

Chronic hepatitis B virus (HBV) infection afflicts approximately 257 million people, who are at high risk of progressing to chronic liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma. Current therapies rarely achieve cure of HBV infection due to the persistence of the HBV episome, the covalently closed circular DNA (cccDNA), in the nuclei of infected hepatocytes. Peripheral markers of cccDNA levels and transcriptional activities are urgently required to guide antiviral therapy and drug development. Serum hepatitis B core-related antigen (HBcrAg) is one such emerging peripheral marker. We have characterized the components of HBcrAg in HBV-infected patients as well as in cell cultures. Our results provide important new quantitative information on levels of each HBcrAg component, as well as their biochemical and biophysical characteristics. Our findings suggest that each HBcrAg component may have distinct functions and applications in reflecting intrahepatic viral activities.
KEYWORDS: hepatitis B virus, hepatitis B core-related antigen, HBcrAg, covalently closed circular DNA, cccDNA, precore/core
ABSTRACT
Current therapies rarely cure chronic hepatitis B virus (HBV) infection due to the persistence of the viral episome, the covalently closed circular DNA (cccDNA), in hepatocytes. The hepatitis B virus core-related antigen (HBcrAg), a mixture of the viral precore/core gene products, has emerged as one potential marker to monitor the levels and activities of intrahepatic cccDNA. In this study, a comprehensive characterization of precore/core gene products revealed that HBcrAg components included the classical hepatitis B virus core antigen (HBc) and e antigen (HBeAg) and, additionally, the precore-related antigen, PreC, retaining the N-terminal signal peptide. Both HBeAg and PreC antigens displayed heterogeneous proteolytic processing at their C termini resulting in multiple species, which varied with viral genotypes. HBeAg was the predominant form of HBcrAg in HBeAg-positive patients. Positive correlations were found between HBcrAg and PreC, between HBcrAg and HBeAg, and between PreC and HBeAg but not between HBcrAg and HBc. Serum HBeAg and PreC shared similar buoyant density and size distributions, and both displayed density and size heterogeneity. HBc, but not HBeAg or PreC antigen, was found as the main component of capsids in DNA-containing or empty virions. Neither HBeAg nor PreC protein was able to form capsids in cells or in vitro under physiological conditions. In conclusion, our study provides important new quantitative information on levels of each component of precore/core gene products as well as their biochemical and biophysical characteristics, implying that each component may have distinct functions and applications in reflecting intrahepatic viral activities.
IMPORTANCE Chronic hepatitis B virus (HBV) infection afflicts approximately 257 million people, who are at high risk of progressing to chronic liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma. Current therapies rarely achieve cure of HBV infection due to the persistence of the HBV episome, the covalently closed circular DNA (cccDNA), in the nuclei of infected hepatocytes. Peripheral markers of cccDNA levels and transcriptional activities are urgently required to guide antiviral therapy and drug development. Serum hepatitis B core-related antigen (HBcrAg) is one such emerging peripheral marker. We have characterized the components of HBcrAg in HBV-infected patients as well as in cell cultures. Our results provide important new quantitative information on levels of each HBcrAg component, as well as their biochemical and biophysical characteristics. Our findings suggest that each HBcrAg component may have distinct functions and applications in reflecting intrahepatic viral activities.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection afflicts an estimated 257 million individuals worldwide, who are at high risk of developing fibrosis, cirrhosis, and hepatocellular carcinoma (1). HBV is the prototype of Hepadnaviridae, with a small partial double-stranded relaxed circular DNA (rcDNA) genome, which is converted into the covalently closed circular DNA (cccDNA) in the nuclei of infected hepatocytes (2). HBV cccDNA, the transcriptional template for all viral mRNAs, exists as a stable minichromosome and is responsible for viral persistence (3, 4). Current therapies rarely cure chronic HBV infection due to the persistence of cccDNA in the nuclei of HBV-infected hepatocytes. Direct measurement of intrahepatic cccDNA levels and transcriptional activities from liver biopsy specimens is limited due to its invasive nature, potential sampling error, and technical difficulties in accurately quantifying cccDNA (4). Thus, peripheral biomarkers capable of reflecting intrahepatic cccDNA levels and transcriptional activities are urgently needed for monitoring intrahepatic viral persistence and guiding antiviral development targeting cccDNA as part of the ongoing HBV cure effort.
Four mRNAs are generated from cccDNA by the host RNA polymerase II. The 3.5-kb HBV pre-genomic RNA (pgRNA) encodes the viral reverse transcriptase (RT) and core protein (HBc), and the slightly longer precore mRNA encodes the precore protein (p25), the precursor to the secreted hepatitis B virus e antigen (HBeAg). The 2.4/2.1-kb PreS/S mRNAs encode three (large, middle, and small) HBV surface (envelope) antigens (HBsAg), and the 0.7-kb mRNA encodes the HBV X protein (2). The RT protein and pgRNA together are encapsidated into a cytoplasmic nucleocapsid (NC) formed by HBc, where pgRNA is reverse transcribed to form rcDNA by RT. The resulting mature NC is either secreted as a complete virion after coating by the viral envelope proteins or recycled back to the nucleus to replenish the cccDNA pool (2). HBV-infected hepatocytes also secrete a large excess of genome-free (empty) virions assembled from cytoplasmic empty capsids and the viral envelope proteins in the absence of pgRNA packaging or DNA synthesis (5–8). In addition, HBsAg, alone, forms spherical and filament subviral particles devoid of either the capsid or genome, which are found in the blood at a 1,000-fold excess over virions (8).
The precore/core open reading frame (ORF) includes two in-frame initiation codons encoding precore (p25) or HBc (p21) (Fig. 1A). HBc, translated from pgRNA, contains an N-terminal domain (NTD), known as the assembly domain, and a C-terminal domain (CTD), linked together by the linker region (Fig. 1A) (9). The precore protein, translated from the precore mRNA, has the same sequence as HBc but has an N-terminal 29 amino acid (aa) extension (Fig. 1A). p25 is further processed in the endoplasmic reticulum (ER) and secreted as HBeAg through the ER-Golgi apparatus secretory pathway, in which two proteolytic cleavage events contribute to HBeAg production (Fig. 1A). First, the N-terminal 19-aa hydrophobic signal peptide is cleaved by the signal peptidase cotranslationally during the translocation of precore into the ER lumen, whereby p25 is converted to the intermediate p22 (10). p22 is thus identical to HBc except for the 10-aa N-terminal extension (10, 11). Subsequently, ca. 30 aa in the CTD of p22 is cleaved by proprotein convertases (e.g., furin) in the secretory pathway to generate multiple species of HBeAg due to heterogeneity in C-terminal processing (Fig. 1B) (12, 13). As detailed below, another precore-derived species, p22cr, distinct from the intracellular p22, has also been reported (14).
FIG 1.

Biogenesis of HBc, HBeAg, and p22cr. (A) Schematic representation of HBcrAg biogenesis. The current HBcrAg assay is thought to detect HBc, HBeAg, and p22cr. HBc, translated from pgRNA, forms the icosahedral capsid inside complete and empty virions. The direct translation product from the precore mRNA is the precore protein (p25), from which HBeAg and p22cr (renamed PreC here) are both derived. Removal of the N-terminal signal peptide (SP) of p25, by the signal peptidase during p25 translocation into the ER lumen, leads to the production of p22, which is further processed at its CTD before being secreted as the dimeric HBeAg (p17). The exact C-terminal processing sites of HBeAg and p22cr appear heterogeneous and not well defined, as indicated by the hashed box. On the other hand, p22cr (starting from Glu −28, in the absence of Met −29) is reported as an incompletely processed product of p25, retaining the N-terminal signal peptide, in contrast to HBeAg, but lacking CTD, similar to HBeAg (14). Another precore-derived protein, similar to p22cr but retaining the initiator Met (−29) in the signal peptide, has also been reported (21). Whereas this remains to be resolved, we use here the nomenclature derived from p22cr, starting from Glu −28, to refer to the PreC proteins we identify here schematically. p22cr was initially reported to form an aberrant capsid inside empty virions, but this has been challenged recently by the detection of HBc in empty virions in the absence of any precore-derived proteins. The diagram shows a comparison of the various components of HBcrAg. Specific epitopes recognized by the following MAbs are indicated with blue braces: MAbs 1A11 and 7E9, specific to precore-derived proteins HBeAg and p22cr (renamed PreC in this study) (the −10 aa region, epitope from aa −10 to 5); MAbs T2221 (epitope from aa 130 to 140), 19C18 (epitope from aa 2 to 5), and 1D8 (epitope from aa 75 to 83), specific to the NTD shared by all HBcrAg components; and MAbs 366-2 (epitope from aa 150 to 164, largely independent of CTD state of phosphorylation), 25-7 (epitope from aa 164 to 182, selective for nonphosphorylated CTD), and 14-2 (epitope from aa 164 to 182, selective for phosphorylated CTD) specific to HBc (CTD). The antibodies HB44 (epitope from aa 31 to 49), HB61 (epitope from aa 131 to 140), HB91 (epitope from aa 1 to 19), H114 (structural epitope from aa 1 to 81, denoted by *), and HB140 (epitope from aa 21 to 40), used in the current HBcrAg commercial assay, and their epitopes are indicated by gray braces and letters (16). (B) Comparison of the CTD sequences retained in the various HBeAg and secreted PreC (p22cr) proteins (as defined in this study) between genotype A and non-A genotypes. Four RXXR motifs, which are potential furin cleavage sites, are underlined (12). Assignment of putative cleavages sites for HBeAg were based on prior publications, except that e2 in genotypes B and C was identified in this study. The arrows indicate the putative cleavage sites to produce HBeAg (referred to as e0, e1, e2, and e3 in this study) and PreC proteins (referred to as PreC0, PreC1, PreC2, and PreC3 in this study), with the preferred cleavage sites indicated by the long arrows.
The hepatitis B virus core-related antigen (HBcrAg) is an emerging surrogate marker in the blood for monitoring intrahepatic cccDNA (15). The current commercial HBcrAg assay, using a panel of antibodies targeting the NTD of precore/core (Fig. 1A), detects a combination of HBc, HBeAg—both free and HBeAg-HBe antibody complex—and the aforementioned p22cr, a 22-kDa precore-derived protein, all found in the blood, after sodium dodecyl sulfate (SDS) and heat treatment (14, 16, 17). Serum HBcrAg has been reported to correlate with intrahepatic cccDNA, viremia (i.e., serum HBV DNA), and response to antiviral treatments (18–20). However, a detailed characterization of HBcrAg, the product of precore/core gene, remains missing. In particular, p22cr, retaining the N-terminal signal peptide, with the removal of N-terminal Met (−29) and N-acetylation of Glu −28, was reported to be an incompletely processed p25 in patient blood, which is processed at the CTD similarly to HBeAg (Fig. 1A) (14). Also, an earlier study identified a precore-derived protein also lacking CTD (designated p20e) but retaining the entire N-terminal signal peptide sequence, including the N-terminal Met (21). It has been claimed that p22cr forms aberrant capsids found in empty virions in the blood of HBV-infected patients (14). However, we have recently shown that the empty capsids found inside cells as well as in empty virions, both formed efficiently in the complete absence of any precore-related proteins, are composed entirely of the authentic HBc with the entire CTD (5, 7). Therefore, the biochemical and biophysical nature of p22cr requires further clarification. In addition, the relative and absolute levels of HBeAg, HBc, and p22cr in HBV-infected patients remain unclear.
The exact functions of precore or HBeAg are currently unknown, but neither protein is required for viral replication (22). Instead, HBeAg is thought to modulate the host immune response to facilitate viral persistence (23, 24). In particular, maternal HBeAg is thought to induce T cell tolerance to HBV to facilitate HBV persistence during vertical (mother-to-child) transmission (25–27). HBeAg is also generally used as a marker for high levels of viral replication. The intracellular p22 has also been reported to regulate innate immune and cancer signaling pathways (28–30). On the other hand, the function of p22cr, if any, remains completely unknown.
In this study, we aimed to comprehensively characterize the composition of HBcrAg, which is defined as the product expressed from HBV precore/core gene, taking advantage of a panel of monoclonal antibodies (MAbs) that specifically recognize different species of HBc and precore-related proteins. We found that precore/core products included HBc and HBeAg, and additionally, the precore-related antigen, p22cr (renamed PreC here) (Fig. 1A), which retains the N-terminal signal peptide. All these could be detected in the sera of HBV-infected patients and in the supernatant of HBV-infected hepatocytes in culture. Both HBeAg and PreC antigens displayed heterogeneous proteolytic processing at their C termini, resulting in multiple species, which further varied with viral genotypes. HBeAg, at ca. 1014 molecules/ml of serum, was the predominant form of HBcrAg in HBeAg-positive patients. Serum HBeAg and PreC shared similar buoyant density and size distributions, both displaying density and size heterogeneity. HBc, but not HBeAg or PreC antigen, was found as the main component of capsids in DNA-containing or empty virions. Neither HBeAg nor PreC protein was able to form capsids in cells or in vitro under physiological conditions. In conclusion, our study provides important new information on the absolute and relative levels of each component of HBcrAg as well as their biochemical and biophysical characteristics, implying that each component may have distinct functions and applications in reflecting intrahepatic viral activities.
RESULTS
HBeAg and another precore-derived protein were secreted from human hepatoma cells expressing the HBV precore gene.
To characterize the components of HBcrAg, which we define here as any proteins produced from the precore/core gene, we employed a panel of MAbs specific to different components of HBcrAg (Fig. 1). These included MAbs 1A11 and 7E9, both recognizing a linear epitope that includes residues within the 10-aa N-terminal extension present in HBeAg and potentially other precore-derived proteins but absent in HBc (31), T2221, similar to HB61 used in the commercial HBcrAg assay (16), recognizing a linear epitope toward the end of NTD shared by HBc and precore-derived proteins (9), and 366-2, recognizing a linear epitope from positions 150 to 164 within precore/core CTD (32). The specificities of these MAbs were first verified using precore (HBeAg) and HBc proteins harvested from Huh7 cell cultures (Fig. 2). As expected, MAb 1A11 specifically detected HBeAg (referred to as e1 [Fig. 1B]) in the culture supernatant of precore-transfected Huh7 cells without cross-reacting with HBc. On the other hand, MAb T2221 detected both HBeAg and HBc (p21), and the CTD-specific MAb 366-2 detected HBc specifically (largely independent of CTD state of phosphorylation) but not HBeAg. In addition to the classical HBeAg (e1) (Fig. 1), we detected another precore species (labeled as PreC1 in Fig. 1B) migrating just above HBc using MAbs 1A11 and T2221, but not 366-2, in the supernatant of precore-transfected cells (Fig. 2, lanes 1, 3, and 5), indicating the presence of the N-terminal precore-specific region but lack of CTD. The slower mobility of PreC1 (ca. 22 kDa), compared to e1 and HBc (p21) (Fig. 2, lane 3 versus 4), could be explained by the retention of the N-terminal signal peptide sequence, as reported for p22cr (14, 21). Thus, we could demonstrate, for the first time, the secretion of a p22cr-like protein in cell culture, as previously reported for human serum (14, 21). We chose to name this precore-derived protein, distinct from HBeAg, as PreC instead of p22cr since it is more related to precore instead of core (HBc) (Fig. 1A). As HBeAg and PreC each showed multiple species, due to different CTD processing (see below), we named them e0/PreC0, e1/PreC1, and so on, with each numeral indicating a particular CTD processing site and the increasing numerals indicating increasing amounts of CTD sequences in both HBeAg and PreC (Fig. 1B). In the absence of any obviously better alternatives, we chose to capitalize P and C in “PreC” as a way to differentiate it from the preC region in the precore/core gene, which is already in use in the literature.
FIG 2.

Secretion of precore and core gene products expressed in human hepatoma cell culture. Supernatants from precore or core (genotype D)-transfected Huh7 cells were concentrated by ultrafiltration and resolved by SDS-PAGE in a 32-cm-tall gel (high-resolution) followed by immunoblotting with MAbs 1A11, T2221, and 366-2 sequentially on the same membrane. Each protein species is represented schematically on the right.
HBeAg, PreC, and HBc (virions) were secreted in HBV-infected PHH cultures.
To examine the secretion of HBcrAg in the context of HBV infection, we analyzed the supernatant of HBV (genotype D [gtD])-infected primary human hepatocytes (PHHs), which are thought to closely mimic HBV-infected hepatocytes in humans, by SDS-PAGE and immunoblotting with the precore- and HBc-specific MAbs as described above. We detected one HBeAg species, e1, and one PreC species, PreC1, besides HBc (p21), in the PHH supernatant (Fig. 3A to C, lanes 4 to 6), as we observed in precore-transfected Huh7 cell culture (Fig. 2). Among the multiple furin cleavage motifs within the precore/core CTD, previous studies have demonstrated that the preferred cleavage in non-A genotypes occurs at the 1st motif (151RRGR154) (Fig. 1B) (12). Thus, e1 and PreC1 were likely cleaved at residue 154, as reported previously (12). We also detected the same components of HBcrAg, including e1, PreC1, and HBc, in the serum of an HBV (gtD)-infected chimpanzee (Fig. 3A to C, lanes 2). Kinetic analysis of the supernatant from HBV-infected PHHs showed that secretion of both HBeAg (e1) and PreC1 peaked earlier than that of HBc (i.e., virions) (Fig. 3B, lane 4 versus lane 5). Our results thus indicated that multiple precore/core products, including HBc (p21), HBeAg, and PreC (i.e., p22cr), were secreted in vivo and in vitro, with the latter two products being processed from the p25 precursor.
FIG 3.

Secretion of precore and core gene products in genotype D HBV-infected PHHs. The culture supernatant (sup) of HBV-infected PHHs was harvested at 7, 10, 15, 19, 24, and 30 days postinfection (dpi) and resolved by high-resolution SDS-PAGE followed by immunoblotting with MAbs 1A11 (A), T2221 (B), and 366-2 (C). HBc (p21) expressed in Huh7 cells was used as a control for HBc, and a genotype D HBV-infected chimpanzee serum was loaded as a reference of HBcrAg in the serum. *, background bands detected by MAb 1A11 in PHH culture supernatant.
HBcrAg consisted of multiple precore-derived proteins in addition to HBc in sera of HBV-infected patients.
Next, we characterized the components of HBcrAg in patient sera by SDS-PAGE and immunoblotting. We detected multiple bands representing HBc and precore-related proteins with different mobilities in patient sera, and the migration patterns of the HBcrAg components varied between genotypes and between individual patients infected by the same genotype (Fig. 4). Alignment of the precore/core protein sequences from genotypes A to D indicated that the epitopes of 1A11, T2221, and 366-2 were conserved across these HBV genotypes (Fig. 1B). The CTD includes multiple furin cleavage motifs, and there is a 2-residue insertion (153DR154) in the genotype A precore/core CTD compared to non-A genotypes (12). Previous studies have demonstrated that the preferred furin cleavage motifs are shifted from the 1st (151RRDR154) to the 2nd (156RSPR159) or even the 3rd (166RRRR169) motif during p22 intermediate processing to generate HBeAg in genotype A strains. However, in non-A genotypes, the preferred cleavage of CTD occurs at the 1st motif (151RRGR154) (Fig. 1B) (12). Cleavage at the most C-terminal motif (174RRRR177) is usually undetectable, possibly due to the competition among potential motifs for proprotein convertase binding and cleavage on p22 (12). In the sera of genotype A HBV-infected patients, the precore-specific MAb 1A11 detected five precore-related proteins all harboring the −10 aa N terminal to the start of HBc (Fig. 4A, lanes 1 to 3). These included three species of HBeAg, referred to as e1(A), e2(A), and e3(A) based on the deduced CTD cleavage sites (Fig. 1B and 4A) and consistent with the previous report (12), and two additional species of precore-derived proteins, PreC2(A) and PreC3(A), which migrated just above HBc (Fig. 4A, lanes 1 to 3), similar to the reported p22cr (14). The NTD-specific MAb T2221 could detect these plus one more band, HBc, as expected (Fig. 4A, lanes 7 to 9 versus 1 to 3). The CTD-specific MAb 366-2 detected HBc, as well as the longest species, e3(A) and PreC3(A), but not e1(A), e2(A), or PreC2(A) (Fig. 4A, lanes 13 to 15). Therefore, both e3(A) and PreC3(A) retained the N-terminal portion of CTD (from positions 150 up to 164, the approximate epitope boundary of MAb 366-2), as reported previously for genotype A HBeAg cleaved at the 166RRRR169 motif (12). Furthermore, these results indicated that both PreC2(A) and PreC3(A) were precore-derived proteins that were processed at the CTD in a manner analogous to HBeAg e2(A) and e3(A), respectively, but retained the N-terminal signal peptide, accounting for their slower mobilities than HBc (p21). Therefore, PreC2(A) and PreC3(A) in these serum samples corresponded to two distinct p22cr-like species (14), differing in CTD processing. We only detected e1(A) as a minor species, consistent with the previous report of less efficient cleavage at the 1st motif in genotype A, 151RRDR154 (12) (Fig. 4A, lanes 2 and 3). Importantly, serum from two patients (number 2 and number 3) after recovery showed no detectable signals in this region of the gel by any of the MAbs used, confirming that no cross-reactive human serum proteins comigrated with the HBV precore/core-related proteins (Fig. 4A, lanes 4, 5, 10, 11, 16, and 17).
FIG 4.

Complex pattern of precore/core gene products (HBcrAg) in the sera of HBV-infected patients. (A) Immunoblot analysis of HBcrAg in the sera of HBV genotype A- and C-infected patients following resolution on SDS-PAGE in a high-resolution gel (with a height of 32 cm), using MAbs 1A11, T2221, and 366-2. Each protein species is represented schematically on the right. Labels 2* and 3* indicate serum samples from patients 2 and 3 at the time point when they were recovered and served as negative controls. Note that the HBeAg and PreC species from the genotype A migrated slightly slower than the corresponding species from the genotype C, due to differential processing in the CTD (Fig. 1B). (B) Immunoblot analysis of HBcrAg components in the serum of genotypes B, C, and D HBV-infected patients by SDS-PAGE in the high-resolution gel. The * in lane 4 and 13 denotes the PreC0 species comigrating with HBc. Each protein species is represented schematically at the bottom. The exact C-terminal processing site(s) of e2 remained uncertain, as indicated by the hatched box. (C) The serum from patient 10 (genotype C) was analyzed by SDS-PAGE followed by Western blotting with MAb 1A11 and another NTD MAb (19C18), which displayed higher sensitivity than MAb T2221, verifying the detection of the various HBeAg and PreC proteins by MAb 1A11.
In the serum of genotype B- and C-infected patients, using the same panel of MAbs, we detected three species of HBeAg (referred to as e0, e1, and e2), HBc, and again at least two additional species of precore-derived proteins (PreC1 and PreC2) (Fig. 4A, lanes 6, 12, and 18, and Fig. 4B). Unlike genotype A, only HBc, but none of the precore-derived proteins, was detected by the MAb 366-2 in non-A genotype-infected patients (Fig. 4A and B), consistent with the predicted CTD cleavage sites to produce HBeAg and PreC proteins in different genotypes (Fig. 1B). Based on the mobilities of each species, e0 (p17), e1, and e2 was likely cleaved at residue 149, 154, and around 159, respectively (Fig. 1B and 4B). e1 was similar to e1(A) in terms of CTD cleavage site selection. Similarly, PreC1 and PreC2, both migrating slower than HBc (p21) and detected by MAbs 1A11 and T2221 but not by 366-2, corresponded to precore-derived proteins equivalent to e1 and e2 in CTD processing but retaining the signal peptide, as in PreC2(A) and PreC3(A) (Fig. 4A). e0 (p17) was detected only occasionally (2 out of 9) as a minor species (Fig. 4B, lanes 2, 4, 11, and 13), which might be processed in the CTD by other proteases (e.g., carboxypeptidase A) possibly in the blood (33). A PreC species called PreC0, corresponding to e0 (p17) in its CTD processing but retaining the signal peptide, was possibly also detected in patient number 7, one of the two patients who were positive for e0 (Fig. 4B, lanes 4 and 13). We noticed that the NTD-specific MAb T2221 was not sufficiently sensitive to detect all precore-derived proteins after stripping and reprobing, especially the PreC2 band, in all the sera, and it also failed to detect HBc in several patients with low viremia (i.e., complete virions) (Fig. 4A and B and Fig. 6), which was usually accompanied by low levels of serum HBc (i.e., empty virions) as we reported before (6). We thus screened for additional NTD-specific MAbs in the hope of finding one that was more sensitive than T2221. Indeed, we found another MAb, 19C18, recognizing a linear epitope at the very N-terminal end of NTD (Fig. 1), that showed enhanced sensitivity and allowed more reliable detection of all precore proteins, especially the PreC2 (Fig. 4C). In the genotype D HBV-infected patient, other than HBc, only one form of HBeAg (e1) and its corresponding precore-derived protein, PreC1, both cleaved at residue 154, were detected (Fig. 4B), as in genotype D precore-transfected cells, genotype D HBV-infected PHHs, and the serum of the HBV (genotype D)-infected chimpanzee (Fig. 2 and 3) (12).
FIG 6.

Analysis of serum virions from HBV-infected patients by native agarose gel electrophoresis. Virions from the sera of HBV-infected patients were resolved by native agarose gel electrophoresis and transferred to a nitrocellulose membrane. HBV DNA (A) and capsid in the virions were detected sequentially using a 32P-labeled HBV DNA probe and the indicated HBc CTD-specific MAbs (B to D), respectively, on the same membrane. V, virions, either containing rcDNA or empty; pCTD, phosphorylated CTD; nonpCTD, nonphosphorylated CTD.
HBeAg was the predominant component of HBcrAg in HBeAg-positive patients.
We estimated the concentration of each precore/core gene product (i.e., each HBcrAg component) using quantitative Western blot analysis and showed that HBeAg contributed predominantly (71.98% ± 10.24%) to total HBcrAg, while HBc and PreC contributed 17.64% ± 8.84% and 15.80% ± 9.13%, respectively (Fig. 5A). In sera of HBeAg-positive patients, the HBeAg concentration was ca. 1 to 5 μg/ml or ca. 1014 molecules/ml of HBeAg monomers, and the PreC concentration was ca. 300 ng/ml or 1013 molecules/ml of PreC (Fig. 5B). The HBc (monomer) concentration was similar to the concentration of PreC (monomers), equivalent to 1010 to 1011 virion particles (both empty and complete)/ml in serum (considering 240 copies of HBc monomer per virion particle) (Fig. 5B), consistent with our previous reports (6, 8). We found positive correlations of HBcrAg with HBeAg and PreC, but not with HBc (Fig. 5C to E), and a positive correlation between PreC and HBeAg (Fig. 5F). However, some exceptions to these general conclusions were noticeable. In patient number 5, the level of serum HBc was much higher than in all the other patients tested, approaching the level of HBeAg (Fig. 4B, lane 11). Interestingly, that same patient showed strong e0 and e1 signals but no detectable PreC0 and PreC1 (Fig. 4B, lanes 2 and 11). We also attempted to detect HBcrAg components in HBeAg-negative patients. However, we could not detect any HBcrAg components using our Western blot analysis due to the much lower levels of HBV replication and gene expression in these patients and the relatively low sensitivity of the Western blot assay (see also Discussion below).
FIG 5.

Quantitative analysis of each component of HBcrAg in chronically HBV-infected patient serum. (A) Relative amounts of each component of HBcrAg (HBc, HBeAg, and PreC) in chronically HBV-infected human sera (means ± SDs; n = 11). Statistical analysis was performed using Student's t test, two-tailed and paired. ns, not significant (P > 0.05); ***, P < 0.001. (B) Concentrations of HBc, HBeAg, PreC, and total HBcrAg (i.e., the sum of HBc, HBeAg, and PreC) in the human sera. The concentration of each component was quantified by immunoblotting using MAb T2221, with known concentrations of recombinant HBc purified from bacterial expression as standards (means ± SDs; n = 11). Statistical analysis was performed using Student’s t test, two-tailed and paired. **, P < 0.01; ***, P < 0.001. (C to F) Correlations of HBeAg, HBc, and PreC with total HBcrAg and correlation of HBeAg with PreC. The correlation coefficient was calculated by Spearman’s correlation test. Two-tailed P value was calculated for a 95% confidence interval.
PreC proteins were not associated with HBV virions and showed density and size heterogeneity in patient sera.
To determine if any of the precore-derived proteins were components of HBV virions in HBV-infected patients, as claimed for the p22cr previously (14), we resolved virions by native agarose gel electrophoresis (NAGE) and detected virion-associated HBc, and precore proteins if any, with the CTD-specific MAb 366-2 (epitope from aa 150 to 164), 14-2 or 25-7 (recognizing a phosphorylated or nonphosphorylated epitope from aa 164 to 183, respectively [Fig. 1]), as well as the NTD-specific MAb T2221. We analyzed the crude human sera by NAGE and showed that all of the virion-associated capsids could be detected by all of the CTD MAbs (Fig. 6), consistent with our previous reports (5, 7). Since MAb 366-2 detected only the PreC3(A) with a relatively long CTD in genotype A and not PreC proteins in any other genotypes (Fig. 4), and neither 25-7 or 14-2 detected any secreted PreC proteins (Fig. 1A), these results indicated that the capsid in virions (empty or complete) contained HBc, as we reported before (7). Furthermore, consistent with our recent report (7), DNA-containing virions harbored dephosphorylated HBc, as evidenced by the correlation of the DNA signals with the 25-7 signals (nonphosphorylated CTD) (Fig. 6A and B, lanes 1, 2, 3, 6, 11, 12, and 14). However, DNA signals, representing DNA-containing virions, did not correlate with the 14-2 (phosphorylated CTD) or 366-2 (total CTD) signals, which reflects total virions (mostly empty virions) (Fig. 6A, lanes 4, 5, 7, and 9, versus Fig. 6C and D, lanes 4, 5, 7, and 9).
We attempted to use the HBeAg/PreC-specific MAb 1A11 or 7E9 to determine if any precore-derived proteins were associated (comigrating) with virions in the human sera after NAGE. However, both MAbs cross-reacted strongly with unknown serum proteins on the agarose gel, precluding specific detection of precore-derived proteins, if any, in the sera (data not shown). To overcome this issue, we first resolved HBV virions from each of the four genotypes (A to D) from serum proteins by CsCl density gradient fractionation and tracked the distribution of virions and precore-derived proteins across the gradient. To localize the virion (both empty and complete) peak fractions, fractions were analyzed by NAGE followed by Southern blotting to detect virion DNA and by immunoblotting using the CTD MAbs (366-2 and 14-2) as well as the NTD MAb T2221 to detect virion HBc (Fig. 7). The gradient fractions were then analyzed by SDS-PAGE followed by immunoblotting using MAbs 1A11, T2221, and 366-2 (Fig. 8) to detect HBeAg, PreC, and HBc. For patient number 3, who had only a low (undetectable by Southern blotting analysis) viremia load from crude serum (Fig. 6A, lane 9), no DNA virion was detected by Southern blot analysis even after CsCl fractionation, but MAbs T2221, 366-2, and 14-2 detected strong signals in fractions 15 and 16, representing empty virions (Fig. 7A, lanes 8, 9, 14, 15, 20, and 21), as we reported (5, 7). For patients 4, 10, and 12, we detected the peak of HBc signals in the fraction with the lighter density than that of the DNA signals (Fig. 7B, lanes 8, 14, and 20 versus lane 3; Fig. 7C, lanes 7, 12, and 17 versus lane 3; and Fig. 7D, lanes 8 and 13 versus lane 4), consistent with our previous findings that empty virions, containing capsids but no DNA or RNA, have a lighter buoyant density than DNA-containing virions (5, 6).
FIG 7.

Analysis of virions from the sera of HBV-infected patients by CsCl density gradient fractionation. Serum samples from HBV-infected patient 3 (genotype A [gtA]) (A), patient 4 (gtB) (B), patient 10 (gtC) (C), and patient 12 (gtD) (D) were fractionated by CsCl gradient ultracentrifugation. The virion particles in the fractions were analyzed by native agarose gel electrophoresis. HBV DNA and core protein were detected, as described in the legend to Fig. 6, using the indicated NTD- or CTD-specific MAbs. The direction of centrifugation (top to bottom) is indicated.
FIG 8.

Separation of HBeAg and PreC proteins from HBV virion particles in patient sera by CsCl density gradient fractionation. Sera from patients infected with the indicated HBV genotype (A, B, C, or D) were fractionated by CsCl density gradient ultracentrifugation. Fractions were resolved by regular SDS-PAGE, followed by immunoblot analysis using MAb 1A11, T2221, or 366-2. Fractions from patient 3 (genotype A or gtA) (A) were resolved by SDS-PAGE in the high-resolution gel, and fractions from patient 4 (gtB) (B), patient 10 (gtC) (C), and patient 12 (gtD) (D) were resolved by SDS-PAGE in a regular gel. V, the virion peak of each gradient as determined by native agarose gel electrophoresis shown in Fig. 6. The direction of centrifugation (top to bottom) is indicated.
To localize the HBeAg and PreC proteins, as well as HBc, in the CsCl density fractions, we analyzed the fractions by SDS-PAGE and Western blotting. For all four genotypes, all of the precore-derived species, PreC, as detected by MAbs 1A11 and T2221, shared the same buoyant densities as HBeAg, and they cofractionated at peaks that were clearly different from the HBc peak representing virions, as detected by MAbs 366-2 and T2221 (Fig. 8A, lane 5 versus lane 7; Fig. 8B, lane 5; Fig. 8C, lane 5 versus lane 7; and Fig. 8D, lane 4 versus lane 6), indicating that none of the PreC or HBeAg proteins were associated with either complete or empty HBV virions in the sera. As with the crude sera, only genotype A-specific PreC3(A) and HBeAg [e3(A)] harboring part of the CTD could be detected by MAb 366-2 (Fig. 8A, lanes 6 to 9); none of the PreC or HBeAg species from genotypes B to D could be detected by MAb 366-2 (Fig. 8B to D). On these isopycnic density gradients, soluble proteins, including HBeAg and PreC, are expected to sediment to dense fractions (ca. 1.3 g/cm3), substantially higher than virions (ca. 1.2 g/cm3). For patients 3 and 12, both HBeAg and PreC indeed sedimented into fractions with this predicted high density (Fig. 8A and D). Interestingly, for patients 4 and 10 (and, to a lesser extent, patient 12), PreC and HBeAg displayed strong density heterogeneity, being distributed to multiple density fractions with buoyant densities higher and lower than virions (Fig. 8B to D). The distribution of HBeAg and PreC proteins to fractions lighter than virions suggested that they might bind to factors with light densities such as lipoproteins or lipids in the serum (see also Discussion).
In addition, we employed sucrose gradient ultracentrifugation to separate the components of HBcrAg in two patient sera based on their sizes, instead of buoyant densities as with CsCl gradients as described above. Our results indicated that serum HBeAg and PreC from both patients cosedimented on the top fractions and peaked in fractions 1 and 2 (Fig. 9A and B, lanes 1 and 2), suggesting that HBeAg and the PreC had similar sizes, the majority of both being much smaller than virions and HBsAg particles detected in the middle of the gradient by the HBsAg-specific antibody (Fig. 9A and B, lanes 8 to 10). These results thus further indicated that most of the PreC and HBeAg were not associated with virions. The small dimeric HBeAg (with a molecular weight of ca. 34 kDa) remaining on top of the sucrose gradient was fully expected. The fact that a portion of HBeAg and the PreC proteins sedimented into the fractions (from fractions 3 to 7) suggested that some of these proteins might be associated with other factors in the serum, a notion also suggested from the buoyant density analysis described above (Fig. 8).
FIG 9.

Analysis of HBeAg and PreC proteins in patient sera by sucrose gradient fractionation. Sera from patient 10 (gtC) (A) and patient 12 (gtD) (B) were fractionated by sucrose density gradient centrifugation. The fractions were resolved by high-resolution SDS-PAGE followed by immunoblotting using the precore MAb 1A11, precore/core MAb T2221, and anti-HBs (Virostat) antibodies sequentially on the same membrane. The concentrated supernatant from HBc (p21)- and precore(p25)-transfected Huh7 cells was loaded as a reference for HBc, HBeAg, and PreC proteins. The direction of centrifugation (top to bottom) is indicated.
PreC proteins were not associated with HBV virions in PHHs.
To further overcome the issue that human serum proteins cross-reacted with the precore-specific MAb 7E9 or 1A11, we analyzed the PreC and HBeAg density profiles relative to virions in the supernatant of HBV-infected PHHs by CsCl gradient ultracentrifugation. We found the HBV DNA, representing complete virions, and HBc, representing mostly empty virions, in fractions 12 to 15 (peak, fraction 13 for HBc/empty virions and fraction 14 for DNA/complete virions), both migrating near the top of the agarose gel after NAGE (Fig. 10A), as the serum virions (Fig. 7). The NTD MAbs T2221 and 1D8 (see Fig. 10C and Table 1 for 1D8 epitope mapping results) also detected a smear signal running faster than the virions on the agarose gel in fractions 14 to 16, which was also detected by the precore-specific MAb 7E9 (Fig. 10A, lanes 24 to 26, 33 to 35, and 42 to –44). 7E9 is another HBeAg- and PreC-specific MAb that is targeted to the −10 region (Fig. 1; Fig. 10C) and showed less background than MAb 1A11 on Western blots after NAGE. Thus, we could detect HBeAg and/or PreC from the PHH supernatant after NAGE, in contrast to those present in the patients’ sera. Indeed, the presence of both HBeAg (e1) and PreC (PreC1) proteins in fractions 14 to 16 was confirmed upon SDS-PAGE and Western blot analysis using MAbs 1A11 and T2221 (Fig. 10B, lanes 7 to 9), clearly below (i.e., having a higher density than) the HBc (virion) peak in fraction 13 detected by MAbs 366-2 and T2221, similar to the NAGE results (Fig. 10A). These results thus demonstrated that virions produced by HBV-infected PHHs in culture, like virions in patients’ sera, contained HBc but not HBeAg or PreC protein. We noticed that there were weak signals detected in fractions 12 to 14 by 7E9 on the agarose gel that comigrated with virions (Fig. 10A, lanes 40 to 42). However, only the HBeAg (e1), and not PreC (PreC1), was detected in these fractions after SDS-PAGE (Fig. 10A, lanes 5 to 7). Since HBeAg is not known to, and is unlikely to, associate with virions, our results indicated that neither HBeAg nor PreC was part of virions, although we could not exclude the possibility of the presence of trace amounts of these precore-derived proteins incorporated into the capsids formed predominantly by HBc.
FIG 10.

Separation of HBeAg and PreC proteins from HBV virion particles secreted by HBV-infected PHH culture by CsCl density gradient fractionation. (A) Concentrated culture supernatant from gtD HBV-infected PHHs harvested 10 days postinfection (dpi) was fractionated by CsCl density gradient centrifugation. The virion particles and antigens in the fractions were analyzed by native agarose gel electrophoresis. HBV DNA and core protein were detected, as described in the legend to Fig. 6, using the indicated NTD- or CTD-specific MAbs. Ca, capsid, containing DNA or RNA or empty; e1/PreC1, secreted HBeAg and PreC protein from HBV genotype D-infected PHHs. (B) Concentrated culture supernatant from mock-infected or gtD HBV-infected PHHs harvested 10 days postinfection, as well as selected fractions from the CsCl gradient shown in panel A, were resolved by high-resolution SDS-PAGE, along with the serum from the gtD HBV-infected patient (no.12) and chimpanzee (no. 1616, week 22) (5; Hong et al., submitted). (C) Epitope mapping for MAbs 1D8 and 7E9 by ELISA using an overlapping peptide library. The linear epitope recognition of the anti-HBV precore/core MAbs 1D8 and 7E9 was interrogated against an overlapping, biotinylated peptide library, numbered 1 to 69, of the linear precore precursor protein (p25, residues −29 to 183 [Table 1]) epitopes, immobilized on a streptavidin-coated ELISA plate. Precore- and HBc-specific regions covered by the overlapping peptide library are indicated. Recombinant HBeAg and HBc were included as assay binding controls. The direction of centrifugation (top to bottom) is indicated in panels A and B.
TABLE 1.
Summary of overlapping peptides that bound to MAb 1D8 or 7E9
| MAb | Peptide | Peptide sequencec | HBc/eAg residues | Epitope residues |
|---|---|---|---|---|
| 1D8 | 36 | ATWVGVNLEDPASRD | 69 to 83 | 75 to 83 (immunodominant loop) |
| 37 | VGVNLEDPASRDLVV | 72 to 86 | ||
| 38 | NLEDPASRDLVVSYV | 75 to 89 | ||
| 39 | DPASRDLVVSYVNTN | 78 to 92 | ||
| 7E9 | 6 | TVQASKLCLGWLWGM | −14 to 1 | −10 to 5 (unique precore sequence) |
| 7 | ASKLCLGWLWGMDID | −11 to 4 | ||
| 8 | SKLCLGWLWGMDIDPa | −10 to 5 | ||
| 9 | SKLCLGWLWGMDIDPb | −10 to 5 | ||
| 10 | CLGWLWGMDIDPYKE | −7 to 8 | ||
| 11 | WLWGMDIDPYKEFGA | −4 to 11 |
N-terminal biotin tag.
C-terminal biotin tag.
Boldface letters denote residues within the respective epitope.
Precore-derived proteins failed to assemble capsids in a cell-free system under physiological conditions.
In order for p22cr or any precore-derived proteins to be enveloped and secreted as virions, they would need to be able to form capsids in infected human hepatocytes. In fact, HBeAg was shown to form capsids under artificial reducing conditions when disulfide bond formation between Cys-7 and Cys61 is prevented, which normally precludes capsid assembly (34). To test whether precore-related proteins can form capsids under physiological conditions, we translated precore-related proteins in the rabbit reticulate lysate (RRL) system, which supports HBV capsid assembly under physiological protein concentration and salt conditions (35). As we reported previously, HBc could be triggered to assemble capsids with phosphatase treatment in RRL (Fig. 11A to C, lane 1), which was verified both by the characteristic capsid mobility on the agarose gel and by immunoblotting using MAb 3120, which recognizes a structural epitope thought to be present only on assembled capsids (33). Also included in this study as another control for the assembly reaction was the HBc mutant 3E, which harbors serine-to-glutamate substitutions at the three SP phosphorylation motifs in CTD and is able to assemble in the absence of exogenous phosphatase treatment (Fig. 11A to C, lanes 6 and 13) (35). In contrast, artificially expressed p17 (corresponding to the secreted HBeAg e0 in Fig. 1A) or p22cr (i.e., PreC0 in Fig. 1A) failed to form capsids (Fig. 11A to C, lanes 2, 4, 9, and 11). Expression of p22, which corresponds to the intracellular intermediate during precore processing, lacking the signal peptide but retaining the −10 region and the entire CTD (36), formed a low level of capsids after phosphatase treatment (Fig. 11A to C, lane 3). However, this was likely attributable to the translation of HBc via internal initiation from the p22 RNA, which was as abundant as p22 as visualized following SDS-PAGE (Fig. 11D, lanes 3 and 10). Indeed, internal initiation from the p22 RNA to produce p21 (HBc) is known to occur (36, 37). Translation of the entire precore precursor, p25, in RRL failed to assemble capsids (Fig. 11A to C, lanes 5 and 12). Although internal initiation of translation from the internal HBc AUG in RRL from p25 RNA was also evident by the production of HBc, any capsids formed by HBc might be below the detection limits. The production of C149 via internal initiation from the p17 RNA (Fig. 11D, lanes 2 and 9) and p22cr RNA (Fig. 11D, lanes 4 and 11) was also evident, but as we have shown previously (35), this truncated HBc (without the CTD) was unable to form capsids under physiological conditions.
FIG 11.

Failure of precore-related proteins to form capsids in cell-free capsid assembly system. The indicated HBc (core) and precore-related proteins were expressed by in vitro translation in the presence of [35S]methionine in RRL, and the translation reaction mixtures were resolved by native agarose gel electrophoresis (A to C) or SDS-PAGE (D). The reaction mixtures were treated with buffer alone (NEB buffer 3) or with CIAP overnight at 37°C. 35S-labeled proteins were detected by autoradiography (A and D). Assembled HBV capsids were detected by MAb 3120 (B), which detects a structural epitope on assembled capsids. The precore/core proteins, with or without assembly, were detected by MAb T2221 (C). pCI-HBc-3E was used as a positive control for assembly without CIAP treatment.
Precore-derived proteins failed to form capsids or virions in human cells.
To further test if ectopic expression of precore-related proteins could lead to the formation of capsids, and virions when the HBV envelope proteins were provided, in human cells, we transfected constructs encoding HBc or precore-related proteins, together with the construct pSVB45H expressing HBV envelope proteins or pSVHBV1.5Core-, which is a full-length HBV replicon construct defective in HBc expression, into HepG2 cells. We found that only HBc (p21) supported capsid formation in the cell (Fig. 12B, lanes 4 and 9). Expression of p22cr and p25 supported the secretion of HBeAg e0 and e1, respectively (Fig. 12A, lanes 6, 7, 11, 12), indicating that the N-terminal signal peptide (−19 aa) was removed from most p22cr during secretion, as for p25. On the other hand, expression of p22 led to its retention inside cells due to the lack of the signal peptide to direct it into the secretory pathway (Fig. 12A, lanes 5 and 10). We could confirm that expression of p25 led to the production of p22 intracellularly, which was identified by its slower migration than HBc (p21) and reactivity with both MAbs 1A11 and T2221 (Fig. 12A, lanes 7 and 12). It was notable that the p22 protein expressed from the p25 construct migrated faster than the p22 protein expressed from the p22 construct (Fig. 12A, lanes 5 versus 7 and 10 versus 12). One possible explanation for the different mobility is that posttranslational modification on p22 expressed from the p22 construct (without the signal peptide coding sequences) versus that from the p25 construct (containing the signal peptide coding sequences) might be different. The p22 protein expressed from the p22 construct is expected to be localized in the cytoplasm, whereas p22 expressed from the p25 construct is expected to be localized to the secretory pathway predominantly, which could affect their posttranslational modification state. Some HBc (p21) could also be expressed from the p25 expression construct (Fig. 12A, lanes 7 and 12), due to internal initiation from the HBc initiation codon, as described above. Artificial expression of p17 produced very low (or undetectable) levels of p17 (designated p17* in Fig. 12A, to be differentiated from p17 processed from p25 or p22cr) either intracellularly or extracellularly (Fig. 12A, lanes 3 and 8), consistent with our previous finding that the truncated HBc (C149) is unstable in human cells (35) and further suggesting that the addition of the −10 region to C149 (i.e., p17) could not rescue its expression. Interestingly, some intracellular HBeAg (e0) was detectable from p22cr expression (Fig. 12A, lanes 6 and 11) but no intracellular HBeAg (e1) was detectable from p25 expression (Fig. 12A, lanes 7 and 12), consistent with the stimulatory effect of CTD on HBeAg secretion (38, 39). In this experiment, we did not detect PreC protein secretion in the concentrated culture supernatant due to the small amounts of PreC proteins in the small-scale cell culture. A large-scale Huh7 cell culture was used to detect the secreted PreC protein in the supernatant above in Fig. 2.
FIG 12.

Failure of precore-related proteins to form capsids or virions in human hepatoma cell culture. HepG2 cells were cotransfected with pSVB45H or pSVHBV1.5Core-, along with pCI-HBc (p21), pCI-p17, pCI-p22, pCI-p22cr, or pCI-precore (p25), as indicated. Cytoplasmic lysate and culture supernatant were harvested at day 5 posttransfection. (A) Cytoplasmic lysate (top two panels) and concentrated supernatant (bottom two panels) were resolved by SDS-PAGE, followed by immunoblotting using the HBeAg/PreC-specific MAb 1A11 or anti-precore/core (NTD) MAb T2221. (B) Cytoplasmic lysate (top two panels) and concentrated cell culture supernatant (bottom three panels) were resolved by native agarose gel electrophoresis. Following transfer to nitrocellulose membrane, HBV RNA associated with intracellular capsids was detected by a plus-strand-specific HBV riboprobe and HBV DNA in extracellular virions and naked capsids was detected by a HBV DNA probe, followed by immunoblotting using the anti-precore/core (NTD) MAb T2221 and anti-HBs polyclonal antibody (in the case of supernatant only). The different viral and subviral particles are represented schematically to the right. The wavy and straight lines inside the capsid (hexagon) indicate the pgRNA and single-stranded DNA, respectively. The small and large circles denotes the HBsAg spheres and HBV virions, respectively. p17*, artificially expressed (not processed from p25 or p22cr); S, HBs subviral particles. *, unassembled precore or core proteins.
Analysis of virion secretion showed that only HBc (p21) supported secretion of DNA-negative (empty) or DNA-containing virions, respectively (Fig. 12B, lanes 4 and 9), as we reported previously (5, 7, 9). In contrast, there was no virion secretion in cells transfected with p17, p22, p22cr, or p25 (Fig. 12B, lanes 3, 5, 6, 7, 8, 10, 11, and 12), consistent with the lack of capsid formation by these constructs. We also performed these transfections in Huh7 cells and obtained the same results as shown in Fig. 12 (data not shown). In conclusion, none of the precore-derived proteins were able to support capsid formation or virion secretion.
DISCUSSION
HBcrAg is an emerging biomarker that is being widely tested in applications such as monitoring intrahepatic cccDNA and predicting outcomes of antiviral treatments (15, 18, 19). However, the detailed biological characterization of HBcrAg (i.e., precore/core gene products) remains largely elusive. In this study, we took advantage of a panel of MAbs that recognize different epitopes of precore/core gene products and characterized the components of HBcrAg in detail. We have demonstrated that HBcrAg included multiple species of HBeAg and PreC proteins derived from the precore ORF, besides HBc encoded by the Core ORF (Fig. 13). Both HBeAg and PreC proteins displayed strong heterogeneity in density and size. We have also demonstrated that HBeAg, at ca. 1014 molecules/ml of serum, was the predominant form of HBcrAg in HBeAg-positive patients. HBc was associated with virion particles but neither HBeAg nor any other precore-derived proteins were.
FIG 13.
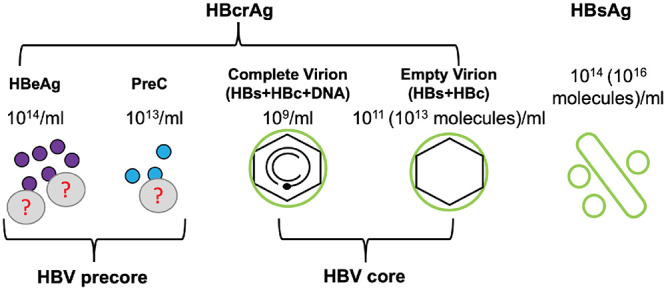
HBcrAg and other serum HBV markers in the blood. HBcrAg is a composite antigen which includes HBeAg, PreC, and HBc. Both HBeAg and PreC proteins are derived from the precore gene and secreted as nonparticulate proteins, at ca. 1014 molecules/ml and 1013 molecules/ml, respectively, in the serum of HBV-infected patients. Some HBeAg and PreC are associated with an unknown low-density factor(s) (?) in the blood. Serum HBc is mostly contributed by empty capsids in empty virions, which are present at a level of 1011 particles/ml (ca. 1013 molecules/ml), ca. 100-fold more than DNA virions (109 particles/ml). In addition, HBsAg, in the form of filament or sphere, is secreted to the blood at a level of 1014 particles/ml (ca. 1016 molecules/ml).
The exact HBeAg and PreC species in the human serum were complex and varied with HBV genotypes, reflecting the differential usage of the multiple proteolytic cleavage sites, the sequence of which varies with genotype, in the CTD during the biosynthesis of both HBeAg and PreC. Using a panel of MAbs targeting different regions of precore, we could confirm the presence of PreC-derived proteins in human serum analogous to p22cr or a 20-kDa HBeAg species with N-terminal signal peptide reported initially many years ago (14, 21). These PreC proteins, like HBeAg, were also derived from the precore precursor p25 (Fig. 1) and lacked most of the CTD in the precore/core but retained the N-terminal signal peptide. In contrast, only one protein species, HBc, is produced from the core ORF. To avoid confusion, we suggest using the name PreC to describe these precore-derived proteins, instead of p22cr or core-related antigen, as these proteins are all encoded by the precore but not core ORF. Obviously, all HBeAg species are also derived from precore and therefore also PreC, but for historical reasons, the name HBeAg is likely to be maintained.
The mechanisms of biogenesis and release of the PreC remain elusive. We found that serum PreC was consistently maintained at ca. 10% of the total serum HBeAg in HBV-infected patients. These results suggest that PreC is secreted using a mechanism similar to that of HBeAg secretion, i.e., through the ER-Golgi secretory pathway. The similar C-terminal processing heterogeneity between the PreC and HBeAg further suggests that both types of precore-derived proteins were processed at the CTD by the same proteases in the secretory pathway (11–13). Unlike HBeAg, the woodchuck hepatitis virus (WHV) eAg is N-glycosylated (40; X. Hong, L. Luckenbaugh, P. Revill, S. F. Wieland, S. Menne, J. Hu, submitted for publication). We have recently demonstrated the existence of N-glycosylated WHV PreC in the serum of chronically WHV-infected woodchuck (Hong et al., submitted), further supporting the notion that PreC biogenesis involves the cellular secretory pathway, like HBeAg (10). How the secreted PreC, in contrast to HBeAg, retains the N-terminal signal peptide sequence remains a mystery. As the secretion of PreC, as well as HBeAg, occurred from precore-expressing-human hepatoma cells in culture at the same proportion as during infection in PHHs or in patients, PreC secretion is apparently independent of any other viral factors. The naturally occurring G1862T mutation in the precore region of the HBV A1 subgenotype results in a valine-to-phenylalanine substitution, which interferes with signal peptide cleavage and leads to impaired HBeAg secretion (41). It would be interesting to test whether such mutations within the signal peptide sequence affect biogenesis and release of PreC identified in this study. A few cellular proteins, including the human apolipoprotein M (ApoM) secreted to the blood by hepatocytes, also retain the signal peptide, but their secretion mechanism also remains unknown (42).
The function of the secreted PreC, if any, remains to be defined. HBeAg, also encoded by the precore ORF, while dispensable for viral replication, is thought to play an important role in modulating the host immune response to facilitate viral persistence (23, 25), although the exact mechanism remains unclear. In addition, another precore-derived protein, the p22 processing intermediate (Fig. 1), which is retained intracellularly, lacks the N-terminal signal peptide (the first 19 aa), but retains the −10 aa as well as the entire CTD, is also thought to play a role in immune modulation (28). p22cr (i.e., PreC) was initially reported to form an aberrant capsid inside the empty virion (14). However, our results reported here and reported earlier (7) have clearly demonstrated that (i) empty virions contain HBc, (ii) none of the precore-derived proteins are required for secretion of empty virions, and (iii) there are no, or very few, precore-derived proteins in virions. Although it was shown that HBeAg could form capsid-like particles under artificial reducing conditions using purified recombinant protein expressed in bacteria (34), our results in this study showed that none of the precore-derived proteins, including HBeAg, PreC (p22cr), or p22, was able to form capsids under physiological conditions in a mammalian cell-free system (RRL) or in human cells. The lack of capsid formation by HBeAg or PreC proteins was in fact anticipated since intramolecular disulfide bond formation between Cys-7 in the precore region with Cys61 in the NTD prevents capsid assembly (37, 43). The lack of capsid formation by p22 in human cells was also reported recently by another group (28). In that study, the authors further found that even the Cys-7 to Glu mutation, which blocks disulfide bond formation, remained defective in capsid formation in human cells (28), even though the same mutation supported capsid formation in Escherichia coli (43). It was suggested that p22, which is thought to be translocated back to the cytosol following signal peptide cleavage in the ER (10), could form mosaic capsids in the cytosol together with HBc and block pgRNA packaging into these mosaic capsids, consequently inhibiting viral replication (36). Although it is possible that a portion of the PreC (p22cr) could also form mosaic capsids with HBc to interfere with HBc functions, it is more difficult to understand, as these proteins would presumably form only in the secretory pathway (needed for CTD processing; see above) and HBc capsids form in the cytosol.
The universally hydrophobic signal peptide, which is retained in the secreted ApoM, was shown to help anchor ApoM to lipoprotein particles and prevent its rapid clearance from plasma by filtration in the kidney (42). The signal peptide retained by HBV PreC, but not HBeAg, could play a similar role. It was reported decades ago that HBeAg showed two different populations in patient sera, with or without binding to immunoglobulin (44). In this study, we found that a population of HBeAg and PreC in some chronic HBV-infected patients were fractionated into a density much lighter than virions and displayed size heterogeneity, suggesting that these HBV proteins may also associate with other factors or structures in the blood, including those with low density such as lipoproteins, which may affect their clearance and/or functions. Interestingly, we also found two different populations of eAg/PreC in chronically, but not in acutely, WHV-infected woodchucks (Hong et al., submitted). Whether the different pattern of eAg/PreC population is associated with disease outcomes remains to be studied. HBeAg is known to form homodimers (43); whether PreC forms homodimers or HBeAg and PreC form heterodimers also remains to be determined. The existence of multiple precore-derived proteins, including extracellular (secreted) HBeAg and PreC (p22cr), as well as the intracellular p22 intermediate, raises the intriguing possibility that each of these products encoded by the precore gene has distinct functions far beyond what has been appreciated so far, which may be further modulated by their associations with other factors or modifications.
The current HBcrAg assay measures products of the precore/core gene, including HBc, HBeAg, and p22cr (i.e., PreC) (14, 16), and HBcrAg is defined, in practice, as a composite antigen. The assay uses a combination of several NTD-specific antibodies (Fig. 1A), similar to the MAbs T2221 and 19C18 used in this study, which can in principle detect all of the core and precore-derived species as we found here, including HBc and the multiple species of HBeAg and PreC that all contain the NTD (14, 16). The relative contribution of each HBcrAg component in the current commercial assay has remained unclear and may vary depending on various viral and host factors, which could affect the accuracy and meaning of HBcrAg as a biomarker. Here, we demonstrated that HBeAg was usually the predominant form of HBcrAg, implying that factors that affect HBeAg levels will also affect HBcrAg levels, such as basal core promoter mutations which reduce HBeAg secretion (and presumably also PreC secretion) (23, 45) and production of antibodies to HBeAg leading to rapid HBeAg clearance from the circulation, as we have demonstrated in another study (Hong et al., submitted). In the case of HBeAg loss due to the precore stop codon mutation, no HBeAg or PreC will be secreted; thus, only HBc will be detected by the current HBcrAg assay. Thus, genetic mutations in cccDNA affecting precore (and consequently, eAg and PreC protein) expression, or development of antibodies to the precore-derived proteins, will lead to decline or loss of these markers in the periphery, without necessarily any decrease or loss of cccDNA in the liver, rendering HBcrAg (mainly HBeAg) unreliable as a marker for intrahepatic cccDNA in these situations. Indeed, in a separate study using matching serum and liver samples from HBV-infected chimpanzees across different stages of infection, we could show that serum HBc (i.e., empty virion) levels correlate better than HBeAg/PreC/HBcrAg levels with intrahepatic cccDNA (Hong et al., submitted). We also attempted to analyze samples from HBeAg-negative patients; however, we could not detect any HBcrAg components using our current Western blot assay, and more sensitive assays for detecting low levels of HBc in these cases will need to be developed. Also, due to the limited sample size of our study, future studies with larger cohorts of patients will be needed to assess the true range in the levels of each precore/core gene product.
Our results from this study and previous studies indicate that naked HBV capsids (i.e., without the viral envelope) are either not released into the blood of HBV-infected patients or chimpanzees or are released only at very low levels below the limit of our detection (Fig. 6) (5–8). Furthermore, an earlier study from a different group also showed that no naked HBV capsids were detectable in immunosuppressed patients, i.e., even in the absence of anti-HBc antibody (46) which could, theoretically, rapidly clear any naked capsids in the circulation released from the liver. Moreover, unlike hepatoma cell lines that release large amounts of naked capsids (e.g., see Fig. 12), HBV-infected PHHs released only very low levels of naked capsids (Fig. 10), which was possibly due to low levels of cell death. In addition, we detected no, or only very low, levels (much lower than capsid levels contained in virions) of naked HBV capsids in the blood of HBV-transgenic mice or HBV-infected humanized chimeric mice (unpublished data), which are immunotolerant to HBV or immunocompromised and do not produce anti-HBc antibodies. As we suggested earlier, the release of naked HBV capsids appears to be an artifact of hepatoma cell cultures; one underlying mechanism for this phenomenon may be the relatively low expression (much lower than in infected humans or chimpanzees) (5, 6) of the HBV envelope proteins relative to capsids in these cultures, since we could show that enhancing envelope protein expression in hepatoma cell cultures could suppress the release of naked capsids (47). Although naked HBV capsids could possibly be released into the blood of HBV-infected patients, e.g., when large number of infected hepatocytes are lysed, and possibly form capsid-antibody complexes (48), we believe that release of naked HBV capsids does not generally occur during natural infection and thus contributes little, if any, to the overall levels of serum HBcAg, which predominantly reflect levels of empty virions (Fig. 13).
In summary, our results here have demonstrated that precore/core gene products (i.e., HBcrAg) in HBV-patient serum included HBc and two types of precore-related proteins, HBeAg and PreC, both heterogeneously processed. HBeAg was the predominant form of HBcrAg in HBeAg-positive patients. HBc, but not HBeAg or PreC, was the main component of capsids in DNA-containing or empty virions. These results deepen our understanding of the composition of HBcrAg and the amount of each component, as well as their biochemical and biophysical characteristics, highlighting the need to monitor HBeAg (or PreC) and HBc (i.e., virions) separately to reflect intrahepatic HBV gene expression and replication and suggesting that each HBcrAg component (i.e., precore/core gene product) may have unique biological functions.
MATERIALS AND METHODS
Serum samples.
Serum samples from HBeAg-positive patients infected with HBV genotype A, B, C, or D were obtained from BioCollections Worldwide, Inc. (Miami, FL), or were obtained from patients enrolled in Gilead Sciences clinical studies GS-US-174-0106 and GS-US-174-0123 (6). Serum samples from HBV (genotype D; GenBank accession number V01460)-infected chimpanzees 1616 (week 22) were described previously (49).
Plasmid constructs.
An HBV DNA sequence spanning the precore/core ORF was amplified from pCIΔA-HBV-HBc-WT (genotype D) (50). The HBc expression construct pCI-HBc was described previously (35), and plasmids for expressing the precore-related proteins, including pCI-p25 (full-length precore), pCI-p22 (deleting signal peptide from aa −2 to −19 of p25), pCI-p17 (deleting signal peptide of aa −2 to −19 and C-terminal aa 150 to 183 of p25), and pCI-p22cr (deleting the C-terminal aa 150 to 183 of p25), were constructed similarly to pCI-HBc in the pCI vector (Promega, WI). pSVB45H expresses the HBV envelope proteins L, M, and S (51) and pSVHBV1.5Core-, which is an HBV replicon construct defective in HBc expression and supports HBV replication upon complementation with HBc expression (52).
Cell cultures, transient transfection, and primary human hepatocyte (PHH) infection.
Human hepatoma cell lines, HepG2 and Huh7, were maintained as described previously (7, 50). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (Gibco) supplemented with 10% fetal bovine serum (FBS) (HyClone). For transient transfection, HepG2 or Huh7 cells seeded in 60-mm dishes were transfected with 4 μg (total) of plasmid using X-tremeGENE HP DNA transfection reagent (Roche) or FuGENE 6 transfection reagent (Promega, WI), respectively. A 1:1 mass ratio of each plasmid was used when two plasmids were used for cotransfections (7). All transfection experiments were repeated three times.
PHHs were purchased from BioreclamationIVT (Westbury, NY). Cryopreserved PHHs were thawed and then seeded in cell plating medium (Life Technologies, CA) in 24-well collagen-coated plates (Corning, NY) and were maintained as described previously (53). Cells were infected with HepAD38 wild-type virions (genotype D) at 400 viral genome equivalents (GE) per cell in medium containing 4% polyethylene glycol 8000 (PEG 8000) for 16 h at 37°C (54, 55). Mock-infected cells were treated with medium containing 4% PEG 8000 in the absence of virus. The cells were cultured in maintenance medium containing 2% dimethyl sulfoxide (DMSO) and 2% FBS. The PHH medium was collected and changed every 2 to 4 days until the end of the study.
Immunoblot analysis.
Serum samples, concentrated cell culture supernatant, and cell lysates were separated using 12.5% SDS-polyacrylamide gel electrophoresis (PAGE) at gel heights of 35 cm (high resolution) or 12.5 cm (regular) and immunoblotting (5). The mouse monoclonal antibody (MAb) clone T2221 (catalog no. 2AHC24) (9) and clone 19C18 (catalog no. 2Z19C18) specific for the HBc NTD were purchased from Tokyo Future Style. The HBc CTD-specific rabbit MAb 366-2 was custom-made (Abcam) using a CTD peptide as an immunogen and has been described before (32). The HBc CTD-specific rabbit MAbs 14-2 and 25-7 (7, 35) were also custom-made (Abcam) and recognize a phosphorylated and dephosphorylated CTD epitope between 164 and 183, respectively. The polyclonal rabbit antibody against HBsAg (Virostat, Portland, ME) was used for the detection of envelope proteins. The mouse anti-precore MAbs, 7E9 and 1A11 (VIDRL, Australia), were used for specific detection of precore-related proteins (31).
Peptide library and epitope mapping by enzyme-linked immunosorbent assay (ELISA).
A library of 67 15-mer peptides with a 12-residue overlap covering the p25 protein (212 residues, labeled residues −29 to 183) was constructed by Mimotopes, Australia. The peptides were produced with an N-terminal biotin tag, and C-terminal amide, and were reconstituted in DMSO (Sigma) prior to phosphate-buffered saline (PBS) dilution for immunoassay. Additional peptides with a C-terminal biotin tag and N-terminal amide were made for peptide 1 (p25 N terminus), peptide 8 (p17 N terminus), and a 10-mer peptide of the unique p17 HBeAg N terminus. The immunoassay was performed as previously described (31). Peptides were detected by 1D8 or 7E9 horseradish peroxidase (HRP)-conjugated antibodies. Additionally, recombinant HBeAg (p17) and truncated (residues 1 to 149) HBc (C149) proteins (0.4 μg/well) were bound to non-streptavidin-coated wells for antibody detection. The overlapping peptides that bound to MAb 1D8 or 7E9 are summarized in Table 1 and Fig. 10C.
Analysis of HBV virion secretion by CsCl density gradient centrifugation.
Virions, subviral particles, and soluble viral antigens in the serum of HBV-infected patients were purified by CsCl density gradient ultracentrifugation (5, 56). Fractions were analyzed by native agarose gel electrophoresis for virion particles and by SDS-PAGE for analysis of viral antigens/proteins.
Analysis of HBV antigens by sucrose gradient ultracentrifugation.
Serum samples from HBV-infected patients were fractionated by sucrose gradient ultracentrifugation as described previously (35), with minor modifications. A total of 1.5 ml of serum was layered on the top of a 15% to 30% linear sucrose gradient containing TNE buffer (10 mM Tris-HCl [pH 8]/100 mM NaCl/1 mM EDTA) in a 12-ml ultracentrifugation tube, and ultracentrifugation was performed at 27,000 rpm for 4 h at 4°C in a Beckman SW40Ti rotor. Fractions were collected from the top to bottom and were analyzed by SDS-PAGE for analysis of viral antigens/proteins.
In vitro translation in the RRL.
A TnT-coupled rabbit reticulocyte lysate (RRL) in vitro translation system (Promega, WI) was used for cell-free expression of HBc or precore-related proteins (35). In vitro-translated proteins were treated with calf intestinal alkaline phosphatase (CIAP) as described previously to initiate capsid assembly (35). In vitro translation and assembly products were resolved by 1% native agarose gel electrophoresis or 12.5% SDS-PAGE and then probed with the desired anti-HBc antibodies. When the translation products were labeled with [35S]methionine, radiolabeled proteins were also detected by phosphorimaging scan following resolution by gel electrophoresis.
Statistical analysis.
Protein signals from Western blot analysis were detected and quantified using the Image Lab system 6.0.1 (Bio-Rad), as described previously (50). The concentration of each HBcrAg component was quantified by immunoblotting using MAb T2221, with known concentrations of recombinant HBc purified from bacterial expression as standards. Subtraction of background signals was done separately for each individual lane. All quantifications were performed within the linear range of the instruments in the absence of signal saturation. Data were analyzed by using Prism 7.0 (GraphPad). Student’s t test, two-tailed and paired, was used when comparing two data sets, and the data are shown as means ± standard deviations (SDs). Correlations between two data sets were determined by linear regression, and the correlation coefficient was calculated by Spearman’s correlation test. Two-tailed P value was calculated for a 95% confidence interval. A P value of <0.05 was considered to be statistically significant.
ACKNOWLEDGMENTS
We thank Volker Bruss (Technical University of Munich) for the HBc-defective genomic plasmid and envelope expression plasmid and Stefan Wieland and Frank Chisari (Scripps Research) for the chimpanzee serum samples.
This work is supported by NIH grants R37AI043453 and R01Al127670 to J.H. and a Royal Melbourne Hospital Keir Fellowship to P.A.R.
REFERENCES
- 1.Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, Levrero M, Li W, Liang TJ, Lim S-G, Lu F, Penicaud MC, Tavis JE, Thimme R, Zoulim F, ICE-HBV Senior Advisors. 2019. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol 4:545–558. doi: 10.1016/S2468-1253(19)30119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5:a021386. doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong X, Kim ES, Guo H. 2017. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology 66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu J, Protzer U, Siddiqui A. 2019. Revisiting hepatitis B virus: challenges of curative therapies. J Virol 93:e01032-19. doi: 10.1128/JVI.01032-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus–single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luckenbaugh L, Kitrinos K, Delaney W, Hu J. 2015. Genome‐free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J Viral Hepat 22:561–570. doi: 10.1111/jvh.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ning X, Basagoudanavar SH, Liu K, Luckenbaugh L, Wei D, Wang C, Wei B, Zhao Y, Yan T, Delaney W, Hu J. 2017. Capsid phosphorylation state and hepadnavirus virion secretion. J Virol 91:e00092-17. doi: 10.1128/JVI.00092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Liu K. 2017. Complete and incomplete hepatitis B virus particles: formation, function, and application. Viruses 9:56. doi: 10.3390/v9030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu K, Luckenbaugh L, Ning X, Xi J, Hu J. 2018. Multiple roles of core protein linker in hepatitis B virus replication. PLoS Pathog 14:e1007085. doi: 10.1371/journal.ppat.1007085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia PD, Ou J-H, Rutter WJ, Walter P. 1988. Targeting of the hepatitis B virus precore protein to the endoplasmic reticulum membrane: after signal peptide cleavage translocation can be aborted and the product released into the cytoplasm. J Cell Biol 106:1093–1104. doi: 10.1083/jcb.106.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Standring DN, Ou J-h, Masiarz FR, Rutter WJ. 1988. A signal peptide encoded within the precore region of hepatitis B virus directs the secretion of a heterogeneous population of e antigens in Xenopus oocytes. Proc Natl Acad Sci U S A 85:8405–8409. doi: 10.1073/pnas.85.22.8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito K, Kim K-H, Lok AS-F, Tong S. 2009. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J Virol 83:3507–3517. doi: 10.1128/JVI.02348-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Lee A, Ou J-H. 1991. Proteolytic conversion of hepatitis B virus e antigen precursor to end product occurs in a postendoplasmic reticulum compartment. J Virol 65:5080–5083. doi: 10.1128/JVI.65.9.5080-5083.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura T, Ohno N, Terada N, Rokuhara A, Matsumoto A, Yagi S, Tanaka E, Kiyosawa K, Ohno S, Maki N. 2005. Hepatitis B virus DNA-negative dane particles lack core protein but contain a 22-kDa precore protein without C-terminal arginine-rich domain. J Biol Chem 280:21713–21719. doi: 10.1074/jbc.M501564200. [DOI] [PubMed] [Google Scholar]
- 15.Cornberg M, Lok AS‐F, Terrault NA, Zoulim F, the 2019 EASL‐AASLD HBV Treatment Endpoints Conference Faculty. 2020. Guidance for design and endpoints of clinical trials in chronic hepatitis B—report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 71:1070–1092. doi: 10.1002/hep.31030. [DOI] [PubMed] [Google Scholar]
- 16.Kimura T, Rokuhara A, Sakamoto Y, Yagi S, Tanaka E, Kiyosawa K, Maki N. 2002. Sensitive enzyme immunoassay for hepatitis B virus core-related antigens and their correlation to virus load. J Clin Microbiol 40:439–445. doi: 10.1128/jcm.40.2.439-445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fanning GC, Zoulim F, Hou J, Bertoletti A. 2019. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov 18:827–844. doi: 10.1038/s41573-019-0037-0. [DOI] [PubMed] [Google Scholar]
- 18.Testoni B, Lebossé F, Scholtes C, Berby F, Miaglia C, Subic M, Loglio A, Facchetti F, Lampertico P, Levrero M, Zoulim F. 2019. Serum hepatitis B core-related antigen (HBcrAg) correlates with covalently closed circular DNA transcriptional activity in chronic hepatitis B patients. J Hepatol 70:615–625. doi: 10.1016/j.jhep.2018.11.030. [DOI] [PubMed] [Google Scholar]
- 19.Campenhout MJH, Rijckborst V, Brouwer WP, Oord GW, Ferenci P, Tabak F, Akdogan M, Pinarbasi B, Simon K, Knegt RJ, Boonstra A, Janssen HLA, Hansen BE. 2019. Hepatitis B core‐related antigen monitoring during peginterferon alfa treatment for HB eAg‐negative chronic hepatitis B. J Viral Hepat 26:1156–1163. doi: 10.1111/jvh.13117. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki F, Miyakoshi H, Kobayashi M, Kumada H. 2009. Correlation between serum hepatitis B virus core‐related antigen and intrahepatic covalently closed circular DNA in chronic hepatitis B patients. J Med Virol 81:27–33. doi: 10.1002/jmv.21339. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Kishimoto S, Ohori K, Yoshizawa H, Machida A, Ohnuma H, Tsuda F, Munekata E, Miyakawa Y, Mayumi M. 1991. Molecular heterogeneity of e antigen polypeptides in sera from carriers of hepatitis B virus. J Immunol 147:3156–3160. [PubMed] [Google Scholar]
- 22.Tong S, Diot C, Gripon P, Li J, Vitvitski L, Trépo C, Guguen-Guillouzo C. 1991. In vitro replication competence of a cloned hepatitis B virus variant with a nonsense mutation in the distal pre-C region. Virology 181:733–737. doi: 10.1016/0042-6822(91)90908-t. [DOI] [PubMed] [Google Scholar]
- 23.Kramvis A, Kostaki E-G, Hatzakis A, Paraskevis D. 2018. Immunomodulatory function of HBeAg related to short-sighted evolution, transmissibility, and clinical manifestation of hepatitis B virus. Front Microbiol 9:2521. doi: 10.3389/fmicb.2018.02521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen HS, Kew MC, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. 1992. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J Virol 66:5682–5684. doi: 10.1128/JVI.66.9.5682-5684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milich D, Liang TJ. 2003. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 38:1075–1086. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 26.Milich DR, Jones JE, Hughes JL, Price J, Raney AK, McLachlan A. 1990. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc Natl Acad Sci U S A 87:6599–6603. doi: 10.1073/pnas.87.17.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y, Kuo C-f, Akbari O, Ou J-hJ. 2016. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 44:1204–1214. doi: 10.1016/j.immuni.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitra B, Wang J, Kim ES, Mao R, Dong M, Liu Y, Zhang J, Guo H. 2019. Hepatitis B virus precore protein p22 inhibits alpha interferon signaling by blocking STAT nuclear translocation. J Virol 93:e00196-19. doi: 10.1128/JVI.00196-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, Li K, Revill P, Locarnini S. 2011. The hepatitis B e antigen suppresses IL‐1β‐mediated NF‐κB activation in hepatocytes. J Viral Hepat 18:e499–e507. doi: 10.1111/j.1365-2893.2011.01484.x. [DOI] [PubMed] [Google Scholar]
- 30.Tran BM, Flanagan DJ, Ebert G, Warner N, Tran H, Fifis T, Kastrappis G, Christophi C, Pellegrini M, Torresi J, Phesse TJ, Vincan E. 2020. The hepatitis B virus pre-core protein p22 activates Wnt signaling. Cancers 12:1435. doi: 10.3390/cancers12061435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walsh R, Nuttall S, Revill P, Colledge D, Cabuang L, Soppe S, Dolezal O, Griffiths K, Bartholomeusz A, Locarnini S. 2011. Targeting the hepatitis B virus precore antigen with a novel IgNAR single variable domain intrabody. Virology 411:132–141. doi: 10.1016/j.virol.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 32.Kornyeyev D, Ramakrishnan D, Voitenleitner C, Livingston CM, Xing W, Hung M, Kwon HJ, Fletcher SP, Beran RK. 2019. Spatiotemporal analysis of hepatitis B virus X protein in primary human hepatocytes. J Virol 93:e00248-19. doi: 10.1128/JVI.00248-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi K, Machida A, Funatsu G, Nomura M, Usuda S, Aoyagi S, Tachibana K, Miyamoto H, Imai M, Nakamura T, Miyakawa Y, Mayumi M. 1983. Immunochemical structure of hepatitis B e antigen in the serum. J Immunol 130:2903–2907. [PubMed] [Google Scholar]
- 34.Eren E, Watts NR, Dearborn AD, Palmer IW, Kaufman JD, Steven AC, Wingfield PT. 2018. Structures of hepatitis b virus core- and e-antigen immune complexes suggest multi-point inhibition. Structure 26:1314–1326.e4. doi: 10.1016/j.str.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, Delaney WEt, Hu J. 2016. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J Virol 90:5830–5844. doi: 10.1128/JVI.00394-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scaglioni PP, Melegari M, Wands JR. 1997. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J Virol 71:345–353. doi: 10.1128/JVI.71.1.345-353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nassal M, Rieger A. 1993. An intramolecular disulfide bridge between Cys-7 and Cys61 determines the structure of the secretory core gene product (e antigen) of hepatitis B virus. J Virol 67:4307–4315. doi: 10.1128/JVI.67.7.4307-4315.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlicht H-J. 1991. Biosynthesis of the secretory core protein of duck hepatitis B virus: intracellular transport, proteolytic processing, and membrane expression of the precore protein. J Virol 65:3489–3495. doi: 10.1128/JVI.65.7.3489-3495.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlier D, Jean-Jean O, Fouillot N, Will H, Rossignol J-M. 1995. Importance of the C terminus of the hepatitis B virus precore protein in secretion of HBe antigen. J Gen Virol 76:1041–1045. doi: 10.1099/0022-1317-76-4-1041. [DOI] [PubMed] [Google Scholar]
- 40.Carlier D, Jean-Jean O, Rossignol JM. 1994. Characterization and biosynthesis of the woodchuck hepatitis virus e antigen. J Gen Virol 75:171–175. doi: 10.1099/0022-1317-75-1-171. [DOI] [PubMed] [Google Scholar]
- 41.Chen CY, Crowther C, Kew MC, Kramvis A. 2008. A valine to phenylalanine mutation in the precore region of hepatitis B virus causes intracellular retention and impaired secretion of HBe‐antigen. Hepatol Res 38:580–592. doi: 10.1111/j.1872-034X.2007.00315.x. [DOI] [PubMed] [Google Scholar]
- 42.Christoffersen C, Ahnström J, Axler O, Christensen EI, Dahlbäck B, Nielsen LB. 2008. The signal peptide anchors apolipoprotein M in plasma lipoproteins and prevents rapid clearance of apolipoprotein M from plasma. J Biol Chem 283:18765–18772. doi: 10.1074/jbc.M800695200. [DOI] [PubMed] [Google Scholar]
- 43.Schödel F, Peterson D, Zheng J, Jones J, Hughes J, Milich D. 1993. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem 268:1332–1337. [PubMed] [Google Scholar]
- 44.Takahashi K, Imai M, Miyakawa Y, Iwakiri S, Mayumi M. 1978. Duality of hepatitis B e antigen in serum of persons infected with hepatitis B virus: evidence for the nonidentity of e antigen with immunoglobulins. Proc Natl Acad Sci U S A 75:1952–1956. doi: 10.1073/pnas.75.4.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson AJV, Nguyen T, Iser D, Ayres A, Jackson K, Littlejohn M, Slavin J, Bowden S, Gane EJ, Abbott W, Lau GKK, Lewin SR, Visvanathan K, Desmond PV, Locarnini SA. 2010. Serum hepatitis B surface antigen and hepatitis B e antigen titers: disease phase influences correlation with viral load and intrahepatic hepatitis B virus markers. Hepatology 51:1933–1944. doi: 10.1002/hep.23571. [DOI] [PubMed] [Google Scholar]
- 46.Possehl C, Repp R, Heermann K-H, Korec E, Uy A, Gerlich W. 1992. Absence of free core antigen in anti-HBc negative viremic hepatitis B carriers. Arch Virol Suppl 4:39–41. doi: 10.1007/978-3-7091-5633-9_8. [DOI] [PubMed] [Google Scholar]
- 47.Ning X, Luckenbaugh L, Liu K, Bruss V, Sureau C, Hu J. 2018. Common and distinct capsid and surface protein requirements for secretion of complete and genome-free hepatitis B virions. J Virol 92:e00272-18. doi: 10.1128/JVI.00272-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai L, Zhang X, Kozlowski M, Li W, Wu M, Liu J, Chen L, Zhang J, Huang Y, Yuan Z. 2018. Extracellular hepatitis B virus RNAs are heterogeneous in length and circulate as capsid-antibody complexes in addition to virions in chronic hepatitis B patients. J Virol 92:e00798-18. doi: 10.1128/JVI.00798-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asabe S, Wieland SF, Chattopadhyay PK, Roederer M, Engle RE, Purcell RH, Chisari FV. 2009. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol 83:9652–9662. doi: 10.1128/JVI.00867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, Xi J, Gao L, Hu J. 2020. Role of hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PLoS Pathog 16:e1008459. doi: 10.1371/journal.ppat.1008459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruss V, Ganem D. 1991. The role of envelope proteins in hepatitis B virus assembly. Proc Natl Acad Sci U S A 88:1059–1063. doi: 10.1073/pnas.88.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ponsel D, Bruss V. 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J Virol 77:416–422. doi: 10.1128/jvi.77.1.416-422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niu C, Livingston CM, Li L, Beran RK, Daffis S, Ramakrishnan D, Burdette D, Peiser L, Salas E, Ramos H, Yu M, Cheng G, Strubin M, Delaney WE, Fletcher SP. 2017. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS One 12:e0169648. doi: 10.1371/journal.pone.0169648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo J, Cui X, Gao L, Hu J. 2017. Identification of intermediate in hepatitis B virus CCC DNA formation and sensitive and selective CCC DNA detection. J Virol 91:e00539-17. doi: 10.1128/JVI.00539-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo J, Luckenbaugh L, Hu H, Yan Z, Gao L, Hu J. 2020. Involvement of host ATR-CHK1 pathway in hepatitis B virus covalently closed circular DNA formation. mBio 11:e03423-19. doi: 10.1128/mBio.03423-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perlman DH, Berg EA, O’Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]