

Summary
Subanesthetic ketamine evokes rapid and long-lasting antidepressant effects in human patients. The mechanism for ketamine’s effects remains elusive, but ketamine may broadly modulate brain plasticity processes. We show that single-dose ketamine reactivates adult mouse visual cortical plasticity and promotes functional recovery of visual acuity defects from amblyopia. Ketamine specifically induces down-regulation of neuregulin-1 (NRG1) expression in parvalbumin-expressing (PV) inhibitory neurons in V1. NRG1 downregulation in PV neurons co-tracks both the fast onset and sustained decreases in synaptic inhibition to excitatory neurons, along with reduced synaptic excitation to PV neurons in vitro and in vivo following a single ketamine treatment. These effects are blocked by exogenous NRG1 as well as PV targeted receptor knockout. Thus, ketamine reactivation of adult visual cortical plasticity is mediated through rapid and sustained cortical disinhibition via downregulation of PV-specific NRG1 signaling. Our findings reveal the neural plasticity-based mechanism for ketamine-mediated functional recovery from adult amblyopia.
Introduction
Ketamine is an anesthetic that acts on N-methyl-D-aspartate receptors (NMDARs). Subanesthetic ketamine is used as an antidepressant [1, 2]. A single subanesthetic dose of ketamine is effective as an antidepressant and evokes behavioral effects up to ~2 weeks [1, 3, 4], well beyond its half-life time of ~2 hours [5]. While the mechanisms underlying ketamine’s acute and sustained effects are unknown, neural circuit plasticity is a potential explanation.
Ketamine alters the phenotype of parvalbumin-expressing (PV) inhibitory interneurons [6], and ketamine and its metabolite, hydroxynorketamine (HNK) modulate cortical circuit activity [7, 8]. Ketamine may reduce inhibition onto excitatory glutamatergic neurons, thus promoting cortical disinhibition [9–11], which initiates neural plasticity [12]. Signaling pathways are implicated in ketamine’s action, including release of brain-derived neurotrophic factor (BDNF) [13, 14], induction of plasticity via eukaryotic elongation factor 2 (eEF2) deactivation, dendrite outgrowth by mammalian target of rapamycin (mTOR) [15–18], and inhibition of glycogen synthase kinase-3 (GSK3) [19, 20]. The mechanism for how ketamine works through neural activity-dependent signaling to induce plasticity is unknown.
Neuregulin-1 (NRG1) is essential for neural development, and its tyrosine kinase receptor ErbB4 regulates visual cortical plasticity [21–23]. Our previous work demonstrates that PVs are the initial locus for visual cortex critical period plasticity [24]. PVs have relatively strong NRG1 expression, and ErbB4 is highly restricted to PVs [21, 25–27]. This may allow for PVs to regulate synaptic plasticity through activity-dependent NRG1/ErbB4 signaling [28]. Critical period monocular deprivation (MD) down-regulates NRG1/ErbB4 signaling in PVs. This evokes retraction of excitatory inputs to PVs, causing cortical disinhibition and critical period visual cortical plasticity [21]. Others show that modulation of NRG1/ErbB4 signaling in PVs enhances adult visual cortex plasticity [22]. We now test if subanesthetic ketamine reactivates adult visual cortical plasticity and promotes visual functional recovery via NRG1 signaling in PVs.
We find that subanesthetic ketamine down-regulates NRG1 expression in PVs, resulting in PV excitatory input loss and sustained cortical disinhibition to enhance cortical plasticity in adult visual cortex. We establish the mechanisms of ketamine-mediated induction of adult visual cortical plasticity to reopen the critical period and promote functional recovery from amblyopia.
Results
Ketamine reactivates adult cortical plasticity
We tested whether subanesthetic ketamine treatment induces visual cortical plasticity during adulthood [29]. Adult mice (P90-P120) had MD for four days, with ketamine treatment (s.c.) after the beginning of MD (Figure 1A,B). After four days, the ocular dominance index (ODI) was assessed using intrinsic signal optical imaging [30–32]. Normally, four days of MD does not change ODIs in adult mice [30]. In comparison, ketamine induces visual cortical plasticity with 4 days of MD (Figure 1C,D. Mann-Whitney U test: p = 0.0078). This resembles the plasticity in critical period animals after MD. We do not observe ketamine-induced visual cortical plasticity in PV-Cre; ErbB4fl/fl mice, where ErbB4 is ablated from PVs [33]( Figure S1).
Figure 1. Ketamine reactivates adult ocular dominance plasticity and restores visual acuity in amblyopic animals.
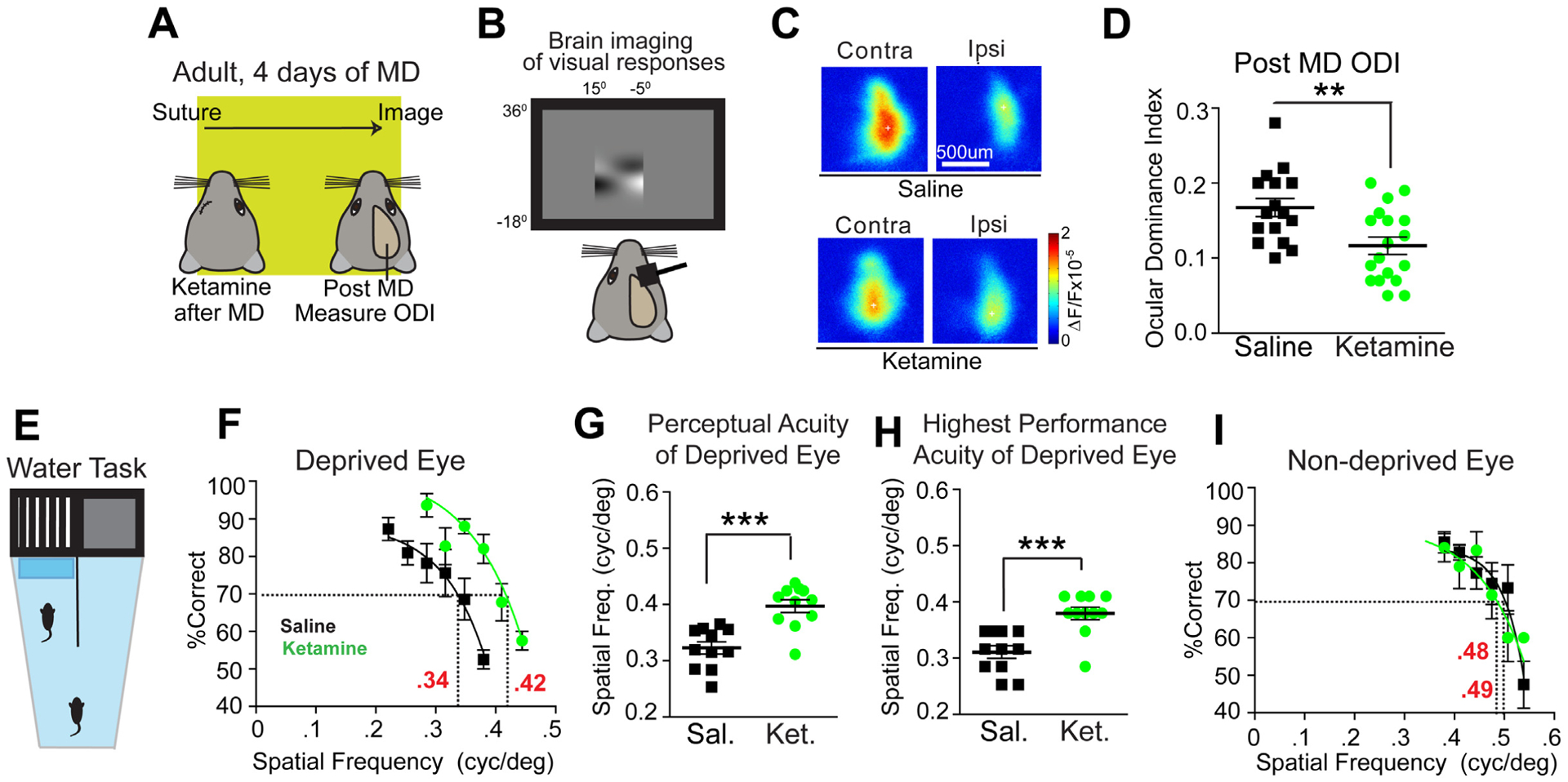
(A-D) Testing the effect of ketamine on ocular dominance plasticity (ODP) during adulthood using intrinsic signal optical imaging. (A) Illustration of the experimental paradigm in adult (P90–120) mice, and the timeline of the protocol. (A, left) After monocular deprivation (MD), saline or ketamine treatments are given, and 4 days later ODP is measured after eyelid suture removal. (B) Intrinsic signal responses of visual cortex to contralateral versus ipsilateral eye stimulation are recorded and the ocular dominance index (ODI) is assessed. The ODI is computed as (C-I)/(C+I) where C and I are the averaged map amplitudes calculated for contralateral and ipsilateral visual stimulation. (C) Representative cortical response maps from a control saline treated animal (top) and a ketamine treated animal (bottom) 4 days after treatment. In adult mice, 4 days of MD does not change ODIs. (D) However, ODI is reduced in ketamine treated adult animals (n=18, green) as compared to saline treated animals (n=16, black) (Mann-Whitney U test, p = 0.0078; mean ± SEM). The data were pooled from cohorts of mice receiving either 10 or 50 mg/kg ketamine treatment, as they shared similar trends. (E-I) Testing the effect of ketamine on restoring functional visual perception in adult amblyopic mice. (E) The visual water maze task (VWMT). Mice had one eye sutured shut from P18-P32. At P32 the eye was re-opened and mice were treated with ketamine (10mg/kg; s.c.) every other day for three total treatments. At P90 mice began training, and were tested at P100. Testing continued until ~P140, with mice doing ~10 trials/day. (F) Performance on the VWMT for all mice treated with either saline (black, n=11) or ketamine (10mg/kg; s.c., green, n=12) using the deprived eye. The performance curve is fit to a sigmoid using the last 6 data points [34]. The x-axis represents the spatial frequency of the visual stimulus gratings. The threshold of perceptual acuity is determined by the spatial frequency (x-axis) corresponding to the 70% value (y-axis, dashed line). For controls the acuity threshold is 0.34 cpd. For ketamine the acuity threshold is 0.42 cpd. (G) Perceptual acuity of deprived eyes across mice using acuity thresholds determined from performance curves of individual eyes. For deprived eyes ketamine (green, n=12) shows significantly improved perceptual acuity compared to saline (black, n=11) (Mann-Whitney U test, p = 0.0008; mean ± SEM). (H) Highest performance acuity at or above the 70% level for the deprived eye on the VWMT for mice treated with saline (black, n=11) or ketamine (green, n=12). Performance acuity is significantly increased with ketamine (Mann-Whitney U test, p = 0.0009; mean ± SEM). (I) Average performance on the VWMT for mice treated with saline (black, n=11) or ketamine (green, n=12) using the non-deprived eye. For saline the acuity threshold is 0.49 cpd. For ketamine the acuity threshold is 0.48 cpd. Average perceptual acuity between groups determined from performance curves of individual non-deprived eyes yielded no significant change (data not shown; Mann-Whitney U test, p = 0.1927; mean ± SEM). Also see related information in Figure S1 and Table S1.
We tested whether ketamine promotes visual functional recovery in adult amblyopic mice, using the visual water maze task (VWMT), a well-established behavior assessment of vision [30, 34]. Young mice had 2 weeks of MD during the critical period (P18–32) to induce amblyopia [30]. After eye opening, mice were treated with ketamine. During adulthood (P90-P120), acuity thresholds of deprived and non-deprived eyes were determined (Figure 1E–I)[30, 34]. Mice were trained to associate a hidden escape platform with a visual grating. Mice were then challenged to find the platform as the spatial frequency of the grating was increased. Acuity was assessed by covering eyes with a patch. Acuities were derived or measured from performance data (Figure 1G,H).
Deprived eye performance is dramatically improved with ketamine (Figure 1F,G. Mann-Whitney U test: p = 0.0008; Figure 1F,H. Mann-Whitney U test: p = 0.0009). The non-deprived eye is unchanged with ketamine (Figure 1I). Thresholds are consistent with publications [34]. This supports that ketamine in vivo promotes recovery of amblyopia in adults. While we focus on V1, multiple brain regions likely contribute to visual recovery [35–39].
Cortical disinhibition evoked by subanesthetic in vivo ketamine
Cortical disinhibition by interneurons is essential for plasticity [21, 24, 40]. We examined the time course of ketamine-evoked cortical disinhibition in adult V1. Ketamine metabolizes in vivo, so we tested bath application of ketamine on changes in inhibitory input to excitatory neurons [24]. We performed whole-cell recording of electrically-evoked inhibitory postsynaptic currents (IPSCs) in L2/3 pyramidal neurons (Figure 2A), by activating L4->L2/3 feedforward projections to L2/3 interneurons through L4 electrical stimulation. There were no acute effects of bath applied ketamine on IPSCs (Figure 2B). We tested the NMDAR antagonist MK-801 and did not observe effects (Figure 2C). This shows that bath applied ketamine or blocking NMDARs does not modulate inhibition to L2/3 pyramidal neurons in V1.
Figure 2. Ketamine evokes sustained cortical disinhibition, which is reversed with exogenous NRG1.
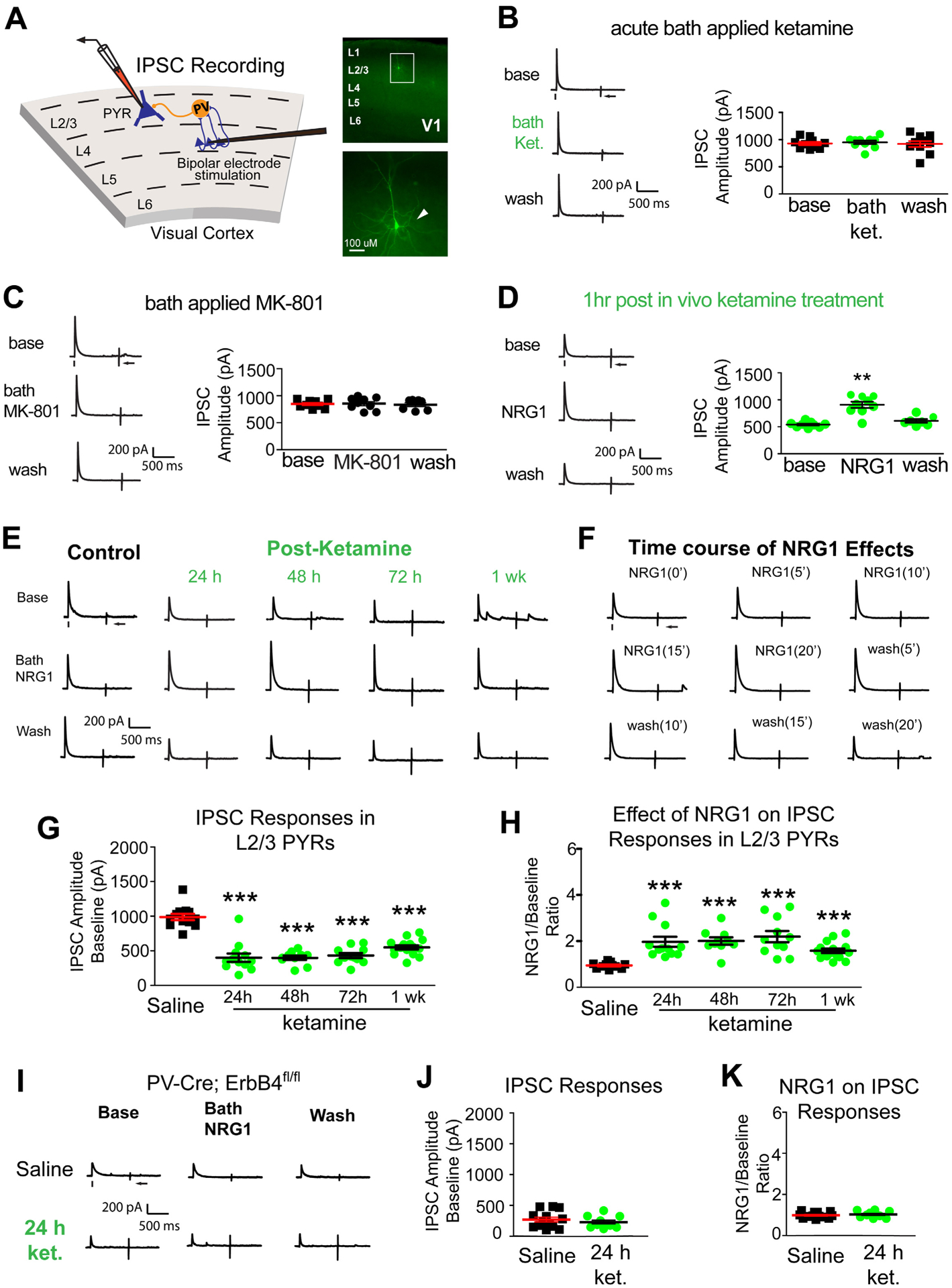
(A) Recording inhibitory postsynaptic currents (IPSCs) in L2/3 pyramidal (PYR) neurons by preferentially activating L4→L2/3 feedforward projections to L2/3 PV neurons through L4 electrical stimulation. Recorded neurons are filled with biocytin for post-hoc confirmation. (B-F) Recordings maintained on the same neuron during baseline, treatment, and washout. For each trial, electrical stimulation (1 ms, 20 μA) was applied, represented by a black tick beneath one example trace. The arrow indicates the current injection response to monitor access resistance during the experiment. (B) Acute bath application of ketamine does not induce any change in inhibitory inputs to L2/3 pyramidal neurons (n=10 same cells for control, drug application, and washout) in adult mouse visual cortex (P56 and above) (Friedman’s test: overall, p = 0.200). The data were obtained from 10 slices (1 cell per slice) of 5 different mice. (C) Acute bath application of MK-801 does not induce any change in inhibitory inputs to L2/3 pyramidal neurons [n=9 cells (7 cells have washout data) from 9 slices of 3 mice] (Friedman’s test: overall, p = 0.600). (D) Ketamine in vivo (10 mg/kg; s.c.) only 1 hour before recording reduces synaptic inhibition to L2/3 pyramidal cells. Bath application of NRG1 (5 nM) then reversed the effect and increased inhibitory inputs [n=9 cells (7 cells have washout data) from 9 slices of 3 mice]. (Friedman’s test: overall, p = 0.0008; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, p = 0.0078; mean ± SEM). (E, G-K) Recordings are from different neurons and different mice at time points that are on different days (24, 48, 72, 1 week). (E) Ketamine in vivo (10 mg/kg; s.c.) reduces evoked IPSCs to PYR neurons at 24 hours (n=12 cells), 48 hours (n=10 cells), 72 hours (n=11 cells), and 1 week (n=12 cells). Ketamine evoked decreases in IPSCs are reversed with bath NRG1. (F) Bath NRG1 increases inhibitory inputs to L2/3 pyramidal neurons with 10–20 minutes and this is washed out within 20 minutes. (G,H) We recorded L2/3 pyramidal neurons: control saline (n=12 cells from 12 slices of 5 mice), 24 hr [n=12 cells (9 cells have washout data) from 12 slices of 9 mice], 48 hr [n=10 cells (all cells have washout data) from 10 slices of 4 mice], 72 hr [n=11 cells (9 cells have washout data) from 11 slices of 5 mice], and 1 week [n=15 cells (13 cells have washout data) from 15 slices of 8 mice] before and after bath NRG1 application. (G) Compared with saline IPSC amplitudes are significantly reduced after in vivo ketamine treatment (Kruskal-Wallis test: overall p = 4.8 × 10−7. Mann-Whitney U test (adjusted for multiple comparisons): in vivo ketamine vs saline, 24h p = 0.0006, 48h p = 0.0004, 72h p= 0.0002, 1wk p = 6.29 × 10−5; mean ± SEM]. (H) NRG1 increases average evoked IPSC amplitudes ratios in L2/3 pyramidal neurons (control saline (n=12 cells), 24 (n=12 cells), 48 (n=10 cells) and 72 hours (n=11 cells) and 1 week (n=15 cells) after NRG1 (Kruskal-Wallis test: overall, p = 1.8×10−6. Mann-Whitney U -test (adjusted for multiple comparisons): in vivo ketamine vs saline, 24h p = 0.0002, 48h p = 0.0006, 72h p = 0.0002, 1wk p = 0.0001; mean ± SEM). (I-K) Removal of ErbB4 in PVs prevents ketamine-induced decreases in IPSCs in L2/3 excitatory neurons: neither ketamine (10mg/kg; s.c.) nor NRG1 affects evoked IPSCs to L2/3 pyramidal neurons of PV-Cre; ErbB4fl/fl mice. Representative IPSC traces shown in (I), and summarized in (J, K) (Figure 2J. Mann-Whitney U test: p = 0.5149) (Figure 2K. Mann-Whitney U test: p = 0.4757). Also see related information in Table S1.
In contrast, in vivo ketamine treatment decreases inhibitory inputs to L2/3 excitatory neurons. At 1 hour IPSC amplitudes were reduced to ~50% of controls (Figure 2D). Motivated by our previous findings that NRG1 increases synaptic inhibition, we tested if ketamine-mediated reductions in inhibition to excitatory neurons is reversed by NRG1 [21, 23]. Recombinant NRG1 containing the epidermal growth factor (EGF) core domain of NRG1-β1 applied to the bath reverses reductions in inhibition to excitatory neurons by ketamine (Figure 2D. Friedman’s test: overall, p = 0.0008; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, p = 0.0078).
We then examined the sustained action of ketamine on the evoked inhibition of L2/3 excitatory neurons at 24, 48, 72 hours, and 1 week after treatment. Inhibitory inputs to excitatory pyramidal neurons are reduced at all time points (Figure 2E,G. Kruskal-Wallis test: overall p = 4.8×10−7. Mann-Whitney U test (adjusted for multiple comparisons): ketamine vs saline, 24h p = 0.0006, 48h p = 0.0004, 72h p = 0.0002, 1wk p = 6.29×10−5). Ketamine-evoked decreases in evoked IPSCs are reversible with bath NRG1 (Figure 2E,H. Kruskal-Wallis test: overall p = 1.8×10−6. Mann-Whitney U test (adjusted for multiple comparisons): ketamine vs saline, 24h p = 0.0002, 48h p = 0.0006, 72h p = 0.0002, 1wk p = 0.0001). Bath acute NRG1 effects are fast and robust but can be washed out (Figure 2F). NRG1 has no effects on IPSCs in control pyramidal neurons.
We then treated PV-Cre; ErbB4fl/fl mice with ketamine, and measured evoked IPSCs in V1 neurons 24 hours after treatment. NRG1’s cognate receptor ErbB4 is removed from PVs in PV-Cre; ErbB4fl/fl mice [33]. We find no significant differences in inhibitory input to L2/3 pyramidal cells after ketamine in PV-Cre; ErbB4fl/fl mice (Figure 2I,J)). NRG1 does not alter evoked IPSCs in either ketamine treated or untreated PV-Cre; ErbB4fl/fl mice (Figure 2I,K). These results support that ketamine-induced effects require PV-specific ErbB4 for NRG1 signaling.
NRG1/ErbB4 signaling in PV neurons reduced by ketamine
To determine if ketamine modulates NRG1 or ErbB4 mRNA expression in PVs or excitatory cells, we used translating ribosome affinity purification (TRAP) [41](Figure 3A). NRG1 and ErbB4 mRNAs were measured from PV+ or Emx1+ neurons (Figure 3B–C). V1 was harvested at 24, 48, 72 hours or 1 week after ketamine. Results are all relative to PV cell-specific NRG1 expression after saline, as indicated by a horizontal tick mark on the y-axis in each panel (Figure 3D,G). Emx1+ neurons express NRG1 mRNA at lower levels than PVs (Figure 3D,F). In PVs, ketamine induces a sustained down-regulation of NRG1 mRNA which returns to saline control baseline only after 1 week (Figure 3D. Kruskal-Wallis test: overall p = 0.0012. Mann-Whitney U test (adjusted for multiple comparisons): ketamine vs saline, 24h p = 0.0348, 48h p = 0.0172, 72h p = 0.0172). ErbB4 mRNA does not change following ketamine treatment in either PV+ or Emx1+ neurons, albeit ErbB4 is expressed at markedly higher levels in PVs (Figure 3E,G).
Figure 3. Ketamine induces sustained down-regulation of NRG1 in visual cortex PV inhibitory neurons.
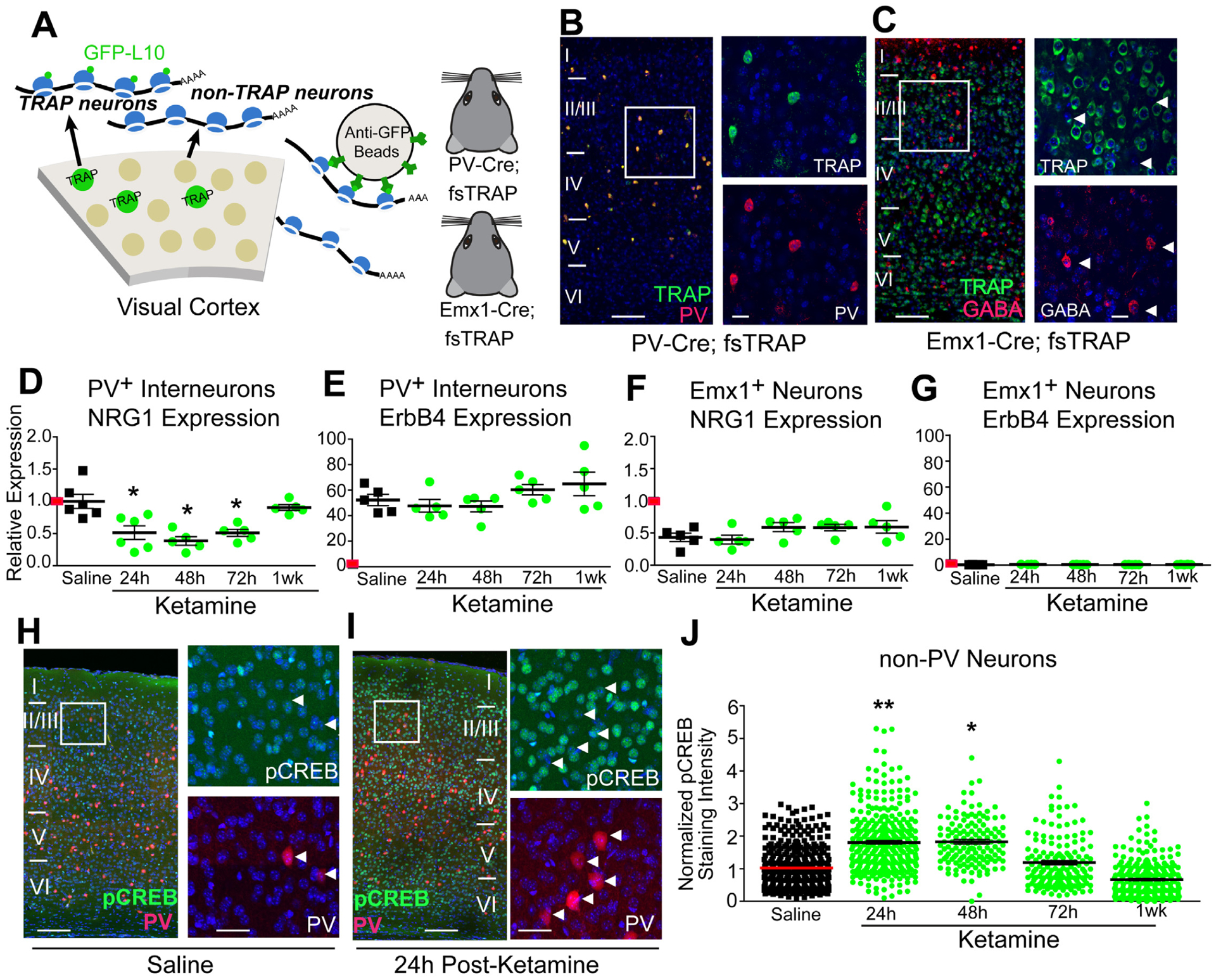
(A-G) Testing the effect of ketamine on cell-type specific NRG1 mRNA expression. (A) The translating ribosome affinity purification (TRAP) strategy. Using PV-Cre; fsTRAP mice, ribosomes (polysomes) from PVs (green cells) have EGFP tags. Lysis of all cells in the PV-Cre; fsTRAP cortex releases polysomes. Tagged polysomes are captured with anti-GFP Abs and are used for purification of PV-specific mRNAs. We also purify mRNA from Emx1+ neurons using Emx1-Cre; fsTRAP mice. Mice were treated with ketamine (10 mg/kg; s.c.) at P56, then sacrificed 24, 48, 72 hours or 1 week later. (B) Confocal images of labeled PVs (green), PV immunolabeling (red) and their overlay in layer I-VI of mouse V1 in PV-Cre; fsTRAP-EGFP mice; scale bar = 200 μm. The white boxes indicate the region of V1 imaged at higher magnification (B, right); scale bar = 50 μm. (C) Confocal images of labeled excitatory cells (green), GABA immunolabeling (red) and their overlay in Emx1-Cre; fsTRAP-EGFP mice. Arrowheads indicate GABA immunopositive cells. (D-G) Results were analyzed using the ddCt method with gapdh as an endogenous control, along with normalization to PV cell-specific nrg1 expression after saline treatment, as indicated by a red horizontal mark on the y-axis, so that all panels (D-G) can be compared [58]. (D) Ketamine treatment significantly lowers PV NRG1 mRNA in visual cortex (n=5–6 samples per treatment group; each sample is pooled from 5 mice for PV cell-specific data) (Kruskal-Wallis test: overall p = 0.0012; Mann-Whitney U test (adjusted for multiple comparisons): ketamine vs saline, 24h p = 0.0348, 48h p = 0.0172, 72h p = 0.0172; mean ± SEM). (E) PV ErbB4 mRNA does not change. (F) Emx1+ cell-specific NRG1 mRNA (n=5 samples per treatment group; each sample is pooled from 2 mice for Emx1+ cell-specific data) does not change. (G) Emx1+ cell-specific ErbB4 mRNA does not change. (H-J) pCREB in excitatory neurons after ketamine. (H) Confocal images of genetically labeled PVs (red), pCREB immunolabeling (green) and their overlay in V1 in P56 PV-Cre; Ai9 treated with saline; scale bar = 200 μm. The white box indicates the region of V1 digitally enlarged (H, right); Scale bar = 50 μm. White arrowheads indicate that PVs have very low pCREB. (I) Confocal images of genetically labeled PVs (red), pCREB immunolabeling (green) and their overlay in V1 in P56 PV-Cre; Ai9 24 hours after ketamine (10 mg/kg; s.c.); scale bar = 200 μm. (J) Quantification of the increase in pCREB immunoreactivity of non-PV, putative excitatory neurons in V1 L2/3 at 24, and 48 hours following ketamine (linear mixed effect model: overall p = 1.0 × 10−4, 24h p = 0.0059, 48h p = 0.0337; mean ± SEM). After 1 week pCREB levels return to control level. The overall normalized values of putative excitatory cells pooled from different mice were compared across treatment groups (n=8 for saline; n=3–7 for ketamine groups). Also see related information in Table S1.
To determine if ketamine increases molecular correlates of cortical disinhibition, the levels of phosphorylated cAMP response element transcription factor (pCREB) were measured (Figure 3H–J). CREB phosphorylation at Ser133 occurs with increased neural activity [42]. Ketamine increases pCREB levels in excitatory neurons in V1 (Figure 3H,I), for at least 48 hours after ketamine; then pCREB returns to baseline at 1 week (Figure 3J: linear mixed effect model: overall p = 1.0×10−4, 24h p = 0.0059, 48h p = 0.0337), showing sustained cortical disinhibition after ketamine treatment.
Sustained loss of excitatory inputs to PV neurons by ketamine
To determine if PV excitatory inputs are the synaptic locus of cortical disinhibition, we tested the effects of ketamine and NRG1 on these inputs. We performed laser scanning photostimulation (LSPS) experiments in adult V1 slices [24, 43] (Figure 4A–D). LSPS involves recording from a single neuron while sequentially stimulating potentially pre-synaptic neurons at surrounding sites; this determines whether there are synaptic inputs from those neurons (Figure S2). LSPS is performed at 24, 48, 72 hours, and 1 week after in vivo ketamine. Control PVs receive excitatory inputs from L4 and upper L5, as well as from L2/3 (Figure 4E,F). In ketamine-treated mice the excitatory drive to PVs is reduced at 24, 48 and 72 hours (Figure 4E, top row). Excitatory input to PVs is attenuated following ketamine for ~1 week.
Figure 4. Ketamine evokes sustained loss of excitatory inputs to PV interneurons, which is restored with NRG1.
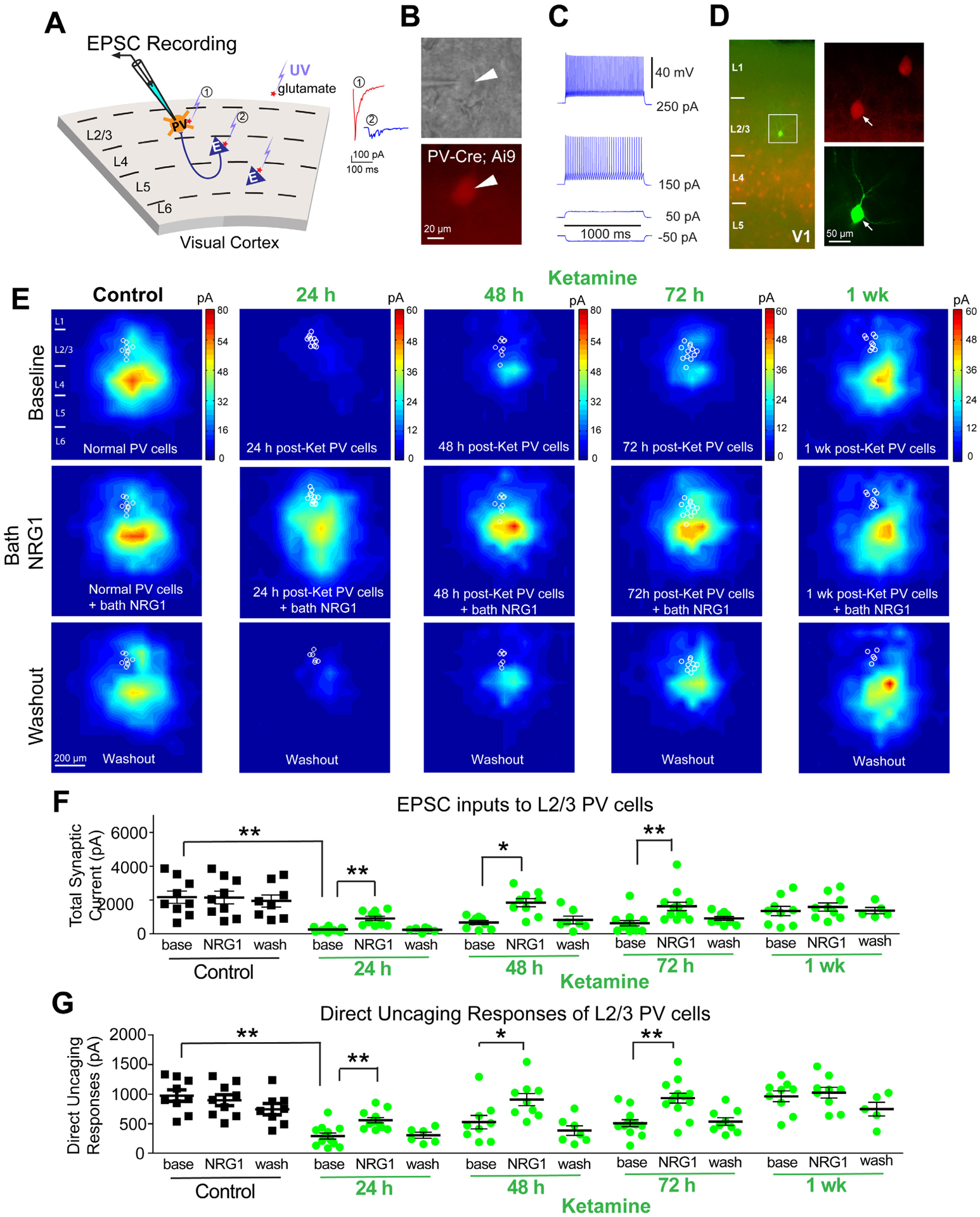
(A) Schematic of laser-scanning photostimulation (LSPS) mapping of cortical synaptic connections to PVs in V1. LSPS maps the spatial pattern of synaptic inputs to a neuron, and distinguishes direct uncaging responses (1, red) to assess glutamate-mediated excitability/responsiveness at perisomatic locations, and synaptically mediated EPSC responses (2, cyan) to assess synaptic inputs from presynaptic neuronal spiking. (B) Targeted recordings of PV neurons are facilitated by tdTomato expression in PV-Cre; Ai9 slices. (C) PVs are further identified by their fast-spiking. (D) Recorded neurons are injected with biocytin for post-hoc morphological analysis. (E-G) Recordings are from different neurons and different mice at time points that are on different days (24, 48, 72, 1 week) after treatment, and (E-G) recordings are maintained on the same neuron during baseline, treatment, and washout. (E) Group-averaged, excitatory input maps of PVs recorded at the specified conditions. White circles in each map represent individual PVs. The color scales code integrated input strength (blue = low, red = high) and applies to all other maps in the same condition. (E, left column) An acute application of NRG1 (5 nM) does not modulate local excitatory synaptic inputs onto PVs. Averaged excitatory input maps of L2/3 PVs are shown before (top), during bath NRG1 (20 min after NRG1 application) (middle), and after washout (bottom). (E, middle columns) Reduced excitatory inputs to PVs are seen at 24, 48 and 72 hours after ketamine (10 mg/kg; s.c.), but not after 1 week. (E, middle row) Excitatory inputs to PVs in ketamine mice are restored by NRG1. (E, bottom row) This acute restoration by NRG1 is eliminated by washout. (F) Summary data of average total synaptic input strength measured for L2/3 PVs under the specified conditions (control saline (n=9 cells), 24 (n=12 cells), 48 (n=9 cells) and 72 hours (n=13 cells) and 1 week (n=9 cells) after ketamine). Ketamine reduces EPSCs and NRG1 increases total EPSCs in PVs (linear mixed effects model (two factors): highly significant; Mann-Whitney U test (adjusted for multiple comparisons): base vs base (control), 24h p = 0.0012, 48h p = 0.0092, 72h p = 0.0067; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, 24h p = 0.0039, 48h p = 0.0312, 72h p = 0.0019; mean ± SEM). (G) Direct uncaging responses before and during bath NRG1 for PVs (control saline (n=9 cells), 24 (n=12 cells), 48 (n=9 cells) and 72 hours (n=13 cells) and 1 week (n=9 cells) after ketamine). Ketamine reduces EPSCs and bath applied NRG1 increases EPSCs (direct) in PVs (linear mixed effects model (two factors): highly significant; Mann-Whitney U test (adjusted for multiple comparisons): base vs base (control), 24h p = 0.0020, 48h p = 0.0624, 72h p = 0.0136; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, 24h p = 0.0039, 48h p = 0.0312, 72h p = 0.0019; mean ± SEM). Also see related information in Figures S2–S4 and Table S1.
We then examined if NRG1 restores excitatory input onto PVs. Bath applied NRG1 does not alter resting membrane potential or intrinsic membrane excitability in PVs (Figure S3). After baseline recording, we applied NRG1 in the bath. In control PVs, neither local excitatory synaptic input currents nor direct uncaging responses are altered after NRG1 application (Figure 4E,F,G). In contrast, both excitatory synaptic input currents and direct uncaging responses of PVs are enhanced by NRG1 in ketamine-treated mice (Figure 4F. Linear mixed effects model (two factors): highly significant; Mann-Whitney U test (adjusted for multiple comparisons): base vs base (control), 24h p = 0.0012, 48h p = 0.0092, 72h p = 0.0067; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, 24h p = 0.0039, 48h p = 0.0312, 72h p = 0.0019) (Figure 4G. Linear mixed effects model (two factors): highly significant; Mann-Whitney U test (adjusted for multiple comparisons): base vs base (control), 24h p = 0.0020, 48h p = 0.0624, 72h p = 0.0136; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, 24h p = 0.0039, 48h p = 0.0312, 72h p = 0.0019). As fast excitatory synaptic currents are mediated by AMPARs in PVs (Figure S2), this result indicates that NRG1 signaling modulates responses through AMPARs.
To confirm the action of ketamine on NRG1/ErbB4 signaling in PVs, we mapped the presynaptic excitatory inputs to L2/3 excitatory neurons in V1 after in vivo ketamine treatment. Ketamine or acute NRG1 does not modulate local excitatory synaptic inputs or direct uncaging responses of L2/3 excitatory neurons (Figure S4). Neither ketamine nor NRG1 alters resting membrane potential or intrinsic membrane excitability of L2/3 pyramidal neurons (Figure S3). This supports that ketamine’s effect is mediated through NRG1/ErbB4 signaling in PVs.
Effects of the ketamine metabolite HNK
Given our contrasting effects of bath-applied ketamine and in vivo ketamine treatment, we consider the metabolism of ketamine to (2R, 6R)-HNK [8] and determined whether HNK effects are similar to ketamine’s effects. Acute bath application of (2R, 6R)-HNK (100 μM) does not change evoked IPSC strengths to L2/3 pyramidal neurons (Figure 5A,B). This is consistent with findings that it takes an hour for HNK to increase AMPAR-mediated excitatory post-synaptic potentials [8]. Inhibitory inputs to L2/3 pyramidal cells are reduced in HNK mice at 24 hours post-treatment in vivo (Figure 5A,C). NRG1 restores those inhibitory inputs (Figure 5A,C. Friedman’s test: overall, p = 0.0007; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, p = 0.0039). We then quantified excitatory inputs to L2/3 PVs in V1 using LSPS. Excitatory inputs onto PVs are largely lost 24 hours following HNK (Figure 5D–G). Counteracting HNK effects, bath application of NRG1 restores PV inputs (Figure 5F. Friedman’s test: overall, p = 0.0034; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, p = 0.0156) (Figure 5G. Friedman’s test: overall, p = 0.0052; Wilcoxon signed-rank test (adjusted for multiple comparisons): NRG1 vs base, p = 0.0156). This indicates that HNK and ketamine require time to impact NRG1/ErbB4 signaling. This agrees with findings that HNK does not bind NMDARs [44], but triggers molecular signaling, such as NRG1/ErbB4 [45].
Figure 5. HNK evokes sustained reduction of cortical inhibition and loss of excitatory inputs to PV inhibitory neurons, both of which are restored with NRG1.
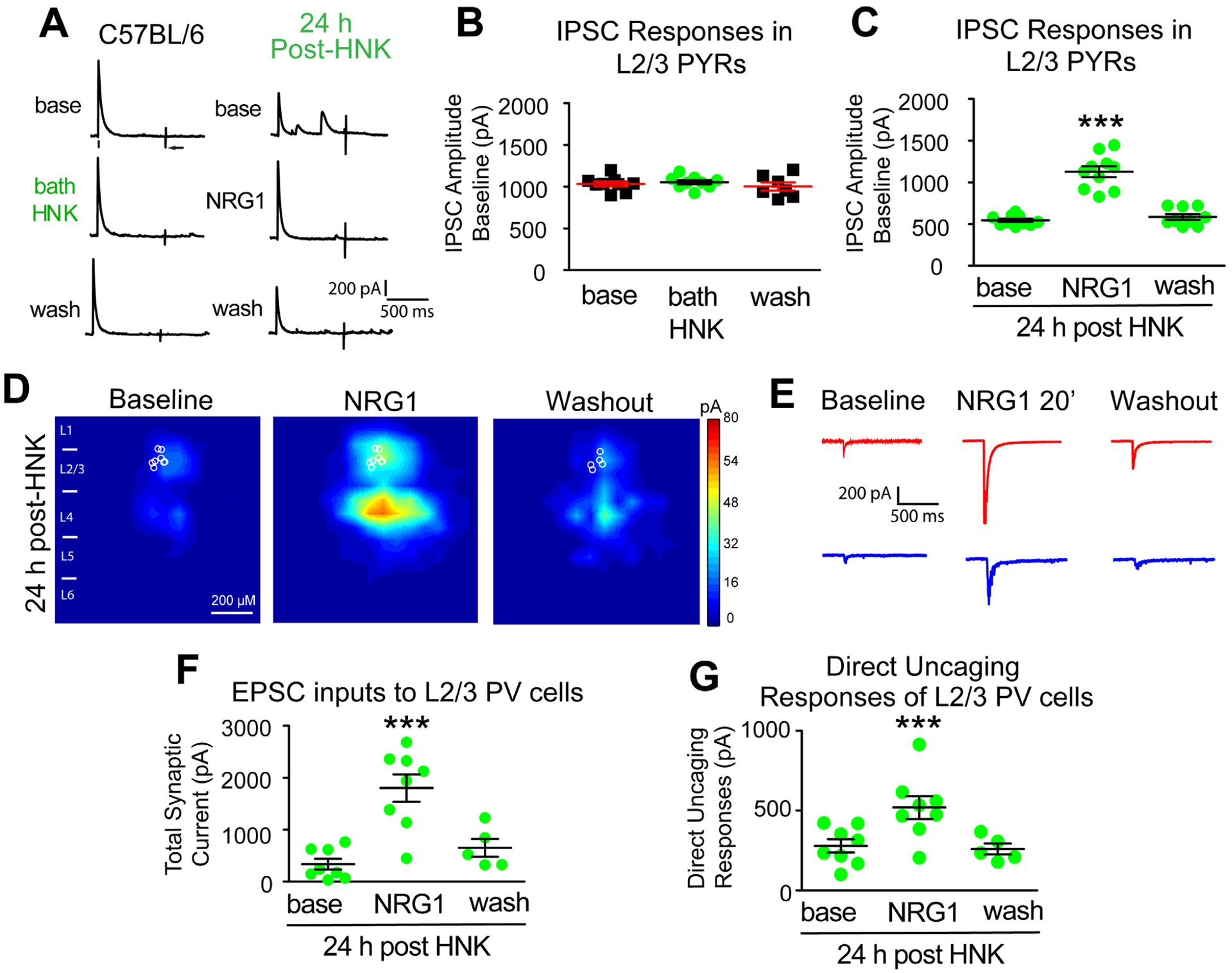
(A-G) Recordings maintained on the same neuron during baseline, treatment, and washout. (A-B) Acute bath application of (2R, 6R)-HNK (100 μM) does not change evoked IPSC strength to L2/3 excitatory pyramidals (n=8 cells). IPSCs were recorded in L2/3 pyramidal (PYR) neurons through L4 electrical stimulation as shown in Figure 2A. (C) HNK treatment (10 mg/kg; s.c.) at 24 hours before recording induces a reduction of IPSCs to L2/3 pyramidals. NRG1 (5 nM) reversed the reduced inhibition induced by HNK treatment (n=10 cells) (Figure 5A,C. Friedman’s test: overall, p = 0.0007; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, p = 0.0039); mean ± SEM). (D) LSPS mapping of local excitatory synaptic inputs to PVs. (E) Illustration of example responses of a PV neuron in HNK treated mouse cortex with LSPS in a perisomatic location (red traces) and in a presynaptic input site (blue traces) at baseline, NRG1 for 20 minutes and washout. (F-G) Both total synaptic inputs (n=8 cells) and direct uncaging (n=8 cells) response are reduced 24 hours after HNK treatment in vivo. NRG1 (5 nM) significantly restores both total excitatory synaptic inputs and direct excitatory synaptic inputs onto PVs in V1 of HNK mice (Figure 5F. Friedman’s test: overall, p = 0.0034; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, p = 0.0156; mean ± SEM) (Figure 5G. Friedman’s test: overall, p = 0.0052; Wilcoxon Signed-Rank test (adjusted for multiple comparisons): for NRG1 vs base, p = 0.0156; mean ± SEM). Also see related information in Table S1.
Longitudinal examination of ketamine effects on population neurons
Extending our slice mapping results to in vivo physiology, we performed 2-photon calcium imaging. We probed GCaMP6-based neural activity as mice viewed a grey screen (Figure 6A). We first performed baseline recordings (Figure S5), and then treated mice with ketamine (Figure 6B). We recorded from the same groups of neurons and images were motion corrected [46]. All extracted neurons were included in the analysis. The activity of excitatory neurons [47], measured by calcium event frequency, significantly increases at 24, 48, and 72 hours following ketamine (Figure 6G. Linear mixed effects model: overall p = 4.0×10−151; 24h p = 1.32×10−88, 48h p = 3.16×10−82, 72h p = 8.8×10−8, 1wk p = 0.005). At 1 week neural activity returns to baseline (Figure 6C,D,G). Thus, ketamine induces sustained cortical disinhibition. This is supported by visually-evoked potential (VEP) recordings [48] from V1 (Figure S6). Ketamine treatment increases the VEP amplitudes 24 and 48 hours, and this is prevented by NRG1 (Figure S6).
Figure 6. In vivo population calcium imaging reveals sustained disinhibition of excitatory neurons and inhibition of PV interneurons by subanesthetic ketamine in V1 of awake head-fixed mice.
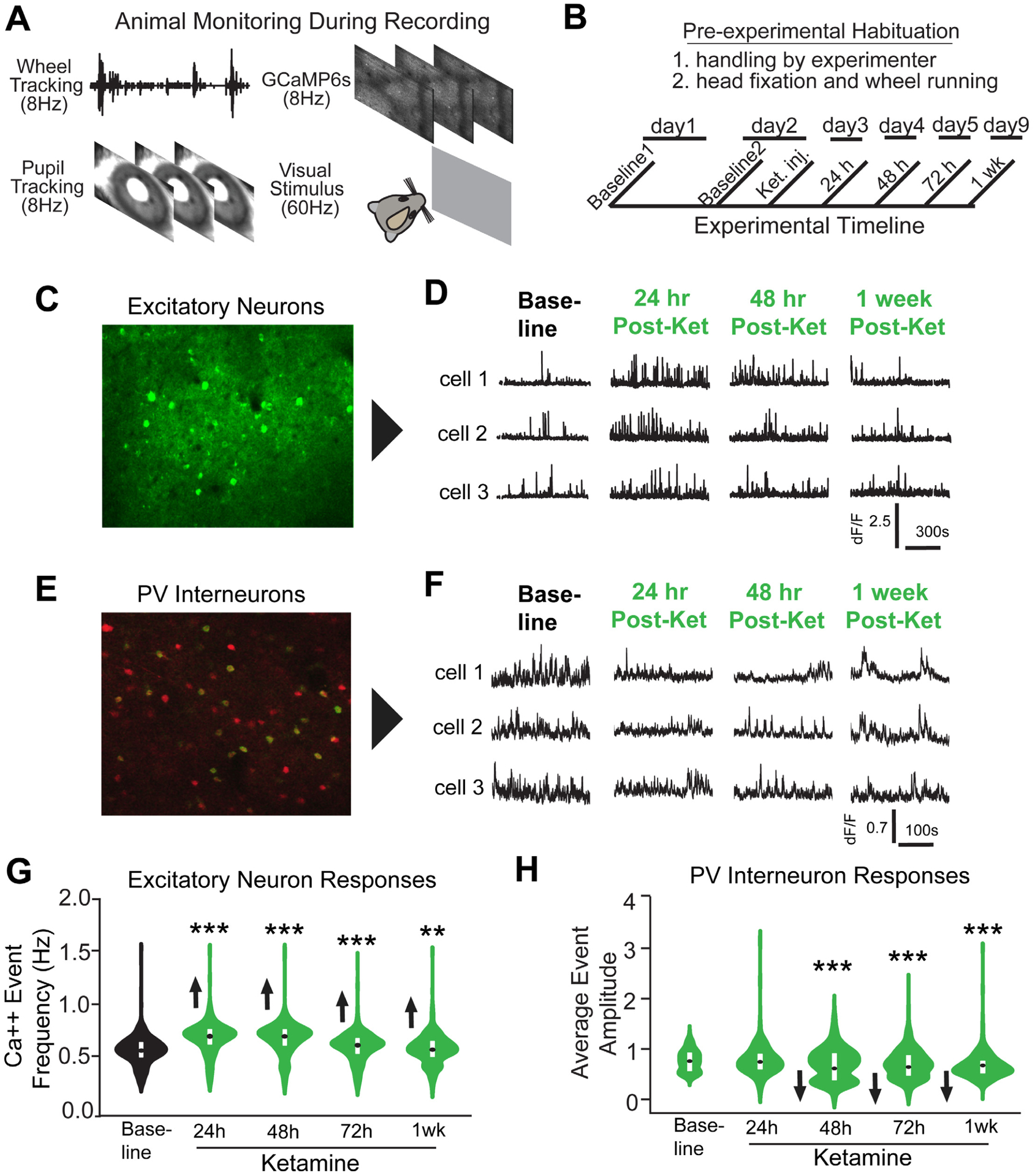
(A) Animal running behavior, GCaMp6s calcium signals, and pupil tracking were acquired. (B) Timeline of the experiment. Baseline neuron activity is recorded and then ketamine is administered (10mg/kg; s.c.) to adult mice expressing GCaMP6s, and subsequent recordings are made at 24, 48, 72 hours and 1 week. The visual display was a grey screen for 15 minutes. (C) A maximum intensity Z-projection image of V1 from a mouse expressing GCaMP6s in excitatory neurons [47]. (D) Calcium transient event data of three representative bV1 excitatory neurons. (E) A maximum intensity Z-projection image of bV1 from a PV-Cre; Ai163 mouse [49] expressing GCaMP6s (green) in PVs (red). (F) Calcium transient event data of three representative bV1 PVs from the experiment described in B. (G) Average calcium event frequencies of excitatory neurons. For excitatory neurons (C,D,G), there are 1255–1396 neurons from 3 mice included for analysis. (Figure 6G. Linear mixed effects model overall: p = 4.0×10−151; 24h p = 1.32×10−88, 48h p = 3.16×10−82, 72h p = 8.8×10−8, 1wk p = 0.005; violin plot with median and interquartile range). (H) Average calcium event amplitudes of PVs. For PVs (E,D,H), there are 383–409 neurons from 4 mice included for analysis. (Figure 6H. Linear mixed effects model: overall p = 1.24×10−12, 24h p = 0.453, 48h p = 0.009, 72h p = 1.31×10−5, 1 week p = 1.26×10−11; violin plot with median and interquartile range). The calcium event frequencies of excitatory neurons or the average amplitudes of PVs was present in a violin plot with their median and interquartile range indicated (G,H). Also see related information in Figures S5 and S6, and Table S1.
Calcium activity of PVs was also measured using GCaMP6 [49] (Figure S5). We treated mice with ketamine (Figure 6B). PVs’ average event amplitudes are reduced at 48, 72 hours and 1 week after ketamine (Figure 6H. Linear mixed effects model: overall p = 1.24×10−12, 24h p = 0.453, 48h p = 0.009, 72h p = 1.31×10−5, 1 week p = 1.26×10−11). This indicates that ketamine-mediated disinhibition of V1 is induced by reducing the activity of PVs.
We then imaged GCaMP6-based neural activity of excitatory neurons in wild type and PV-Cre; ErbB4fl/fl V1, using miniscopes [50–52](Figure 7A–C; Figure S7). We selectively expressed GCaMP6 in excitatory neurons in V1. No differences in behavior are observed for PV-Cre; ErbB4fl/fl and control mice (Figure 7D,E,H). Increases in the amplitudes of excitatory neurons are found in PV-Cre; ErbB4fl/fl mice compared to controls (Figure 7F,G,I,J: Figure 7I. Linear mixed effects model p = 1.1×10−8) (Figure 7J. Linear mixed effects model p = 3.2×10−19). This supports the idea that ketamine acts on PV NRG1/ErbB4 signaling to regulate cortical disinhibition and plasticity in adults.
Figure 7. NRG1/ErbB4 signaling in PV interneurons is implicated in regulation of cortical inhibition.
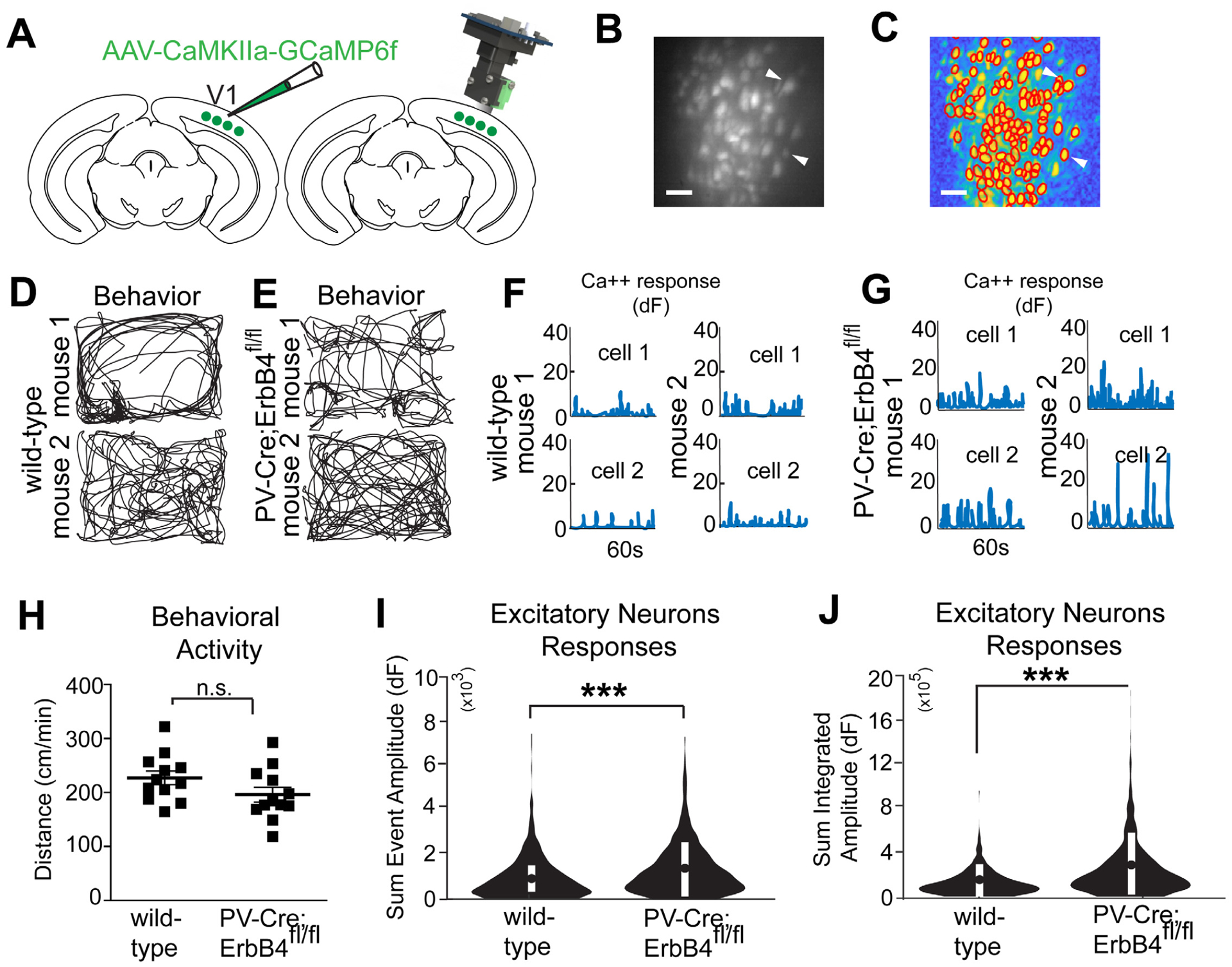
(A) Illustration of AAV1 injection for targeted expression of GCaMP6f in bV1 excitatory neurons. Illustration of a miniscope used to image in vivo calcium signals in bV1 excitatory neurons. The GRIN lens implanted over bV1 at 2 weeks after an AAV1-Camk2a-GCaMP6f injection into bV1, is shown in grey. Recording of neural and behavioral activity was performed during 5–10 minute sessions. (B) Maximum intensity Z-projection image showing recorded bV1 neurons from a combined recording session, scale bar = 50 μm. (C) A spatial profile image shows extracted neurons and their contours (see white arrows) using the CNMF-E algorithm (see Materials and Methods for details) based on the concatenated video in (B), scale bar = 50 μm. (D) Behavioral tracking data (black lines) of two representative wild type mice. (E) Two representative PV-Cre; ErbB4fl/fl mice. (F) Calcium transient event data of two representative bV1 neurons from 2 different wild type mice (top, neuron 1; bottom, neuron 2). (G) Two neurons from two PV-Cre; ErbB4fl/fl mice (top, neuron 1; bottom, neuron 2). (H) There is no difference in behavior (normalized distance; cm/min) between wild type and PV-Cre; ErbB4fl/fl mice. (I-J) Sum event amplitudes are the sum of the peak values of calcium events; total integrated amplitudes are the summation of the integrated area under the calcium event. (I) Sum event amplitudes (Figure S7; see Methods for details), for neurons from wild type mice (n = 360 cells) as compared to PV-Cre; ErbB4fl/fl mice (n = 627 cells) (linear mixed effects model: overall, p = 1.1×10−8; modified violin plot showing mean ± SEM). (J) Sum integrated amplitudes for neurons from wild type mice (n=260) as compared to PV-Cre; ErbB4fl/fl mice (n=627) (linear mixed effect model: overall, p = 3.2×10−19; modified violin plot showing mean ± SEM). Also see related information in Figure S7 and Table S1.
Discussion
Ketamine reactivates adult visual cortical plasticity and promotes functional recovery from amblyopia. Re-opening adult visual cortical plasticity depends on PV-mediated NRG1/ErbB4 signaling. In contrast to chronic fluoxetine administration that requires weeks of treatment [53], ketamine’s effects are rapid. The ketamine metabolite (2R, 6R)-HNK has rapid and long-lasting effects, mimicking ketamine treatment. These pre-clinical findings provide motivation for testing ketamine and its metabolite HNK for treating amblyopia in adults.
Previously, we showed that activity-dependent NRG1 signaling and sensory experience shape circuits in V1 during the critical period [21]. We now find that ketamine reduces synaptic inhibition to excitatory neurons in adult V1 in a rapid and sustained manner. The mechanism is independent of acute NMDAR antagonism as bath-applied ketamine or MK-801 does not modulate synaptic inhibition to pyramidal cells. A reduction in evoked IPSC inputs to excitatory neurons occurs within 24 hours of ketamine. Ketamine increases pCREB in excitatory neurons, indicating increased neural activity.
Ketamine treatment downregulates NRG1/ErbB4 signaling in PVs, which correlates with the decreases in synaptic inhibition to excitatory neurons. NRG1 rapidly restores excitatory input onto PVs and enhances cortical inhibition in ketamine-treated cortex. In accordance with our previous study [21], rapid NRG1 potentiation of glutamate-evoked responses of PVs in ketamine-treated cortex is consistent with insertion of AMPARs on PVs. Ketamine-induced effects require PV-specific ErbB4 for NRG1 signaling. Ketamine modulation of cortical disinhibition through PV-specific ErbB4 may be present in other brain regions (such as the superior colliculus and V2). Our findings offer insights into previous studies showing that within an hour, (2R, 6R)-HNK induces increases in AMPAR-mediated EPSPs, which is sustained after washout [8]. Acute and sustained cortical disinhibition via downregulated NRG1 may account for NMDAR inhibition-independent antidepressant actions of ketamine metabolites [8].
Our findings focus on PVs as the initial locus underlying ketamine’s effects. The PV-cell specific NRG1/ErbB4 mechanism identified in our work unifies previous proposals regarding cortical plasticity. The cortical disinhibition evoked by PV-specific NRG1 downregulation increases pCREB activity in excitatory neurons, which promotes BDNF production [42]. The BDNF receptor TrkB can activate further downstream pathways, including the serine-threonine kinase mTOR, which is also involved in neural plasticity [54]. Activation of mTOR in excitatory cells is involved in promoting ketamine-induced dendrite spine growth [7, 18]. mTOR signaling inhibits GSK3, which is the target of many mood stabilizers and antidepressants [55]. This PV-directed mechanism for ketamine’s effects on cortical plasticity is informative for treating depression.
While this study has focuses on PVs, ketamine may modulate non-PV interneurons. PVs can be inhibited by somatostatin-expressing interneurons and VIP interneurons preferentially inhibit somatostatin-expressing cells [56]. Also further studies will be required to determine what role PVs play in re-opening critical period plasticity for sensitivity to deprivation versus recovery of closed eye inputs [39]. In addition, our experimental controls were saline-treated adult animals with 4 days of MD in adulthood. These mice did not exhibit significant changes in their ODI’s. In contrast, we found that ketamine and MD induced a significantly decreased ODI compared with saline-treated controls. While we believe that ocular dominance plasticity and changes in the ODI do not deviate from normal development in critical period-aged and adult animals without MD or a similar manipulation that alters Hebbian competition between eye inputs [57], further control could be to treat the mice with ketamine but without MD and measure the effect on ODI after 4 days, which can help to rule out the possibility that ketamine treatment affects the OD by itself.
In summary, ketamine treatment promotes adult cortical plasticity and functional recovery from amblyopia by downregulating PV-specific NRG1 signaling, which results in PV excitatory input loss and cortical disinhibition. Fast and sustained ketamine actions show promise for therapeutic applications that rely on re-activating adult cortical plasticity.
Star Methods
Resource Availability
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xaingmin Xu (xiangmix@hs.uci.edu).
Materials Availability:
This study did not generate new unique reagents.
Data and Code Availability:
The datasets/code supporting the current study have not been deposited in a public repository but are available from the corresponding author upon reasonable request.
Experimental Model and Subject Details
Animals:
All experiment were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine. See Table S1 for detailed experimental and animal information. Male and female mice were used. The possible role of gender was tested, though there were no differences, in agreement with the literature. Mice were age P18 to produce amblyopia; all other mice were P90-P140.
To enable PV specific labeling and mRNA expression analysis, PV-IRES-Cre knock-in (Jackson Laboratory, stock #008069) mice were crossed to fsTRAP mice [41](Jackson Laboratory, stock #022367) to generate PV-Cre+/−; fsTRAP mice. To enable excitatory neuron labeling and mRNA expression analysis, Emx1-Cre mice (Jackson Labortory, stock #005628) were crossed to fsTRAP mice to generate Emx1-Cre+/−; fsTRAP mice [59]. For all experiments, mice were hemizygous for both transgenes. To genetically label PV cells PV-Cre mice were crossed with Ai9 tdTomato reporter knock-in mice (Jackson Laboratory, stock #007905). To generate ErbB4 conditional knockout mice, mice homozygous for loxP-flanked alleles [33] were crossed with PV-Cre mice to produce PV-Cre+/−; ErbB4flx/+ mice. These mice were then crossed back to the homozygous loxP-flanked ErbB4 mice to produce PV-Cre+/−; ErbB4 flx/flx mice. To selectively express GCaMP6s in PV cells, PV-Cre mice were crossed to Ai163 mice (Allen Institute) to generate PV-Cre; Ai163 mice.
Animals (2–5 mice per cage) were housed in a vivarium room with a 12-h light/dark cycle with access to food and water ad libitum. They were randomly assigned to groups with treatment of either saline or ketamine. Unless specified otherwise in some Figure 1 related experiments, a single treatment of ketamine (10 mg/kg; ketamine hydrochloride; VedCo., Inc.) was used. The (2R, 6R)-HNK dosage (6094; 10 mg/kg; Tocris Bioscience) of the published study [8] was used for our in vivo HNK treatment. We performed in vivo exogenous NRG1 treatment (1 μg per mouse) via subcutaneous administration of recombinant NRG1 containing only the EGF core domain of NRG1-β1 (396-HB-050; R&D systems) [21, 60].
Method Details
Monocular Deprivation:
For ocular dominance plasticity experiments in adults (~P90), one eye lid was closed for 4 days using two mattress sutures (7–0 silk, Ethicon) and checked daily. To produce visual deficits, closure of the one eye was maintained for the duration of the normal critical period (~P18-P32) and was checked daily. If the suture had signs of fraying, a small drop of tissue adhesive (Vetbond) was applied to the suture knot. If an animal was found to have an eye that opened or if the eye was damaged, it was excluded from the experiments. At P32 the eye was re-opened and mice are treated with either saline or subanesthetic ketamine (10mg/kg; s.c.) every other day for three treatments.
Intrinsic Signal Optical Imaging:
Monocular deprivation was initiated at ~P90. After 4 days, the sutured eyelid was opened and the skull over the visual cortex was exposed and then covered with agarose (1.5% w/v in 1X PBS) and a coverslip. The agarose and edges of the coverslip were sealed using sterile ophthalmic ointment (Rugby) to prevent drying. A recording session was then initiated.
The primary visual cortex was mapped in lightly anesthetized mice with 0.7% isoflurane and a dose of chlorprothixene (2.5mg/kg, i.p.) using intrinsic signal optical imaging through the intact skull as described previously [30]. A custom-designed macroscope (Nikon 135 × 50 mm lenses) equipped with a Dalsa 1M30 CCD camera was used to collect 512 × 512 pixel images sampled at 7.5 Hz (2.2 × 2.2 mm image area). The surface vasculature was first visualized using a 530 nm LED light and the intrinsic signal was then visualized with a 617 nm LED light (Quadica). The camera was focused ~600 μm beneath the pia surface. Custom written Matlab (Mathworks) code was used to acquire images and to stream them to disk. Visual stimuli (described below) were presented and response data was collected in 5-min session for a total of 3–4 imaging sessions per eye.
Responses to stimulation of the contralateral and ipsilateral eye were recorded from the visual cortex. A visual stimulus was periodically presented and swept within −18 to 36 degrees of the visual field elevation. This stimulus was created by multiplying a band limited (<0.05 cyc/degree; >2 Hz) spatiotemporal noise movie with a one dimensional Gaussian spatial mask (20 degrees) that was phase modulated at 0.1 Hz. Stimuli were presented on an Acer V193 monitor (30 × 37 cm, 60 Hz refresh rate, 20 cd/m2 mean luminance). Stimuli were alternatively presented for 5 minutes to each eye. Recordings were made at 0 degrees and 180 degrees. Maps of amplitude and phase of cortical responses were extracted from optical imaging movies via Fourier analysis of each pixel at the frequency of stimulus repetition (0.1 Hz) using custom written Matlab code. Overall map amplitude was computed by taking the maximum of the Fourier amplitude map smoothed with a 5 × 5 Gaussian kernel. Ocular dominance index (ODI) was computed as ODI = (C-I)/(C+I) where C and I are the averaged map amplitudes calculated for contralateral and ipsilateral visual stimulation respectively.
Visual Water Maze Task:
Visual acuity was assessed using a forced choice, two alternative discrimination task in a visual water maze [34]. By testing each eye independently, visual acuity in that eye was determined. A post was surgically implanted onto the skull of the mouse using dental cement. The post allowed for the attachment of a removable custom-made eye occluder to test each eye independently. A week after surgery, mice were trained and then tested. Mice were motivated to swim toward a hidden platform whose location was indicated by the vertical sine wave gratings displayed on a screen at a spatial frequency of 0.063 cpd when viewed from the water maze choice plane. Mice continued training until they reached 90%−100% correct performance levels. Testing was then completed one eye at a time. During testing, the stimulus was increased 0.032 cpd when a mouse achieved at least 70% correct over 10 trials. Mice were required to complete 3 consecutive trials at frequencies under 0.28 cpd in order to automatically advance. For frequencies over 0.28 cpd mice were required to complete 5 consecutive trials in order to automatically advance. Mice were required to complete a block of 10 if any error was made, and the 70% rule was enforced. If performance fell below the 70% correct rate the stimulus was decreased by at least 0.096 cpd. Testing took roughly 200–250 trials per mouse to complete. The perceptual acuity (Figure 1F,I) was determined by measuring the spatial frequency corresponding to the 70% value of a sigmoidal fit to performance data using the last 6 data points (Prusky et al., 2000). We also report analyses (Figure 1H) which does not rely on interpolation, where the highest spatial frequency achieved at or above the 70% level is shown.
Translating Ribosome Affinity Purification (TRAP):
Purification of polysomally bound mRNA from visual cortical lysate was performed as described with modifications [41]. Briefly, visual cortex was dissected in ice-cold ACSF (in mM: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 glucose). Pooled visual cortex from two to five mice was grinded to powder on dry ice, followed with sonication for 5 seconds in ice-cold lysis buffer [20 mM HEPES (pH 7.4), 150 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol, 100 μg/ml cycloheximide (Sigma-Aldrich), protease inhibitors (Roche) and 40 U/mL recombinant RNase inhibitor (Promega, Madison, WI)]. Homogenates were centrifuged for 10 minutes at 2,000× g, 4 °C, to pellet nuclei and large cell debris, and NP40 (Invitrogen, Carlsbad, CA) and DHPC (Avanti Polar Lipids, Alabaster, Alabama) were added to the supernatant at final concentrations of 1% (vol/vol) and 30 mM, respectively. After incubation on ice for 5 minutes, the lysate was centrifuged for 10 minutes at 20,000× g to pellet insoluble material. Two mouse monoclonal anti-GFP antibodies, Htz-GFP19C8 and Htz-GFP19F7 (50 μg each; RRID:AB_2716737 & RRID:AB_2716736 Memorial Sloan-Kettering Monoclonal Antibody Facility, New York, NY) were added to bind to 375 μL protein G Dyna magnetic beads (Invitrogen). Alternatively, 300 μL of the streptavidin Myone T1 dynabeads were binding to 120 μg biotinylated protein L first, then followed with two mouse monoclonal anti-GFP antibodies incubation. After being washed twice with the polysome extraction buffer, the beads were then added to the cell-lysate supernatant, and the mixture was incubated at 4 °C with end-over-end rotation for 30 minutes. The beads were subsequently collected on a magnetic rack and washed four times with high-salt polysome wash buffer [20 mM HEPES (pH 7.4), 350 mM KCl, 5 mM MgCl2, 1% NP-40, 0.5 mM dithiothreitol and 100 μg/mL cycloheximide]. RNA was eluted from the beads by incubating beads in RLT buffer (74004; RNeasy Micro Kit, Qiagen, Venlo, Netherlands) with β-mercaptoethanol (10 μL/mL) for 5 minutes at room temperature. Eluted RNA was purified using RNeasy Micro Kit (Qiagen) per the manufacturer’s instructions including in-column DNase digestion. Immunoprecipitated RNA yield for each sample was approximately 20 ng/μL.
For PV-Cre; fsTRAP experiments, both cortices (V1) from 5 mice were pooled together to generate n=1 before mRNA extraction. For Emx1-Cre; fsTRAP experiments both cortices (V1) from 2 mice were pooled together to generate n=1 before mRNA extraction.
Quantitative Real-Time Polymerse Chain Reaction (qPCR):
Purified RNA (30ng) was converted to cDNA using Superscript® III reverse transcriptase (18080093; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Quantitative changes in cDNA levels were determined by real-time PCR using the Power SYBR Green Master Mix (4368577; Thermo Fisher Scientific, Waltham, MA, USA), using primers at a concentration of 500nM. Primers were custom designed for mouse NRG1, ErbB4, and for the endogenous control GAPDH. PCR was carried out for 2 min 50 °C, 5 min 95 °C, 40 cycles (15 seconds for 95°C, 30 seconds for 50°C), followed by a melt curve. Technical triplicates were used. GAPDH was used to normalize gene expression and data presented at mean ± SEM. Cycling and quantitation were performed on a ViiA™ 7 Real-Time PCR System instrument (Thermo Fisher Scientific, Waltham, MA, USA) using the ViiA 7 software v1.2. The following primers were used: NRG1-F GCAAGTGCCCAAATGAGTTTAC; NRG1-R GCTCCTCCGCTTCCATAAAT; ErbB4-F CATGGCCTTCCAACATGACTCTGG; ErbB4-R GGCAGTGATTTTCTGTGGGTCCC; GAPDH-F TGCACCACCAACTGCTTAGC; GAPDH-R CAGTCTTCTGAGTGGCAGTGATG.
Immunohistochemistry:
For immunochemical staining experiments, animals were first deeply anesthetized with Uthasol (sodium pentobarbital, 100 mg/kg, i.p.) and were then perfused transcardially with 5mL of 1X phosphate buffered saline (PBS, pH 7.3–7.4), followed by 20 mL 1X PBS containing 4% paraformaldehyde (PFA) and phosphatase inhibitor (PhosSTOP, 1 tablet for 20 mL, Roche, Switzerland). Brains were removed and maintained in 4% PFA for 24 hours, and then transferred to 30% sucrose in 1X PBS for 24 hours. Then, using a freezing microtome (Leica SM2010R, Germany), coronal sections of the brain were taken at a 30 μm thickness. Mouse V1 coronal sections from bregma −3.40 to −3.80 mm were used for immunohistochemical staining and analysis.
Free floating sections were rinsed 5 times with 1X PBS, and incubated in a blocking solution for 1 hour at room temperature on a shaker. The blocking solution contained 5% normal donkey serum and 0.25% Triton X in 1X PBS. Sections were then incubated with the primary antibody diluted in blocking solution for 36 hours at 4 °C. After incubation with the primary antibody, brain sections were rinsed thoroughly with 1X PBS, and then incubated with the secondary antibody diluted in blocking solution for 2 hours at room temperature. After the secondary antibody was rinsed off, sections were counterstained with 10 μM 4’−6-diamidino-2- phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO) for 5 minutes to help distinguish cortical laminar structure and neuronal nuclei. Lastly, sections were rinsed and then mounted on microscope slides. Sections were cover-slipped with Vectashield mounting medium (H-1000,Vector, Burlingame, CA). To identify PV positive neurons in PV-Cre; fsTRAP mice, the primary goat anti-PV antibody (PVG-213, Swant, Switzerland; RRID:AB_10000345; 1:1000) and a Cy3-conjugated donkey anti-goat antibody (Jackson ImmunoResearch; RRID: AB_2307351; 1:200) were used. To identify GABA positive neurons in Emx1-Cre; fsTRAP mice, the primary rabbit anti-GABA antibody (A2052; SigmaAldrich; RRID:AB_477652; 1:1000) and a Cy3-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch; RRID: AB_2307443; 1:200) was used. To identify pCREB positive neurons, the primary rabbit anti-pCREB antibody (87G3; Cell Signaling; RRID:AB_2561044; 1:1000) was used.
Immunostained sections were examined, and 10X and 40X image stacks were acquired using a confocal microscope (LSM 780, Carl Zeiss Microscopy, Germany). Image tiles, overlaying, maximum projections, and subset z-stack selections were performed using the Zeiss image processing software. For fluorescent imaging, all sections of a staining series were acquired using the same settings (laser power, pinhole size, line scans), and data images were digitally processed identically.
Electrophysiology and Laser Scanning Photostimulation:
Coronal sections (400uM thick) of V1 were cut from mouse with a vibratome (VT1200S, Leica Biosystems, Buffalo Grove, IL) in sucrose containing ACSF (85 mM NaCl, 75 mM sucrose, 2.5 mM KCl, 25 mM glucose, 1.25 mM NaH2PO4, 4 mM MgCl2, 0.5 mM CaCl2, and 24 mM NaHCO3). Slices were incubated for at least 30 minutes in sucrose-containing ACSF at 32°C before being transferred into slice-recording chambers with standard ACSF (126 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 2 mM CaCl2, 2 mM MgCl2, 1.25 mM NaH2PO4, and 10 mM glucose). Throughout the cutting, incubation and recording, the solutions were continuously supplied with 95% O2–5% CO2.
We have previously described our methods for electrophysiological recording, imaging, and photostimulation in detail, including the definitions of all reported parameters [61, 62]. For our more recent publications using these same methods see [15, 24, 43, 63]. Briefly, whole-cell recordings were performed in oxygenated ACSF at room temperature under a differential interference contrast (DIC)/fluorescent Olympus microscope (BX51WI). Oxygenated ACSF was fed into the slice recording chamber through a custom-designed flow system, driven by pressurized 95% O2–5% CO2 (3 PSI) with a perfusion flow rate of about 2 mL/minute. Slices were first carefully examined under a 4x objective for targeting either L2/3 PV interneurons that express red fluorescent protein (RFP) or tdTomato or pyramidal neurons within the binocular regions of mouse V1, using landmarks defined in the previous study [64]. To perform whole-cell recordings, neurons were visualized at high magnification (60x objective, 0.9 NA; LUMPlanFl/IR, Olympus America Inc). The cell bodies of recorded neurons were at least 50uM below the surface of the slice. Patch pipettes (4–6 MΩ resistance) made of borosilicate glass were filled with an internal solution containing 126 mM K-gluconate, 4 mM KCl, 10 mM HEPES, 4 mM ATP-Mg, 0.3 mM GTP-Na, and 10 mM phosphocreatine (pH 7.2, 300 mOsm) when measuring excitatory postsynaptic currents (EPSCs) and action potentials (APs). In separate experiments, a cesium-based internal solution containing 130 mM CsOH, 130 mM D-gluconic acid, 0.2 mM EGTA, 2 mM MgCl2, 6 mM CsCl, 10 mM HEPES, 2.5 mM ATP-Na, 0.5 mM GTP-Na, and 10 mM phosphocreatine (pH 7.2, 300 mOsm) was used to voltage clamp pyramidal neurons at the excitatory reversal potential (0–5mV) and measure inhibitory postsynaptic currents (IPSCs). The electrodes also contained 0.1% biocytin for post-hoc cell labeling and further morphological labeling. Once stable whole-cell recordings were achieved with good access resistance (<30 MΩ), basic electrophysiological properties were examined through depolarizing and hyperpolarizing current injections. Electrophysiological data were acquired with a Multiclamp 700B amplifier (Molecular Devices), data acquisition boards (models PCI MIO 16E-4 and 6713, National Instruments), and a custom-modified version of Ephus software 5 (Ephus, available at https://openwiki.janelia.org/). Data were digitized at 10 kHz. Any recordings in which the access resistance changed by >20% during the course of the experiment were excluded from analysis. For a great majority of eletrophysiological experiments, recordings were obtained from the same neurons during baseline, treatment, and washout. Recordings were from different neurons and different mice for different time points (24 hr, 48 hr, 72 hr, 1 week) after treatment.
During photostimulation experiments, the microscope objective was switched from 60x to 4x for Laser scanning photostimulation (LSPS). The same low-power objective lens was used for delivering ultraviolet flash stimuli. Stock solution of MNI-caged-l-glutamate (Tocris Bioscience) was added to 20 mL of circulating ACSF for a concentration of 0.2 mM caged glutamate. The cortical slice image, acquired through the 4x objective, was visualized using a high-resolution digital CCD camera, and this was in turn was used for guiding and registering photostimulation sites. A laser unit (DPSS Lasers) was used to generate 355 nm UV laser pulses for glutamate uncaging. Short pulses of laser flashes (1 ms, 20 mW) were delivered using an electro-optical modulator and a mechanical shutter. Focal laser spots approximated a Gaussian profile with a diameter of ~50 μm.
Voltage clamping the recorded neuron allowed determination of sites contributing direct synaptic input. By systematically surveying synaptic inputs from hundreds of different sites across a large region, aggregate synaptic input maps were generated for individual neurons. For our mapping experiments, a standard stimulus grid (16×16 stimulation sites, 65 μm2 spacing) was used to tessellate V1 from pia to white matter. The LSPS site spacing was empirically determined to separate adjacent stimulation sites by the smallest predicted distance in which photostimulation differentially activated adjacent neurons. Glutamate uncaging laser pulses were delivered sequentially in a nonraster, nonrandom sequence, following a “shifting-X” pattern designed to avoid revisiting the vicinity of recently stimulated sites [65]. Because glutamate uncaging agnostically activates both excitatory and inhibitory neurons, we empirically determined the excitatory and inhibitory reversal potentials in L2/3 pyramidal cells to properly isolate EPSCs and IPSCs. We voltage clamped PV and pyramidal cells at −70 mV to determine LSPS evoked EPSCs.
Photostimulation induces two kinds of excitatory responses: (1) responses that result from direct activation of the recorded neuron’s glutamate receptors, and (2) synaptically mediated responses (EPSCs) resulting from the suprathreshold activation of presynaptic excitatory neurons. Responses that occur within 10 ms of the laser pulse onset are considered direct; these responses exhibit a distinct waveform and occur immediately after glutamate uncaging. Synaptic currents with such short latencies are not possible because they would have to occur before the generation of action potentials in photostimulated neurons. Therefore, direct responses are excluded from local synaptic input analysis, but they are used to assess glutamate-mediated excitability/responsiveness of recorded neurons. At some locations, synaptic responses over-ride the relatively small direct responses, and these responses are identified and included in synaptic input analysis. For data map analysis, we implement the approach for detection and extraction of photostimulation-evoked postsynaptic current responses described in reference [62]. LSPS evoked EPSCs are quantified across the 16×16 mapping grid for each cell, and 2 to 4 individual maps are averaged per recorded cell, reducing the likelihood of incorporating noise events in the analysis window. The EPSC input from each stimulation site is the measurement of the sum of individual EPSCs within the analysis window (>10 ms to 160 ms post photostimulation), with the baseline spontaneous response subtracted from the photostimulation response of the same site. The value is normalized with the duration of the analysis window (i.e., 150 ms) and expressed as average integrated amplitudes in picoamperes (pA). The analysis window is used because photostimulated neurons fire most of their action potentials during this time. For the color-coded map displays, data are plotted as the average integrated EPSCs amplitude per pixel location (stimulation site), with the color scale coding input strength. For the group maps obtained across multiple cells, the individual cell maps were first aligned by their slice images using laminar cytoarchitectonic landmarks. Then a new map grid is created to re-sample and average input strength at each site location across cell maps; a smooth version of color-coded map is presented for overall assessments. To further quantitatively compare input strength across cell groups or different conditions, we measure the total ESPC inputs across all map sites (total synaptic input strength) for individual cells.
For the experiments that examine the effects of bath application of NRG1 (R&D systems), MK801 or (2R,6R)-hydroxynorketamine (HNK) (Tocris Bioscience), the reagent(s) are added into the recording solution with specified concentrations. The drug application for 20 minutes was estimated to produce full effects, while the washout of 20–30 minutes was considered to remove the added drug from the recording solution.
Miniscope Imaging Experiments:
At 2 weeks after AAV1-CaMKII-GCaMP6f injection (100834-AAV1; Addgene; AAV.CamKII.GCaMP6f.WPRE.SV40; RRID:Addgene_100834), a gradient refractive index (GRIN) lens was implanted at the injection site in the bV1. A 1.8-mm diameter circular craniotomy was centered at the coordinates for the bV1 injections site. ACSF was repeatedly applied to the exposed tissue, and the dura was carefully removed. No cortex below the craniotomy was aspirated. The GRIN lens (0.25 pitch, 0.55 numerical aperture, 1.8-mm diameter and 4.31 mm in length, Edmund Optics) was slowly lowered using a stereotaxic arm to the surface of bV1. Next, a skull screw was used to anchor the GRIN lens to the skull. Both the GRIN lens and skull screw were fixed with cyanoacrylate and dental cement. Kwik-Sil (World Precision Instruments) was used to cover the lens. Two weeks later, a small aluminum baseplate was cemented onto the head of the animal atop the existing dental cement. A miniscope was fitted into the baseplate and locked in a position so that the field of view was in focus to visualize GCaMP6f-expressing neurons and visible landmarks, such as blood vessels.
For behavioral experiments involving bV1, mice were first handled for 10 minutes per day for a week in order to allow the animals to become comfortable with the behavioral experimenter. Then animals were habituated to the behavioral arena for 10 minutes per day for 2 days without a miniscope. Behavior was performed in a quiet designated behavioral room under low lighting conditions (<20 lux). The behavioral arena was 35cm × 25 cm in size with bedding which was changed between animals. Animals were then habituated to the behavioral arena with the miniscope fixed onto their head while they behaved freely for 10 minutes per day for 3 days, so that animals were acclimated and habituated to the behavioral experimenter, arena, and miniscope gear. After preparations were completed, in vivo GCaMP6-based calcium imaging of population bV1 neurons was performed in awake freely behaving mice. Behavioral recording was made by tracking the location of the LED on the miniscope through the experimental sessions. Sessions were 5–10 minutes in length. Using the behavioral tracking video data, the position and speed of the animal was determined using a custom Matlab script.
Please refer to a previous publication [52] and www.miniscope.org/ for technical details of our custom-constructed miniscopes. The head-mounted scope has a mass of about 3 g and uses a single, flexible coaxial cable to carry power, control signals and imaging data to custom open-source data acquisition (DAQ) hardware and software. Under our experimental conditions, the miniscope has a 700 μm × 450 μm field of view with a resolution of 752 pixels × 480 pixels (~1 μm per pixel). The electronics packaged the data to comply with the USB video class protocol and then transmitted the data over SuperSpeed USB to a PC running custom DAQ software. The DAQ software was written in C++ and uses Open Computer Vision libraries for image acquisition. Images were acquired at ~30 frames per s and recorded to uncompressed .avi files. The DAQ software simultaneously records the behavior of an animal through a high-definition webcam (Logitech) at ~30 frames per s, with time stamping of both video streams for offline alignment.
Miniscope videos were first concatenated and down-sampled by a factor of two using NIH ImageJ software, and then motion-corrected using the NoRMCorre MATLAB package [46]. Next, we visually inspected the maximum intensity projection images of individual video sessions and manually performed minor linear translations to align videos. Then, we created a large combined dataset by concatenating all aligned video images using NIH ImageJ. While this large data concatenation required significant computer resources (that is, 28 cores, 128 GB dynamic random-access memory, 1 TB solid state drive, 8 TB hard disk drive), it greatly enhanced our ability to obtain cell tracking from different days. Subsequent analysis was performed using custom Matlab scripts. We adopted the newly developed method of extended constrained non-negative matrix factorization for endoscopic data (CNMF-E) [66] to extract the calcium activity of individual neurons. The CNMF-E is based on the CNMF framework [67], which enables simultaneously denoising, deconvolving and demixing of calcium imaging data. Its key features include modeling the large rapidly fluctuating background that has a low spatial-frequency structure and allows good separation of single-neuron signals from this background. After iteratively solving a constrained matrix factorization problem, CNMF-E extracts the spatial footprints of neurons and their associated temporal calcium signal traces. Specifically, the first step of estimating the temporal activity of a neuron (neuron.C is a denoised version of dF, which is the change of the fluorescence intensity over time) is computing the weighted average of fluorescence intensities after subtracting the temporal activity of other neurons within the region of interest of that neuron. Then, a deconvolution algorithm, OASIS [68], was applied to obtain the deconvolved calcium event activity (neuron.S), as illustrated in Figure S7. Sum event amplitudes were calculated as follows: (1) For each cell, the peak values of calcium events that exceed 0.1*max (calcium peak values) were found, (2) the peak values (neuron.S (dF)) were summed. Total integrated amplitudes were calculated as the summation of the integrated area under the calcium event (neuron.C (dF)).
Visually Evoked Potential (VEP) Recording:
The general procedure for surgery has been described previously [48]. A steel head bar was affixed to the skull anterior to bregma using cyanoacrylate glue. Burr holes (< 0.5 mm) were then drilled into the skull over binocular V1 (~3.1 mm lateral of lambda) over the region identified as binocular visual cortex using intrinsic signal optical imaging. Tapered tungsten recording electrodes (FHC, Bowdoinham, ME, US), 50 μm in diameter at their widest point, were implanted in each hemisphere, ~350 μm below the visual cortical surface into deep L2/3. Silver wire (A-M systems, Sequim, WA, US) reference electrodes were implanted over prefrontal cortex. Mice were allowed to recover for at least 2 weeks prior to experimentation.
Visual stimuli were generated using software (http://bearlab-s1.mit.edu/supp6/PlxStimOne_Opto.zip) written and shared by Jeffrey Gavornik in either C++ for interaction with a VSG2/2 card (Cambridge Research systems, Kent, U.K.) or Matlab (MathWorks, Natick, MA, U.S.) using the PsychToolbox extension (http://psychtoolbox.org) to control stimulus drawing and timing. The display was positioned 20 cm in front of the mouse and centered, thereby occupying 92° × 66° of the visual field. Visual stimuli consisted of full-field bright to dark screen reversal at a frequency of 2 Hz. Bright screen stimulus luminance was X cd/m2. Experiments were fully automated and each session consisted of 400 phase reversals and included two 30-s intervals during which the screen was gray for spontaneous response data collection.
VEP recordings were conducted in awake, head-restrained mice. Prior to recording, mice were habituated to the restraint apparatus by sitting in situ in front of a gray screen for a 15-minute session on each of three consecutive days. On stimulus presentation days, VEP magnitudes were quantified by measuring trough-baseline response magnitude averaged over all phase reversals. Recordings were amplified and digitized using the Neuralynx electrode adaptor interface board (EIB-16) (Neuralynx., Bozeman, MT), and the Intan headstage preamplifier (RHD-2132) and Intan RHD2000 USB Interface Board (Intan, Los Angeles, CA). A recording channel was dedicated to recording VEPs from bV1 in each implanted hemisphere and another recording channel was reserved for collecting reference information. Local field potentials were recorded from V1 with 20 kHz sampling rate and a 250 Hz low-pass filter, and a notch filter (59–61 Hz). Data was extracted from the binary storage files generated by the Intan RHD2000 Evaluation software package and analyzed using custom software (http://bearlab-s1.mit.edu/supp6/VEP_Analysis.zip) written in C++, Matlab and Labview. VEPs were averaged across all stimulation reversals within a session of 400 trials, and trough-baseline differences were measured within a 100-millisecond period from bright screen start.
Two-Photon Calcium Imaging:
The general procedure for surgery has been described previously [24, 30]. After the skull was exposed, it was dried and covered by a thin layer of Krazy glue. After the Krazy glue dried (~15 minutes), it provided a stable and solid surface on which to affix an aluminum headplate with dental acrylic. The headplate was affixed to the skull and the margins sealed with dental acrylic to prevent infections. A 4-mm-diameter region of skull overlying the visual cortex was removed. The craniotomy opened at center coordinates 3mm lateral, and 1.7mm anterior to the lambda suture. Care was taken to leave the dura intact. A sterile, 4-mm-diameter cover glass was then placed directly on the dura and sealed at its edges with Krazy glue. When dry, the edges of the cover glass were further sealed with dental acrylic. At the end of the surgery, all exposed skull and wound margins were sealed with VetBond and dental acrylic. Mice were then removed from the stereotaxic apparatus, given a subcutaneous bolus of warm sterile saline and carprofen, and allowed the mouse to recover on the heating pad. When fully alert, they were placed back in their home cages, and gave carprofen daily for at least 3 days. Typically, the mouse was permitted to recover for at least seven days and conditioned to the head restraint and running wheel for several days, 20-minutes per day before mapping. And wait for 10–14 days after surgery to perform imaging.
Two-photon calcium imaging was performed using a resonant scanning, two-photon microscope (Neurolabware, Los Angeles, CA) controlled by Scanbox acquisition software (Scanbox, Los Angeles, CA). The light source was a Coherent Chameleon Ultra II laser (Coherent Inc, Santa Clara, CA) running at 920nm. The objective was an 16X water immersion lens (Nikon, 0.8NA, 3mm working distance). Usually we used the bidirectional scanning mode for the imaging. the microscope frame rate was 16.05 Hz (494 lines with a resonant mirror at 8kHz). The imaging field covered ~700 μm × 500 μm. Eye movements and pupil size of both eye were recorded via a Dalsa Genie M1280 camera (Teledyne Dalsa, Ontario, Canada) fitted with a UV cut-off filter (wavelength range 350 – 2200 nm). Images were captured at layer 2/3 (150–300um below pia). During imaging a substantial amount of light exits from the brain through the pupil. Thus, no additional illumination was required to image the pupil. Mice were head-fixed on a spherical treadmill on which they had been trained to balance and run. Movement of the spherical treadmill was recorded via a ball bearing option tracker (H5, US digital). Both locomotion and eye movement data were synchronized to the microscope frames. We used the wavelength 920nm to image GCaMP6s activities. For in vivo two-photon imaging sessions, the mice were presented a grey screen (mean luminance, 20 cd/m2) at all times, and we then recorded for 15 minutes. The responses we recorded were sustained spontaneous activity. This experimental design is sufficient to quantify stable baseline neural activity levels and avoids introducing artifacts associated with visual experience dependent plasticity [48]. All extracted neurons from each imaging session were included in the analysis.
Custom-written Matlab pipelines using the Constrained Nonnegative Matrix Factorization (CNMF)-based CalmAn [66, 69], which is an open-source library for calcium imaging data analysis, were used to remove motion artifact, extract neurons and neural activity identification and perform analyses. During recordings, the positions of neurons may change slightly due to cortex movements. To correct for translation and rotation differences between each condition of imaging, we aligned each imaging session with several reference neurons, yielding the locations of spatial footprints from different days recording in a single coordinate system. The cells generally maintained their spatial footprints over long time periods. Movements were corrected with the established motion correction pipeline NormCorre [46]. The frames are first smoothed with a 12*12-pixel Gaussian kernel (std=4) to suppress the background noise, then non-rigid translation is applied to every two successive frames by dividing each frame of multiple 128*128 pixel patches, then the software estimates the motion between corresponding patches and translates them for alignment. After motion correction, neuron footprints (i.e., weighted regions of interest consisting of each pixel’s contribution to the cell’s fluorescence) and their corresponding temporal calcium signal traces were extracted from the recordings using the CNMF-based CalmAn pipeline [66, 69]. The pipeline models the calcium signal in the following:
Where Y is the observed calcium recording, A is the matrix containing the “spatial footprint” of each neuron soma; C is the matrix containing each neuron’s calcium dynamic, B is the background, and E is the Gaussian noise matrix that compensates the estimation errors. A, C, B, E are first initialized with the greedy ROI method described in [46]. Then the CNMF iteratively applies this to update and refine the spatial footprint and calcium dynamics. The neuropil signal is contained in B and is discarded after the processing. The peak amplitudes of calcium events were measured by subtraction of baseline from the peak values (ΔF), and converted to ΔF/F in the following steps: (1) For each neuron i, the baseline fluorescence signal, F0 is estimated by Yi-Ai*Ci, where its extracted temporal fluorescence trace is Ci, its spatial footprint is Ai, and the original calcium signal trace in the footprint is Yi. F0 is a matrix that includes the baseline fluorescence signal of all pixels in the footprint Ai. (2) The average F0 is calculated; F0_avg=mean(F0) (3) The normalized ΔF /F equals (Ci/F0_avg). The calcium events were detected by applying sparse non-negative deconvolution to the extracted ΔF/F signals [46], and for each calcium event, its peak amplitude was assigned as the closet peak amplitude of the ΔF/F signals. For each neuron, the calcium signal event threshold was set to 10% of the maximum event amplitude across the recording duration. Calcium events that exceed the threshold were included for further analysis. The calcium event frequency was calculated by normalizing the total number of detected events with the session length (15 minutes each). After locating all the events from each neuron with their amplitudes that exceed the threshold, we calculated the average event peak amplitude of each neuron. For further data analysis, the average calcium event rates and average event amplitudes are only included when animals were not running. The average calcium event rate, and average event amplitude were measured for 15 minutes while animals viewed a gray screen.
GCaMP6-based Epi-fluorescence Retinotopic Mapping:
Prior to two-photon calcium imaging of binocular visual cortical activities, retinotopic maps were acquired by epi-fluorescence imaging in 4X/0.16 objective. The surface vasculature and GCaMP6s signal were visualized from White Mounted LEDs (400 – 700 nm) (MCWHLP1, Thorlabs), via a bandpass filter (482/18nm, Thorlabs) and fluorescence was detected by a sCMOS camera (PCO.edge 4.2 LT) via a 520/28nm bandpass filter (Thorlabs). The focal plane of the microscope was at 300–400 μm below the surface vasculature. During retinotopic mapping, mice were awake and the head was restrained via the implanted headplate and eyes were on a horizontal plane. Mice were well-trained and habituated to the spherical treadmill 3 days before the recording. Furthermore, in off-line analyses, recorded data were first motion-corrected, before additional retinotopy map analysis is performed. In our experiments, GCaMP6-based epi-fluorescence signal is stronger than intrinsic signal. Furthermore, retinotopic maps are widefield, and are largely unaffected by small eye movements. Eye movements occur during all phases of the mapping stimulus and mean eye displacement is less than 1 degree [70]. We used the same visual stimulation as Zhuang et. al. did; they found that visual field maps are not affected by small eye movements. Furthermore, eye movement-related effects are largely eliminated during map data processing by averaging 10 repetitions of visual stimulation [70].
Visual stimuli were generated in real-time with Processing using OpenGL shaders. The stimulation was displayed on a 27’’ LCD Monitor (Asus vg 278) refreshed at 60 Hz, placed 20 cm perpendicular from the mouse midline. The monitor subtending approximately −56 to 56 degrees in azimuth and −40 to 40 degrees in altitude. A TTL pulse was generated with an Arduino at each stimulus transition and the pulse was recorded by the microscope and time-stamped which used for synchronizing between visual stimulation and imaging data.
Retinotopic maps were generated by sweeping a bar across the monitor (Kalatsky and Stryker, 2003). The bar contained black and white checkerboard pattern on it, on a grey background with 50% of the maximum luminance, with checker size 4 degree. It was swept across the screen 8 times in each of four cardinal directions with a period of 10s. There is a 10s interval of blank gray screen was inserted between sweeps. Azimuth and elevation retinotopic maps were used to generate a visual field sign maps (Garrett et al., 2014). Binocular V1 was confined to regions adjacent to the intersection of the horizontal and vertical meridians at the border of V1 and LM. In vivo two-photon imaging was in the binocular area of V1 identified by retinotopic mapping.
Quantification and Statistical Analysis
Data analysis was conducted using Matlab and R. Because the data presented in this article were collected using different experimental designs, several appropriate statistical tests were applied in the data analysis. This included Kruskal-Wallis test, Friedman’s test, Mann–Whitney U test, Wilcoxon signed rank test, linear mixed-effect model, and Bonferroni correction for multiple comparisons.
Linear Mixed-Effect Model (LME):
LME has been widely used to analyze correlated data such as longitudinal data (also known as panel data) and clustered data [71–74]. The main idea of LME (“fitlme” in Matlab) is to take the inherent correlations in correlated data, such as the neurons from the same mouse, into consideration when conducting statistical modeling and hypothesis testing. The LME test includes paired t-test and repeated measures ANOVA as two special cases. In addition, compared with a paired t-test or repeated measures ANOVA, LME can handle unbalanced designs and missing values, and has greater statistical power in the presence of missing values. The importance of LME and its more generalized versions has been increasingly recognized in recent studies involving large cell sample data collected from a relatively small number of animals [18, 75–77]. In this article, we used LME for several figures. For example, in Figure 3J, presented are observations of normalized pCREB staining intensity based on 1,200 measures from 24 mice. We fitted an LME by using time for a fixed effect and mouse id for a random effect. The overall p for the time effect is 0.0001. Then two significant comparisons were revealed: p = 0.0015 for 24h and p = 0.0084 for 48h. For data comparisons in Figure 4 F and G, we used LME tests to simultaneously determine the effects of treatment and time.
Normality and Nonparametric Tests:
For statistical comparisons across and between groups, data were checked for normality distribution. For statistics used for slice experiments (e.g., Figures 2 and 5), we preferred to use Friedman’s test (the “muStat” R package), the nonparametric counterpart of One-way ANOVA with repeated measures, or Kruskal-Wallis test, the nonparametric counterpart of One-way ANOVA for ≥ 2 group comparisons. We used Wilcoxon signed rank test (the nonparametric version of paired t test) or Mann-Whitney U test (the nonparametric version of two-sample t test) for between group comparisons (with adjustment for multiple comparisons as needed). This is in line with routine guidelines for a relatively small sample size, and does not require assumptions of normality or equal variance required for parametric tests.
Multiple Comparisons:
The data comparisons in many figures involve multiple comparisons. If left unadjusted, type I error rates would be inflated. We used the Bonferroni correction, which is a simple and stringent method to handle multiple comparisons. P values reported in the manuscript have been adjusted by using Bonferroni’s correction.
Supplementary Material
Acknowledgements
This work was supported by US National Institutes of Health (NIH) grants R01EY028212, R01EY027407, R01 MH105427, R35GM127102 and R01EY029490.
Footnotes
Declaration of Interests
All authors disclose no conflict of interests for this work.
References
- 1.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, and Krystal JH (2000). Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47, 351–354. [DOI] [PubMed] [Google Scholar]
- 2.Duman RS, and Aghajanian GK (2012). Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, and Manji HK (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63, 856–864. [DOI] [PubMed] [Google Scholar]
- 4.Price RB, Nock MK, Charney DS, and Mathew SJ (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry 66, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, and Monteggia LM (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, and Dugan LL (2007). Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318, 1645–1647. [DOI] [PubMed] [Google Scholar]
- 7.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, and Duman RS (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, et al. (2016). NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Browne CA, and Lucki I (2013). Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front Pharmacol 4, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krystal JH, Sanacora G, and Duman RS (2013). Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73, 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Homayoun H, and Moghaddam B (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27, 11496–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller OH, Moran JT, and Hall BJ (2016). Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: Direct inhibition and disinhibition. Neuropharmacology 100, 17–26. [DOI] [PubMed] [Google Scholar]
- 13.Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, and Baudry M (2009). Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci 29, 8688–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepack AE, Fuchikami M, Dwyer JM, Banasr M, and Duman RS (2014). BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X, Olivas ND, Levi R, Ikrar T, and Nenadic Z (2010). High precision and fast functional mapping of cortical circuitry through a novel combination of voltage sensitive dye imaging and laser scanning photostimulation. J Neurophysiol 103, 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, and Nawa H (2004). Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci 24, 9760–9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, and Yang JJ (2014). Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry 29, 419–423. [DOI] [PubMed] [Google Scholar]
- 18.Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, Witztum J, Shaver DC, Rosenthal DL, Alway EJ, et al. (2019). Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beurel E, Song L, and Jope RS (2011). Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry 16, 1068–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beurel E, Grieco SF, Amadei C, Downey K, and Jope RS (2016). Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord 18, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y, Ikrar T, Davis MF, Gong N, Zheng X, Luo ZD, Lai C, Mei L, Holmes TC, Gandhi SP, et al. (2016). Neuregulin-1/ErbB4 Signaling Regulates Visual Cortical Plasticity. Neuron 92, 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu Y, Tran T, Murase S, Borrell A, Kirkwood A, and Quinlan EM (2016). Neuregulin-Dependent Regulation of Fast-Spiking Interneuron Excitability Controls the Timing of the Critical Period. J Neurosci 36, 10285–10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grieco SF, Holmes TC, and Xu X (2019). Neuregulin directed molecular mechanisms of visual cortical plasticity. J Comp Neurol 527, 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, and Trachtenberg JT (2013). A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, and Rico B (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380. [DOI] [PubMed] [Google Scholar]
- 26.Yang JM, Zhang J, Chen XJ, Geng HY, Ye M, Spitzer NC, Luo JH, Duan SM, and Li XM (2013). Development of GABA circuitry of fast-spiking basket interneurons in the medial prefrontal cortex of erbb4-mutant mice. J Neurosci 33, 19724–19733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grieco SF, Wang G, Mahapatra A, Lai C, Holmes TC, and Xu X (2019). Neuregulin and ErbB expression is regulated by development and sensory experience in mouse visual cortex. J Comp Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei L, and Xiong WC (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci 9, 437–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon JA, and Stryker MP (1996). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci 16, 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis MF, Figueroa Velez DX, Guevarra RP, Yang MC, Habeeb M, Carathedathu MC, and Gandhi SP (2015). Inhibitory Neuron Transplantation into Adult Visual Cortex Creates a New Critical Period that Rescues Impaired Vision. Neuron 86, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalatsky VA, and Stryker MP (2003). New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron 38, 529–545. [DOI] [PubMed] [Google Scholar]
- 32.Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, and Gandhi SP (2010). Cortical plasticity induced by inhibitory neuron transplantation. Science 327, 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long W, Wagner KU, Lloyd KC, Binart N, Shillingford JM, Hennighausen L, and Jones FE (2003). Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development 130, 5257–5268. [DOI] [PubMed] [Google Scholar]
- 34.Prusky GT, West PW, and Douglas RM (2000). Behavioral assessment of visual acuity in mice and rats. Vision Res 40, 2201–2209. [DOI] [PubMed] [Google Scholar]
- 35.Stephany CE, Ma X, Dorton HM, Wu J, Solomon AM, Frantz MG, Qiu S, and McGee AW (2018). Distinct Circuits for Recovery of Eye Dominance and Acuity in Murine Amblyopia. Curr Biol 28, 1914–1923 e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaepel J, Hubener M, Bonhoeffer T, and Rose T (2017). Lateral geniculate neurons projecting to primary visual cortex show ocular dominance plasticity in adult mice. Nat Neurosci 20, 1708–1714. [DOI] [PubMed] [Google Scholar]
- 37.Sommeijer JP, Ahmadlou M, Saiepour MH, Seignette K, Min R, Heimel JA, and Levelt CN (2017). Thalamic inhibition regulates critical-period plasticity in visual cortex and thalamus. Nat Neurosci 20, 1715–1721. [DOI] [PubMed] [Google Scholar]
- 38.Hooks BM, and Chen C (2020). Circuitry Underlying Experience-Dependent Plasticity in the Mouse Visual System. Neuron 106, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hensch TK, and Quinlan EM (2018). Critical periods in amblyopia. Vis Neurosci 35, E014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hensch TK (2005). Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6, 877–888. [DOI] [PubMed] [Google Scholar]
- 41.Zhou P, Zhang Y, Ma Q, Gu F, Day DS, He A, Zhou B, Li J, Stevens SM, Romo D, et al. (2013). Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci U S A 110, 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen S, and Greenberg ME (2008). Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol 24, 183–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X, Olivas ND, Ikrar T, Peng T, Holmes TC, Nie Q, and Shi Y (2016). Primary visual cortex shows laminar-specific and balanced circuit organization of excitatory and inhibitory synaptic connectivity. J Physiol 594, 1891–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, Lovett J, Kim S, Wang FH, Schmidt S, et al. (2019). Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci U S A 116, 5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukumoto K, Fogaca MV, Liu RJ, Duman C, Kato T, Li XY, and Duman RS (2019). Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A 116, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pnevmatikakis EA, and Giovannucci A (2017). NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J Neurosci Methods 291, 83–94. [DOI] [PubMed] [Google Scholar]
- 47.Wekselblatt JB, Flister ED, Piscopo DM, and Niell CM (2016). Large-scale imaging of cortical dynamics during sensory perception and behavior. J Neurophysiol 115, 2852–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaplan ES, Cooke SF, Komorowski RW, Chubykin AA, Thomazeau A, Khibnik LA, Gavornik JP, and Bear MF (2016). Contrasting roles for parvalbumin-expressing inhibitory neurons in two forms of adult visual cortical plasticity. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480 e422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, and Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat Methods 8, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Y, Jin S, Lin X, Chen L, Qiao X, Jiang L, Zhou P, Johnston KG, Golshani P, Nie Q, et al. (2019). CA1-projecting subiculum neurons facilitate object-place learning. Nat Neurosci 22, 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cai DJ, Aharoni D, Shuman T, Shobe J, Biane J, Song W, Wei B, Veshkini M, La-Vu M, Lou J, et al. (2016). A shared neural ensemble links distinct contextual memories encoded close in time. Nature 534, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O’Leary OF, Castren E, and Maffei L (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 320, 385–388. [DOI] [PubMed] [Google Scholar]
- 54.Duman RS, Li N, Liu RJ, Duric V, and Aghajanian G (2012). Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 62, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beurel E, Grieco SF, and Jope RS (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kepecs A, and Fishell G (2014). Interneuron cell types are fit to function. Nature 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rauschecker JP, and Hahn S (1987). Ketamine-xylazine anaesthesia blocks consolidation of ocular dominance changes in kitten visual cortex. Nature 326, 183–185. [DOI] [PubMed] [Google Scholar]
- 58.Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 59.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abe Y, Namba H, Kato T, Iwakura Y, and Nawa H (2011). Neuregulin-1 signals from the periphery regulate AMPA receptor sensitivity and expression in GABAergic interneurons in developing neocortex. J Neurosci 31, 5699–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu X, Olivas ND, Levi R, Ikrar T, and Nenadic Z (2010). High precision and fast functional mapping of cortical circuitry through a combination of voltage sensitive dye imaging and laser scanning photostimulation. J Neurophysiol 103, 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Y, Nenadic Z, and Xu X (2010). Novel use of matched filtering for synaptic event detection and extraction. PLoS One 5, e15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Nguyen AQ, Nguyen JP, Le L, Saur D, Choi J, Callaway EM, and Xu X (2014). Cell-type-specific circuit connectivity of hippocampal CA1 revealed through Cre-dependent rabies tracing. Cell Rep 7, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antonini A, Fagiolini M, and Stryker MP (1999). Anatomical correlates of functional plasticity in mouse visual cortex. J Neurosci 19, 4388–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shepherd GM, Pologruto TA, and Svoboda K (2003). Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron 38, 277–289. [DOI] [PubMed] [Google Scholar]
- 66.Zhou P, Resendez SL, Rodriguez-Romaguera J, Jimenez JC, Neufeld SQ, Giovannucci A, Friedrich J, Pnevmatikakis EA, Stuber GD, Hen R, et al. (2018). Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, Reardon T, Mu Y, Lacefield C, Yang W, et al. (2016). Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 89, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLoS Comput Biol 13, e1005423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giovannucci A, Friedrich J, Gunn P, Kalfon J, Brown BL, Koay SA, Taxidis J, Najafi F, Gauthier JL, Zhou P, et al. (2019). CaImAn an open source tool for scalable calcium imaging data analysis. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhuang J, Ng L, Williams D, Valley M, Li Y, Garrett M, and Waters J (2017). An extended retinotopic map of mouse cortex. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fisher R (1918). The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh 52, 399–433. [Google Scholar]
- 72.Laird N, Ware (1982). Random-Effects Models for Longitudinal Data. Biometrics. International Biometric Society. 38, 963–974. [PubMed] [Google Scholar]
- 73.Henderson C (1973). Sire evaluation and genetic trends. Journal of Animal Science. American Society of Animal Science., 10–41. [Google Scholar]
- 74.McLean RAS, William L; Stroup Walter W. (1991). A Unified Approach to Mixed Linear Models. The American Statistician. American Statistical Association. [Google Scholar]
- 75.Indersmitten T, Schachter MJ, Young S, Welty N, Otte S, Nassi JJ, Lovenberg T, Bonaventure P, and Wyatt RM (2019). In vivo Calcium Imaging Reveals That Cortisol Treatment Reduces the Number of Place Cells in Thy1-GCaMP6f Transgenic Mice. Front Neurosci 13, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stobart JL, Ferrari KD, Barrett MJP, Stobart MJ, Looser ZJ, Saab AS, and Weber B (2018). Long-term In Vivo Calcium Imaging of Astrocytes Reveals Distinct Cellular Compartment Responses to Sensory Stimulation. Cereb Cortex 28,184–198. [DOI] [PubMed] [Google Scholar]
- 77.Stobart JL, Ferrari KD, Barrett MJP, Gluck C, Stobart MJ, Zuend M, and Weber B (2018). Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 98, 726–735 e724. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets/code supporting the current study have not been deposited in a public repository but are available from the corresponding author upon reasonable request.