

Abstract
INTRODUCTION:
Smoking and genetic predisposition are established risk factors for colorectal cancer (CRC). We aimed to assess and compare their individual and joint impact on CRC risk using the novel approach of genetic risk equivalent (GRE).
METHODS:
Data were extracted from the Darmkrebs: Chancen der Verhütung durch Screening study, a large population-based case-control study in Germany. A polygenic risk score (PRS) based on 140 CRC-related single nucleotide polymorphisms was derived to quantify genetic risk. Multiple logistic regression was used to estimate the individual and joint impact of smoking and PRS on CRC risk, and to quantify the smoking effect in terms of GRE, the corresponding effect conveyed by a defined difference in PRS percentiles.
RESULTS:
There were 5,086 patients with CRC and 4,120 controls included. Current smokers had a 48% higher risk of CRC than never smokers (adjusted odds ratio 1.48, 95% confidence interval 1.27–1.72). A PRS above the 90th percentile was significantly associated with a 3.6-, 4.3-, and 6.4-fold increased risk of CRC in never, former, and current smokers, respectively, when compared with a PRS below the 10th percentile in never smokers. The interaction between smoking and PRS on CRC risk did not reach statistical significance (P = 0.53). The effect of smoking was equivalent to the effect of having a 30 percentile higher level of PRS (GRE 30, 95% confidence interval 18–42).
DISCUSSION:
Both smoking and the PRS carry essentially independent CRC risk information, and their joint consideration provides powerful risk stratification. Abstinence from smoking can compensate for a substantial proportion of genetically determined CRC risk.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer and the second most common cause of cancer-related death worldwide, with more than 1.8 million new cases and about 850,000 deaths in 2018 (1). In line with other multifactorial diseases, the development of CRC is a result of complex interplay between lifestyle and genetic factors (2–4).
Smoking is one of the best established lifestyle risk factors for colorectal adenoma and CRC (5–8). It is unclear, however, to what extent it interacts with genetic predisposition in its impact on CRC risk. Although previous studies have explored interactions of smoking with specific single CRC susceptibility loci (9–12), data are lacking on the individual and joint contribution of smoking and overall genetic risk, as summarized by a polygenic risk score (PRS) based on large numbers of CRC susceptibility loci that have been identified by genomewide association studies (GWAS) in the past 2 decades (13–16). Detailed knowledge of such individual and joint contributions would be of high relevance for enhanced risk stratification and targeted efforts of prevention. Furthermore, for effective risk communication in preventive efforts, it would be helpful to quantify how much genetically determined CRC risk could be lowered by abstinence from smoking.
The aims of this study were therefore twofold (i) to assess CRC risk by categories of smoking behavior and various levels of genetic risk and (ii) to estimate to what extent genetically determined CRC risk could be lowered by abstinence from smoking. For the second aim, a new measure entitled “genetic risk equivalent” (GRE) was introduced and estimated, which provides quantitative information about how much predetermined genetic predisposition to CRC could be compensated for by abstinence from smoking.
METHODS
Study design and study population
Data for the analyses were drawn from the Darmkrebs: Chancen der Verhütung durch Screening (DACHS) study, which has been described in detail elsewhere (17,18). Briefly, DACHS is an ongoing population-based case-control study initiated in 2003 and performed in the Rhine-Neckar region in southwest Germany. All 22 hospitals in this area offering first-line treatment to patients with CRC are involved in recruitment. German-speaking patients aged 30 years and older with a first diagnosis of CRC and physically and mentally able to participate in a 1-hour interview are informed about the study by their clinicians, usually a few days after surgery. The study center is notified on receipt of informed consent. Patients who cannot be recruited during their in-patient stay are contacted by mail after discharge. Overall, the recruited patients represent about 50% of all eligible patients in the study area. The controls are randomly selected from population registries using frequency matching by age (5-year groups), sex, and county of residence. Controls with a history of CRC are excluded. The current analysis is based on data from patients with CRC and controls recruited from 2003 to 2017 for whom detailed risk factor data and genetic data from genomewide arrays are available. The DACHS study was approved by the ethics committees of Heidelberg University and the state medical boards of Baden-Wuerttemberg and Rhineland-Palatinate. Written informed consent was obtained from each participant.
Data collection
Standardized in-person interviews were conducted with both cases (typically during their hospital stay) and controls (at their homes) by trained interviewers. In these interviews, data were collected on demographics, medical history, family history of CRC, and various lifestyle factors. In addition, blood or buccal samples were taken. For all cases, hospital discharge letters and pathology reports were collected. A minority of control participants who were not willing to participate in a personal interview provided key information in a self-administered questionnaire. However, as they have not been genotyped, these participants were excluded from this analysis.
Assessment of smoking behavior
At baseline, participants provided information on their current and previous smoking behavior and the year in which they stopped smoking, if applicable. We classified participants as current smokers if they were still smoking at the time of diagnosis (cases) or interview (controls) or reported to stop smoking in the year before, as former smokers if they had stopped smoking 2 or more years before the diagnosis/interview, or as never smokers if they had never smoked regularly.
As a measure of smoking intensity, pack-years of active smoking were calculated for both current and former smokers from the average number of cigarettes smoked daily, multiplied by the duration of smoking in years, divided by 20 (e.g., smoking 20 cigarettes per day for 1 year corresponds to 1 pack-year).
Derivation of the polygenic risk score
DNA was extracted from blood samples (in 99.1% of participants) or from buccal cells (in 0.9% of participants) using conventional methods. Details regarding genotyping and imputation are provided in Supplementary Table S1 (see Supplementary Digital Content 1, http://links.lww.com/CTG/A517). A total of 140 single nucleotide polymorphisms (SNPs) identified to be associated with CRC risk in populations of European descent in a recent GWAS (16) were extracted from our data set. We calculated the PRS as the sum of risk alleles of the respective variants (0, 1, or 2 copies of the risk allele for genotyped SNPs; imputed dosages for imputed SNPs), and categorized based on the distribution of the PRS among controls (cutoffs at the 10th, 25th, 75th, and 90th percentile, respectively).
Statistical analysis
The distribution of the demographic and other characteristics of the study population was assessed by descriptive statistics and compared between cases and controls using χ2 tests for categorical data.
Multivariable logistic regression models with various degrees of adjustment were used to estimate odds ratios (ORs) and 95% confidence intervals (CIs) for the individual association of smoking exposure and the PRS with CRC risk. Model 1 was adjusted for the matching factors age and sex. Model 2 was additionally adjusted for education, body mass index, physical activity, alcohol consumption, red meat consumption, history of colonoscopy, history of diabetes, family history of CRC in a first-degree relative (FH), use of statins and use of nonsteroidal anti-inflammatory drugs (NSAIDs), and PRS (for the analysis of smoking status) or smoking status (for the analysis of PRS).
We stratified the population by smoking status to allow for investigating potentially differential effects of the PRS on CRC risk between current, former, and never smokers. Potential interaction of smoking with the PRS was tested for statistical significance by additionally including a cross-product term of the PRS as a categorical variable with smoking status or as a continuous variable with pack-years of smoking (continuous variable) in the models. Furthermore, we assessed CRC risk after joint classification of participants by smoking and PRS categories, using never smokers with a PRS below the 10th percentile as the uniform reference group.
To explore potential variation of associations across population subgroups, we performed additional analyses stratified by age (≤55/>55 years), sex (male/female), history of colonoscopy (yes/no), and FH (yes/no). Interactions of smoking and these stratification variables on CRC risk were tested by including a cross-product term along with the main effect terms in the models. Besides, we also assessed the association of smoking with CRC risk by cancer sites (colon/rectum).
All analyses were conducted using R software version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria). Statistical tests were 2-sided, and P values less than 0.05 were considered statistically significant. Statistical tests for interaction between smoking and PRS were confirmatory. Additional statistical tests for interaction between smoking and covariates were exploratory, and no adjustment for multiple testing was performed.
Derivation of the genetic risk equivalent
GREs were calculated from logistic regression models as ratios of the regression coefficients of smoking and PRS percentiles, using an approach previously developed for the well-established concept of risk and rate advancement periods (19). In brief, consider an analysis based on a multivariable logistic regression:
where ln(R) reflects the log odds of the disease risk, and a, b1, b2, and ci (i = 1,…, n) refer to the intercept and the model parameters for S (smoking exposure, categorized as 1 for the respective smoking group and 0 for never smokers), P (PRS percentile according to distribution among controls), and F (other covariates), respectively. Using this approach, the estimated effect of smoking may be directly compared with the estimated impact of an increase in PRS by 1 percentile among the controls (which reflect the source population in which the cases arose from). Specifically, the ratio of regression coefficients b1/b2 provides an estimate of the smoking impact in terms of the equivalent difference in background genetic risk, expressed in difference in genetic risk percentiles in the population. Confidence intervals for GREs were calculated in analogy with previously described methods for risk and rate advancement periods (19) as outlined in the Supplementary Methods (see Supplementary Digital Content 2, http://links.lww.com/CTG/A518).
RESULTS
A total of 5,086 cases and 4,120 controls for whom genetic and smoking data were available were included in this analysis (see Supplementary Figure, Supplementary Digital Content 3, http://links.lww.com/CTG/A519). Table 1 shows the distribution of sociodemographic factors and risk and protective factors of CRC among cases and controls. Approximately 60.2% of cases and 61.3% of controls were men. The median age was 69 years for cases and 70 years for controls. Generally, patients with CRC were more likely to have a lower level of education, to smoke, to consume more alcohol or red meat, to have a higher level of physical activity, to be overweight or obese, to have a history of diabetes and FH. They were less likely to use NSAIDs and statins or to have participated in a colonoscopy examination before diagnosis compared with control participants.
Table 1.
Distribution of characteristics in patients with colorectal cancer and controls
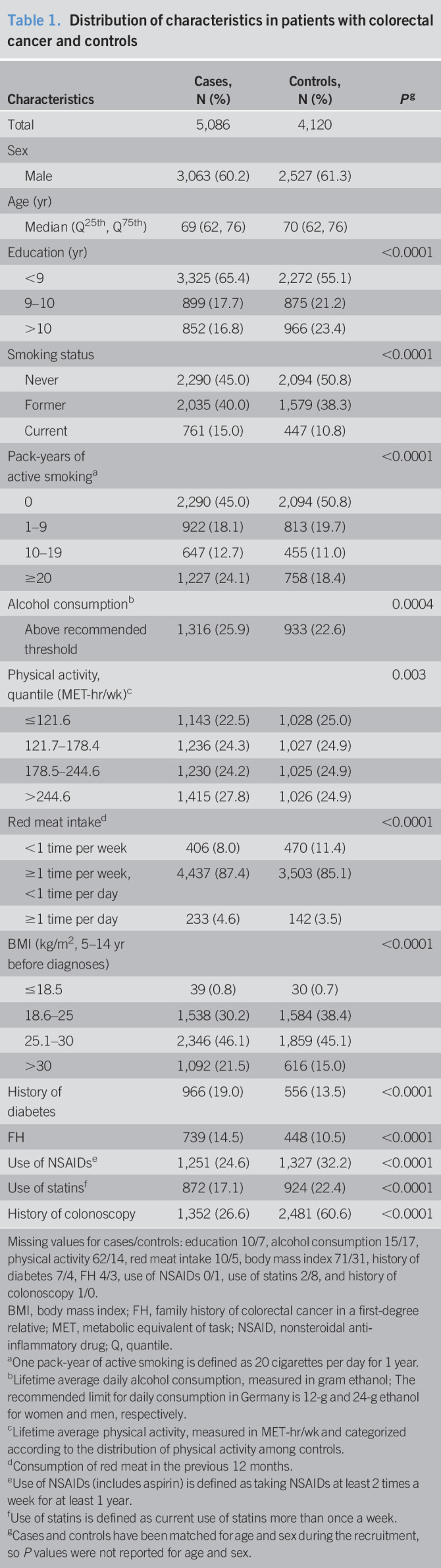
| Characteristics | Cases, N (%) | Controls, N (%) | Pg |
| Total | 5,086 | 4,120 | |
| Sex | |||
| Male | 3,063 (60.2) | 2,527 (61.3) | |
| Age (yr) | |||
| Median (Q25th, Q75th) | 69 (62, 76) | 70 (62, 76) | |
| Education (yr) | <0.0001 | ||
| <9 | 3,325 (65.4) | 2,272 (55.1) | |
| 9–10 | 899 (17.7) | 875 (21.2) | |
| >10 | 852 (16.8) | 966 (23.4) | |
| Smoking status | <0.0001 | ||
| Never | 2,290 (45.0) | 2,094 (50.8) | |
| Former | 2,035 (40.0) | 1,579 (38.3) | |
| Current | 761 (15.0) | 447 (10.8) | |
| Pack-years of active smokinga | <0.0001 | ||
| 0 | 2,290 (45.0) | 2,094 (50.8) | |
| 1–9 | 922 (18.1) | 813 (19.7) | |
| 10–19 | 647 (12.7) | 455 (11.0) | |
| ≥20 | 1,227 (24.1) | 758 (18.4) | |
| Alcohol consumptionb | 0.0004 | ||
| Above recommended threshold | 1,316 (25.9) | 933 (22.6) | |
| Physical activity, quantile (MET-hr/wk)c | 0.003 | ||
| ≤121.6 | 1,143 (22.5) | 1,028 (25.0) | |
| 121.7–178.4 | 1,236 (24.3) | 1,027 (24.9) | |
| 178.5–244.6 | 1,230 (24.2) | 1,025 (24.9) | |
| >244.6 | 1,415 (27.8) | 1,026 (24.9) | |
| Red meat intaked | <0.0001 | ||
| <1 time per week | 406 (8.0) | 470 (11.4) | |
| ≥1 time per week, <1 time per day | 4,437 (87.4) | 3,503 (85.1) | |
| ≥1 time per day | 233 (4.6) | 142 (3.5) | |
| BMI (kg/m2, 5–14 yr before diagnoses) | <0.0001 | ||
| ≤18.5 | 39 (0.8) | 30 (0.7) | |
| 18.6–25 | 1,538 (30.2) | 1,584 (38.4) | |
| 25.1–30 | 2,346 (46.1) | 1,859 (45.1) | |
| >30 | 1,092 (21.5) | 616 (15.0) | |
| History of diabetes | 966 (19.0) | 556 (13.5) | <0.0001 |
| FH | 739 (14.5) | 448 (10.5) | <0.0001 |
| Use of NSAIDse | 1,251 (24.6) | 1,327 (32.2) | <0.0001 |
| Use of statinsf | 872 (17.1) | 924 (22.4) | <0.0001 |
| History of colonoscopy | 1,352 (26.6) | 2,481 (60.6) | <0.0001 |
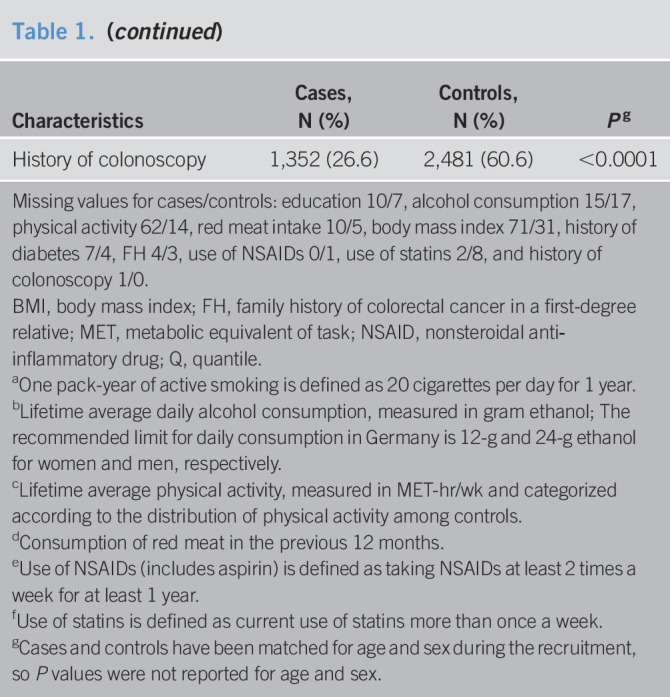
Missing values for cases/controls: education 10/7, alcohol consumption 15/17, physical activity 62/14, red meat intake 10/5, body mass index 71/31, history of diabetes 7/4, FH 4/3, use of NSAIDs 0/1, use of statins 2/8, and history of colonoscopy 1/0.
BMI, body mass index; FH, family history of colorectal cancer in a first-degree relative; MET, metabolic equivalent of task; NSAID, nonsteroidal anti‐inflammatory drug; Q, quantile.
One pack-year of active smoking is defined as 20 cigarettes per day for 1 year.
Lifetime average daily alcohol consumption, measured in gram ethanol; The recommended limit for daily consumption in Germany is 12-g and 24-g ethanol for women and men, respectively.
Lifetime average physical activity, measured in MET-hr/wk and categorized according to the distribution of physical activity among controls.
Consumption of red meat in the previous 12 months.
Use of NSAIDs (includes aspirin) is defined as taking NSAIDs at least 2 times a week for at least 1 year.
Use of statins is defined as current use of statins more than once a week.
Cases and controls have been matched for age and sex during the recruitment, so P values were not reported for age and sex.
Individual associations of smoking status and the PRS with CRC risk are presented in Table 2. Smoking was significantly associated with an increased risk of CRC in the whole study population. Compared with never smokers, adjusted ORs (95% CIs) were 1.48 (1.27–1.72) and 1.26 (1.14–1.40) for current and former smokers, respectively. Having a PRS in the top decile was associated with a 4.0-fold (95% CI 3.3–4.9) increased risk of CRC compared with the lowest decile in age- and sex-adjusted analysis. After controlling for all other covariates, the association remained robust (OR 4.23, 95% CI 3.40–5.29).
Table 2.
Individual association of smoking status and polygenic risk score with colorectal cancer risk

| Exposure | Cases, N (%) | Controls, N (%) | Model 1a, OR (95% CI) | Model 2b, OR (95% CI) |
| Smoking status | ||||
| Never | 2,193 (44.5) | 2,057 (50.9) | Ref. | Ref. |
| Former | 1,990 (40.4) | 1,544 (38.2) | 1.25 (1.14–1.37) | 1.26 (1.14–1.40) |
| Current | 745 (15.1) | 440 (10.9) | 1.61 (1.14–1.85) | 1.48 (1.27–1.72) |
| Per 20 pack-years | 1.21 (1.13–1.29) | 1.16 (1.08–1.24) | ||
| PRSc | ||||
| Very low | 214 (4.3) | 405 (10.0) | Ref. | Ref. |
| Low | 499 (10.1) | 613 (15.2) | 1.54 (1.26–1.89) | 1.57 (1.26–1.96) |
| Medium | 2,428 (49.3) | 2,018 (49.9) | 2.28 (1.91–2.72) | 2.40 (1.99–2.91) |
| High | 928 (18.8) | 602 (14.9) | 2.91 (2.40–3.54) | 3.05 (2.47–3.77) |
| Very high | 859 (17.4) | 403 (10.0) | 4.03 (3.29–4.94) | 4.23 (3.40–5.29) |
| Per 20 percentiles | 1.28 (1.24–1.32) | 1.28 (1.24–1.33) |
CI, confidence interval; OR, odds ratio; PRS, polygenic risk score; Ref., reference.
Adjusted for age and sex.
Additionally adjusted for education, body mass index, physical activity, alcohol consumption, red meat consumption, history of colonoscopy, history of diabetes, family history of colorectal cancer in a first-degree relative, use of statins and use of nonsteroidal anti-inflammatory drugs, PRS (for the analysis of smoking status), and smoking status (for the analysis of PRS).
Classification of PRS: very low, ≤10th percentile; low, 11th–25th percentile; medium, 26th–75th percentile; high, 76th–90th percentile; and very high, >90th percentile.
A PRS in the top decile was associated with 3.6-, 4.8-, and 5.6-fold increased risk of CRC compared with a PRS in the lowest decile in never, former, and current smokers, respectively (Table 3). Although the magnitude of the association of the PRS with CRC risk seemed stronger among current smokers than among never or former smokers, a test for interaction did not reach statistical significance (P = 0.53, in model 2). Similarly, there was no interaction (P = 0.58, in model 2) between pack-years of smoking (continuous variable) and the PRS (continuous variable, per 20 percentiles).
Table 3.
Association of the polygenic risk score with colorectal cancer risk stratified by smoking status

| Smoking status | PRSa | Cases, N (%) | Controls, N (%) | Model 1b, OR (95% CI) | Model 2c, OR (95% CI) |
| Never | Very low | 113 (5.2) | 211 (10.3) | Ref. | Ref. |
| Low | 226 (10.3) | 327 (15.9) | 1.29 (0.97–1.72) | 1.25 (0.92–1.71) | |
| Medium | 1,071 (48.8) | 1,018 (49.5) | 1.97 (1.54–2.52) | 2.03 (1.56–2.65) | |
| High | 410 (18.7) | 294 (14.3) | 2.62 (1.99–3.45) | 2.66 (1.98–3.58) | |
| Very high | 373 (17.0) | 207 (10.1) | 3.40 (2.56–4.53) | 3.59 (2.64–4.90) | |
| Per 20 percentiles | 1.27 (1.21–1.32) | 1.28 (1.22–1.34) | |||
| Former | Very low | 74 (3.7) | 151 (9.8) | Ref. | Ref. |
| Low | 202 (10.2) | 234 (15.2) | 1.75 (1.26–2.46) | 1.91 (1.33–2.76) | |
| Medium | 1,000 (50.3) | 771 (49.9) | 2.64 (1.98–3.56) | 2.95 (2.15–4.07) | |
| High | 379 (19.0) | 233 (15.1) | 3.31 (2.41–4.59) | 3.67 (2.59–5.23) | |
| Very high | 335 (16.8) | 155 (10.0) | 4.39 (3.14–6.17) | 4.83 (3.35–7.00) | |
| Per 20 percentiles | 1.29 (1.23–1.36) | 1.30 (1.23–1.37) | |||
| Current | Very low | 27 (3.6) | 43 (9.8) | Ref. | Ref. |
| Low | 71 (9.5) | 52 (11.8) | 2.17 (1.20–3.98) | 2.27 (1.18–4.45) | |
| Medium | 357 (47.9) | 229 (52.0) | 2.48 (1.50–4.17) | 2.61 (1.49–4.62) | |
| High | 139 (18.7) | 75 (17.0) | 2.95 (1.70–5.20) | 3.16 (1.72–5.89) | |
| Very high | 151 (20.3) | 41 (20.3) | 5.86 (3.27–10.72) | 5.64 (2.97–10.90) | |
| Per 20 percentiles | 1.26 (1.15–1.37) | 1.26 (1.14–1.38) | |||
| P interactiond | — | — | — | 0.46 | 0.53 |
CI, confidence interval; OR, odds ratio; PRS, polygenic risk score; Ref., reference.
Classification of PRS: very low, ≤10th percentile; low, 11th–25th percentile; medium, 26th–75th percentile; high, 76th–90th percentile; and very high, >90th percentile.
Adjusted for age and sex.
Additionally adjusted for education, body mass index, physical activity, alcohol consumption, red meat consumption, history of colonoscopy, history of diabetes, family history of colorectal cancer in a first-degree relative, use of statins, and use of nonsteroidal anti-inflammatory drugs.
Interactions were tested by additionally including a cross-product term of smoking status and PRS categories in model 1 and model 2.
The joint association of smoking and the PRS with CRC risk is presented in Table 4. A PRS above the 90th percentile was significantly associated with a 3.6-, 4.3-, and 6.4-fold increased risk of CRC in never, former, and current smokers, respectively, when compared with a PRS below the 10th percentile in never smokers. There was no significant difference in CRC risk between current, former, and never smokers among individuals with a PRS below the 10th percentile.
Table 4.
Joint association of smoking status and polygenic risk score with colorectal cancer risk

| Smoking status | OR (95% CI) | ||||
| PRS | PRS | PRS | PRS | PRS | |
| Very low | Low | Medium | High | Very high | |
| Never | Ref. | 1.26 (0.93–1.72) | 2.03 (1.56–2.65) | 2.67 (1.99–3.60) | 3.60 (2.64–4.91) |
| Former | 0.90 (0.61–1.33) | 1.73 (1.25–2.39) | 2.67 (2.04–3.50) | 3.30 (2.43–4.49) | 4.34 (3.15–6.02) |
| Current | 1.11 (0.62–1.97) | 2.45 (1.55–3.90) | 2.91 (2.14–3.96) | 3.50 (2.37–5.21) | 6.40 (4.15–10.05) |
Adjustment variables included age, sex, education, body mass index, physical activity, alcohol consumption, red meat consumption, history of colonoscopy, history of diabetes, family history of colorectal cancer in a first-degree relative, use of statins, and use of nonsteroidal anti-inflammatory drugs; classification of PRS: very low, ≤10th percentile; low, 11th–25th percentile; medium, 26th–75th percentile; high, 76th–90th percentile; and very high, >90th percentile.
CI, confidence interval; OR, odds ratio; PRS, polygenic risk score; Ref., reference.
GREs estimated for different smoking categories in the whole population are presented in Figure 1 and Supplementary Table S2 (see Supplementary Digital Content 1, http://links.lww.com/CTG/A517). Current smokers had a GRE of 29.9 (95% CI 17.7–42.1) compared with never smokers, which can be interpreted as the effect of smoking on CRC risk was equivalent to the effect of having a 30 percentile higher level of PRS. The GRE for 20 or more pack-years of smoking was 31 PRS percentiles (95% CI 20–42).
Figure 1.

Genetic risk equivalent for comparisons between smoking status in the whole population and different subgroups. *Interactions were tested by additionally including the multiplicative term of stratification factors (sex, age, history of colonoscopy, or FH) and smoking status in the model 2 but with a polygenic risk score included as percentiles (per 10 percentiles, continuous factor). Never smokers were used as reference in each subgroup. FH, family history of colorectal cancer in a first-degree relative.
Results for the stratified analyses are summarized in Figure 1 and Supplementary Tables S3–S7 (see Supplementary Digital Content 1, http://links.lww.com/CTG/A517). GREs for colon cancer were much higher for current (33.0, 95% CI 19.2–46.7) than for former smokers (15.2, 95% CI 6.0–24.4), whereas they were quite similar between both groups for rectum cancer (26.2, 95% CI 10.5–41.9, and 22.9, 95% CI 11.4–34.4, respectively). GREs were consistently higher for current smokers than for former smokers in all of the examined subgroups. GREs ranged from 25 to 35 PRS percentiles for current smokers in all subgroups except the small subgroup of those with a family history in a first-degree relative for which the GRE was higher (GRE = 45.6), but the confidence interval was very broad (95% CI 17.2–74.0). Tests for interaction did not reach statistical significance for any of the subgroup comparisons.
Stratified analyses for pack-years of smoking generally confirmed dose-response relationships with GRE across subgroups (see Supplementary Figure S2, Supplementary Digital Content 3, http://links.lww.com/CTG/A519). A particularly strong dose-response relationship with GRE was seen among those with a family history of CRC (GRE for ≥20 pack-years = 53.1, 95% CI 27.4–78.9, unadjusted P value for interaction = 0.04), but again confidence intervals of GREs in this relatively small group were wide.
DISCUSSION
In this large case-control study, smoking and a PRS were strongly associated with an increased risk of CRC. Both predictors independently contributed to the risk of CRC which enabled much better risk stratification through joint consideration. The effect of smoking on CRC risk was equivalent to that of having a 30 percentile higher level of PRS. Associations were stronger, and GREs were higher for current than for former smokers, and dose-response relationships were seen with pack-years of smoking, underlining the benefits of smoking cessation.
Individual associations between smoking and the PRS on CRC risk are in line with previous findings. Several studies have synthesized a large amount of accumulated evidence on the association of smoking exposure with CRC risk and have demonstrated that smoking, especially long-term and heavy smoking, modestly elevates the risk of CRC (5,7,8). Jenkins et al. (20) estimated that a PRS in the highest decile was associated with a 5-fold increase in 5-year CRC risk compared with a PRS in the lowest decile, based on GWAS data available in 2016. They also validated the results in a separate population in 2019 (21). Other risk prediction models incorporating common genetic variants to estimate incidence of CRC consistently showed a strong positive relationship between polygenic risk and CRC risk, which were summarized in a recent systematic review (22). To date, most studies have focused on adding the PRS into traditional prediction models for more personalized risk assessment (22,23). However, little is known about the interactions between established environmental risk factors, most of which are modifiable, and predetermined genetic profiles evaluated by the PRS on CRC risk, which might also serve as an important step in developing individualized prevention strategies for CRC.
Cigarette smoke with more than 60 different carcinogenic compounds could influence disease risk through various pathways involving tumor-related genes and other regulatory elements, which provides biological rationality for smoking-gene interaction (24,25). Nevertheless, studies (9–12) addressing potential interaction of smoking with specific CRC susceptibility variants and the genomewide gene-smoking interaction study for CRC risk mostly yielded null results (10–12). These studies might have been underpowered because of weak main effects of single genetic variants. The 2020 study by Yang et al. (12) also examined potential interaction between smoking and a PRS in CRC patients and controls from the United Kingdom and did not find statistically significant results. The weak association between smoking status and CRC risk (ever vs never smokers: OR 1.12, 95% CI 1.04–1.21) in their study might have limited the ability to detect interaction. In our study, a stronger effect of current smoking was seen, and results pointed to potentially stronger effects of the PRS among current smokers, but the test for interaction likewise did not reach statistical significance. Even larger studies are needed to clarify potential interaction between smoking and CRC polygenic risk. Testing and disproving interactions are essential for correct joint modeling of genes and environment for risk prediction (26). Our findings underline the importance of nonsmoking and smoking cessation regardless of the individual genetic profile and provide an insight into joint modeling of the PRS and smoking (an easy-to-collect risk factor) for CRC risk stratification.
Another important aspect in cancer prevention research is to develop metrics that could be easily understood by the public. Traditional approaches such as odds ratios might be hard to comprehend and communicate by themselves (27,28), and risk communication becomes even more complex with respect to individual and joint effects of modifiable and genetic risk factors. The novel concept of GRE introduced in the current study, and applied to quantify the effect of smoking by a defined difference in PRS percentiles, might be a useful approach in this context. In particular, it may help to convey the message that the risk of CRC is not a fate that is predetermined by genetics, but that the background genetic risk can be substantially reduced by healthy lifestyles. The large GRE for current smokers indicates that a substantial proportion of genetically determined CRC risk could be compensated for by abstinence from smoking. The GRE may be a useful metric for comparing risks from lifestyle and genetic risk factors in general, and may have great potential to promote risk communication and thus improve adherence to healthy guidelines, especially among individuals with high genetic predisposition.
Our study also included detailed stratified analyses across subgroups of a variety of covariates. In general, a dose-response relationship of smoking exposure with GRE was seen across those subgroups. The subgroup analyses also suggested a particularly strong effect of smoking among those with a family history of CRC, possibly reflecting interaction of smoking with shared genes, shared environmental factors, or both of familial cases (29,30). However, confidence intervals of GREs in this small group were wide, and the apparently significant interaction between pack-years and family history of CRC needs to be interpreted with caution as we did not adjust for multiple testing. Nevertheless, this finding should be followed up in studies with an even larger sample size.
Our study has several strengths. First, the DACHS study is one of the largest population-based case-control studies on CRC in the world with both GWAS data and comprehensive information provided by the study participants. Thus, thorough adjustment for and stratification by important covariates was possible. The size of the study also enabled assessment of potential interaction between smoking and the PRS. To the best of our knowledge, this was the first study providing estimates of GREs as a novel tool to quantify and communicate the impact of smoking and genetic risk in a comparative manner.
There are also some limitations that deserve careful consideration. First, information about potential risk and protective factors was gathered retrospectively through personal interviews using standardized questionnaires, thus recall bias could not be ruled out, potentially leading to underestimation of the effects. In particular, it is difficult to retrospectively assess the number of cigarettes smoked at various ages over a lifetime. Second, our study exclusively consists of a white population; thus, the results should be validated in other ethnic groups. Third, although multiple potential confounding factors were considered in the analyses, residual confounding could not be ruled out. Fourth, despite the overall large number of participants, confidence intervals for GREs were rather broad for some of the subgroup specific analyses. Also, adjustment for multiple testing was not performed for the exploratory tests, and suggested interactions between smoking and family history need to be confirmed in other, ideally even larger studies. Finally, null results in overall interaction analyses do not mean null results in tumor subtype-specific analyses. However, given that tumor subtype information was only available for a subgroup of the study population, our study lacked reasonable power to address potential interactions between smoking and the polygenic risk for specific CRC subtypes. Further studies are needed to follow-up and address this important issue.
Despite these limitations, our study provides a detailed insight into individual and joint effect of a PRS and smoking on CRC risk. Smoking contributes to a higher risk of CRC irrespective of genetic risk. Abstinence from smoking can compensate for a substantial proportion of genetically determined CRC risk. The novel metric of GRE may be useful for comparing risk based on lifestyle and genetic factors, providing a possible avenue for improving risk communication to the public and targeting prevention at individuals with high genetic predisposition. Further research should follow-up the suggested interactions, in particular the interaction of smoking with family history of CRC and provide more precise estimates of GREs for the high-risk group with family history of CRC.
CONFLICTS OF INTEREST
Guarantor of the article: Hermann Brenner, MD, MPH.
Specific author contributions: H.B.: study concept and design. X.C.: analysis of data. X.C., L.J., F.G., M.H., J.C.-C., and H.B.: interpretation of data. X.C., L.J., and H.B.: drafting the manuscript. All authors provided comments, revised the draft, and approved the final version of the manuscript.
Financial support: The first author (X.C.) was supported by the Guangzhou Elite Project (GEP). The DACHS study was supported by grants from the German Research Council (BR 1704/6-1, BR1704/6-3, BR 1704/6-4, BR 1704/6-6, CH 117/1-1, BR 1704/17-1, and HO 5117/2-1) and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, and 01GL1712). The sponsors had no role in the study design and in the collection, analysis, and interpretation of data.
Potential competing interests: None to report.
Study Highlights.
WHAT IS KNOWN
✓ Polygenic risk scores (PRSs) have been developed and are increasingly used for colorectal cancer (CRC) risk stratification. The combination of multiple risk loci in the PRS enables more powerful risk stratification for CRC.
✓ Smoking is associated with increased risk of colorectal adenoma and CRC, but little is known about its potential interaction with overall genetic risk.
✓ Traditional metrics such as odds ratios are hard to comprehend for the public and thus may have limited use in cancer prevention and risk communication.
WHAT IS NEW HERE
✓ Smoking and polygenic risk, quantified by a PRS, contribute independently to CRC risk. The use of both information enhances CRC risk stratification.
✓ Abstinence from smoking can compensate for a substantial proportion of genetically predetermined CRC risk.
✓ The novel approach of genetic risk equivalent is useful for comparing risks from lifestyle factors and genetic predisposition.
TRANSLATIONAL IMPACT
✓ Our results contribute to enhanced quantification and communication of smoking-related CRC risk.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge helpful language editing by Hannah Stocker.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A517, http://links.lww.com/CTG/A518, http://links.lww.com/CTG/A519
Contributor Information
Xuechen Chen, Email: xuechen.chen@dkfz-heidelberg.de.
Lina Jansen, Email: l.jansen@dkfz-heidelberg.de.
Feng Guo, Email: f.guo@dkfz-heidelberg.de.
Michael Hoffmeister, Email: m.hoffmeister@dkfz-heidelberg.de.
Jenny Chang-Claude, Email: j.chang-claude@dkfz-heidelberg.de.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Heavey PM, McKenna D, Rowland IR. Colorectal cancer and the relationship between genes and the environment. Nutr Cancer 2004;48:124–41. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed FE. Gene-gene, gene-environment & multiple interactions in colorectal cancer. J Environ Sci Heal C 2006;24:1–101. [DOI] [PubMed] [Google Scholar]
- 4.Ogino S, Chan AT, Fuchs CS, et al. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011;60:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannucci E. An updated review of the epidemiological evidence that cigarette smoking increases risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev 2001;10:725–31. [PubMed] [Google Scholar]
- 6.Botteri E, Iodice S, Raimondi S, et al. Cigarette smoking and adenomatous polyps: A meta-analysis. Gastroenterology 2008;134:388–95. [DOI] [PubMed] [Google Scholar]
- 7.Liang PS, Chen T-Y, Giovannucci E. Cigarette smoking and colorectal cancer incidence and mortality: Systematic review and meta-analysis. Int J Cancer 2009;124:2406–15. [DOI] [PubMed] [Google Scholar]
- 8.Cheng J, Chen Y, Wang X, et al. Meta-analysis of prospective cohort studies of cigarette smoking and the incidence of colon and rectal cancers. Eur J Cancer Prev 2015;24:6–15. [DOI] [PubMed] [Google Scholar]
- 9.Song N, Shin A, Jung HS, et al. Effects of interactions between common genetic variants and smoking on colorectal cancer. BMC Cancer 2017;17:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song N, Lee J, Cho S, et al. Evaluation of gene-environment interactions for colorectal cancer susceptibility loci using case-only and case-control designs. BMC Cancer 2019;19:1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang T, Li X, Montazeri Z, et al. Gene–environment interactions and colorectal cancer risk: An umbrella review of systematic reviews and meta‐analyses of observational studies. Int J Cancer 2019;145:2315–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang T, Li X, Farrington SM, et al. A systematic analysis of interactions between environmental risk factors and genetic variation in susceptibility to colorectal cancer. Cancer Epidemiol Biomarkers Prev 2020;29:1145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters U, Jiao S, Schumacher FR, et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 2013;144:799–807.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiffin N, Hosking FJ, Farrington SM, et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet 2014;23:4729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huyghe JR, Bien SA, Harrison TA, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 2019;51:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas M, Sakoda LC, Hoffmeister M, et al. Genome-wide modeling of polygenic risk score in colorectal cancer risk. Am J Hum Genet 2020;107:432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner H, Chang-Claude J, Seiler CM, et al. Protection from colorectal cancer after colonoscopy: A population-based, case-control study. Ann Intern Med 2011;154:22–30. [DOI] [PubMed] [Google Scholar]
- 18.Brenner H, Chang–Claude J, Jansen L, et al. Reduced risk of colorectal cancer up to 10 years after screening, surveillance, or diagnostic colonoscopy. Gastroenterology 2014;146:709–17. [DOI] [PubMed] [Google Scholar]
- 19.Brenner H, Gefeller O, Greenland S. Risk and rate advancement periods as measures of exposure impact on the occurrence of chronic diseases. Epidemiology 1993;4:229–36. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins MA, Makalic E, Dowty JG, et al. Quantifying the utility of single nucleotide polymorphisms to guide colorectal cancer screening. Futur Oncol 2016;12:503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins MA, Win AK, Dowty JG, et al. Ability of known susceptibility SNPs to predict colorectal cancer risk for persons with and without a family history. Fam Cancer 2019;18:389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGeoch L, Saunders CL, Griffin SJ, et al. Risk prediction models for colorectal cancer incorporating common genetic variants: A systematic review. Cancer Epidemiol Biomarkers Prev 2019;28:1580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balavarca Y, Weigl K, Thomsen H, et al. Performance of individual and joint risk stratification by an environmental risk score and a genetic risk score in a colorectal cancer screening setting. Int J Cancer 2020;146:627–34. [DOI] [PubMed] [Google Scholar]
- 24.Schaal C, Chellappan SP. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol Cancer Res 2014;12:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grando SA. Connections of nicotine to cancer. Nat Rev Cancer 2014;14:419–29. [DOI] [PubMed] [Google Scholar]
- 26.Chatterjee N, Shi J, García-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet 2016;17:392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Cohen P, Chen S. How big is a big odds ratio? Interpreting the magnitudes of odds ratios in epidemiological studies. Commun Stat Simul Comput 2010;39:860–4. [Google Scholar]
- 28.Norton EC, Dowd BE, Maciejewski ML. Odds ratios-current best practice and use. JAMA 2018;320:84–5. [DOI] [PubMed] [Google Scholar]
- 29.Jasperson KW, Tuohy TM, Neklason DW, et al. Hereditary and familial colon cancer. Gastroenterology 2010;138:2044–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoffel EM, Kastrinos F. Familial colorectal cancer, beyond lynch syndrome. Clin Gastroenterol Hepatol 2014;12:1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.