

Previous studies identified sorting nexins 17 and 27, as well as the retromer complex, as playing a role in HPV infection. This study shows that the newly identified retriever complex also plays an important role and begins to shed light on how both sorting nexins contribute to retromer and retriever recruitment during the infection process.
KEYWORDS: human papillomavirus, retriever, retromer, sorting nexins, viral trafficking
ABSTRACT
Previous studies have identified an interaction between the human papillomavirus (HPV) L2 minor capsid protein and sorting nexins 17 and 27 (SNX17 and SNX27) during virus infection. Further studies show the involvement of both retromer and retriever complexes in this process since knockdown of proteins from either complex impairs infection. In this study, we show that HPV L2 and 5-ethynyl-2′-deoxyuridine (EdU)-labeled pseudovirions colocalize with both retromer and retriever, with components of each complex being bound by L2 during infection. We also show that both sorting nexins may interact with either of the recycling complexes but that the interaction between SNX17 and HPV16 L2 is not responsible for retriever recruitment during infection, instead being required for retromer recruitment. Furthermore, we show that retriever recruitment most likely involves a direct interaction between L2 and the C16orf62 subunit of the retriever, in a manner similar to that of its interaction with the VPS35 subunit of retromer.
IMPORTANCE Previous studies identified sorting nexins 17 and 27, as well as the retromer complex, as playing a role in HPV infection. This study shows that the newly identified retriever complex also plays an important role and begins to shed light on how both sorting nexins contribute to retromer and retriever recruitment during the infection process.
INTRODUCTION
A wide variety of different viruses infect mammalian cells by exploiting and manipulating the cellular endocytic pathways. Whether this involves the endoplasmic reticulum or endosomal sorting pathways, viral proteins must interact with cellular components of either or both networks to transport their genomes to suitable sites for replication (1). In the case of human papillomaviruses (HPVs), capsid entry is a highly complex process, with both the major (L1) and minor (L2) capsid components playing essential roles (for a general review on L2 function, see reference 2). Following capsid binding to a primary receptor on the cell surface, the L2 protein is cleaved by cell surface furin. There is then subsequent cleavage of the L1 capsid protein by kallikrein-8, and the capsid binds to a secondary surface receptor whose identity is unknown but which may involve growth factor receptors, and previous studies have shown that the endocytosis of viral particles stimulates the production of tubular recycling endosomes in a VAP- and epidermal growth factor receptor (EGFR)-dependent manner (3, 4). Acidification of the endosomal compartments further loosens the capsid structure, allowing at least a partial separation of L1 and L2. Further disassembly is accomplished through the interactions of cyclophilins and ESCRT components (5–7), ensuring that the majority of L1 is sorted to lysosomes, where it is degraded, while L2 and the viral genome avoid destruction by engaging with endosomal sorting complexes. Current models suggest that the viral genome is bound in a sequence-specific manner by L1 pentamers, which in turn are tethered to the amino terminus of L2 in the lumen of sorting endosomes (8). L2 passes through the endosomal membrane by means of a transmembrane domain (9), and the remainder of the molecule lies in the cytosol, where it interacts with a variety of vesicle coat proteins that mediate its retrograde transport to the trans-Golgi apparatus. Following nuclear membrane dissolution during mitosis, which is required for nuclear entry (10), L2 binds to mitotic chromosomes, most likely through its chromatin-binding residues (11), before moving to nuclear promyelocytic leukemia protein (PML) bodies (12).
Studies using small interfering RNA (siRNA) screening and siRNA-mediated knockdown of individual subunits identified retromer as one of the endosomal coat proteins required for viral trafficking (13), and further work showed that retromer could bind to the carboxy terminus of HPV16 L2 (14). Our previous studies indicated that L2 proteins from a variety of different HPV types interact with sorting nexin 27 (SNX27), a component and cargo carrier of the retromer complex, through a noncanonical interaction with the SNX27 PDZ domain (15), thus indicating a further means by which L2 could interact with the retromer complex. However, L2 also interacts with SNX17 through an NPAY motif conserved in a variety of HPV L2 proteins, and this interaction is highly important for infection, although SNX17 is not considered to be a retromer component (16, 17). The interaction with SNX17 can be detected as early as 2 h postinfection (18), but it was not clear whether other sorting complex components are similarly bound at this time. siRNA-mediated knockdown of either SNX17 or SNX27 has been shown to reduce infection efficiency in a pseudovirion (PsV) infection model (15–17). However, their relative contributions to viral trafficking appear to differ, with SNX27 knockdown having less of an effect on infection efficiency than SNX17 knockdown.
Recently, a new endosomal sorting complex was identified from a series of human proteomic interaction networks and originally termed “commander” (19–23). It has similarities to the retromer complex, with a basic core trimer formed from C16orf62 (also termed VPS35L), DSCR3 (also termed VPS26c), and VPS29. However, unlike retromer, commander has additional subunits, including CCD22, CCD93, and a variety of COMMD proteins. McNally et al. (24), who also identified this complex and termed it “retriever,” showed that siRNA-mediated knockdown of several of its subunits impairs HPV infection efficiency, thus implicating commander/retriever as being important for viral infection. Previous proteomic studies identified SNX27 as a component of the retromer complex and SNX17 as a component of retriever. The two complexes appear to control the fates of different cargo groups, and although they can be present simultaneously in the same vesicular compartment, they are recruited to endosomal sites by different mechanisms (24). There is, however, some evidence that SNX17, although not considered to be a retromer component, can enhance the sorting of at least one cellular cargo in a retromer-dependent manner (25).
In this study, we demonstrate that both complexes may be recruited by HPV L2 during viral trafficking but that retromer is recruited more frequently than retriever. We show that HPV16 L2 can simultaneously interact with both SNX17 and SNX27 and also that both sorting nexins may interact with each other. However, the requirement for L2-SNX17 binding may be separate from the SNX17-retriever interaction, since infection efficiency reduced by SNX17 knockdown can be rescued by the expression of SNX17 mutants that do not interact with retriever. We show here that SNX17 may interact with retromer subunits, while SNX27 may interact with retriever subunits. We also show that the cytosolic region of L2 interacts directly with the VPS35 subunit of the retromer complex, as previously demonstrated (14), as well as with the C16orf62 (VPS35L) subunit of retriever.
RESULTS
HPV16 L2 binds to both VPS35 of the retromer complex and C16or62 of the retriever complex.
Because siRNA knockdown experiments had previously shown that the retriever complex is important for viral trafficking (24) (Fig. 1), we reasoned that HPV16 L2 might recruit retriever as well as retromer to accomplish this. Previous studies suggested that HPV16 L2 binds directly to retromer through residues in the cytoplasmic portion of the L2 molecule (14). We predicted that, because the VPS35 subunit of retromer and the C16orf62 subunit of retriever are structurally similar, L2 might interact directly with both retromer and retriever in a similar manner. To examine this hypothesis, we expressed an 86-amino-acid C-terminal portion of HPV16 L2 as a glutathione S-transferase (GST) fusion protein, introducing substitution mutations at residues FYL and YYMD and both sets of residues together (Fig. 2A). We incubated these with HEK 293T cell lysates, and the levels of endogenous VPS35 and C16orf62 binding were monitored by Western blotting. The results in Fig. 2B show that despite the relative abundance of both endogenous VPS35 and C16orf62, the interactions between them and the wild-type (WT) L2 C-terminal fragment are extremely weak. However, L2 appears to bind C16orf62 more strongly than VPS35, and in both cases, the mutant L2 fragments bind less efficiently than the wild type.
FIG 1.

As previously shown by McNally et al. (24), siRNA-mediated knockdown of several retriever complex subunits reduces PsV infection efficiency. We also examined the levels of sorting nexin 27 in response to siRNA-mediated knockdown of the retriever subunits. (A) Histograms showing reduced infection efficiency in HaCaT cells of HPV16 pseudovirions upon siRNA-mediated knockdown of retriever complex components, as measured by luminometry. Error bars represent standard deviations. (B) Western blotting of the cell extracts used for luminometry showing the efficiency of siRNA knockdowns. Of note, the knockdown of the retriever subunit CCDC93 also leads to reduced SNX27 levels. Scr., scrambled siRNA.
FIG 2.
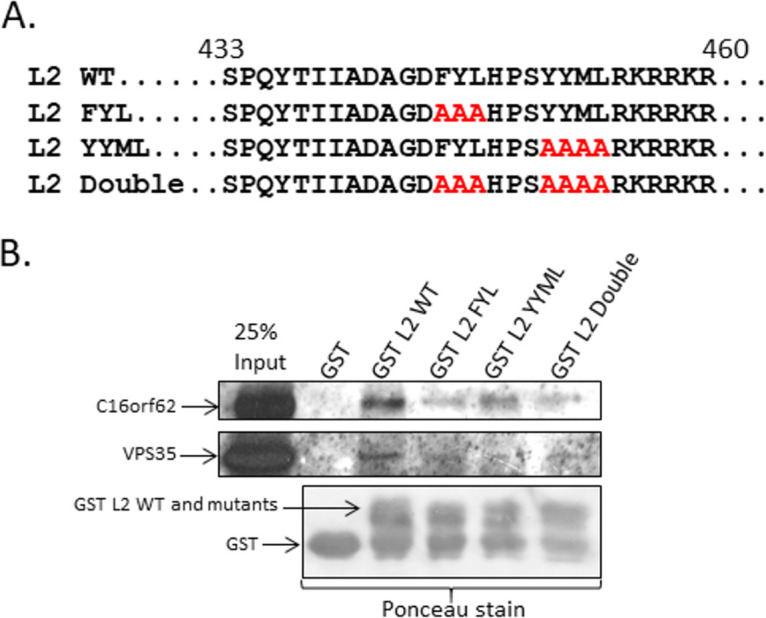
The carboxy terminus of HPV16 L2 interacts with C16orf62 of the retriever complex via the same residues used for its interaction with VPS35 of the retromer complex. (A) We cloned a 106-amino-acid C-terminal fragment of HPV16 L2 into the GST tag vector pQE9 and then made mutations in the same sites previously demonstrated to be required for binding to VPS35 (14), FYL to AAA and YYML to AAAA, and the double mutation. (B) We incubated the wild-type and single and double mutant proteins bound to nickel agarose with the HEK 293T whole-cell lysate and then analyzed bound VPS35 and C16orf62 by Western blot analysis.
Retriever and retromer components can independently colocalize with HPV16 L2 during infection.
To examine the relative contributions of retromer and retriever sorting complexes to retrograde trafficking of the viral genome during HPV infection, we compared the colocalizations of HPV16 L2 with VPS35, as a marker of the retromer complex, and with C16orf62, as a marker of the retriever complex. HeLa cells were infected with HPV16 PsVs, and at 4 h postinfection, cells were fixed and stained for VPS35, C16orf62, and HPV16 L2. The results are shown in a series of selected z-stacks from a single cell in Fig. 3A. A separate cell, color separated and color combined to more easily see the colocalization, is shown in Fig. 3B, and these images confirm apparent colocalizations between retromer and HPV16 L2 and between retriever and L2. Interestingly, in a number of cases, when we examined z-stacked cells, we observed colocalization of all three signals. This was more frequently visible toward the cell apex and appeared to occur in larger structures, visible in at least 3 consecutive slices at a separation of 150 nm, that might be endosomal membranes, as shown in Fig. 3C. These data suggest that retriever and retromer can be simultaneously recruited by the viral L2 protein during viral trafficking. No evidence of retromer and retriever colocalization was seen in uninfected control cells at the same time point. This is consistent with previous observations that retriever and retromer do not occupy the same regions of endosomal membranes (24); indeed, we observed that they do not typically localize even to the same parts of the cell. Both interactions are also readily visible by fluorescence microscopy as early as 2 h and as late as 7 h postinfection, as shown in Fig. 4A and B. Although we did not quantitate the apparent colocalization of VPS35, L2, and VC16orf62 together, we measured separately the frequencies of L2 colocalization with VPS35 and C16orf62 at 2, 4, and 7 h postinfection using Manders’ coefficient; L2’s recruitment and binding of retromer occur at a higher frequency than its interaction with retriever. We also observed that the frequencies of interaction with both complexes are similar at 2 and 4 h postinfection, but that by 7 h postinfection there is a slight decrease in the L2 signal overlapping VPS35 and a slight increase in the L2 signal overlapping C16orf62 (Fig. 4C). Interestingly, when we examined separately the interactions of HPV16 L2 with SNX17 and SNX27, we also observed numerous cases of both sorting nexins colocalizing with L2 at the 2-h time point, as seen in Fig. 5, suggesting that both sorting nexins may be required for the recruitment of sorting/recycling complexes early in infection.
FIG 3.

Retriever and retromer complexes colocalize together with HPV16 L2 during viral infection. (A) A series of six enlarged images from a z-stacked uninfected HeLa cell (top row) and an HPV16 PsV-infected cell (bottom row), at 4 h postinfection, probed for VPS35, C16orf62, and HPV16 L2 (16L2). In uninfected cells, there is no indication of retromer and retriever colocalization, but when HPV16 L2 is present, clear examples of L2 colocalization with VPS35 or C16orf62 or of all three proteins colocalizing together are visible, in structures that may be endosomal membranes or tubules. (B) Enlarged image of the topmost slice of a different z-stacked HeLa cell at 4 h postinfection showing fluorescence for VPS35, C16orf62, and HPV16 L2 separately and color combined. (C) Enlarged images of consecutive slices, 150 nm apart, of the same z-stacked cell shown in panel B demonstrating that the structures to which C16orf62, VPS35, and HPV16 L2 all colocalize are sufficiently large as to be visible in descending microscopic slices.
FIG 4.
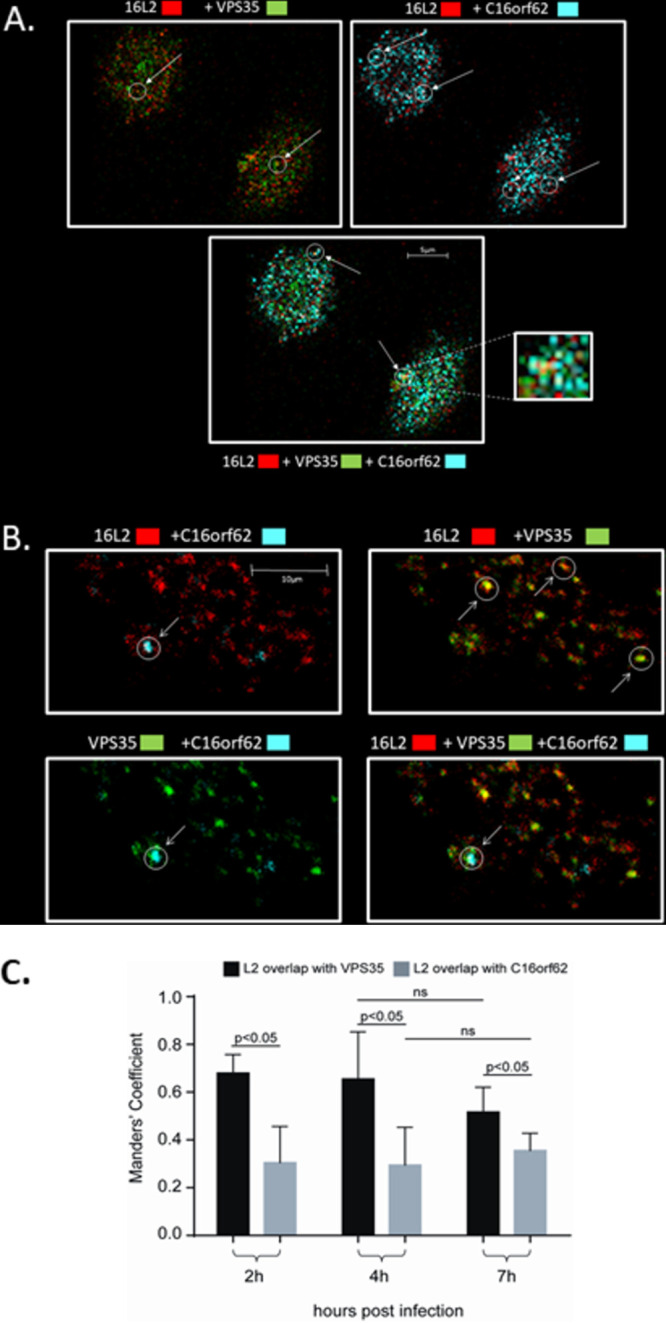
(A) Enlarged images from a single HeLa cell at 2 h postinfection with separated color channels showing levels of colocalization between L2 and VPS35, L2 and C16orf62, VPS35 and C16orf62, and all three proteins together. (B) Enlarged images from a single slice of a z-stacked HeLa cell at 7 h postinfection with separated color channels showing that HPV16 L2 can still be observed associating separately with both retriever and retromer and with both complexes together. (C) Manders’ coefficient analysis showing the frequency of association between HPV16 L2 and VPS35 or C16orf62 at 2, 4, and 7 h postinfection. ns, not significant.
FIG 5.
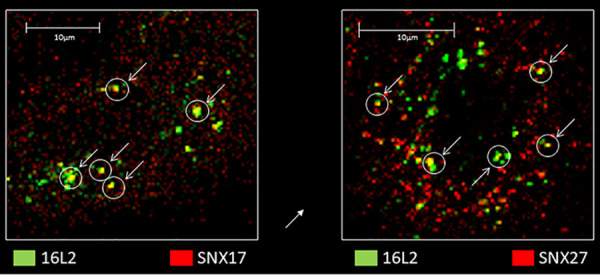
Engagement of HV16 L2 with SNX17 (left) and SNX27 (right) at 2 h postinfection.
Sorting nexins 17 and 27 can bind simultaneously to HPV16 L2 but may also interact with each other.
Previous studies have shown that HPV16 L2 interacts with SNX17 and SNX27 through two independent mechanisms (15, 16). The above-described results indicate that L2 can recruit both sorting nexins to the same location within the cell, indicating that this may occur through simultaneous interactions. To determine whether both SNX17 and SNX27 can indeed be bound by L2 at the same time, GST-L2 pulldown assays were performed using increasing amounts of whole-cell lysates in which either SNX17 or SNX27 was expressed until L2 was saturated. We then incubated the saturated GST-L2 agarose beads with a lysate containing the other sorting nexin. The results, shown in Fig. 6A, indicate that even when GST-L2 has bound one sorting nexin to saturation, it can still bind to the second sorting nexin. This suggests that both sorting nexins can interact simultaneously with a single L2 molecule. However, when we looked at cells constitutively expressing green fluorescent protein (GFP)-tagged SNX27, we observed strong colocalization with SNX17, as shown in Fig. 6B, suggesting that the two sorting nexins may be very closely associated with each other. When we transfected HEK 293T cells with constructs expressing myc-tagged SNX27 and Flag-tagged SNX17, immunoprecipitation of SNX27 with anti-myc antibodies allowed the detection of bound Flag-tagged SNX17 by Western blot analysis (Fig. 6C). Although neither the immunofluorescence nor coimmunoprecipitation experiments are definitive proof of a direct interaction between SNX17 and SNX27, they show that both sorting nexins may be closely associated during certain endosomal sorting/recycling events and further suggest a means by which both might interact simultaneously with L2.
FIG 6.

Sorting nexins 17 and 27 can bind simultaneously to a single L2 molecule but may also interact directly with each other. (A) GST-fused HPV16 L2 bound to glutathione-agarose beads was incubated with increasing amounts of an HEK 293T cell extract in which either SNX17 or SNX27 was expressed until saturation was achieved. At this point, the cell extract expressing the alternative sorting nexin was added. After washing, bound proteins were detected by probing the Western blot (WB) with anti-SNX17 or anti-SNX27 antibodies. (B) Anti-SNX17 immunofluorescence of HeLa cells constitutively expressing GFP-tagged SNX27 showing very high levels of colocalization between both sorting nexins. (C) Immunoprecipitation (IP) of myc-tagged SNX27 pulls down detectable levels of Flag-tagged SNX17 from extracts of HEK 293T cells expressing both tagged proteins.
Infection by HPV16 requires SNX17, but SNX17 does not require interaction with retriever.
We have previously demonstrated the important role played by SNX17 during infection with HPV pseudovirions (16–18) and that this role requires a direct interaction with HPV L2 protein. Consequently, we now asked whether this interaction was required for the recruitment of retriever during viral infection. We examined this by measuring the infection efficiency in H1299 cells in which SNX17 levels were reduced by siRNA treatment and then replaced with siRNA-resistant wild-type SNX17 and two SNX17 mutants that fail to interact with the retriever complex. As shown in Fig. 7, the infection efficiency is rescued by both wild-type SNX17 and the mutants that fail to bind to retriever. Nearly identical results were obtained after the expression of the same constructs in HeLa cells CRISPR engineered to knock out SNX17 expression (a kind gift of Kerrie McNally) (data not shown). These findings argue that although the interaction of SNX17 with HPV L2 is essential for viral infection, SNX17 does not need to bind to the retriever complex and is therefore probably not the means by which retriever is recruited by HPV L2.
FIG 7.

Sorting nexin 17 binding to the retriever complex is not required for HPV PsV trafficking. H1299 cells were transfected with siRNA against SNX17, and after 48 h they were then transfected with constructs expressing siRNA-resistant wild-type SNX17 and two mutants of SNX17 that are unable to bind to the retriever complex. After a further 24 h, the cells were infected with PsVs, and 48 h later, the infection efficiency was determined by luminometry. (A) Western blot showing siRNA-mediated knockdown of endogenous SNX17 and expression levels of ectopically expressed siRNA-resistant WT and mutant SNX17. Scrambled siRNA (siScr.) was used as a negative control. (B) Histogram of infection efficiency, based on data from 4 separate experiments. Error bars represent standard deviations.
HPV16 PsV infection increases SNX17 colocalization with VPS35.
If the function of SNX17 for trafficking during HPV16 infection does not require its direct interaction with retriever, we asked whether it might instead be involved in the recruitment of the retromer. To answer this question, we analyzed the colocalization of SNX17 and VPS35 in cells infected with HPV16 PsVs and compared this with that found in uninfected cells. To do this, HeLa cells, seeded on coverslips, were infected with HPV16 PsVs that contained 5-ethynyl-2′-deoxyuridine (EdU)-labeled pseudogenomes. After 4 h, cells were fixed and permeabilized, and the EdU-labeled genomes were stained using a Click-iT assay (Invitrogen). SNX17 and VPS35 were detected by immunofluorescence, and the cells were analyzed for the colocalization of SNX17 and VPS35 in the presence or absence of EdU-labeled viral pseudogenomes. The results shown in Fig. 8A and B indicate first that SNX17 and VPS35 colocalize with a frequency that remains more or less constant, in both uninfected and infected cells. However, during pseudovirion infection, the frequency with which they colocalize together with EdU-labeled pseudogenomes is far higher than that in the absence of EdU-labeled pseudogenomes. In contrast, when the assay was repeated with pseudovirions where the SNX17-binding motif of L2 had been mutated from NPAY to AAAA to abolish SNX17 interaction (17), the colocalization of SNX17, VPS35, and EdU-labeled viral genomes was greatly reduced, to the level of SNX17/VPS35 colocalization observed in uninfected cells. These results demonstrate that HPV16 L2, through association with SNX17, promotes the recruitment of the retromer complex following HPV infection.
FIG 8.

Sorting nexin 17 and VPS35 colocalize more frequently in the presence of EdU-labeled pseudogenomes than in their absence but at a lower frequency when the NPAY SNX17-binding motif of L2 is mutated to AAAA. (A) Representative immunofluorescence images showing the colocalization of SNX17, VS35, and EdU-labeled pseudogenomes. (B) The colocalization between SNX17 and VPS35 was analyzed by Pearson’s correlation coefficients using Fiji software on 30 individual cells under each condition. Statistical significance between uninfected cells and cells infected with the NPAY-AAAA mutant and WT HPV16 was determined by one-way analysis of variance (ANOVA). NS, not significant. ****, P < 0.0001; statistically significant.
Sorting nexins 17 and 27 can interact with subunits of both retriever and retromer complexes.
Although no previous proteomic studies had identified any connection between SNX17 and the retromer or between SNX27 and the retriever, we wanted to understand how SNX17 binding by L2 appears necessary for retromer rather than retriever recruitment and how SNX27 function might relate to the retriever. To study this, we carried out two parallel experiments, expressing Flag-tagged SNX17 and untagged SNX27 in HEK 293T cells and then incubating whole-cell lysates with GST alone and GST-fused VPS26, VPS35, VPS29, C16orf62, and DSCR3. The results, shown in Fig. 9A and B, suggest that, despite the lack of previous evidence, SNX17 may interact with the retromer through binding the VPS35 and VPS29 subunits and that SNX27 may be able to interact weakly with all the GST-fused subunits, although we cannot rule out that endogenous sorting complex proteins in the cell lysates may be contributing to the pulldown of ectopically expressed proteins. We note that Ponceau staining of the two membranes does not reveal expressed GST-C16orf62 despite its predicted molecular mass being around 140 kDa. To confirm that it was expressed, we probed one of the two membranes with antibodies against C16orf62, and the extra panel in Fig. 9B confirms this; extra bands below the predicted molecular mass are probably breakdown products.
FIG 9.

SNX17 and -27 may interact with retromer and retriever complexes, respectively. Purified GST and GST-fused VPS26, VPS35, VPS29, C16orf62, and DSCR3 were incubated with HEK 293T whole-cell lysates in which Flag-tagged SNX17 or SNX27 was ectopically expressed. After PAGE separation and Western blot transfer, bound proteins were detected by probing with anti-Flag antibodies (for SNX17) (A) or anti-SNX27 antibodies (B). To confirm the expression of GST-fused C16orf62, one of the two membranes was probed with anti-C16orf62 antibodies.
DISCUSSION
Our interest in the involvement of the retriever complex in HPV trafficking stems from initial observations showing that siRNA-mediated knockdown of some retriever complex subunits reduces virus infection efficiency (24). Consequently, we sought to examine HPV’s requirement for both retromer and retriever to identify their relative contributions to efficient viral trafficking during infection. To explain how the viral L2 protein might interact with both retromer and retriever complexes, we looked for direct interactions with these complexes. Popa et al. (14) reported the direct binding of L2 to the retromer complex and identified two stretches of residues in the carboxy terminus that might be required for VPS35 interaction. Our approach consisted of expressing the 86-amino-acid C-terminal stretch of HPV16 L2 as a GST fusion protein, binding it to glutathione-agarose beads, and incubating it with a whole-cell extract. Although in our hands, both interactions are rather weak, this approach showed that endogenous C16orf62 binds to the L2 C terminus more strongly than does VPS35 and that mutations made in the same residues that eliminate VPS35 binding (14) in our assay reduced binding to either endogenous protein. To examine the contribution of retromer and retriever during viral trafficking, we carried out immunofluorescence experiments, examining colocalization 2, 4, and 7 h after infection with HPV16 pseudovirions. We observed that the VPS35 and VPS35L (C16orf62) subunits of the retromer and retriever complexes, respectively, independently colocalize with the HPV16 L2 protein but that L2 recruits retromer more frequently than retriever, as shown by the Manders’ coefficient results. The relative levels of interaction with either complex are more or less the same at 2 and 4 h postinfection, but by 7 h, there appears to be a slight decrease in retromer association and a slight increase in retriever association. Additionally, we observed that in a number of cases, L2 colocalizes with both proteins at distinct structures that we suspect are endosomal membranes. All these data strongly suggest that both retromer and retriever complexes are involved in viral trafficking and that both are brought into proximity with each other in a way that does not occur in uninfected cells. To examine how both complexes might be recruited, we looked at the interactions of HPV16 L2 with SNX17 and SNX27 since these cargo carriers associate with retriever and retromer complexes, respectively, and both have previously been shown to play a role in trafficking (15–18). By fluorescence microscopy, we observe an association between HPV16 L2 and both sorting nexins as early as 2 h postinfection, implying that both are involved in the recruitment of sorting complexes. To examine how L2 might utilize sorting nexins in conjunction with sorting/recycling complexes, we show here, using binding saturation experiments, that both sorting nexins can most likely bind simultaneously to the same L2 molecule. However, it seems unlikely that L2 recruits the retriever complex through SNX17 since we show that infection is equally effective in cells expressing mutants of SNX17 that cannot interact with retriever. Instead, we show that during infection, colocalization between SNX17 and VPS35 occurs far more frequently when EdU-labeled, HPV-encapsidated DNA is also present, showing that the presence of PsVs brings SNX17 and retromer together more frequently than when PsVs are not present. We invariably observe a consistently low level of colocalization between SNX17 and the VPS35 subunit of the retromer complex, even though hitherto, SNX17 was not considered to interact directly with retromer. This suggests that SNX17 may play a general role in the retrograde trafficking of certain cargoes. Interestingly, another study demonstrated the SNX17-enhanced trafficking of the JAG-1 receptor along a retromer-specific route (25). These data demonstrate that interaction between L2 and SNX17 is required for the recruitment of retromer, rather than retriever, as shown by the greatly reduced colocalization between the EdU-labeled pseudogenome, SNX17, and VPS35 when the SNX17-binding motif of L2 is mutated from NPAY to AAAA. Although previous studies have not shown a direct relationship between SNX17 and retromer, we show that GST-fused subunits of retromer may interact with SNX17, and GST-fused subunits of retriever may interact with SNX27, with the caveat that this particular experimental approach using ectopically expressed sorting nexins in whole-cell lysates may be affected by endogenous sorting complex subunits. Further studies are aimed at characterizing the interactions in more detail. However, such interactions begin to provide some explanation of our previous observation that siRNA-mediated knockdown of some retriever subunits, which may impair the overall stability of the complex, also reduces levels of SNX27 (24), as also shown in Fig. 1. These findings suggest multiple interactions between the two sorting nexins and the subunits of either recycling complex so that the dual usage of retromer and retriever might occur early in infection, when a single, partly disassembled virus particle containing multiple L2 molecules protruding through the endosomal membrane might interact with either or both sorting complexes. A speculative model is shown in Fig. 10. It is also possible that the dual binding of both complexes might be the result of L2 forming oligomers within the endosomal lumen or at the point of passing through the membrane, with the C terminus of each L2 interacting with different complexes. Two recently reported models of retromer structure and function, although different, suggest how both sorting complexes might be assembled (26, 27). More structural details of how retriever is assembled and recruited to endosomal membranes will shed light on this aspect of its function, but it should be noted that the dual recruitment of both sorting complexes during infection might also modulate their normal functions and have implications for the recycling of other cellular cargoes, thus affecting aspects of cellular biology relevant to viral infection.
FIG 10.

Putative model for the recruitment of retriever and retromer complexes by HPV L2 protein early during trafficking. Loosening of the virion structure allows multiple L2 molecules to penetrate the endosomal membrane and thus recruit both retromer and retriever through interaction of their C-terminal residues with VPS35 and C16orf62, respectively, interactions which might be strengthened through L2 binding of the PDZ domain of SNX27 and interactions between SNX17 and SNX27 themselves. SNX17 binding may also be involved in the interaction with retromer, but a direct interaction of SNX17 with retriever is not required.
MATERIALS AND METHODS
Cell lines.
HEK 293TT cells were used to produce wild-type HPV16 pseudovirions. HaCaT cells were used for infection studies where the infection efficiency was measured by luminometry. HeLa cells were utilized for infection experiments to visualize protein colocalization by immunofluorescence. For SNX17 knockout experiments, we used H1299 cells and siRNA against SNX17 (Dharmacon). The WT and SNX17 knockout HeLa cells were kind gifts of Kerrie McNally (P. Cullen laboratory). HEK 293T cells were used as a source for endogenous VPS35 and C16orf62 for His-tagged L2 pulldowns. All cells were maintained in Dulbecco’s modified Eagle medium supplemented with glutamine and penicillin/streptomycin, with 10% fetal bovine serum (Gibco). HeLa cells stably expressing GFP-tagged SNX27 were produced by transfection of a GFP-SNX27 construct (a kind gift of Martin Playford) followed by extended G418 treatment and selection of resistant colonies.
Plasmid constructs.
Plasmid constructs pEBB-2HA-VPS35L-HA, expressing hemagglutinin (HA)-tagged human VPS5L, and pEBB-HA-DSCR3, expressing HA-tagged DSCR3, were kind gifts of Ezra Burstein and were subcloned into pGEX4. Constructs pGEX6P VPS35, pGEX6P VPS29, and pGEX6P VPS26A, expressing GST-fused components of the retromer complex, were kind gifts of Peter Cullen, as were the constructs expressing siRNA-resistant wild-type and mutant SNX17. Constructs expressing Flag-tagged SNX17 and myc-tagged SNX27 were described previously (15).
Pseudovirion production.
HPV16 pseudovirions packaged with a pGL3Luci construct expressing firefly luciferase were produced in HEK 293TT cells by the method developed by Buck et al. (28), using construct pShell16, where L2 carries an N-terminal HA tag, or construct pXuLL16, where L2 is untagged.
5-Ethynyl-2′-deoxyuridine labeling of pseudogenomes during pseudovirion production.
PsVs containing a packaged 5-ethynyl-2′-deoxyuridine (EdU)-labeled plasmid were prepared as previously described (18). Briefly, 25 μM EdU was added to the HEK 293TT cells at 12 h posttransfection. Harvesting and purification of the EdU-labeled PsVs were then performed as described above. Visualization of Edu-labeled pseudogenomes during infection was performed after fixation of the cells on coverslips, using a Click-iT assay (Invitrogen) according to the manufacturer’s protocol.
siRNA-mediated knockdown of retriever complex proteins.
Knockdown experiments were performed as previously described (24).
Immunofluorescence.
HeLa cells were plated at approximately 105 cells per dish onto 4-cm petri dishes containing a coverslip. After 24 h, the cells were infected with approximately 8 × 107 Edu-labeled PsVs for 1 h on ice, followed by 2 washes with phosphate-buffered saline (PBS) before replacement of the medium and incubation at 37°C. Infected cells and uninfected controls were harvested at 2, 4, and 8 h postinfection by washing with PBS, fixing with 37% paraformaldehyde for 20 min, and then washing twice with PBS. Cells were permeabilized with PBS–0.5% Triton X-100 for 20 min and then washed twice with PBS–0.1 M glycine. For the colocalization of VPS35 and SNX17 together with the viral pseudogenome, immunofluorescence was carried out using goat anti-VPS35 antibody (catalog no. ab10099; Abcam) and mouse anti-SNX17 (catalog no. sc-166957; Santa Cruz) at 37°C for 2 h, and cells were washed twice with PBS, followed by the fluorescent secondary antibodies donkey anti-goat (Alexa Fluor 488, catalog no. A11055) and donkey anti-mouse (Alexa Fluor 647, catalog no. A31571), for 30 min at 37°C. After washing with PBS, the viral DNA was labeled using a Click-iT kit (Invitrogen) according to the manufacturer’s protocol. When visualizing HPV16 L2 together with C16orf62 and VPS35, we used mouse anti-HA (catalog no. 12CA5; Roche) together with rabbit anti-C16orf62 antibody (catalog no. PA5-28553; Invitrogen) and goat anti-VPS35 antibody (catalog no. ab10099; Abcam) at 37°C for 2 h, and cells were washed twice with PBS, followed by the fluorescent secondary antibodies donkey anti-goat (Alexa Fluor 488, catalog no. A11055), donkey anti-rabbit (Alexa Fluor 647, catalog no. A31573), and goat anti-mouse rhodamine X (catalog no. R6393; Invitrogen), for 30 min at 37°C. For visualizing L2 colocalization with sorting nexin 17 or 27, we used rat anti-HA antibodies (Sigma) and mouse anti-SNX17 (catalog no. sc-166957; Santa Cruz) or mouse anti-SNX27 (catalog no. ab77799; Abcam) antibodies, followed by donkey anti-mouse (Alexa Fluor 647, catalog no. A31571) and chicken anti-rat (Alexa Fluor 488), incubated as indicated above. Slides were visualized with a Zeiss LSM 510 confocal microscope at an original magnification of ×630. z-stacked images were captured at 150-nm separation. The colocalization of L2 protein with either VPS35 of retromer or C16orf62 of retriever was carried out by Manders’ coefficient analysis using the JACoP plug-in for ImageJ.
GST-binding saturation experiments.
GST-fused HPV16 L2 bound to glutathione-agarose beads was produced as previously described (29). HEK 293 cells were transfected with constructs expressing SNX17 or SNX27 by calcium phosphate-mediated transfer (30), and after 24 h, cells were lysed in a buffer containing 50 mM HEPES (pH 7.0), 250 mM NaCl, and 0.1% NP-40. Increasing amounts of cell lysates containing either ectopically expressed SNX17 or SNX27 were incubated with GST-L2 to determine empirically at what point the GST-L2 was saturated for binding. Next, at this maximum input, an equal amount of the lysate containing the other sorting nexin was added and coincubated with GST-L2. After washing, bound proteins were eluted by boiling in loading buffer, separated by PAGE, transferred to nitrocellulose membranes, and probed with antibodies against SNX17 and SNX27.
Binding of HPV16 L2 to C16orf62 and VPS35.
We cloned the open reading frame encoding an 86-amino-acid C-terminal portion of HPV16 L2 into bacterial expression vector pGEX4t to produce an N-terminal GST fusion protein. Mutations in residues previously reported to be required for binding to VPS35 were made using the Gene-Tailor site-directed mutagenesis system (Invitrogen) according to the manufacturer’s protocol and were verified by sequencing. Purified GST-fused L2 C-terminal fragments, the wild type, and mutants were purified from bacterial cultures after isopropyl-β-d-thiogalactopyranoside (IPTG) induction, as previously described (29); bound to glutathione-agarose beads; and incubated with the whole-cell lysate produced after lysing 4- by 10-cm petri dishes of HEK 293T cells in a buffer containing 50 mM HEPES (pH 7.0), 250 mM NaCl, and 0.1% NP-40. After several washes, bound proteins were eluted, separated by PAGE, transferred to membranes, and probed for bound C16orf62 and VPS35 using goat anti-VPS35 antibodies (catalog no. ab10099; Abcam) and rabbit anti-C16orf62 (catalog no. PA5-28553; Invitrogen).
ACKNOWLEDGMENTS
We are very grateful to Kerrie McNally and Peter Cullen for reagents and advice. We express our thanks to Om Basukala and Martina Bergant for advice and help with data analysis. We are also grateful to Miranda Thomas for comments on the manuscript.
This work was supported in part by a research grant from the Associazione Italiana per la Ricerca sul Cancro (no. IG2019 Id23572). A.S. was the recipient of an ICGEB Arturo Falaschi postdoctoral fellowship. J.B. gratefully acknowledges support from the Umberto Veronesi Foundation (postdoctoral fellowship, years 2018 and 2020).
REFERENCES
- 1.Bugnon Valdano M, Pim D, Banks L. 2019. Choosing the right path: membrane trafficking and infectious entry of small DNA tumour viruses. Curr Opin Cell Biol 59:112–120. doi: 10.1016/j.ceb.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 2.Campos SK. 2017. Subcellular trafficking of the papillomavirus genome during initial infection: the remarkable abilities of minor capsid protein L2. Viruses 9:370. doi: 10.3390/v9120370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqa A, Massimi P, Pim D, Broniarczyk J, Banks L. 2018. Human papillomavirus 16 infection induces VAP-dependent endosomal tubulation. J Virol 92:e01514-17. doi: 10.1128/JVI.01514-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiqa A, Massimi P, Pim D, Banks L. 2019. Diverse papillomavirus types induce endosomal tubulation. Front Cell Infect Microbiol 9:175. doi: 10.3389/fcimb.2019.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86:9875–9887. doi: 10.1128/JVI.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broniarczyk J, Bergant M, Goździcka-Józefiak A, Banks L. 2014. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 460–461:83–90. doi: 10.1016/j.virol.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Broniarczyk J, Pim D, Massimi P, Bergant M, Goździcka-Józefiak A, Crump C, Banks L. 2017. The VPS4 component of the ESCRT machinery plays an essential role in HPV infectious entry and capsid disassembly. Sci Rep 7:45159. doi: 10.1038/srep45159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiGiuseppe S, Bienkowska-Haba M, Guion LGM, Keiffer TR, Sapp M. 2017. Human papillomavirus major capsid protein L1 remains associated with the incoming viral genome throughout the entry process. J Virol 91:e00537-17. doi: 10.1128/JVI.00537-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bronnimann MP, Chapman JA, Park CK, Campos SK. 2013. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87:464–473. doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. 2014. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog 10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aydin I, Villalonga-Planells R, Greune L, Bronnimann MP, Calton CM, Becker M, Lai KY, Campos SK, Schmidt MA, Shelhaas M. 2017. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog 13:e1006308. doi: 10.1371/journal.ppat.1006308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, DiMaio D. 2015. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog 11:e1004699. doi: 10.1371/journal.ppat.1004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pim D, Broniarczyk J, Bergant M, Playford MP, Banks L. 2015. A novel PDZ domain interaction mediates the binding between human papillomavirus 16 L2 and sorting nexin 27 and modulates virion trafficking. J Virol 89:10145–10155. doi: 10.1128/JVI.01499-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergant Marušič M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergant M, Banks L. 2013. SNX17 facilitates infection by diverse papillomavirus types. J Virol 87:1270–1273. doi: 10.1128/JVI.01991-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergant M, Peternel S, Pim D, Broniarczyk J, Banks L. 2017. Characterizing the spatio-temporal role of sorting nexin 17 in human papillomavirus trafficking. J Gen Virol 98:715–725. doi: 10.1099/jgv.0.000734. [DOI] [PubMed] [Google Scholar]
- 19.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Berzginov A, Chessman K, Pal S, Cromar G, Papoulas O, Ni Z, Boutz DR, Stoilova S, Havugimana PC, Guo X, Malty RH, Sarov M, Greenblatt J, Babu M, Derry WB, Tillier ER, Wallingford JB, Parkinson J, Marcotte EM, Emili A. 2015. Panorama of ancient metazoan macromolecular complexes. Nature 525:339–344. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dey G, Jaimovich A, Collins SR, Seki A, Meyer T. 2015. Systematic discovery of human gene function and principles of modular organization through phylogenetic profiling. Cell Rep 10:993–1006. doi: 10.1016/j.celrep.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M. 2015. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 22.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wühr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. 2015. The BioPlex network: a systematic exploration of the human interactome. Cell 162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Calvo SE, Gutman R, Liu JS, Mootha VK. 2014. Expansion of biological pathways based on evolutionary inference. Cell 158:213–225. doi: 10.1016/j.cell.2014.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNally KE, Faulkner R, Steinberg F, Gallon M, Ghai R, Pim D, Langton P, Pearson M, Danson CM, Nägele H, Morris LL, Singla A, Overlee BL, Heesom KJ, Sessions R, Banks L, Collins BM, Berger I, Billadeau DD, Burstein E, Cullen PJ. 2017. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nat Cell Biol 19:1214–1225. doi: 10.1038/ncb3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin W, Liu D, Liu N, Xu L, Li S, Lin S, Shu X, Pei D. 2012. SNX17 regulates Notch pathway and pancreas development through the retromer-dependent recycling of Jag1. Cell Regen 1:4. doi: 10.1186/2045-9769-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kovtun O, Leneva N, Bykov YS, Ariotti N, Teasdale RD, Schaffer M, Engel BD, Owen DJ, Briggs JAG, Collins BM. 2018. Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature 561:561–564. doi: 10.1038/s41586-018-0526-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, Hierro A. 2016. Structural mechanism for cargo recognition by the retromer complex. Cell 167:1623–1635. doi: 10.1016/j.cell.2016.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462. doi: 10.1385/1-59259-982-6:445. [DOI] [PubMed] [Google Scholar]
- 29.Smith DB, Johnson KS. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 30.Graham FL, van der Eb AJ. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]