs) (18, 31, 32, 36, 40, 41), and in silico sequence analysis using MEME (multiple expected maximization for motif elicitation) has predicted other VpsR binding sites upstream of the following genes: vpsR, VCA0075, rbmA, vpsA, vpsU, bap1, cdgC, and rbmC (18). Using DNase I footprinting, two VpsR binding sites were also determined upstream of vpsL at positions −31 to −53 (proximal site) and −297 to −336 (distal site) relative to the vpsL promoter, PvpsL (30). In previous work, we demonstrated that VpsR binding to the proximal site directly activates transcription in vitro from PvpsL, while the presence of the distal site has no effect on PvpsL transcription in the in vitro system (31). Consequently, given the location of the distal site just upstream of the divergent gene rbmF (Fig. 1), we speculated that this site might be needed for VpsR activation of rbmF transcription.
To extend this work, we investigated the possible binding of VpsR to the rbmA, vpsU, and rbmF sites using EMSAs (Fig. 2). At all three promoters, we observed the formation of multiple complexes, with larger complexes forming as more VpsR was added. This behavior can be indicative of protein-protein interactions or the ability of multiple VpsR proteins to bind to the DNA fragment. As DNase I footprints (detailed below) indicated only a single site of VpsR binding, it seems likely that these larger species represent protein-protein binding or aggregation. Consequently, it is important to note that we determined the apparent dissociation constant [Kd(app)] as the amount of VpsR needed to shift 50% of the free DNA. Thus, the Kd(app) was calculated by quantifying the amount of free DNA that has been lost rather than the amount that has been shifted.
FIG 2.

VpsR binds PvpsU, PrbmA, and PrbmF with and without c-di-GMP. Representative EMSAs of 32P-labeled DNA harboring −130 to +120 of PvpsU (A), −223 to +84 of PrbmA (B), or −228 to +112 of PrbmF (C) relative to the +1 TSS with increasing VpsR concentrations from 0 to 2.5 μM (lanes 1 to 5; lanes 6 to 10) either in the absence of c-di-GMP (lanes 1 to 5) or in the presence of 50 μM c-di-GMP (lanes 6 to 10). Gray arrows indicate free DNA, while black arrows represent shifted protein-DNA complexes. Samples were incubated at room temperature for 10 min prior to electrophoresis on 5% TBE polyacrylamide gels. Apparent DNA-binding dissociation constants Kd(app) (bottom) were determined as the concentration of VpsR needed to retard 50% of the free DNA. Values from at least three EMSAs were analyzed using one-way analysis of variance (ANOVA) with Tukey’s HSD post hoc analysis (ns, not significant; *, P < 0.05).
As we had observed previously for the vpsL proximal site (31), the presence of c-di-GMP does not significantly affect VpsR binding at the vpsU site (Fig. 2A). However, for the rbmA and rbmF promoters, the presence of c-di-GMP improves VpsR binding affinity modestly (50%) and significantly (∼5-fold), respectively (Fig. 2B and C). Relative to the EMSAs for rbmA and vpsU promoters, the binding of VpsR to the rbmF promoter appeared to generate complexes that were less discrete, resulting in a smearing pattern. No kinetic data can be determined from our EMSAs, but we speculate that at this specific promoter, VpsR may be forming a less discrete complex due to a faster off rate.
Primer extension analyses define the 5′ ends of rbmA, rbmF, and vpsU transcripts in vivo.
Previously, differential RNA-sequencing (dRNA-seq) has been used to determine TSSs throughout the V. cholerae genome under low and high cell density (42). The TSSs for rbmA, rbmF, and vpsU were identified as the T, A, and G that are 110 bp upstream, 60 bp downstream, and 52 bp upstream from the translation start site, respectively (42). For vpsU, an additional TSS was identified as the A that is 315 bp upstream of the translation start site.
To compare and determine the 5′ ends of rbmA, rbmF, and vpsU in vivo transcripts, we performed primer extension analyses of RNA isolated from wild type (WT) or ΔvpsR grown in the presence or absence of c-di-GMP. We obtained major primer extension products corresponding to the A 18 bp upstream of the TTG start codon of rbmA (Fig. 3A, lane 3), to the A 45 bp upstream of the ATG start codon of rbmF (Fig. 3B, lane 3), and to the T 53 bp upstream of the ATG start codon of vpsU (Fig. 3C, lane 3). These products were not seen using RNA isolated from the ΔvpsR mutant or under unaltered c-di-GMP levels (Fig. 3, lanes 1, 2, and 4), indicating that their presence is dependent on the presence of both VpsR and c-di-GMP. As detailed below, further in vitro analyses were consistent with assigning these ends as TSSs for the promoters for rbmA (PrbmA), rbmF (PrbmF), and vpsU (PvpsU). In each case, analysis of the DNA sequence upstream of the determined TSS indicated the presence of a suitable −10 element (consensus, −12TAtaaT−7 [capital letters indicate a greater level of consensus]) or extended −10 element for PrbmF (consensus, −15TGnTAtaaT−7) at a reasonable distance from the TSS (Fig. 1B, boxes). Not surprisingly, given their need for a transcriptional activator, none of the promoters contained a strong match to consensus within the −35 region. All lacked the very highly conserved “TT” within the −35TTGaca−30 sequence. (Consensus sequences represent matches to the derived E. coli σ70-RNAP consensus [25]; −10 and −35 element sequences derived from chromatin immunoprecipitation sequencing [ChIP-seq] analyses of 497 σ70-binding sites in V. cholerae [43] are nearly identical [−12TAnAAT−7 and −35TTGnaa−30, respectively].) Though our primer extensions identified TSSs that are different from those determined from dRNA-seq (42), we are confident in our analyses, as our in vivo TSSs are consistent with our in vitro TSSs (below). We suggest that the differences observed for rbmF and vpsU could be due to RNA processing in vivo.
FIG 3.

Determination of +1 TSS in vivo and in vitro for PrbmA (A), PrbmF (B), and PvpsU (C). Primer extension analyses were performed with 32P-labeled primer that hybridized 84, 112, and 120 bp downstream of the rbmA, rbmF, and vpsU TSSs, respectively. RNA was isolated from V. cholerae (lanes 1 to 4) or in vitro transcriptions (lanes 5 to 8). GATC lanes are sequencing lanes performed using the end-labeled DNA. Black arrows designate the major 5′ ends seen in vivo and in vitro; the gray arrow designates an alternate 5′ end observed for vpsU both in vivo and in vitro. The assigned TSS for each gene is shown in Fig. 1B.
Another prominent vpsU 5′ end dependent on both VpsR and c-di-GMP was also observed in vivo, 10 bp upstream of the PvpsU start. This upstream TSS is preceded by a good match to an extended −10 promoter sequence (designated by the green rectangle in Fig. 1B), suggesting that another PvpsU promoter may also be active in cells.
In the presence of c-di-GMP, VpsR activates transcription from PrbmA, PrbmF, and PvpsU in vitro.
A previous microarray study demonstrated that rbmA, rbmF, and vpsU were downregulated 7.6-fold, 7.3-fold, and 10.6-fold, respectively, in a V. cholerae vpsR deletion strain compared to the rugose parent strain, indicating in vivo regulation by VpsR (44). However, such regulation could be direct or indirect. Direct regulation by VpsR and c-di-GMP at these genes has not been demonstrated (29). To determine if VpsR and c-di-GMP directly activate transcription of rbmA, rbmF, and vpsU, we constructed supercoiled in vitro transcription templates containing PrbmA, PrbmF, and PvpsU located upstream of the rrnBP1 terminator. In vitro transcription analyses indicated that in the presence of both VpsR and c-di-GMP, a unique transcript was significantly elevated for each template (Fig. 4, lanes 4). The activated transcription from PrbmA, PrbmF, and PvpsU was ∼3-fold, 4-fold, and 17-fold over the basal level obtained with RNAP alone (lanes 1) or RNAP with either 25 μM c-di-GMP (lanes 2) or VpsR (lanes 3). Since H-NS represses basal level transcription in vivo (39), we predict these fold increases to be lower in vitro due to the absence of H-NS. This prediction is further supported by our observation that there is no detectable basal level of transcription from these promoters in vivo as observed in the primer extension analyses (Fig. 3).
FIG 4.

VpsR and c-di-GMP activate transcription from PrbmA, PrbmF, and PvpsU in vitro. Representative images of single round in vitro transcription from supercoiled plasmid templates containing PrbmA, PrbmF, and PvpsU with RNAP alone (lanes 1), RNAP and 25 μM c-di-GMP (lanes 2), RNAP and VpsR (lanes 3), and RNAP and VpsR and 25 μM c-di-GMP (lanes 4). The activation fold change (average from 3 reactions) for RNAP, VpsR, and 25 μM c-di-GMP relative to basal level expression of RNAP alone is indicated below the image.
Besides PrbmA, PrbmF, and PvpsU, we also investigated whether VpsR and c-di-GMP could activate σ70-RNAP transcription at other putative VpsR-regulated promoters. Previous studies using EMSAs have shown VpsR binding sites upstream of the TSSs at PaphA, PvpsT, and PepsC (32, 36, 41). However, for these three promoters, we were unable to detect transcription above the basal level with VpsR and c-di-GMP (see Fig. S1 in the supplemental material).
To compare the 5′ ends of the in vitro RNAs generated from the PrbmA, PrbmF, and PvpsU templates to the ends observed in vivo, we again used primer extension analyses. In each case, we obtained major products that were identical to those found in vivo (Fig. 3, lanes 8, black arrows). In addition, a minor product observed using vpsU in vitro RNA corresponded to the alternate TSS seen in vivo (Fig. 3C, lanes 3 and 5 to 8, gray arrow). The difference in intensity of this band when using in vitro RNA versus what was observed using in vivo RNA suggests that perhaps different conditions or other factors may contribute to its activity in vivo, allowing for more regulation at this promoter.
To determine the position of VpsR binding relative to the TSS, we performed DNase I footprints. We found that VpsR protected rbmA from −83 to −57, rbmF from −100 to −66, and vpsU from −52 to −31 relative to the TSS of each promoter (Fig. 1B and Fig. 5A). Although the presence of c-di-GMP improved the DNA binding affinity of VpsR to PrbmA and PrbmF (Fig. 2), the DNase I footprints did not reveal any differences in the protein-DNA contacts in the presence or absence of c-di-GMP. Analysis of the VpsR protection sites relative to the TSS for each promoter indicated that for PrbmA and PrbmF, the sites occur >50 bp upstream of the TSS, while for PvpsU, the VpsR binding site is closer to the core promoter, like PvpsL (Fig. 1B) (31).
FIG 5.

DNase I footprinting of VpsR with and without c-di-GMP at PrbmA, PrbmF, and PvpsU. (A) DNase I footprints of the template strand were obtained using DNA alone or DNA incubated with VpsR without or with 50 μM c-di-GMP as indicated. VpsR protection is indicated to the right of each image with a dotted line. (B) Sequences of determined VpsR protection sites at PrbmA, PrbmF, PvpsU, and PvpsL aligned to indicate sequence similarity. The consensus sequence underneath was determined from the presence of a base in at least 3 out of the 4 binding sites. A previously reported Logo sequence (30) is shown for comparison.
Alignment of the protection sites for these four promoters revealed a conserved sequence of 5’ tGtcTtanaaTTGA 3′ (Fig. 5B), in which the lowercase letters correspond to a 3 out of 4 match at a particular base among the sites, and the capital letters correspond to a perfect match. We assigned this sequence as the VpsR binding site for these promoters. While this 5′→3′ sequence is found on the nontemplate strand for PrbmA, PrbmF, and PvpsL, it occurs on the template strand for PvpsU and thus in the opposite direction relative to the promoter. Comparison of the consensus to a previously determined Logo, compiled from both DNase I footprinting at PvpsL and in silico data (30), indicated a sequence similarity (Fig. 5B). However, the Logo sequence match begins two bases downstream of the consensus sequence determined from our footprints and extends two bases farther downstream. Because of this, the Logo implies an imperfect palindrome of tcTcAnnnnTgAga, whereas our consensus lacks such a palindrome. A clearer elucidation of the specific bases required for VpsR binding awaits the determination of more binding sites and biochemical/structural analyses.
To determine the protein-DNA contacts within the activated transcription complex at these promoters, we performed DNase I footprints of the RNAP/VpsR/c-di-GMP complexes. To ensure that we were observing footprints of the activated transcription complex, we challenged complexes with heparin to eliminate unstable complexes, treated complexes with DNase I, and then isolated the stable transcription complexes from EMSAs prior to purifying the DNA. This procedure enabled us to also remove all uncut background DNA.
In the presence of both VpsR and c-di-GMP, both PrbmF and PvpsU generated footprints expected for an activated transcription complex. At PrbmF, the footprint extended from approximately −85 to +22 with hypersensitivity sites at −36 and −55, consistent with the VpsR binding site centered at −78.5 together with RNAP binding at the core promoter sequence (Fig. 6). At PvpsU, protection extended from −57 to +15, which included the VpsR binding site centered at −40.5 and protection by RNAP downstream; hypersensitivity sites were observed at −26 and −61 (Fig. 6). At either of these promoters, we also observed protection in the presence of RNAP alone (lane 2), consistent with basal transcription that was observed from these promoters in the absence of VpsR. However, within these regions, the patterns obtained by RNAP alone (lane 2) differed from those obtained in the presence of VpsR/c-di-GMP (lane 3). In particular, the strong hypersensitivity sites observed in the presence of VpsR were not seen with RNAP alone. These results indicate that the activated complex contains a different protein-DNA architecture than what is formed by RNAP alone. Unlike PrbmF and PvpsU, we were unable to obtain clear DNase I footprints for VpsR/c-di-GMP and RNAP at PrbmA despite multiple attempts using either template-labeled or non-template-labeled DNA. This result suggests that the activated transcription complex at PrbmA may be unusually unstable.
FIG 6.
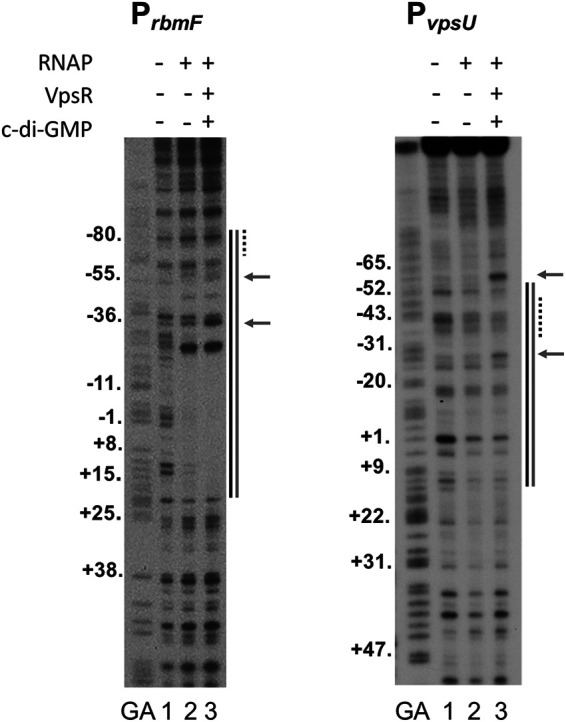
DNase I footprints (template strand) of complexes containing RNAP alone or RNAP with VpsR and c-di-GMP at PrbmF and PvpsU. GA represents G+A ladder. Lanes 1 contain DNA only, lanes 2 contain RNAP, and lanes 3 contain RNAP, VpsR, and 25 μM c-di-GMP. Protection sites of RNAP alone are indicated by a black line to the right of each image. Protection sites of the activated transcription complexes (RNAP, VpsR/c-di-GMP, and DNA) are indicated by a gray line, and hypersensitivity sites are represented by arrows, both to the right of each image. VpsR binding sites (Fig. 1B and Fig. 5B) are indicated by the dashed lines to the right of each image.
DISCUSSION
Not surprisingly, regulation of biofilm genes is complex, requiring multiple transcriptional activators (VpsR, VpsT, and AphA) and repressors (H-NS and HapR). The activators VpsR and VpsT function in conjunction with the small signaling molecule c-di-GMP, whose level increases when biofilm formation is warranted. VpsR induces expression of vpsT, and both transcription factors jointly regulate downstream genes that are necessary for generating the biofilm matrix in a feed-forward network (32). It is thought that VpsT functions through anti-repression of H-NS. Not only do EMSAs show that both H-NS and VpsT/c-di-GMP bind rbmA and vpsU promoters as well as the intergenic region between vpsL and rbmF, but chromatin immunoprecipitation (ChIP) assays have demonstrated that deletion of vpsT results in higher H-NS promoter occupancy, while overexpression of c-di-GMP leads to a decrease in H-NS promoter occupancy at these promoters (18, 38, 39, 45). In addition to VpsR and VpsT, AphA, the master regulator of virulence expression, also upregulates vpsT and vps-I (32). Providing additional repression is HapR, which inhibits both aphA expression and biofilm formation under high-cell density conditions (33, 46). Further regulation is provided by the small molecules guanosine 3′-diphosphate 5′-triphosphate and guanosine 3′,5′-bis (diphosphate) [(p)ppGpp] and cAMP (through the cAMP receptor protein, CRP) (47, 48).
As the master regulator of the c-di-GMP-regulated transcriptional network, VpsR binds c-di-GMP with a Kd of 1.6 μM using unknown residues. Alignment of VpsR with other c-di-GMP binding proteins has not revealed a conserved c-di-GMP binding motif (49), and the crystal structure of VpsR remains unsolved. Recently, we have shown that VpsR directly activates σ70-RNAP transcription from PvpsL, the promoter at the head of the vps-II operon (31). In the work here, we demonstrate that VpsR directly activates σ70-RNAP transcription from the other promoters within the biofilm formation cluster, including PvpsU, at the beginning of the vps-I operon, PrbmA, at the start of the rbm cluster, and PrbmF, which lies upstream of the divergent rbmF and rbmE genes. Thus, despite its sequence similarity to EBPs that work with σ54-RNAP, VpsR functions to directly activate σ70-RNAP transcription for biofilm biogenesis regulation.
A variety of different mechanisms are used to direct RNAP to specific promoters to activate transcription in response to environmental cues and growth signals. Activators of σ70-RNAP can be generally categorized into different classes based on promoter binding location and RNAP contacts. In class I activation, the activator binds upstream of RNAP (typically around −60) and interacts with one or both of the C-terminal domains of the α subunit (αCTD) (22). In class II activation, the activator binds adjacent to or overlapping the −35 element, typically interacting with the C-terminal region of σ70 (region 4) and/or the N-terminal domain of α (αNTD) (22). Furthermore, some activators, such as the Bordetella pertussis response regulator BvgA at PfhaB, use a combination class I/class II, contacting both σ70 region 4 as well as the αCTDs (50). Another set of bacterial regulators, such as MerR, alter the promoter DNA conformation to shorten the suboptimal distance between the −35 and −10 elements, allowing RNAP to bind (51). As shown here, VpsR can be classified as both a class I and a class II activator (Fig. 7). At PvpsL and PvpsU, VpsR functions as a class II activator by binding immediately adjacent/overlapping the core promoter sequence, and at PrbmA and PrbmF, VpsR functions as a class I activator by binding to sequences farther upstream (22, 28).
FIG 7.

VpsR uses at least three different mechanisms to activate transcription. With σ70-RNAP, VpsR and c-di-GMP (cdG) together activate PrbmA and PrbmF using a class I mechanism and activate PvpsL and PvpsU using a class II mechanism. In vivo ChIP-seq analyses suggest a third mechanism: VpsR is an antirepressor of H-NS at PvpsT (38). Promoters, which we tested but did not activate transcription in vitro under our conditions, are labeled with an asterisk. Though direct VpsR binding has been demonstrated for PvpsT, PtfoY, PepsC, and PaphA in EMSAs and or DNase I footprinting (30, 32, 36, 40, 41), transcription mechanisms for these four promoters have not been recapitulated and determined in vitro.
The ability of a regulator to function as both a class I and a class II activator is not unusual and is also seen with Escherichia coli CRP. At the class I lac promoter, CRP binds upstream of the core promoter at a site centered at −61.5, and transcription activation is mediated by protein-protein interactions between a surfaced-exposed α-turn of CRP and one of the αCTDs (52). This increases the affinity of RNAP for promoter DNA, activating transcription through polymerase recruitment. At the class II galP1 promoter, CRP binds to a site centered at −41.5, overlapping the −35 element, and transcription activation is mediated by three protein-protein interactions between CRP and the αCTD, αNTD, and σ70 region 4 portions of RNAP (52). These interactions both recruit RNAP and facilitate the transition from RPc to RPo (52).
We have shown that the presence of c-di-GMP affects the affinity of VpsR for the DNA differently depending on the promoter. While the presence of c-di-GMP increases the affinity of VpsR for its site at the class I rbmA and rbmF promoters (Fig. 2), c-di-GMP does not alter VpsR affinity for its binding site at the class II vpsU and vpsL promoters (Fig. 2 and reference 31, respectively). Based on what is known about CRP as a class I and class II activator, we speculate that VpsR’s mechanism of transcription initiation might work similarly, but in a manner that involves c-di-GMP (Fig. 7). In this scenario, we hypothesize that at the class I promoters, VpsR uses c-di-GMP to enhance DNA binding and then recruit RNAP, whereas at the class II promoters, VpsR uses c-di-GMP to affect its interaction with RNAP, stimulating RPo formation. However, as of now, the contacts between VpsR and RNAP as well as the precise mechanisms are still unknown.
Besides its involvement in biofilm regulation, VpsR also directly binds to several other genes, including the transcription factor genes aphA, vpsT, and tfoY and the promoter upstream of the type II secretion system operon (32, 36, 40, 41). Although EMSAs and/or DNase I footprinting have demonstrated the binding of VpsR to sites upstream of these promoters (32, 36, 40, 41), we did not observe activation at aphA, vpsT, and epsC with σ70-RNAP under our in vitro transcription conditions. These results suggest that VpsR may have additional mechanisms for activating these genes (Fig. 7). In addition, previous ChIP-seq studies of H-NS occupancy in V. cholerae have indicated that VpsR functions as an antirepressor of H-NS (either directly or indirectly) at the vpsT promoter (Fig. 7) (38). Future studies, such as ChIP assays to globally identify VpsR binding sites and reconstruction of vpsT transcription with VpsR and H-NS in vitro, will help determine the mechanism by which VpsR regulates other promoters.
Finally, previous work also indicated that the intergenic region between the divergently transcribed rbmF and vpsL genes contains two VpsR binding sites and suggested that the distal VpsR binding site (relative to the +1 TSS of vpsL) functions as an H-NS antirepressor site for VpsR (31). While this may still be true, it is now also clear that this site is directly involved in activating the divergent rbmF promoter. These two VpsR binding sites are ∼240 bp apart. Our previous work has shown that VpsR forms a dimer in the presence and absence of c-di-GMP in solution (31), leading to the speculation that in the presence of DNA containing both the vpsL and the rbmF binding sites, VpsR might be able to form higher-order structures and generate DNA looping as has been seen for the E. coli regulator GalR (53). Such DNA looping would further prevent H-NS binding and repression while simultaneously bolstering transcription.
In summary, our data support a complex model of vps and rbm regulation. While VpsT/c-di-GMP is important in releasing the DNA from negative regulation by H-NS, VpsR/c-di-GMP is important for positive regulation with σ70-RNAP. Because the various affected promoters vary in their VpsR-DNA binding affinities in the absence and presence of c-di-GMP (Fig. 2) (31), we speculate that the relative activity of each promoter will be different depending on the concentration of VpsR and the level of c-di-GMP. In addition to regulating biofilm matrix genes, VpsR and VpsT concomitantly control, in a c-di-GMP manner, several other phenotypes, including type II secretion, DNA repair, and catalase production, suggesting these transcription factors induce an adaptive program for a sessile lifestyle (41, 54, 55). This two-tiered type of regulation should allow for fine-tuning of gene expression, promoting the exquisite control needed for this sophisticated process.
MATERIALS AND METHODS
DNA.
In vitro transcription templates, pMLH04 containing the vpsT promoter from −188 to +154 relative to the +1 transcriptional start site (TSS), pMLH05 containing the aphA promoter from −127 to +154 relative to the TSS, pMLH06 containing the vpsL promoter from −97 to +213 relative to the TSS, pMLH40 containing the rbmA promoter from −223 to +119 relative to the TSS, pMLH41 containing the rbmF promoter from −228 to +145 relative to the TSS, pMLH42 containing the vpsU promoter from −130 to +155 relative to the TSS, and pMLH43 containing the epsC promoter from −100 to +188 relative to the TSS were cloned into the EcoRI and HindIII restriction enzyme sites of pRLG770 (56). This position places the inserts upstream of the rrnBP1 terminator (56). Inserts were first obtained as PCR products which had been amplified with primers (Table 1) from V. cholerae genomic DNA (BH1514) (Table 2) using Pfu turbo polymerase (Stratagene). Vectors were digested with the appropriate restriction enzymes, ligations were performed using standard Gibson techniques (57), and resulting products were transformed into DH5α. Fragments used for EMSAs and DNase I footprinting were obtained as PCR products using Pfu turbo polymerase and upstream and downstream PCR primers (Table 1) annealing to V. cholerae regions listed above. To radiolabel the DNA, template primer was treated with T4 polynucleotide kinase (Affymetrix) in the presence of [γ-32P] ATP prior to PCR. Radiolabeled PCR products were purified as described (58). pMLH11 is a pET-28b(+) derivative (Novagen) that contains vpsR cloned within the NdeI and XhoI restriction sites constructed as previously described (31).
TABLE 1.
Plasmids and primers used in this study
| Plasmid or primer | Description | Reference no. or sequence(s) |
|---|---|---|
| pCMW75 | IPTG-inducible V. harveyi diguanylate cyclase gene qrgB | 33 |
| pCMW98 | IPTG-inducible V. harveyi diguanylate cyclase gene qrgB mutant (GG→AA) | 33 |
| pMLH04 | vpsT promoter −188 to +154 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, ATTAAGCAACTTGGCTTATATGTG; reverse, GGGTCAGGTGGGACCCAAGCTT, CAAGCCAGAGCTCAGAAAATGGTGT |
| pMLH05 | aphA promoter −127 to +154 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, CTAAAAGGTCACAACTTTGTGGCC; reverse, GGGTCAGGTGGGACCCAAGCTT, GTGCTTCTAATGGCACTCATATCAAC |
| pMLH06 | vpsL promoter −97 to +213 relative to +1 TSS cloned into pRLG770 | 31 |
| pMLH11 | vpsR cloned into pET28b(+) | 31 |
| pMLH17 | vpsR cloned into pHERD20T | 31 |
| pMLH40 | rbmA promoter −223 to +119 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, CTATGCTTGGTTATTGCTCAATGAA; reverse, GGGTCAGGTGGGACCCAAGCTT, AATCCACTTCCGCATAAGAAGCCG |
| pMLH41 | rbmF promoter −228 to +145 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, CAAATAAAACCTTGAGACTCACTTATTAAC; reverse, GGGTCAGGTGGGACCCAAGCTT, GATCAAATGAAGCGGTAGTG |
| pMLH42 | vpsU promoter −130 to +155 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, GATTCAAATCGATGGCTTTTAATAC; reverse, GGGTCAGGTGGGACCCAAGCTT, GCGTTTTTGCCGAATCTTATCGCG |
| pMLH43 | epsC promoter −100 to +188 relative to +1 TSS cloned into pRLG770 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, TCATGCTTAACAATGGTGTTTGCAG; reverse, GGGTCAGGTGGGACCCAAGCTT, GTAGCGTTAATCCTTCGCTGATCGG |
| Reverse primer for primer extension products of rbmA | 84 nucleotides upstream of +1 TSS and 66 nucleotides upstream of +1 GTG translation start site of rbmA | CAATGCCAAGCATGAGGCCA |
| Reverse primer for primer extension products of rbmF | 112 nucleotides upstream of +1 TSS and 67 nucleotides upstream of +1 ATG translation start site of rbmF | CTGCGAATGCATTCATGCTAGG |
| Reverse primer for primer extension products of vpsU | 120 nucleotides upstream of +1 TSS and 67 nucleotides upstream of +1 ATG translation start site of vpsU | CTGCCATTGGCGAACGACA |
| Radiolabeled DNA for EMSA and DNase I footprinting | rbmA promoter −223 to +84 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, CTATGCTTGGTTATTGCTCAATGAA; reverse, CAATGCCAAGCATGAGGCCA |
| Radiolabeled DNA for EMSA and DNase I footprinting | rbmF promoter −228 to +112 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, CAAATAAAACCTTGAGACTCACTTATTAAC; reverse, CTGCCATTGGCGAACGACA |
| Radiolabeled DNA for EMSA and DNase I footprinting | vpsU promoter −130 to +120 | Forward, GAGGCCCTTTCGTCTTCAAGAATTC, GATTCAAATCGATGGCTTTTAATAC; reverse, CTGCCATTGGCGAACGACA |
TABLE 2.
Strains used in this study
| Strain | Description | Source or reference no. |
|---|---|---|
| DH5α | E. coli strain used for cloning | NEBa |
| BL21(DE3) | E. coli strain used for protein purification | NEB |
| Rosetta 2(DE3)/pLysS | E. coli strain used for protein purification | NEB |
| BH1514 | V. cholerae El Tor strain C6707str2 (WT) | 59 |
| WN310 | ΔvpsL ΔvpsR | 32 |
NEB, New England BioLabs.
Strains and growth conditions.
E. coli DH5α was used for cloning, and BL21(DE3) or Rosetta 2(DE3)/pLysS (New England Biolabs) were used for protein purification. The V. cholerae strains used in this study were derived from El Tor biotype C6707str2 (59). Growth conditions for primer extensions and protein purifications are listed below under sections labeled primer extensions and proteins, respectively.
Proteins.
E. coli RNAP core was purchased from New England Biolabs. E. coli σ70 was purified as previously described (60). VpsR protein was isolated from Rosetta 2(DE3)/pLysS (Novagen) containing pMLH11 and grown and purified as previously described (31). Protein concentrations were determined by comparison of known amounts of core RNAP after SDS-PAGE and gel staining with colloidal Coomassie blue (Invitrogen).
In vitro transcriptions.
Single round in vitro transcriptions were assembled first in 5 μl containing 0.02 pmol of supercoiled template, 3.0 pmol of VpsR, 0 or 25 μM c-di-GMP, reconstituted RNAP (0.2 pmol of σ70 plus 0.05 pmol of core), and transcription buffer (40 mM Tris-acetate [pH 7.9], 150 mM potassium glutamate, 4 mM magnesium acetate, 0.1 mM EDTA [pH 7.0], 0.01 mM dithiothreitol [DTT], and 100 μg/ml bovine serum albumin [BSA]). Samples were incubated at 37°C for 10 min prior to the addition of 1 μl ribonucleoside triphosphates (rNTPs) containing 1.43 mM ATP, GTP, CTP, and 36 μM [α-32P] UTP at 5 × 104 dpm/pmol and 500 ng heparin. After incubation for 10 min at 37°C, reactions were collected on dry ice, load solution (15 μl; 9.4 mM EDTA [pH 7], 0.9% bromophenol blue, and 0.9% xylene cyanol dissolved in deionized formamide) was added, and aliquots were electrophoresed on 4% (wt/vol) polyacrylamide, 7 M urea denaturing gels for 2,500 V/h in 0.5× Tris-borate-EDTA (TBE) buffer. After electrophoresis, gels were exposed to X-ray films and films were scanned using a GS-800 calibrated densitometer and analyzed with Quantity One software (Bio-Rad).
Primer extensions.
Primer extension products generated from RNA isolated from in vitro transcriptions were obtained as described above and processed according to the manufacturer’s instructions (Promega) and as described previously (31). Briefly, 5 μl of the in vitro transcription reaction (made using nonradioactive UTP) was added to 6 μl of a primer mixture containing 2× avian myeloblastosis virus (AMV) primer extension buffer, AMV reverse transcriptase, and 2 pmol of 32P-labeled primer (rbmA primer, which anneals 84 bp downstream of the TSS of rbmA, rbmF primer, which anneals 112 bp downstream of the TSS of rbmF, or vpsU primer, which anneals 120 bp downstream of the TSS of vpsU). Samples were electrophoresed on 8% (wt/vol) polyacrylamide, 7 M urea denaturing gels for 5,000 V/h in 0.5× TBE. Imaging, densitometry, and quantification were performed as described above.
In vivo V. cholerae RNA was obtained from strain WN310 (59) containing pMLH17 (31) and either pCMW75 or pCMW98. pCMW75 contains an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible Vibrio harveyi diguanylate cyclase enzyme, QrgB, used to synthesize elevated intracellular concentrations of c-di-GMP, while pCMW98 contains an inactive version of the enzyme used as a negative control to generate unaltered c-di-GMP concentrations (33). Cells were grown and harvested, and RNA was extracted as previously described (31). Primer extension products were performed as described previously (31).
EMSAs.
VpsR-DNA complexes and transcription complexes with RNAP were formed in transcription buffer using 0.05 pmol of DNA as previously described (31). To ensure observation of specific complexes, a 1–μl solution of competitor containing 1 mg/ml of poly(dI-dC) or 500 μg/ml heparin was added to VpsR-DNA complexes (20 μl) during binding or to transcription complexes (10 μl) after binding, respectively. After addition of competitor, VpsR-DNA complexes were loaded onto a 5% (wt/vol) nondenaturing polyacrylamide gel already running at 100 V/h in 1× TBE buffer and subsequently electrophoresed for 1.5 h at 100 V/h. Transcription complexes were loaded onto a 4% (wt/vol) nondenaturing polyacrylamide gel already running at 100 V/h in 1× TBE buffer and electrophoresed for 3 h at 380 V/h. After autoradiography, films were scanned as described above. Kd(app) values were calculated as the concentration of VpsR needed to shift 50% of the free DNA.
DNase I footprinting.
Solutions in transcription buffer were assembled as described above for EMSAs using 0.04 μM DNA and, as indicated, 1.4 μM VpsR, 50 μM c-di-GMP, and/or 0.16 μM reconstituted RNAP. After incubation at 37°C for 10 min, a 1-μl solution of 1 mg/ml of poly(dI-dC) or 500 μg/ml heparin was added to VpsR-DNA complexes and transcription complexes with RNAP, respectively, for 15 s at 37°C. To initiate the cleavage reaction, 0.33 U of DNase I was added to the VpsR-DNA complexes, and 0.375 U of DNase I was added to the transcription complexes with RNAP. Solutions were incubated for an additional 30 s at 37°C, immediately loaded onto a 4% (wt/vol) nondenaturing, polyacrylamide gel already running at 100 V/h in 1× TBE buffer, and electrophoresed for 3 h at 380 V/h. After autoradiography, the protein/DNA complexes were excised, and extracted DNA was electrophoresed on denaturing gels as described (31, 61).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Melissa Arroyo-Mendoza, Abraham Correa-Medina, Leslie Knipling, Kyung Moon, Jennifer Patterson-West, Bokyung Son, and members of the Waters laboratory for helpful discussions and comments on the manuscript.
We received funding from the National Institutes of Health (NIH) (grant no. F30GM123632 to M.L.H. and R01GM109259 to C.M.W.), National Science Foundation (grant no. MCB1253684 to C.M.W.), Michigan State University DO/PhD Program (to M.L.H.), and intramural research program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (to M.L.H. and D.M.H.).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Kaper JB, Morris JG, Jr, Levine MM. 1995. Cholera. Clin Microbiol Rev 8:48–86. doi: 10.1128/CMR.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faruque SM, Albert MJ, Mekalanos JJ. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol Mol Biol Rev 62:1301–1314. doi: 10.1128/MMBR.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alam M, Islam A, Bhuiyan NA, Rahim N, Hossain A, Khan GY, Ahmed D, Watanabe H, Izumiya H, Faruque AS, Akanda AS, Islam S, Sack RB, Huq A, Colwell RR, Cravioto A. 2011. Clonal transmission, dual peak, and off-season cholera in Bangladesh. Infect Ecol Epidemiol 1:1–13. doi: 10.3402/iee.v1i0.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Islam MS, Jahid MI, Rahman MM, Rahman MZ, Islam MS, Kabir MS, Sack DA, Schoolnik GK. 2007. Biofilm acts as a microenvironment for plankton-associated Vibrio cholerae in the aquatic environment of Bangladesh. Microbiol Immunol 51:369–379. doi: 10.1111/j.1348-0421.2007.tb03924.x. [DOI] [PubMed] [Google Scholar]
- 5.Faruque SM, Biswas K, Udden SM, Ahmad QS, Sack DA, Nair GB, Mekalanos JJ. 2006. Transmissibility of cholera: in vivo-formed biofilms and their relationship to infectivity and persistence in the environment. Proc Natl Acad Sci U S A 103:6350–6355. doi: 10.1073/pnas.0601277103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conner JG, Teschler JK, Jones CJ, Yildiz FH. 2016. Staying alive: Vibrio cholerae's cycle of environmental survival, transmission, and dissemination. Microbiol Spectr 4. doi: 10.1128/microbiolspec.VMBF-0015-2015. [DOI] [Google Scholar]
- 7.Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. 2010. A bistable switch and anatomical site control Vibrio cholerae virulence gene expression in the intestine. PLoS Pathog 6:e1001102. doi: 10.1371/journal.ppat.1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alam M, Sultana M, Nair GB, Siddique AK, Hasan NA, Sack RB, Sack DA, Ahmed KU, Sadique A, Watanabe H, Grim CJ, Huq A, Colwell RR. 2007. Viable but nonculturable Vibrio cholerae O1 in biofilms in the aquatic environment and their role in cholera transmission. Proc Natl Acad Sci U S A 104:17801–17806. doi: 10.1073/pnas.0705599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colwell RR, Huq A, Islam MS, Aziz KM, Yunus M, Khan NH, Mahmud A, Sack RB, Nair GB, Chakraborty J, Sack DA, Russek-Cohen E. 2003. Reduction of cholera in Bangladeshi villages by simple filtration. Proc Natl Acad Sci U S A 100:1051–1055. doi: 10.1073/pnas.0237386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huq A, Small EB, West PA, Huq MI, Rahman R, Colwell RR. 1983. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Appl Environ Microbiol 45:275–283. doi: 10.1128/AEM.45.1.275-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Absalon C, Van Dellen K, Watnick PI. 2011. A communal bacterial adhesin anchors biofilm and bystander cells to surfaces. PLoS Pathog 7:e1002210. doi: 10.1371/journal.ppat.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong JC, Syed KA, Klose KE, Yildiz FH. 2010. Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiology 156:2757–2769. doi: 10.1099/mic.0.040196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yildiz FH, Schoolnik GK. 1999. Vibrio cholerae O1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci U S A 96:4028–4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong JC, Karplus K, Schoolnik GK, Yildiz FH. 2006. Identification and characterization of RbmA, a novel protein required for the development of rugose colony morphology and biofilm structure in Vibrio cholerae. J Bacteriol 188:1049–1059. doi: 10.1128/JB.188.3.1049-1059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong JC, Yildiz FH. 2007. The rbmBCDEF gene cluster modulates development of rugose colony morphology and biofilm formation in Vibrio cholerae. J Bacteriol 189:2319–2330. doi: 10.1128/JB.01569-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moorthy S, Watnick PI. 2005. Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol Microbiol 57:1623–1635. doi: 10.1111/j.1365-2958.2005.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berk V, Fong JC, Dempsey GT, Develioglu ON, Zhuang X, Liphardt J, Yildiz FH, Chu S. 2012. Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337:236–239. doi: 10.1126/science.1222981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teschler JK, Zamorano-Sanchez D, Utada AS, Warner CJ, Wong GC, Linington RG, Yildiz FH. 2015. Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat Rev Microbiol 13:255–268. doi: 10.1038/nrmicro3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol 187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh M-L, Hinton DM, Waters CM. 2020. Cyclic di-GMP regulation of gene expression, p 379–394. In Chou S-H, Guiliani N, Lee VT, Römling U (ed), Microbial cyclic di-nucleotide signaling. Springer International Publishing, Cham, Switzerland. doi: 10.1007/978-3-030-33308-9_23. [DOI] [Google Scholar]
- 22.Lee DJ, Minchin SD, Busby SJ. 2012. Activating transcription in bacteria. Annu Rev Microbiol 66:125–152. doi: 10.1146/annurev-micro-092611-150012. [DOI] [PubMed] [Google Scholar]
- 23.Feklistov A, Darst SA. 2011. Structural basis for promoter-10 element recognition by the bacterial RNA polymerase sigma subunit. Cell 147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH. 2012. Structural basis of transcription initiation. Science 338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hook-Barnard IG, Hinton DM. 2007. Transcription initiation by mix and match elements: flexibility for polymerase binding to bacterial promoters. Gene Regul Syst Bio 1:275–293. doi: 10.1177/117762500700100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saecker RM, Record MT, Jr, Dehaseth PL. 2011. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J Mol Biol 412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker KB, Hinton DM. 2013. Transcription regulation at the core: similarities among bacterial, archaeal, and eukaryotic RNA polymerases. Annu Rev Microbiol 67:113–139. doi: 10.1146/annurev-micro-092412-155756. [DOI] [PubMed] [Google Scholar]
- 28.Browning DF, Busby SJ. 2016. Local and global regulation of transcription initiation in bacteria. Nat Rev Microbiol 14:638–650. doi: 10.1038/nrmicro.2016.103. [DOI] [PubMed] [Google Scholar]
- 29.Yildiz FH, Dolganov NA, Schoolnik GK. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPS(ETr)-associated phenotypes in vibrio cholerae O1 El Tor. J Bacteriol 183:1716–1726. doi: 10.1128/JB.183.5.1716-1726.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamorano-Sanchez D, Fong JC, Kilic S, Erill I, Yildiz FH. 2015. Identification and characterization of VpsR and VpsT binding sites in Vibrio cholerae. J Bacteriol 197:1221–1235. doi: 10.1128/JB.02439-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh ML, Hinton DM, Waters CM. 2018. VpsR and cyclic di-GMP together drive transcription initiation to activate biofilm formation in Vibrio cholerae. Nucleic Acids Res 46:8876–8887. doi: 10.1093/nar/gky606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava D, Harris RC, Waters CM. 2011. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J Bacteriol 193:6331–6341. doi: 10.1128/JB.05167-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waters CM, Lu W, Rabinowitz JD, Bassler BL. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J Bacteriol 190:2527–2536. doi: 10.1128/JB.01756-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beyhan S, Bilecen K, Salama SR, Casper-Lindley C, Yildiz FH. 2007. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J Bacteriol 189:388–402. doi: 10.1128/JB.00981-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. 2010. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science 327:866–868. doi: 10.1126/science.1181185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin W, Kovacikova G, Skorupski K. 2007. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64:953–967. doi: 10.1111/j.1365-2958.2007.05693.x. [DOI] [PubMed] [Google Scholar]
- 37.Casper-Lindley C, Yildiz FH. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J Bacteriol 186:1574–1578. doi: 10.1128/jb.186.5.1574-1578.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayala JC, Wang H, Silva AJ, Benitez JA. 2015. Repression by H-NS of genes required for the biosynthesis of the Vibrio cholerae biofilm matrix is modulated by the second messenger cyclic diguanylic acid. Mol Microbiol 97:630–645. doi: 10.1111/mmi.13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ayala JC, Wang H, Benitez JA, Silva AJ. 2015. RNA-Seq analysis and whole genome DNA-binding profile of the Vibrio cholerae histone-like nucleoid structuring protein (H-NS). Genom Data 5:147–150. doi: 10.1016/j.gdata.2015.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pursley BR, Maiden MM, Hsieh ML, Fernandez NL, Severin GB, Waters CM. 2018. Cyclic di-GMP regulates TfoY in Vibrio cholerae to control motility by both transcriptional and posttranscriptional mechanisms. J Bacteriol 200:578–617. doi: 10.1128/JB.00578-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sloup RE, Konal AE, Severin GB, Korir ML, Bagdasarian MM, Bagdasarian M, Waters CM. 2017. Cyclic Di-GMP and VpsR induce the expression of Type II secretion in Vibrio cholerae. J Bacteriol 199:e00106-17. doi: 10.1128/JB.00106-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papenfort K, Forstner KU, Cong JP, Sharma CM, Bassler BL. 2015. Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc Natl Acad Sci U S A 112:E766–E775. doi: 10.1073/pnas.1500203112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manneh-Roussel J, Haycocks JRJ, Magan A, Perez-Soto N, Voelz K, Camilli A, Krachler AM, Grainger DC. 2018. cAMP receptor protein controls Vibrio cholerae gene expression in response to host colonization. mBio 9:e00966-18. doi: 10.1128/mBio.00966-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yildiz FH, Liu XS, Heydorn A, Schoolnik GK. 2004. Molecular analysis of rugosity in a Vibrio cholerae O1 El Tor phase variant. Mol Microbiol 53:497–515. doi: 10.1111/j.1365-2958.2004.04154.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Ayala JC, Silva AJ, Benitez JA. 2012. The histone-like nucleoid structuring protein (H-NS) is a repressor of Vibrio cholerae exopolysaccharide biosynthesis (vps) genes. Appl Environ Microbiol 78:2482–2488. doi: 10.1128/AEM.07629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hammer BK, Bassler BL. 2007. Regulatory small RNAs circumvent the conventional quorum sensing pathway in pandemic Vibrio cholerae. Proc Natl Acad Sci U S A 104:11145–11149. doi: 10.1073/pnas.0703860104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson J, Balsalobre C, Wang SY, Urbonaviciene J, Jin DJ, Sonden B, Uhlin BE. 2000. Nucleoid proteins stimulate stringently controlled bacterial promoters: a link between the cAMP-CRP and the (p)ppGpp regulons in Escherichia coli. Cell 102:475–485. doi: 10.1016/s0092-8674(00)00052-0. [DOI] [PubMed] [Google Scholar]
- 48.Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J Mol Biol 293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 49.Matsuyama BY, Krasteva PV, Baraquet C, Harwood CS, Sondermann H, Navarro MV. 2016. Mechanistic insights into c-di-GMP-dependent control of the biofilm regulator FleQ from Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 113:E209–18. doi: 10.1073/pnas.1523148113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Decker KB, Chen Q, Hsieh ML, Boucher P, Stibitz S, Hinton DM. 2011. Different requirements for sigma Region 4 in BvgA activation of the Bordetella pertussis promoters P(fim3) and P(fhaB). J Mol Biol 409:692–709. doi: 10.1016/j.jmb.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. 2003. The MerR family of transcriptional regulators. FEMS Microbiol Rev 27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 52.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. 2004. Catabolite activator protein: DNA binding and transcription activation. Curr Opin Struct Biol 14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qian Z, Dimitriadis EK, Edgar R, Eswaramoorthy P, Adhya S. 2012. Galactose repressor mediated intersegmental chromosomal connections in Escherichia coli. Proc Natl Acad Sci U S A 109:11336–11341. doi: 10.1073/pnas.1208595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernandez NL, Srivastava D, Ngouajio AL, Waters CM. 2018. Cyclic di-GMP positively regulates DNA repair in Vibrio cholerae. J Bacteriol 200:e00005-18. doi: 10.1128/JB.00005-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez NL, Waters CM. 2019. Cyclic di-GMP increases catalase production and hydrogen peroxide tolerance in Vibrio cholerae. Appl Environ Microbiol 85:e01043-19. doi: 10.1128/AEM.01043-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ross W, Thompson JF, Newlands JT, Gourse RL. 1990. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J 9:3733–3742. doi: 10.1002/j.1460-2075.1990.tb07586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 58.March-Amegadzie R, Hinton DM. 1995. The bacteriophage T4 middle promoter PuvsX: analysis of regions important for binding of the T4 transcriptional activator MotA and for activation of transcription. Mol Microbiol 15:649–660. doi: 10.1111/j.1365-2958.1995.tb02374.x. [DOI] [PubMed] [Google Scholar]
- 59.Thelin KH, Taylor RK. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect Immun 64:2853–2856. doi: 10.1128/IAI.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsieh ML, James TD, Knipling L, Waddell MB, White S, Hinton DM. 2013. Architecture of the bacteriophage T4 activator MotA/promoter DNA interaction during sigma appropriation. J Biol Chem 288:27607–27618. doi: 10.1074/jbc.M113.475434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boulanger A, Moon K, Decker KB, Chen Q, Knipling L, Stibitz S, Hinton DM. 2015. Bordetella pertussis fim3 gene regulation by BvgA: phosphorylation controls the formation of inactive vs. active transcription complexes. Proc Natl Acad Sci U S A 112:E526–E535. doi: 10.1073/pnas.1421045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.