

Chikungunya virus (CHIKV) is a mosquito-borne RNA virus that causes febrile illness and debilitating arthralgia in humans. CHIKV causes explosive outbreaks but there are no approved therapies to treat or prevent CHIKV infection. The CHIKV genome contains functional RNA secondary structures that are essential for proper virus replication. Since RNA secondary structures have only been defined for a small portion of the CHIKV genome, we used a chemical probing method to define the RNA secondary structures of CHIKV genomic RNA. We identified 23 highly specific structured regions of the genome, and confirmed the functional importance of one structure using mutagenesis. Furthermore, we defined the RNA secondary structure of three CHIKV 3′UTR variants that differ in their ability to replicate in mosquito cells. Our study highlights the complexity of the CHIKV genome and describes new systems for designing compensatory mutations to test the functional relevance of viral RNA secondary structures.
KEYWORDS: RNA structure, SHAPE-MaP, chikungunya, plus-strand RNA virus
ABSTRACT
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus associated with debilitating arthralgia in humans. RNA secondary structure in the viral genome plays an important role in the lifecycle of alphaviruses; however, the specific role of RNA structure in regulating CHIKV replication is poorly understood. Our previous studies found little conservation in RNA secondary structure between alphaviruses, and this structural divergence creates unique functional structures in specific alphavirus genomes. Therefore, to understand the impact of RNA structure on CHIKV biology, we used SHAPE-MaP to inform the modeling of RNA secondary structure throughout the genome of a CHIKV isolate from the 2013 Caribbean outbreak. We then analyzed regions of the genome with high levels of structural specificity to identify potentially functional RNA secondary structures and identified 23 regions within the CHIKV genome with higher than average structural stability, including four previously identified, functionally important CHIKV RNA structures. We also analyzed the RNA flexibility and secondary structures of multiple 3′UTR variants of CHIKV that are known to affect virus replication in mosquito cells. This analysis found several novel RNA structures within these 3′UTR variants. A duplication in the 3′UTR that enhances viral replication in mosquito cells led to an overall increase in the amount of unstructured RNA in the 3′UTR. This analysis demonstrates that the CHIKV genome contains a number of unique, specific RNA secondary structures and provides a strategy for testing these secondary structures for functional importance in CHIKV replication and pathogenesis.
IMPORTANCE Chikungunya virus (CHIKV) is a mosquito-borne RNA virus that causes febrile illness and debilitating arthralgia in humans. CHIKV causes explosive outbreaks but there are no approved therapies to treat or prevent CHIKV infection. The CHIKV genome contains functional RNA secondary structures that are essential for proper virus replication. Since RNA secondary structures have only been defined for a small portion of the CHIKV genome, we used a chemical probing method to define the RNA secondary structures of CHIKV genomic RNA. We identified 23 highly specific structured regions of the genome, and confirmed the functional importance of one structure using mutagenesis. Furthermore, we defined the RNA secondary structure of three CHIKV 3′UTR variants that differ in their ability to replicate in mosquito cells. Our study highlights the complexity of the CHIKV genome and describes new systems for designing compensatory mutations to test the functional relevance of viral RNA secondary structures.
INTRODUCTION
Chikungunya virus (CHIKV) is an arthropod-borne alphavirus that causes febrile illness associated with severe acute and persistent arthralgia. Since its identification in 1952, CHIKV has caused sporadic outbreaks in Africa, Asia, and the Indian subcontinent. However, recent outbreaks in the countries surrounding the Indian Ocean in 2005, as well as the 2013 introduction of the virus into the Americas, illustrate CHIKV’s reemergence as a global threat to public health (1, 2). Despite its status as a significant emerging disease threat, there are currently no approved vaccines or virus-specific therapies for treating acute or chronic CHIKV disease. Therefore, it is important to understand the factors that contribute to CHIKV pathogenesis, since this information may inform the development of safe and effective vaccines and therapies.
The alphavirus genome is a positive sense, single-stranded RNA that encodes two polyproteins. The first polyprotein encodes the four nonstructural proteins (nsP1 to nsP4), which together comprise the RNA replication machinery. The second, an internally encoded polyprotein encompassing the 3′ third of the viral genome, encodes the virion structural proteins from a subgenomic RNA. While the role of viral proteins in the alphavirus life cycle has been extensively studied, a growing body of evidence suggests that coding and noncoding RNA structural elements (e.g., stem loops) are also critical determinants of alphavirus replication and pathogenesis. These include structures in the 5′UTR that prevent host innate immune recognition, RNA packaging signals, and RNA elements that regulate viral transcription and translation (3–7). However, the full complement of RNA secondary structures in the CHIKV genomic RNA has not been determined. Given the importance of RNA secondary structure in alphavirus biology, a better understanding of CHIKV RNA secondary structures is likely to provide new insights into the viral factors that contribute to the CHIKV life cycle and CHIKV disease pathogenesis.
We attempted to identify conserved alphavirus RNA secondary structures using Sindbis virus (SINV), Venezuelan equine encephalitis virus (VEEV), and CHIKV but found little of the RNA secondary structure landscape is conserved across these viruses (8). Retrospectively, this is not entirely surprising, since some recognized functional RNA secondary structures, such as RNA packaging signals, are found at different locations of the genomes of different alphaviruses, while other structures are only found in a subset of viruses in the genus (9, 10). Furthermore, alphaviruses have very low signals of nucleotide covariation, the traditional “gold standard” for identifying conserved, functional RNA secondary structures (8, 11–15).
Because the RNA secondary structure landscape of alphaviruses is not highly conserved (8), we used selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) to perform de novo RNA structural analysis on the CHIKV genome to identify potentially functional RNA secondary structures. SHAPE-MaP is an RNA structure probing technique that combines chemical probing of unpaired nucleotides of the genome with next generation sequencing to identify highly flexible, or unstructured, regions in long RNAs (16). The mutational profiling is combined with rigorous thermodynamic free energy modeling to generate experimentally derived, high-confidence models of RNA secondary structure (11, 16). This method has been applied to several other RNA viruses to identify important RNA structural features (8, 17–20). We hypothesized that functionally important RNA secondary structures specific to CHIKV would fold into a single, specific conformation relative to the rest of the genome. With this approach, we identified the four known functional RNA elements in the CHIKV coding sequence, as well as 19 previously unidentified elements. We confirmed the functional importance of one element through structure disrupting mutagenesis strategies. Furthermore, three variants of the CHIKV 3′UTR have been reported (21, 22), and our studies defined the RNA structure of each variant. We further characterized the impact of each of these variants on CHIKV host range. Together, these studies provide important new information on the location and stability of RNA structures distributed throughout the CHIKV genome.
RESULTS
SHAPE-MaP analysis of the CHIKV genomic RNA.
RNA structure plays an important role in alphavirus biology and contributes to functions ranging from regulation of RNA and protein synthesis to immune evasion (3–7). However, despite CHIKV’s importance as an emerging pathogen, our understanding of how viral RNA structure impacts the CHIKV life cycle has largely been inferred from analysis of the RNA secondary structure in other alphaviruses (5, 6). Extensive analysis of functional RNA secondary structures has been performed on Sindbis virus (SINV), Semliki Forest virus (SFV), and Venezuelan equine encephalitis virus (VEEV). These analyses identified specific regions within alphavirus genomes where RNA secondary structure plays important functional roles in RNA packaging, RNA and protein synthesis, and immune evasion (3, 4, 23, 24). We recently determined the full genome RNA structure of the Girdwood S.A. strain of SINV and the ZPC738 strain of VEEV using SHAPE-MaP (selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling). This analysis found that the genomes of both SINV and VEEV are highly structured beyond the 5′UTRs of both the genomic and subgenomic RNAs. This finding suggests that in addition to the previously defined functional RNA structures, additional RNA secondary structures may play a role in SINV or VEEV replication. However, the structure profiles are highly divergent between the two viruses, with little correlation between SHAPE-MaP profiles, few conserved RNA secondary structures, and low structure compatibility among conserved structures (8). We used structural conservation as a method to identify functionally important RNA structures in SINV, but this method failed to identify all the known RNA secondary structures important for SINV replication. Therefore, the field needs alternative methods to identify potentially important structured regions in alphaviruses.
We hypothesized that RNA structures likely to fold into a single specific conformation had an increased likelihood of being functionally important. Therefore, to test this idea, we set out to determine the RNA secondary structure of the genomic RNA of a human CHIKV isolate from the 2013 outbreak on the Caribbean island of St. Martinique. We treated purified CHIKV genomic RNA isolated from cell-free virions with the 1M7 SHAPE reagent, and SHAPE-MaP was performed to generate a SHAPE reactivity profile for the entire CHIKV genome (Fig. 1). SHAPE reactivity indicates the relative flexibility of a nucleotide, and nucleotide flexibility correlates with base-pairing likelihood. The SHAPE-MaP technique measures SHAPE reactivities with single nucleotide precision (25). SHAPE reactivities above 0.8 indicate likely unpaired bases (shown in red), as illustrated by an unstructured region spanning nucleotides 6350 to 6550 (Fig. 1A, left). SHAPE reactivities below 0.4 indicate likely paired and therefore unreactive bases (colored black), as illustrated by a representative structured region spanning nucleotides 10550 to 10750 (Fig. 1A, right). There are large-scale fluctuations in the SHAPE reactivity across the genome, which are best visualized as a median windowed SHAPE reactivity, as shown in Fig. 1B, where we plotted the median windowed SHAPE (called regional SHAPE). These data indicate there are specific regions in the CHIKV genome that have low median SHAPE or are more likely to form RNA secondary structures.
FIG 1.

SHAPE-MaP indicates specific RNA secondary structures are found throughout the CHIKV genome. (A, left) Example of SHAPE reactivities in ahigh-SHAPE, unstructured region. (Right) Example of SHAPE reactivities in a low-SHAPE, very structured region. Although individual reactivities do not reveal local structure, the pattern of reactivities in a region provides information about structuredness within that region. (B, top) Cartoon of the CHIKV genome. (Bottom) Regional median SHAPE reactivities across the CHIKV genome compared to the global median SHAPE. Regions below the x axis indicate more structure than average, while regions above the x axis indicate less structure than average. The histogram is colored according to the regional conservation score using representative full-genome sequences across the alphavirus genus (8, 27). Yellow indicates highly conserved regions, and purple indicates less conserved regions. HVD, hypervariable domain. (C, top) Base pairs within CHIKV genome. The color indicates base pairing probability. (Bottom) Windowed Shannon entropy across the genome. Low Shannon entropy values correspond to regions that form a single structure. (D, top) Boxes along the genome indicate highly structured regions, as determined by both computational prediction and experimental reactivities. Red boxes indicate structured regions with previously known functional importance. Black boxes indicate novel structured regions. (Bottom) SHAPE-MaP informed secondary structure models of highly structured regions. Nucleotide color corresponds to SHAPE reactivity scale in panel A.
While RNA secondary structures are not highly conserved across alphaviruses, it is unknown if overall viral RNA structured-ness correlates with sequence conservation. Figure 1B shows the regional SHAPE of CHIKV colored by the sequence conservation scores from our previous work (8). We observed very little pattern for highly conserved regions and their overall structured-ness within CHIKV. Regions with high sequence conservation scores (Fig. 1B, in yellow) are found with low- and above-average SHAPE reactivity values. Likewise, the least conserved regions of the genome (dark purple and blue) also fluctuate between very low and very high SHAPE reactivity values. For example, the 3′UTR of CHIKV, which differs within CHIKV strains, contains some of the least and most reactive nucleotides in the genome (26). Interestingly, the hypervariable domain near the end of nsP3, which is often omitted from alignments of alphavirus genomes due to drastic divergence in sequence, has very low SHAPE reactivity in CHIKV, SINV, and VEEV (8, 27). However, this shared low SHAPE reactivity does not result in similar secondary structures (8).
In order to identify the likely RNA secondary structures across the CHIKV genome, we computed base-pairing probabilities for the entire genome from the SHAPE reactivity data (Fig. 1C, top arches). From these base-pairing probabilities, we computed the Shannon entropy of base pairing for each nucleotide using a 55-nucleotide window (Fig. 1C, bottom), where low-entropy suggests a single, well-defined conformation (13, 14, 16, 28, 29). We used these base pairing probabilities and entropy values to generate SHAPE-MaP-derived structural predictions, which represent the most likely structural conformation for a specific region of the genome. We included the SHAPE-MaP derived RNA secondary structures for the entire CHIKV genome, with nucleotides colored according to SHAPE reactivity in Fig. S1 (https://drive.google.com/file/d/1ZlrFGYxsrUeRE0QcUB_rWPirpLki4C6F/view?usp=sharing) in the supplemental material.
Identification of specific RNA secondary structures within the CHIKV genomic RNA.
SHAPE-MaP analysis of the CHIKV genome indicates that RNA secondary structure is distributed throughout the viral genome (Fig. 1; see also Fig. S1). While SHAPE-MaP-derived structural predictions represent the most likely structural conformation for any region of the genome, it is likely that many regions of the genome are capable of adopting several different conformations. We hypothesized that functionally important RNA secondary structures would be both highly structured (SHAPE reactivity of <0.3) and highly specific (Shannon entropy of <0.04) compared to the rest of the genome and therefore likely to adopt a single RNA secondary structure. Low SHAPE reactivity indicates the region is often involved in base pairing interactions. While low SHAPE indicates likely structure, the RNA sequence can be involved in many different conformations to achieve this low SHAPE reactivity (11). Regions with low Shannon entropy indicate that there are few possible conformations; therefore, regions with low SHAPE reactivity and low Shannon entropy are likely to have a single well-determined secondary structure (16). By using cutoffs that were shown to yield specific structures in prior studies (8), we identified 23 regions that meet these criteria (Fig. 1D). Four of these 23 regions contain RNA structures known to be functionally important in CHIKV replication or pathogenesis: a stem-loop of the 5′ conserved sequence element (CSE), a region within the CHIKV packaging signal, a stem-loop just 3′ of the opal termination codon that is involved in opal termination codon readthrough, and an RNA structure involved in ribosome frameshifting to produce the viral TF protein (3, 5, 7, 23, 30).
5′ conserved sequence element.
Our analysis identified nucleotides 70 to 195 as being highly specific and highly structured. This region includes stem-loop 3 (SL3) of the genome, which contains the nonstructural polyprotein start codon, and SL4, the first stem-loop of the 5′CSE. The 5′CSE (nucleotides 165 to 216 in CHIKV) is one of the few structurally conserved motifs across alphaviruses and is important for proper alphavirus genome replication during infection (5, 6, 8). Figure 2A expands this structured region in CHIKV to include the entire 5′CSE. Both stem loops of the 5′CSE are predicted to be composed of 9 bp each, with the first stem-loop having seven unpaired nucleotides in the terminal loop and the second stem-loop having four unpaired nucleotides composing the terminal loop. The SINV 5′CSE second stem-loop has more than 4 unpaired nucleotides in the apical loop and only 8 bp making up the stem (5, 8, 31). The CHIKV 5′CSE secondary structure is more similar to that of VEEV, which is also predicted to have 9 bp making up each stem-loop and only four unpaired nucleotides of the second stem-loop (6, 8).
FIG 2.
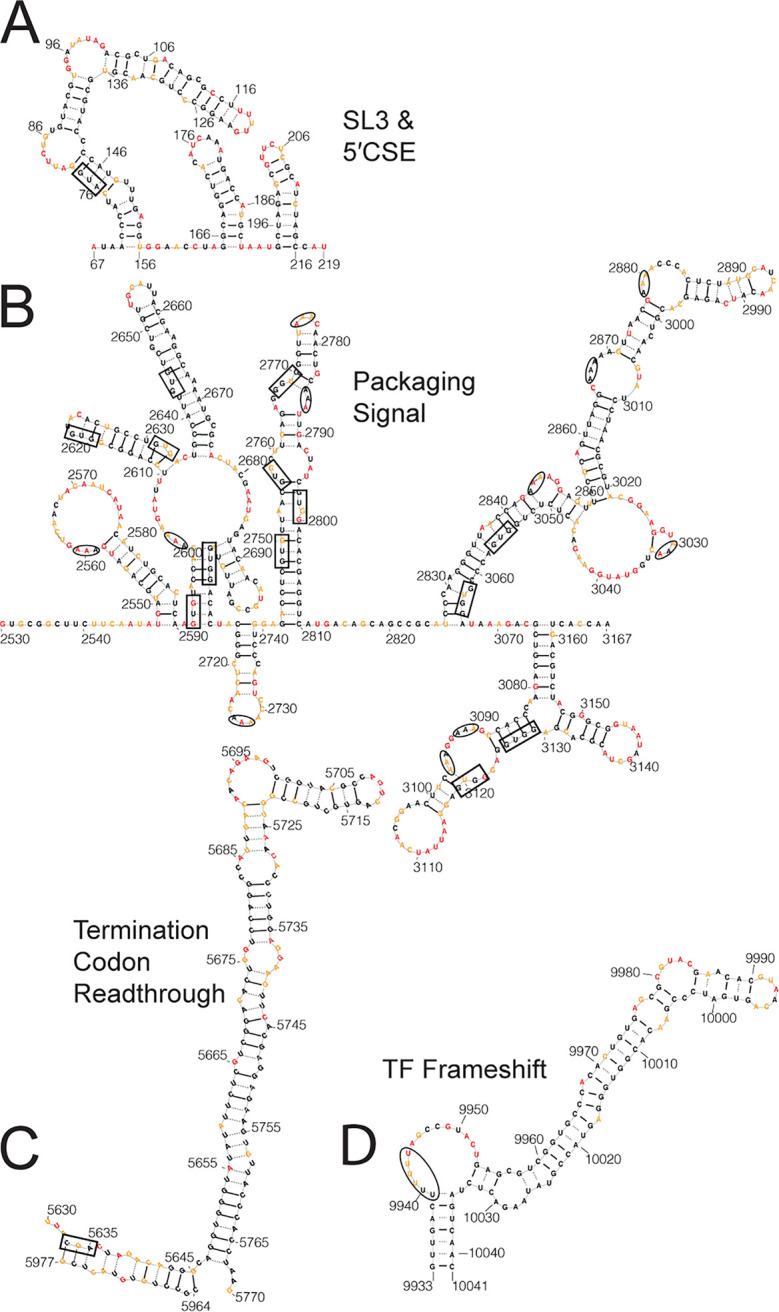
SHAPE-MaP analysis identifies previously known functional RNA secondary structures. (A) SL3 of the CHIKV genome and the 5′ conserved sequence element modeled using SHAPE-MaP data. The nonstructural polyprotein start codon is boxed. (B) Putative CHIKV packaging signal, as identified by Firth et al. (3). Triple adenosine motifs within loops and bulges are circled; the predicted GUG(G) motifs from Kim et al. are boxed (3). (C) The CHIKV TCR with the canonical opal stop codon position boxed. The CHIKV strain used had predominantly Arg at this position and is modeled as such. (D) The CHIKV TF frameshift element is plotted with the slippery U motif circled. Nucleotide color corresponds to the SHAPE reactivity key in Fig. 1A.
Packaging signal.
The putative packaging signal for CHIKV is in nsP2 (nucleotides 2501 to 3079) (3), but an RNA structure had yet to be determined for this region (Fig. 2B). Our analysis identified a portion of this region (nucleotides 2590 to 2713) as one of the 23 highly specifically structured regions. For SINV and VEEV, the packaging signal is composed of four to six stem loops with triple G motifs in the loops. The CHIKV packaging signal was predicted to have a similar multistem motif but instead of GGG, the stems would be topped with a GUG(G) motif (3). The SHAPE-MaP model indicates that the region encompassing the CHIKV packaging signal is composed of eight stem loops with the predicted GUG(G) motifs being predominantly nonreactive nucleotides contained entirely within stems (Fig. 2B, boxed). Instead we identified multiple instances of a triple A motif located in loops and bulges following a similar reactivity pattern to that of the triple G motif in SINV and VEEV (8).
Opal termination readthrough element.
The third known functional RNA secondary structure is a stem-loop found at the start of nsP4, or just 3′ of the canonical opal stop codon (Fig. 2C). We found a highly specific stem-loop at nucleotides 5672 to 5742 that is part of the termination codon readthrough (TCR) element, which increases readthrough of the opal stop codon in order to translate the full-length nonstructural polyprotein (nsP1 to nsP4) (7, 30). It should be noted that our virus contained a mixed population at the opal stop codon itself (nsP3 aa520), with the majority coding for an arginine (codon CGA) at this position and a minor population containing the canonical opal stop codon (UGA) (32). While this region was modeled with the majority Arg residue codon (520R), it should be noted that the structure of the TCR is not dependent on the codon at nsP3 aa520. Our SHAPE-MaP derived structural prediction indicates that the 520R codon is contained in a stem structure adjacent to the TCR, where the two stems are separated by a one nucleotide space (Fig. 2C). However, many of these nucleotides are moderately reactive and therefore flexible during treatment. This suggests these nucleotides likely adopt an open conformation which would create a spacer of 11 nucleotides between the 520R codon and the base of the TCR, which is consistent with the TCR model previously proposed by Firth et al. (30).
TF frameshift element.
The final known functionally important RNA secondary structure identified was the TF frameshift element (nucleotides 9933 to 10041) (Fig. 2D). This element is located in the 6K coding region and causes a −1 frameshift due to a slippery UUUUUU motif followed by a hairpin. The new reading frame encodes the TF protein. The UUUUUU element is present in other alphavirus genomes but secondary structure following this motif is not predicted to be present in all alphaviruses (7, 8, 23). The general motif predicted for this element in CHIKV was the UUUUUU motif, followed by a spacer of five to nine nucleotides, and then a structured region, which was based largely on comparison to other viruses (23). Our model, generated from data gathered from the full-length genomic RNA, indicates the UUUUUU motif is followed by nine nucleotides, the majority of which are highly reactive supporting the prediction that these nucleotides are unpaired. A long stem follows the unpaired nucleotides, which agrees with the general motif predicted for other frameshift elements (23). Furthermore, all nucleotides predicted to participate in base pairing are supported by very low reactivity scores.
Overall, comparisons between our SHAPE-MaP data and prior computational RNA structure predictions are largely consistent (7, 8, 23), though we did identify subtle, but potentially functional important differences. This illustrates the utility of combining structure probing techniques combined with high-throughput sequencing like SHAPE-MaP for both identifying and providing more refined information of RNA secondary structures in RNA virus genomes.
CHIKV SL3 enhances genome transcription.
As noted above, identification of the 5′CSE by SHAPE-MaP validated the accuracy of our approach. However, we were intrigued by the identification of SL3, the stem-loop immediately upstream of the 5′CSE that contains the initiating AUG, as a highly specific structure. Previous studies of the 5′CSE in other alphaviruses predicted this stem-loop and disrupted it when probing the functional importance of the 5′CSE itself. However, the function of SL3 alone has never been studied (5, 6). Disruption of the stem loops in the 5′CSE in combination with the upstream stem-loop results in a decrease in SINV replication in mammalian cells and severe defects in replication within mosquito cells, while analogous mutations in VEEV are lethal to the virus (5, 6). However, during serial passaging of the disrupted VEEV, Michel et al. reported that compensatory mutations were generated in the large stem-loop 5′ of the 5′CSE, analogous to SL3 of CHIKV (6), and these compensatory mutations were predicted to stabilize the large stem-loop. This suggests that SL3 is functionally important, at least in the context of a structurally disrupted 5′CSE.
To assess the impact of disruption of this region on CHIKV replication, we mutated nucleotides 67 to 216, which encompasses SL3 and the 5′CSE (∂SL3-5). We also designed two mutants that disrupted SL3 alone (∂SL3) and the 5′CSE alone (∂5′CSE) (Fig. 3A, red stars; see also Table S1 [https://drive.google.com/file/d/1ZlrFGYxsrUeRE0QcUB_rWPirpLki4C6F/view?usp=sharing]). To avoid affecting coding capacity, our mutagenesis strategy used wobble-base codon shuffling to maximally disrupt base pairing in the RNA secondary structure, while maintaining both the coding capacity and dinucleotide frequency (8). In vitro-transcribed genomic CHIKV RNA was electroporated into BHK-21 cells and successful infection was measured by the number of resulting infectious centers. Disrupting SL3 alone had no impact on infectious center production compared to the wild-type (WT) control (Fig. 3B). In contrast, the ∂5′CSE mutant produced significantly fewer infectious centers, confirming the importance of this region for alphavirus replication (5, 6). The ∂SL3-5 mutant was nonviable, yielding no infectious centers. This suggests that both SL3 and the 5′CSE are necessary for optimal CHIKV RNA infectivity.
FIG 3.

CHIKV SL3 enhances RNA transcription. (A) Secondary structure models of CHIKV SL3-5, SL3, and the 5′CSE alone, SL4-5. Starred nucleotidesindicate nucleotides mutated to disrupt RNA secondary structure and sequence using CodonShuffle (15). (B) Infectious centers assay of mutant viruses. The data represent aggregates of three independent experiments. (C and D) Mutant virus growth in mammalian Vero81 cells (C) and mosquito C6/36 cells (D). The data are means of nine biological replicates across three independent experiments. (E) SL3 genome transcription was assessed by qRT-PCR in mammalian Vero81 cells. The data shown represent one of three independent experiments, each performed with three biological replicates. (F) Viral protein synthesis was assessed by Western blotting. The blot is representative of three independent experiments. (G) Densitometry was performed for nsP3 and E2 using ImageJ software. The data are representative of two independent experiments analyzed. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The symbols in panels C and D indicate the P value for the following comparisons: *, WT versus ∂SL3; #, WT versus ∂5′CSE; and +, ∂SL3 versus ∂5′CSE.
We next tested whether ∂SL3 and ∂5′CSE were impaired for replication in mammalian and mosquito cells. Both mutants exhibited slower replication kinetics compared to WT in mammalian cells (Fig. 3C). In mosquito cells, the ∂SL3 mutant had an intermediate phenotype between that of WT and the ∂5′CSE mutant (Fig. 3E). This suggested that SL3 is required for efficient virus replication in both mosquito and mammalian cells, but that the magnitude of effect of SL3 is host dependent, while the 5′CSE is important for CHIKV replication for both host cell types.
We next defined the stage in the CHIKV replication cycle that requires SL3. Genome and protein accumulation were measured early in infection by qRT-PCR and Western blotting, respectively (Fig. 3D and F). Disrupting SL3 resulted in a delay in accumulation of genomic RNA compared to WT. However, the protein nsP3, a component of the viral replicase complex, and E2 glycoprotein accumulated to similar levels in cells infected with the WT or mutant viruses. Densitometry analysis indicated that viral proteins were slightly more abundant in WT infected cells than in ∂SL3 infected cells by 12 h postinfection (Fig. 3G). This is likely due to a greater abundance of WT RNA present at 8 and 12 h postinfection and not due to impaired translation of the ∂SL3 RNA since there are similar levels of viral protein accumulation at 8 h postinfection. This suggests that SL3 functions to enhance genomic RNA replication and is not necessary for proper viral protein synthesis in mammalian cells.
While our mutations were designed to disrupt RNA secondary structure in SL3 and the 5′CSE, this method also disrupts sequence. In order to test whether ∂SL3 and ∂5′CSE mutants were attenuated due to structure disruption or sequence disruption, we generated three additional mutants. Using the same wobble-base codon shuffle algorithm, we chose sequences that maintained the secondary structure of the region but used a different sequence to maintain coding capacity. The CodonShuffle program predicts a minimum free energy structure for each codon shuffled sequence generated (15). We mutated nearly all available nucleotides possible that would also maintain the predicted secondary structure. These “fixed” structure mutants (scrSL3, scr5′CSE, and scrSL3-5) differ in sequence from WT but are predicted to be structurally the same (Fig. 4A; see also Table S1). When these fixed structure mutants are assessed for infectivity as before, all mutants produce the same number of infectious centers as WT RNA (Fig. 4B). These data indicate that structure within SL3 plays an important role in promoting efficient CHIKV replication in combination with the 5′CSE stem loops.
FIG 4.
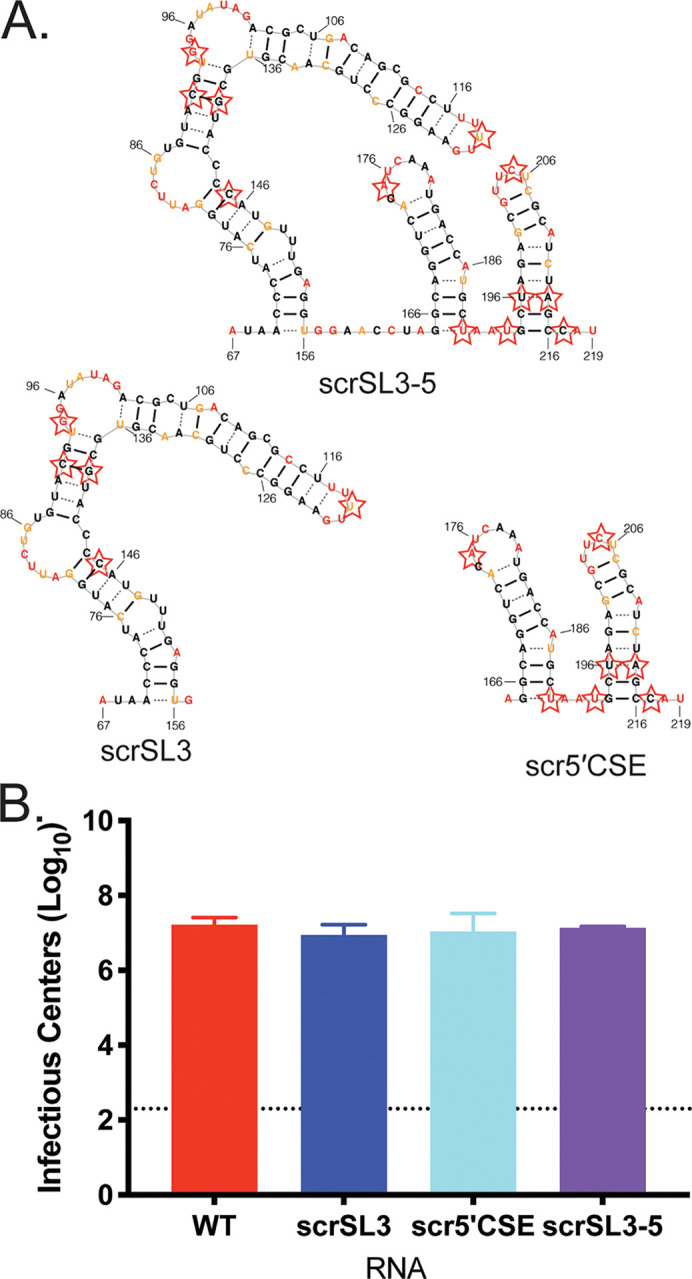
Preservation of RNA secondary structure when primary sequence is disrupted complements full structure disruption phenotypes. (A) Secondary structure models of CHIKV SL3-5, SL3, and the 5′CSE alone, SL4-5. Starred nucleotides indicate nucleotides mutated to disrupt primary sequence and maintain RNA secondary structure using CodonShuffle (15). (B) Infectious centers assay of mutant viruses. The data are aggregated from three independent experiments.
Identification of 3′UTR variants in CHIKV.
SHAPE-MaP analysis requires high-throughput sequencing of RNA treated with a SHAPE chemical probe to detect SHAPE adduct induced mutations. These sequencing results are compared to an untreated control, therefore providing deep sequencing results of the viral genomic RNA. The sequencing results for the negative-control portion of our SHAPE-MaP analysis found that the 3′UTR of the CHIKV isolate used in our study was 738 nucleotides in length and had repeat element nucleotide sequences and pattern consistent with those found in Asian CHIKV strains (26, 33). However, we also noted the read depth increased in the (1 + 2) repeat element regions of the 3′UTR (Fig. 5A), similar to a previously described duplication in this region within Caribbean CHIKV isolates (21, 22). To further define the 3′UTR of our isolate, we performed 3′RACE on RNA isolated from the virus stock. This analysis revealed three distinct isoforms of the viral 3′UTR (Fig. 5B): (1) the 738-nucleotide canonical 3′UTR (Fig. 5A, top), (2) a 912-nucleotide variant with a partial 3′UTR duplication (Fig. 5A, middle, solid red box) which has been previously identified (21, 22), and (3) a novel 583-nucleotide variant containing a 152-nucleotide deletion that removes the 3′ end of the first copy of the (1+2) repeat element and the majority of the second (1+2) repeat element (Fig. 5A, bottom, dotted red box and line).
FIG 5.

St. Martinique CHIKV isolate contained three 3′UTR variants. (A) Cartoon representations of the CHIKV 3′UTR. Colored boxes indicate unique repeat elements (RE) that are labeled within the diagrams. (Top) Duplication variant 3′UTR. (Middle) Canonical Asian genotype 3′UTR. (Middle bottom) Deletion variant 3′UTR and nucleotide length ruler. Solid red boxes indicate the location of the duplicated sequence. The additional sequence is both boxed and shaded in the duplication 3′UTR cartoon. A dashed red box indicates the sequence that was deleted in reference to the canonical 3′UTR, and the dashed red line indicates where this sequence would be in the deletion 3′UTR. (B) 3′RACE products were subcloned into blunt vectors for sequencing and clarification of 3′UTR sizes. Clones containing one of each 3′UTR were digested and separated by gel electrophoresis.
We constructed three CHIKV infectious clones, each containing one of the three 3′UTR variants. Since the 3′UTR duplication has been shown to enhance CHIKV replication in mosquito cells and deletion mutants were observed to be attenuated, we initially tested the three viruses for their ability to replicate in C6/36 mosquito cells (21, 22, 34, 35). We observed three distinct phenotypes from the three 3′UTRs (Fig. 6A). The 3′UTR duplication clone (3′dup) exhibited faster kinetics and achieved an overall higher peak titer than the canonical (3′canon) or deletion (3′del) 3′UTR variant. The 3′del virus was severely attenuated for growth in mosquito cells, achieving a peak titer 100- to 1,000-fold lower than the other 3′UTR variants. However, all three viruses exhibited similar growth kinetics in Vero81 mammalian cells (Fig. 6B). These data show that duplications in the CHIKV 3′UTR are beneficial for virus replication in mosquito cells and are unimportant for virus replication in mammalian cells.
FIG 6.

Variation in CHIKV 3′UTR impacts virus replication in mosquito cells but not the vertebrate host. Growth curves of 3′UTR variant infectious clones in mosquito C6/36 cells (A) and mammalian Vero81 cells (B) are shown. The data are aggregated from three independent experiments with nine total biological replicates. (C) Inoculated footpad swelling of C57BL/6J mice after infection with 3′UTR variants. Data for days 0 to 7: n = 30 for 3′canon, n = 30 for 3′dup, and n = 31 for 3′del. Data for days 8 to 14: n = 16 for 3′canon, n = 15 for 3′dup, and n = 16 for 3′del. The data are aggregated from four independent experiments. Male and female mice were used. (D) Infectious virus load of C57BL/6J mice at 3 days postinfection. Open symbols indicate samples with undetectable virus and are plotted at the limit of detection for that tissue (dictated by tissue weight). Dashed lines indicate limit of detection for liquid samples. n = 13 for 3′canon, n = 14 for 3′dup, and n = 14 for 3′del. Male and female mice were used. Symbols indicate the following comparisons: *, 3′ canonical versus 3′ duplication; #, 3′ canonical versus 3′ deletion; and +, 3′ duplication versus 3′ deletion. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
To determine how 3′UTR variation impacts virus replication and CHIKV-induced pathology in vivo, we used the C57BL/6J mouse model of CHIKV pathogenesis. Previous studies of CHIKV 3′UTR variation and its impact on replication in vivo have used infant mice and artificial 3′UTR constructs (26). The impact of naturally occurring 3′UTR variants on CHIKV-induced disease has not yet been assessed. We infected 6-week-old C57BL/6J mice with 100 PFU of virus in the left hind footpad and monitored CHIKV-induced footpad swelling using our established methods (32). As shown in Fig. 6C, each of the three variants induced a similar degree of swelling in the footpad. We also found no differences in viral replication between the three viruses at 3 days postinfection in the left foot (inoculation site), right foot, sera, or spleen (Fig. 7D). Therefore, we did not detect a role for the 3′UTR variation in modulating CHIKV replication or disease in a mouse model of CHIKV disease. Rather, consistent with prior results, our studies suggest that variants in this region of the CHIKV 3′UTR are primarily affecting viral fitness in mosquito cells (21, 26, 34, 35).
FIG 7.
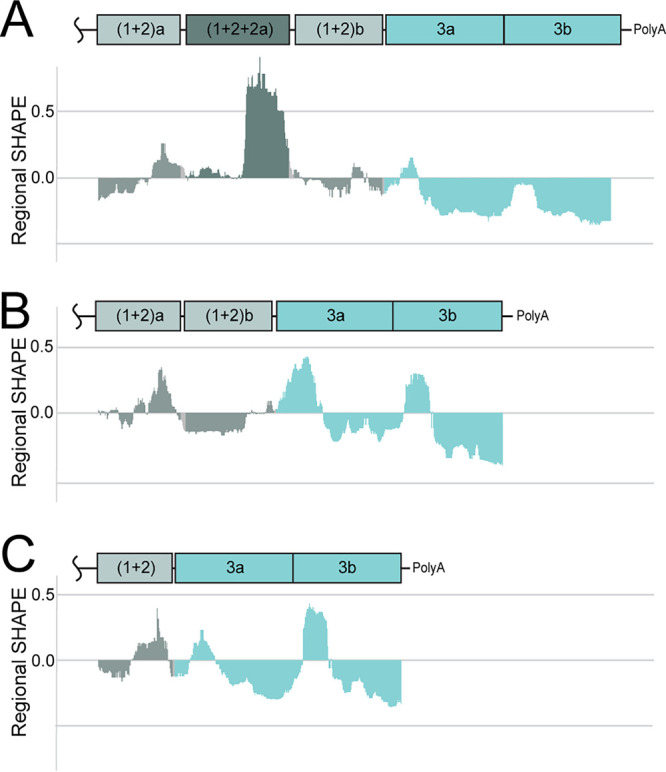
3′UTR variants are sequence similar but distinct in reactivity. Median SHAPE reactivity profiles for the duplication (A), canonical (B), and deletion (C) 3′UTRs are shown beneath cartoons depicting the repeat regions of the 3′UTR. Histograms are colored according to repeat element. Gray, (1+2) repeat elements; dark gray, hybrid (1+2) repeat element; light turquoise, repeat element 3a and 3b.
RNA structure differences between CHIKV 3′UTR variants.
The differences in sequence of the 3′UTR variants suggested that each 3′UTR might assume a different RNA secondary structure. The 3′UTR variants primarily differ in sequence at the start of the 3′UTR where the duplication and deletion occurred, creating one, two, or two and a hybrid copy of a repeat element. The 3′ ends of the 3′UTR variants are identical in sequence and repeat composition. Therefore, we hypothesized that if the RNA structure of the three 3′UTRs were to differ, it would be at the start of the 3′UTR, while the 3′ end of the UTR would have a similar SHAPE reactivity pattern and RNA secondary structure model.
The duplication, deletion, and canonical 3′UTRs were in vitro transcribed from the respective infectious clone. SHAPE-MaP was performed on the in vitro transcribed 3′UTR RNA using the same conditions applied to the full-length RNA genome. Figure 7 shows the median SHAPE reactivity profiles for each 3′UTR. The color of the reactivity profiles corresponds to the repeat elements in the 3′UTR. The SHAPE reactivity profile of the duplication variant contains highly reactive nucleotides that correspond to the hybrid sequence repeat element unique to the duplication 3′UTR (Fig. 7A, dark gray). These nucleotides reach much higher reactivity levels than any of the nucleotides in the canonical or deletion 3′UTR (Fig. 7B and C, respectively). This suggests that the additional sequence in the duplication 3′UTR is less structured than any portion of the canonical or deletion 3′UTRs.
This highly reactive extra sequence supported our hypothesis that RNA secondary structure at the start of the 3′UTR would be different among the three variants, but the end of each 3′UTR would be similar. We modeled the RNA secondary structure for each 3′UTR based on SHAPE data and found all three 3′UTRs share a secondary structure at the end of the 3′UTR (see Fig. S1, end), while the major differences in secondary structure occur at the start of the 3′UTR (Fig. 8). Each 3′UTR begins with the same hairpin, but the structures that follow are slightly different. The duplication 3′UTR is predicted to be largely single stranded beyond some shared secondary structure at the start and a few other hairpins. The start of the deletion 3′UTR also lacks significant structure compared to the canonical and duplication 3′UTR. Interestingly, we noticed a two-hairpin motif that occurs once in the canonical 3′UTR and twice in the duplication 3′UTR and is absent in the deletion 3′UTR (Fig. 8, boxes). Of note, the duplication virus has the most copies of this structure motif and replicates faster and to higher titers in mosquito cells, while the deletion virus lacks this structure motif and replicates slower and to lower titers (Fig. 6A and 8).
FIG 8.
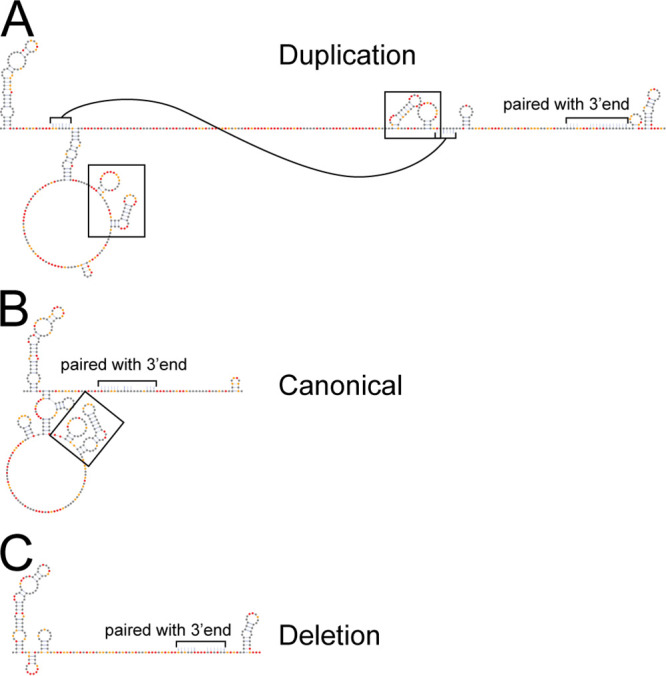
Variation in 3′UTR reactivity corresponds to distinct models of secondary structure. SHAPE informed secondary structure models of the 5′ ends of the (A) duplication, (B) canonical, and (C) deletion 3′UTRs are shown. Secondary structure models are for the (1+2) repeat element region for each 3′UTR where the duplication and deletion events occurred. Nucleotides that pair with sequence not shown have been indicated by brackets and a note. In the duplication 3′UTR one stem-loop is indicated with connected brackets for clarity. Black boxes indicate two stem-loop motif that was duplicated. Nucleotides are colored according to SHAPE reactivity key in Fig. 1A.
DISCUSSION
RNA secondary structure plays a major role in multiple aspects of RNA virus biology. Given the importance of CHIKV as a re-emerging pathogen, we generated a whole-genome RNA secondary structure model for a human isolate of CHIKV to identify potentially functional novel RNA secondary structures. Our single-nucleotide resolution model both confirmed and refined past structural analyses of CHIKV RNA motifs and identified novel RNA structures in the CHIKV genome. We found RNA secondary structures are distributed throughout the genome and identified 23 regions across the genome that are predicted to adopt a single RNA conformation due to their high structural stability. These 23 regions include four previously identified functional RNA elements (5′CSE, packaging signal, TCR, and the TF slippery site) (3, 5–7, 23). This suggests that high structural stability may provide some predictive value for identifying additional functionally relevant RNA structures in alphavirus genomes. We also demonstrate that a stem-loop adjacent to the 5′CSE was functionally important in combination with the 5′CSE. In the process of performing SHAPE-MaP analysis of the CHIKV genome, we analyzed several previously identified variants of the viral 3′UTR. SHAPE-MaP analysis of these 3′UTR variants indicates a duplication in the 3′UTR results in more unstructured RNA at the start of the 3′UTR as well as the duplication of a two-stem-loop motif. Using infectious clones, we showed that the variation in the CHIKV 3′UTR had host-specific effects. The duplication 3′UTR enhances virus replication and the deletion 3′UTR inhibits virus replication in mosquito cells. However, variation in the CHIKV 3′UTR had no effect on virus replication or swelling in our mouse model of pathogenesis.
Validation and discovery of highly specific structures.
Several RNA secondary structures were previously shown to be functionally important for CHIKV replication. These structures provided us an opportunity to both validate our SHAPE-MaP results and to test our new method for identifying important RNA secondary structures for CHIKV replication and pathogenesis. The 5′CSE is the most conserved RNA secondary structure element across the alphavirus genus. It can be found by sequence conservation or structure conservation analysis and unsurprisingly is one of the most specifically structured regions in CHIKV (Fig. 2A) (8). Most prior analyses of this region focused on SINV and VEEV showing variable necessity depending on the virus (5, 6). It is interesting that our analysis also identified the large stem-loop 5′ of the 5′CSE, SL3, as being highly specific and structured. SL3, and its homolog across alphaviruses, contains the start codon for the nonstructural polyprotein. Early structure disruption studies of the 5′CSE included the SL3 homolog in their disruption strategies, but the role of this specific stem-loop had not been studied (5, 6).
Recently, Kendall et al. released a SHAPE-informed RNA secondary structure model for the CHIKV 5′end of the genome (36). Our model of the 5′CSE and the base of SL3 agree with the Kendall model. Our models diverge after the base of SL3. The top of the Kendall et al. model diverges into a Y-shaped structure, while the model we propose continues from the base as a single stem with bulges. The 5′ arm of the “Y” has a number of reactive nucleotides placed in a stem with many unreactive nucleotides placed in a single-stranded loop. These reactivity data are somewhat contradictory to the model. They propose the shorter arm of the “Y” may actually form a pseudoknot with the start codon and surrounding nucleotides to explain this discrepancy, but attempts to confirm this structure were inconclusive (36). While we cannot rule out a pseudoknot, the Kendall et al. reactivity data corresponds well with our reactivity data and would support our model of a single long stem-loop. Regardless of this difference in modeling, our functional analyses and the Kendall et al. functional analyses of this stem-loop demonstrate the importance of SL3 in regulating CHIKV replication (Fig. 3). Our data and Kendall et al. data support that RNA secondary structure is necessary for SL3 and 5′CSE enhancement of virus replication (Fig. 4). Therefore, these data refine and support recent advancements in understanding the role RNA structure plays in the CHIKV life cycle. Furthermore, our identification of SL3 as one of the 23 most specifically structured regions of the genome, along with the previously known functional RNA elements, suggests the remaining 19 specifically structured regions may play functional roles as well.
Our analysis also identified a region of the putative packaging signal in CHIKV and provides an RNA secondary structure model for this region (3). The packaging signal of CHIKV, and likely very closely related alphaviruses, is located in the coding region of nsP2, while SINV and other New World alphaviruses have a packaging signal further upstream in nsP1 (3). Our data suggest the CHIKV packaging signal has multiple stem loops in close proximity and is more specifically structured compared to the rest of the genome, which is consistent with the packaging signal motifs identified for SINV and VEEV. However, it does not have the triple G motif reported in other alphaviruses or the hypothesized GUG(G) motif located in the loops (3). Instead, our secondary structure model of the region contains a triple A motif in five of the loops with the same reactivity pattern observed for the triple G motif in SINV (Fig. 2B, circled) (8). The lack of a triple G motif in the loops of the CHIKV packaging signal, combined with the data that CHIKV capsid can recognize and use the CHIKV, VEEV, and SINV packaging signals suggest that CHIKV capsid is capable of recognizing a triple pyrimidine motif in order to package genomic RNA (3).
The third known structure our analysis identified was the top of the TCR, a large stem-loop 3′ of the nsP3 coding region, canonically following an opal stop codon. This RNA secondary structure was predicted to be conserved across alphaviruses and can be found in other viruses and organisms in general that contain an opal stop codon (30). The CHIKV TCR was modeled recently by Kendra et al. from SHAPE data generated from in vitro transcribed RNA segments (7). This model differed slightly from past predictions that used sequence analysis of closely related alphaviruses (23, 30). Models generated in silico for the TCR of VEEV, EEEV, and SINV clade viruses predicted an 8- to 12-nucleotide spacer between the opal stop codon and the base of the TCR stem-loop followed by an 11- to 12-bp stem with a one nucleotide asymmetric bulge (30). The Kendra et al. CHIKV TCR model contains a spacer of five nucleotides between the opal stop codon and the base of the TCR element with only a 3-bp stem before a large bulge, followed by a 9-bp stem (7). Our model, generated from full-length genomic RNA probed with the SHAPE-MaP technique, places the opal stop codon in a stem 11 nucleotides away from the base of the TCR, which itself contains a one-nucleotide asymmetric bulge in the first 12 bp of the stem. We propose the true base of the CHIKV TCR be with the first A:U, as shown in our model, which corresponds to the base of the second helix in the Kendra et al. model, with the 11 intervening nucleotides between the opal codon and the TCR base either single-stranded and flexible or in transitory base pairs with neighboring sequence. We believe this would bring the three CHIKV TCR models into concordance.
The final known functional RNA secondary structure in CHIKV identified by our analysis was the frameshift element in the 6K coding region to generate the TF protein. This structure has been validated as functional in CHIKV and SFV (7, 23). Our frameshift element model differs from that proposed by Kendra et al. but is in accordance with the general model proposed previously for secondary structures following slippery sites (7, 23). The Kendra et al. SHAPE data were generated from a CHIKV genome fragment and predicts the spacer region involved in a helix at the base of a stem-loop. The discrepancies in this model and the previously discussed TCR model are likely a reflection of the methods used (SHAPE versus SHAPE-MaP), the sample RNA probed (in vitro-transcribed RNA fragments versus genomic RNA), and the length of the sequence modeled (78-nucleotide fragment versus the 109-nucleotide fragment composite generated from multiple windows) (7).
There are two RNA secondary structures found in other alphaviruses we either did not identify or which failed to meet our criteria for highly structured and specific in our analysis: the 5′IFIT stem-loop and the subgenomic downstream loop (DLP). The 5′IFIT stem-loop was first shown to be functionally important for evading detection by IFIT1 in VEEV (4). Later studies showed that this stem-loop is likely present and serves the same purpose, albeit with varied efficacy, in other alphaviruses including CHIKV (37). Our SHAPE-MaP data support the presence of this stem-loop as the first stem-loop of the CHIKV genome (see Fig. S1). Since this structure appears in the first 28 nucleotides of the genome, and our analysis relied on calculations over a rolling window of 55 nucleotides of the genome it was not captured in our analysis of highly structured and specific regions. While the subgenomic DLP has been shown to aid in replication of the SINV sgRNA during infection, this structure was not predicted to be present in CHIKV (38, 39). We also do not find an RNA secondary structure similar to the SINV DLP within the first 200 nucleotides of the CHIKV capsid coding region. This supports the hypotheses that CHIKV has an alternative mechanism for translating the sgRNA during infection.
Cocirculation of 3′UTR variants.
When CHIKV was introduced to the Western hemisphere in 2013, a bottle-necking event occurred in which a duplication in the 3′UTR of CHIKV became fixed in the population (21, 22). The additional duplication in the 3′UTR set the American strain of CHIKV apart from the parental Asian strain, which itself harbored multiple duplications from the predicted ECSA parental strain (26). We confirmed by 3′RACE that our virus stock consisted of a mixed population of viruses that possessed the duplication 3′UTR, as well as the canonical and a deletion 3′UTR variant (Fig. 5).
We confirmed previous reports that the 3′UTR duplication event enhanced replication in mosquito cells but found that, conversely, the deletion inhibited replication compared to the canonical and duplication 3′UTR (Fig. 6A) (21, 26, 34). It was known that the 3′UTR variants had no effect on virus replication in mammalian tissue culture, but the impact of these specific 3′UTRs on in vivo replication and pathogenesis had not been assessed. We saw no effect on virus replication, dissemination, or pathogenesis in our mouse model of CHIKV disease (Fig. 6C and D). This differs from previous studies that suggested deletions of the 3′UTR increased virus fitness compared to the WT 3′UTR isoform. However, those studies evaluated virus RNA persistence in a 12-day-old CD1 mouse model (26), while our studies evaluated acute viral replication and pathogenesis in 6-week-old adult C57BL/6J mice.
3′UTR variant structure.
Given the impact of the 3′UTR variants on replication in mosquito cells, we used SHAPE-MaP to analyze the RNA secondary structure of each of these variants. Of note, the extra sequence found in the duplication 3′UTR is composed of highly reactive nucleotides. Instead of creating an additional 177 nt with similar reactivity pattern, these nucleotides are more reactive, or flexible, than any other nucleotides found in the other two 3′UTRs or elsewhere in the duplication 3′UTR. Differing reactivities of individual repeat elements, which reflect different levels of structured versus unstructured RNA in the 3′UTR (Fig. 7 and 8) may affect RNA accessibility to host factors in the context of infection. Importantly, the additional sequence in the duplication 3′UTR created a second copy of a two-hairpin motif found only once in the canonical 3′UTR. This same motif is completely absent in the deletion 3′UTR. The copy number of this secondary structure corresponds with the replicative fitness observed in mosquito cells by each 3′UTR variant virus. The repeat of this two-stem-loop structure is reminiscent of the 3′UTR structures found in SINV that have been implicated in enhanced SINV replication in insect cells (40). However, future work will need to be done to assess whether the mosquito cell replication phenotypes in CHIKV are due to nucleotide sequence, copy number of novel RNA secondary structures, or the length and flexibility of unstructured RNA present in the 3′UTR. This may influence accessibility to host factors interacting with the primary sequence motifs within the duplicated 3′UTR.
The variability in 3′UTR structure and the consequences of this variation on virus replication in one host but not the other suggests that the 3′UTR is a flexible part of the genome used to aid in host switching. A similar phenomenon has been reported in dengue virus and related flaviviruses (41). The 3′UTR of mosquito-borne flaviviruses contain stem loops resistant to Xrn1 degradation, termed xrRNAs (41–43). Flaviviruses produced from mosquito cells have highly heterogenous 3′UTR sequences, with often mutated or deleted xrRNAs. These mutated variants exhibit replication advantages in mosquito cells. However, when these viruses are transmitted to the vertebrate host, the 3′UTR diversity collapses to a nearly singular 3′UTR variant with multiple stable xrRNAs. This is because at least one functional xrRNA significantly enhances virus replication in vertebrate cells (44, 45).
The opposite appears to be true for CHIKV. Replication in vertebrate cells generates 3′UTR diversity, often through deletions of repeat elements by copy-choice recombination, but these deletions are deleterious for replication in mosquito cells (35). Our analysis confirms these observations and provides experimental evidence of previously predicted RNA secondary structures associated with specific repeat elements in the 3′UTR: SL-a, SL-b, and SL-Y (35, 46). Our chemical probing data support that presence of SL-a, a large stem-loop at the start of the 3′UTR present in all CHIKV 3′UTR variants, and SL-b, the second stem-loop of the two-hairpin motif found to be deleted or duplicated, and the forked stem loops predicted for SL-Y (Fig. 8; see also nucleotides 11715 to 11753 and 11899 to 11937 in Fig. S1). Finally, our data also suggests these structures are separated by large spans of unstructured regions (35, 46). Our data in combination with prior studies of the 3′UTR, strengthens the hypothesis that the 3′UTR contains RNA secondary structures and sequences that are functionally important for efficient host switching. Duplicated RNA secondary structures and repeat sequence elements are found in other alphavirus 3′UTRs, and there is some evidence that other alphaviruses generate heterogenous 3′UTRs in a host-dependent manner like CHIKV (40, 47). Future studies should focus on the host specific function of each structure and begin to tease apart the relationship between the RNA secondary structure and underlying sequence.
CHIKV RNA structure considerations.
We now understand that RNA secondary structures are not conserved across the alphavirus family aside from a few important structures (8), and it remains difficult to identify novel functional RNA secondary structures in the broader context of RNA viruses generally (17, 19, 20, 48). We proposed identification of low Shannon entropy regions within a specific virus genome as a method to identify novel functional RNA elements. Our study supports the idea that functionally important RNA secondary structures can be identified by determining the most stably structured regions of a genome after SHAPE-MaP analysis, as we identified the four known important secondary structures and 19 novel structures in CHIKV with this strategy. Future studies will focus on assessing the 19 uncharacterized structured regions for functional importance using structure disruption strategies similar to those done in this study to assess the 5′ end of the genome (Fig. 3). However, these types of analyses will require multiple mutagenesis and phenotyping strategies for each structure and are beyond the scope of this study.
New methods assessing the tertiary structure of RNAs, such as RING-MaP and SPLASH, offer additional strategies when looking for functional RNA elements (20, 49). These strategies can more accurately identify longer range nucleotide interactions than SHAPE-MaP, where our analysis was limited to structures with a maximum pairing distance of 500 nucleotides. Tertiary structure and long-distance RNA-RNA interactions within a single RNA molecule are also likely to be more transient and, in terms of RNA viruses, highly dependent on the stage of replication and interacting proteins (50–52). These considerations also illustrate the need for further refinement of methods to assess RNA secondary structure in cells. Therefore, while our models of RNA secondary structures for virion-derived genomic RNA provide an important resource, there are likely other viral RNA conformations that occur within the infected cell (20, 53). Using sequence-based strategies like synonymous site conservation, which was designed to identify functional elements in RNA with coding constraints, in combination with experimentally informed RNA secondary structure models may also aid in identification of functional RNA elements (54). However, sequence-based methods often require dozens of sequences of the same RNA to be reliable. This would not be useful for newly emerged viruses, where few sequences are available, or for regions of viral genomes with little to no coding capacity, or with extensive overlapping reading frames.
The ability to generate experimentally informed RNA secondary structure models of long RNAs is advantageous for known and recently emerged viruses. Future work should be done to identify characteristics of known functional secondary structures so that they can be used to predict functional importance in novel structures. These characteristics can help prioritize novel structures for experimental testing. This will be especially helpful among structurally divergent but related RNAs, like virus genomes, and newly emerged viruses for which few genome sequences are available.
MATERIALS AND METHODS
SHAPE-MaP of CHIKV genomic and 3ʹ UTR RNA.
CHIKV genomic RNA was extracted from sucrose-purified virions produced from Vero81 cells with TRIzol according to the manufacturer’s protocol. Individual 3ʹUTRs were transcribed in vitro. For SHAPE modification, 2 μg of virion RNA was incubated at 37°C for 15 min in folding buffer (110 mM HEPES [pH 8], 10 mM MgCl2, 111 mM KCl) and then treated with 100 nM 1-methyl-7-nitroisatoicanhydride (1M7) for 5 min at 37°C. Negative-control RNA was incubated with 5 μl of dimethyl sulfoxide in place of 1M7. The denatured control RNA sample was incubated at 95°C for 2 min and then treated with 100 nM 1M7 for 2 min at 95°C. The treated RNA was purified over Zymo RNA Clean and Concentrator-5 columns (Zymo). A 500-ng portion of Random Primer 9 (NEB) was added, followed by incubation at 65°C for 5 min, and then the sample was placed on ice. Reverse transcriptase buffer (10 mM deoxynucleoside triphosphates, 0.1 M dithiothreitol, 500 mM Tris [pH 8.0], 750 KCl, and 500 mM MnCl2) was added, and the reaction mixture was incubated at 42°C for 2 min before the addition of 200 U of SuperScript II (Thermo Fisher Scientific), followed by incubation at 42°C for 180 min. The reaction was heat inactivated at 70°C for 15 min, and RNA was purified over G-50 columns (GE Healthcare). The NEBNext mRNA second strand synthesis module (NEB) was used to generate double-stranded cDNA according to the manufacturer’s protocol. The double-stranded cDNA was fragmented, tagged, amplified, and barcoded using a Nextera XT DNA library preparation kit (Illumina) according to the manufacturer’s directions. Excess oligonucleotides, primer dimers, small library fragments, and nucleotides were removed with Agencourt AMPure XP beads (Beckman Coulter) at a DNA-to-bead ratio of 0.6:1. Library size was determined by using a 2100 Bioanalyzer (Agilent Technologies) and quantified with a Qubit fluorimeter (Thermo Fisher Scientific). Sequencing was performed on a MiSeq desktop sequencer (Illumina).
SHAPE data processing.
Reads from the untreated RNA control were aligned to CHIKV reference sequence AF369024.2 using bowtie2 (v2.2.3) to generate a reference sequence (12). The ShapeMapper pipeline (v1.2) was used to map SHAPE reactivities to the CHIKV genome (16). Because of low coverage for nucleotides 11400 to 12012 with the CHIKV genomic RNA, the SHAPE reactivities for this region were calculated with additional data from in vitro-transcribed RNA. Default parameters for the ShapeMapper pipeline were used except for a maximum insert size of 1,000, and for the 3′ end, a minimum map quality of 20. The mean SHAPE reactivity and standard error for each nucleotide of the genomic RNA and individual 3ʹUTRs tested are reported in Data Set S1 in the supplemental material. Median SHAPE values were calculated over a rolling 55-nucleotide window. Generally, SHAPE reactivities below 0.4 indicate likely paired bases, and SHAPE reactivities above 0.8 indicate likely unpaired bases. We were unable to determine a quantitative odds ratio for paired and unpaired nucleotides at these specific SHAPE reactivity thresholds (55, 56). Odds ratio calculations require a known RNA secondary structure, such as rRNA which was not present due to using purified virion particles for our genomic RNA source. Base pairing probabilities for the whole genome and each individual 3ʹ UTR were obtained with Superfold v1.0 using the RNA structure software suite (v5.8.1), with a maximum pairing distance of 500 nucleotides (14, 16). SHAPE data were used as a folding restraint. Superfold was also used to find Shannon entropies of base pairing at each position. Regional entropies were generated by finding the median Shannon entropy over a 55-nucleotide rolling window. Highly structured regions were defined as regions with low median Shannon entropy and low SHAPE, as in Siegfried et al. and Smola et al. (11, 16). In total, 23 structured regions were found within the CHIKV genome. Structures for these regions were extracted from the whole-genome structure obtained with Superfold.
To compare the SHAPE reactivity among the 3′UTR variants in Fig. 7, the rolling median SHAPE values were calculated in reference to the median SHAPE reactivity of the whole-genome CHIKV SHAPE excluding the 3′UTR to create a common reactivity scale. Consequently, the denominator when calculating the median SHAPE reactivity for each 3′UTR was the average SHAPE reactivity of the CHIKV genome from positions 1 to 11,301 (the 3′UTR begins at position 11,302).
Sequence conservation.
Sequence conservation analysis used the multiple sequence alignment generated in Kutchko and Madden et al., as well as the same method of calculating conservation scores (8). Data were smoothed by calculating the median conservation score over a rolling 55-nucleotide window.
3′RACE analysis of CHIKV genomic RNA.
3′RACE of the CHIKV genome was performed on purified genomic RNA using the RLM-RACE kit (Ambion) according to the manufacturer’s directions. Briefly, RNA was reverse transcribed using the 3′RACE adapter (5′-GCGAGCACAGAATTAATACGACTCACTATAGGT12VN-3′). 3′UTR-specific PCR was performed with the 3′RACE adapter outer primer (5′-GCGAGCACAGAATTAATACGACT-3′) and 3′UTR gene-specific forward primer (5′-CTTGACAACTAGGTATGAAG-3′) recognizing CHIKV genome position 11302. PCR products were resolved on a 1% agarose gel and gel purified before cloning into the pCR-Blunt vector (Thermo Fisher). Multiple transformants of each PCR amplicon were sequenced, and the 3′UTR variants were cloned into the infectious clone of the Caribbean CHIKV isolate using Gibson Assembly. In each case, the infectious clone was resequenced to ensure the absence of unintended mutations.
Generation of an infectious clone of the CHIKV Caribbean isolate.
The full-length cDNA clone of the early outbreak Caribbean CHIKV isolate virus was assembled from the consensus nucleotide sequence of purified genomic viral RNA (MG208125) (32). Michael Diamond (Washington University) provided the clinical isolate sequenced for construction of the infectious clone. The isolate was originally obtained on the island of St. Martin during the 2013 outbreak and was banked at the World Reference Center for Emerging Viruses and Arboviruses (University of Texas Medical Branch). This virus was amplified on Vero cells three times prior to receipt by our lab and amplified once on C6/36 cells before sequencing. Briefly, 14 dsDNA gBlocks were synthesized by IDT to span the entire 12,012nt genome. gBlocks were assembled into 5 overlapping genomic fragments, and each cloned into the pCR-Blunt vector (Thermo Fisher). Each fragment was PCR amplified and assembled by ligation using unique restriction sites in the CHIKV genome. A unique SacI restriction site was included upstream of the SP6 promoter in fragment 1, and a unique NotI site was included in the fragment downstream of a poly(A) sequence. The assembled CHIKV genome was inserted into the SacI and NotI sites of plasmid pSinRep5 (Invitrogen). The sequence of the full-length clone and each 3′UTR variant clone was confirmed by Sanger sequencing (GenBank accession no. MT228631, MT228632, and MT228633).
Cells and viruses.
Vero81 cells were cultured in DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 0.2 mM l-glutamine (Gibco). BHK-21 cells were cultured in αMEM (Gibco) supplemented with 10% FBS and 0.2 mM l-glutamine. The mosquito cell line C6/36 was cultured in Leibovitz L-15 media (Corning/Cellgro) supplemented with 10% FBS, 10% tryptose phosphate broth (Sigma), and 0.2 mM l-glutamine. A low-passage-number isolate of Caribbean CHIKV was propagated in C6/36 cells, and infectious supernatants were collected and purified over a 20% sucrose cushion.
The St. Martin CHIKV infectious clone was used for 3′UTR replication and pathogenesis studies (GenBank accession no. MG208125) (32), and the 181/25 infectious clone was used for 5′ end structure studies (GenBank accession no. EF452494) (57). Clonal virus pools made from infectious clones were generated by linearizing the infectious clone plasmid and in vitro transcribing full-length capped genomic RNA using mMessage mMachine SP6 transcription kits (Ambion).
1.0 × 107 BHK-21 cells were electroporated (850 V, 25 μF, three pulses) in a 4-mm gap cuvette (Bio-Rad) with 10 μg of RNA after being washed three times with PBS lacking Ca2+ and Mg2+. Cells were recovered in maintenance media. Supernatants with virus were harvested 24 h later at peak titer. Cell debris were pelleted by centrifugation at 1,000 rpm for 10 min at 4°C. Single-use aliquots were made and stored at –80°C.
Structure-disrupting mutations.
CodonShuffle was used to generate mutant sequences within the coding portion of the virus genome that maintain amino acid sequence and nucleotide composition (15). The dn231 algorithm was used, which also preserves dinucleotide frequency. Because CodonShuffle generates many possible mutant sequences, the final mutant sequence for a region was selected to maximize structural disruption in that region while maintaining similar codon usage frequencies within the virus. Synthetic DNA fragments (IDT) containing selected mutations and two unique restriction sites were incorporated into an infectious clone of the 181/25 (TSI-GSD-218) vaccine strain of CHIKV (57) by Gibson assembly (NEB) (see Table S1).
In vitro analysis of virus replication.
C6/36 mosquito cells and Vero81 monkey kidney cells were infected at a multiplicity of infection (MOI) equal to 0.01 in biological triplicate. Supernatant samples were collected at indicated times and stored at –80°C until titering. Virus titers of cell culture supernatants were quantified by plaque assay on Vero81 cells after samples were diluted in 1× PBS (Gibco) with 1% FBS and Ca2+/Mg2+. Cells were overlaid with 1× αMEM with 5% FBS, 0.2 mM l-glutamine (Gibco), 1 mM HEPES (Corning), 1% penicillin-streptomycin (Gibco), and 1.25% carboxymethylcellulose sodium (Sigma). Virus was allowed to plaque for 48 h (SM CHIKV) or 72 h (181/25) before monolayers were fixed with 4% paraformaldehyde, rinsed, and stained with crystal violet (0.25%; VWR). Data were analyzed by two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test in Prism8 (GraphPad Software).
Infectious centers assay.
A total of 1.0 × 107 BHK-21 cells were electroporated (850 V, 25 μF, three pulses) in a 4-mm gap cuvette (Bio-Rad) with 10 μg of virus genomic RNA after being washed three times with PBS lacking Ca2+ and Mg2+. Cells were recovered in maintenance media and serially diluted in maintenance media. Cell dilutions were plated over Vero81 monolayers overlaid with 1× αMEM with 5% FBS, 0.2 mM l-glutamine (Gibco), 1 mM HEPES (Corning), 1% penicillin-streptomycin (Gibco), and 1.25% carboxymethylcellulose sodium (Sigma). Plaques were allowed to form for 72 h before monolayers were fixed with 4% paraformaldehyde, rinsed, and stained with crystal violet (0.25%; VWR). Data were analyzed by one-way ANOVA with Tukey’s multiple-comparison test in Prism8 (GraphPad Software).
Western blotting for viral proteins during infection.
Vero81 cells were infected at an MOI of 5 with either 181/25 or mutant virus and incubated for 1 h with rocking every 15 min. After the incubation, cells were washed three times with PBS, and the medium was replaced. Cellular lysates were made at indicated times in radioimmunoprecipitation assay buffer. First, 5 μg of total protein was loaded and separated on a 4 to 20% gradient TGX precast protein gel and transferred to a polyvinylidene difluoride membrane. The membrane was then blocked overnight with 5% milk in PBST and probed with primary antibodies for 1 h to overnight (mouse anti-nsP3 1:1,000 and mouse anti-E2 1:500 in 5% milk PBS-T). Secondary antibodies were incubated for 1 h at room temperature in 5% milk with 0.01% SDS in 1× TBST on a rocker. Membranes were washed three times with 1× TBST for 10 min each wash. The membranes were then washed three times in 1× TBS for 10 min each time. The membranes were visualized with the Odyssey infrared Imaging system (Li-Cor).
Densitometry analysis.
Bands were quantified for E2, nsP3, and actin using ImageJ software (National Institutes of Health; v1.53a). Band densities for individual proteins were normalized to actin loading control densities. Fold change in expression was measured relative to WT expression of E2 or nsP3 at 8 h postinfection.
qRT-PCR for detecting virus genome during infection.
Vero81 cells were infected at an MOI of 5 with either 181/25 or mutant virus for 1 h with rocking every 15 min. Following the incubation, cells were washed three times with PBS, and the medium was replaced. At the indicated times postinfection, the medium was removed, and the cells were washed once in PBS before being lysed in TRIzol (Life Technologies) for total RNA isolation. RNA was purified according to the manufacturer’s protocol. qPCR was performed on RNA using an iTaq Universal Probes one-step kit (Bio-Rad) and primers and probe specific to either the 18S rRNA gene or the CHIKV nsP1 gene. Standard curves of both mammalian 18S cDNA and 181/25 infectious clone were run in parallel with samples for absolute quantification of the gene copy number. All reactions were run in 96-well plates on an ABI 7300 real-time PCR machine in technical duplicate. Data were analyzed by multiple t tests with Holm-Sidak correction in Prism8.
In vivo analysis of virus replication and pathogenesis.
All animal studies were done following IACUC-approved protocols under the supervision and scrutiny of University of North Carolina veterinarians. The C57BL/6J mice that were utilized in this study were bred at UNC after breeding pairs were purchased from Jackson Laboratories. Animals were allowed to age to 6 weeks before use. Animals were inoculated with 100 PFU of virus in 10 μl of vehicle (PBS with 1% FBS and Ca2+/Mg2+). The inoculation was given as a subcutaneous injection in the left hind footpad. For analysis of footpad swelling, the footpad width was measured daily for 1 week using calipers (58). Data were analyzed by two-way ANOVA with Tukey’s correction for multiple comparisons using Prism8 (GraphPad Software). To quantify infectious virus levels in tissues, infected animals were sacrificed on day three after infection, and tissues were harvested into Vero81 media containing sterile glass beads. After weighing, the tissues were homogenized, and infectious virus was quantified by plaque assay on Vero81 cells as done for analysis of virus replication in vitro. Data within each tissue were analyzed by one-way ANOVA with Tukey’s correction for multiple comparisons using Prism8.
Data availability.
Viruses and materials used in this study will be provided upon request. Nucleotide sequences for the CHIKV 3′UTR variant infectious clones have been deposited in GenBank under accession numbers MT228631, MT228632, and MT228633. SHAPE-MaP data are available online in SNRNASM format (https://docs.google.com/spreadsheets/d/1OrnU4lmvytfHhv-nh47PHyUxMdiHc-hDe0OS0r4o9dA/edit?usp=sharing). Figure S1 and Table S1 are available online in a single PDF document (https://drive.google.com/file/d/1ZlrFGYxsrUeRE0QcUB_rWPirpLki4C6F/view?usp=sharing).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Heise, Moorman, and Laederach laboratories for reviews of the manuscript. We thank Kevin Weeks for his advice and thoughtful conversations on SHAPE-MaP analysis. We thank Michael Diamond of Washington University and Robert Tesh of the World Reference Center for Emerging Viruses and Arboviruses at the University of Texas Medical Branch for kindly sharing the CHIKV Caribbean isolate with us. We also thank the reviewers of our paper for their thoughtful and constructive comments in the midst of a pandemic. Their attention to detail and commitment to improving our paper was greatly appreciated.
This study was supported by the National Institutes of Health (U19 AI142759 to M.T.H., U19 AI 107810 to M.T.H., U19 AI 109761 to M.T.H., R21 AI 138056 to M.T.H. and N.J.M., AI 123811 to N.J.M., T32 AI 007151-36A1 to E.A.M., and F32 AI 126730 to K.S.P.). Funding for the open access charge was provided by the National Institutes of Health.
We report no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Weaver SC, Forrester NL. 2015. Chikungunya: evolutionary history and recent epidemic spread. ANTIVIRAL Res 120:32–39. doi: 10.1016/j.antiviral.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Morrison CR, Plante KS, Heise MT. 2016. Chikungunya virus: current perspectives on a reemerging virus, p 143–161. In Emerging infections 10. American Society of Microbiology, Washington, DC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim DY, Firth AE, Atasheva S, Frolova EI, Frolov I. 2011. Conservation of a packaging signal and the viral genome RNA packaging mechanism in alphavirus evolution. J Virol 85:8022–8036. doi: 10.1128/JVI.00644-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, Klimstra WB, Amarasinghe GK, Diamond MS. 2014. A viral RNA structural element alters host recognition of nonself RNA. Science 343:783–787. doi: 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fayzulin R, Frolov I. 2004. Changes of the secondary structure of the 5′ end of the Sindbis virus genome inhibit virus growth in mosquito cells and lead to accumulation of adaptive mutations. J Virol 78:4953–4964. doi: 10.1128/jvi.78.10.4953-4964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michel G, Petrakova O, Atasheva S, Frolov I. 2007. Adaptation of Venezuelan equine encephalitis virus lacking 51-nt conserved sequence element to replication in mammalian and mosquito cells. Virology 362:475–487. doi: 10.1016/j.virol.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kendra JA, Advani VM, Chen B, Briggs JW, Zhu J, Bress HJ, Pathy SM, Dinman JD. 2018. Functional and structural characterization of the chikungunya virus translational recoding signals. J Biol Chem 293:17536–17545. doi: 10.1074/jbc.RA118.005606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutchko KM, Madden EA, Morrison C, Plante KS, Sanders W, Vincent HA, Cruz Cisneros MC, Long KM, Moorman NJ, Heise MT, Laederach A. 2018. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res 46:3657–3670. doi: 10.1093/nar/gky012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrasco L, Sanz MA, González-Almela E. 2018. The regulation of translation in alphavirus-infected cells. Viruses 10:70. doi: 10.3390/v10020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ventoso I. 2012. Adaptive changes in alphavirus mRNA translation allowed colonization of vertebrate hosts. J Virol 86:9484–9494. doi: 10.1128/JVI.01114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smola MJ, Rice GM, Busan S, Siegfried NA, Weeks KM. 2015. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10:1643–1669. doi: 10.1038/nprot.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soldatov RA, Vinogradova SV, Mironov AA. 2014. RNASurface: fast and accurate detection of locally optimal potentially structured RNA segments. Bioinformatics 30:457–463. doi: 10.1093/bioinformatics/btt701. [DOI] [PubMed] [Google Scholar]
- 14.Reuter JS, Mathews DH. 2010. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorge D. M d M, Mills RE, Lauring AS. 2015. CodonShuffle: a tool for generating and analyzing synonymously mutated sequences. Virus Evol 1:vev012. doi: 10.1093/ve/vev012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM. 2014. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 11:959–965. doi: 10.1038/nmeth.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mauger DM, Golden M, Yamane D, Williford S, Lemon SM, Martin DP, Weeks KM. 2015. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc Natl Acad Sci U S A :201416266–201416218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirakitikulr N, Kohlway A, Lindenbach BD, Pyle AM. 2016. The coding region of the HCV genome contains a network of regulatory RNA structures. Mol Cell 62:111–120. doi: 10.1016/j.molcel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dethoff EA, Boerneke MA, Gokhale NS, Muhire BM, Martin DP, Sacco MT, McFadden MJ, Weinstein JB, Messer WB, Horner SM, Weeks KM. 2018. Pervasive tertiary structure in the dengue virus RNA genome. Proc Natl Acad Sci U S A 115:11513–11518. doi: 10.1073/pnas.1716689115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stapleford KA, Moratorio G, Henningsson R, Chen R, Matheus S, Enfissi A, Weissglas-Volkov D, Isakov O, Blanc H, Mounce BC, Dupont-Rouzeyrol M, Shomron N, Weaver S, Fontes M, Rousset D, Vignuzzi M. 2016. Whole-genome sequencing analysis from the chikungunya virus Caribbean Outbreak reveals novel evolutionary genomic elements. PLoS Negl Trop Dis 10:e0004402. doi: 10.1371/journal.pntd.0004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R, Puri V, Fedorova N, Lin D, Hari KL, Jain R, Rodas JD, Das SR, Shabman RS, Weaver SC. 2016. Comprehensive genome scale phylogenetic study provides new insights on the global expansion of chikungunya virus. J Virol 90:10600–10611. doi: 10.1128/JVI.01166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol J 5:108. doi: 10.1186/1743-422X-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toribio R, Díaz-López I, Boskovic J, Ventoso I. 2018. Translation initiation of alphavirus mRNA reveals new insights into the topology of the 48S initiation complex. Nucleic Acids Res 46:4176–4187. doi: 10.1093/nar/gky071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kutchko KM, Laederach A. 2017. Transcending the prediction paradigm: novel applications of SHAPE to RNA function and evolution. Wiley Interdiscip Rev RNA 8:e1374. doi: 10.1002/wrna.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen R, Wang E, Tsetsarkin KA, Weaver SC. 2013. Chikungunya virus 3’ untranslated region: adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog 9:e1003591. doi: 10.1371/journal.ppat.1003591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forrester NL, Palacios G, Tesh RB, Savji N, Guzman H, Sherman M, Weaver SC, Lipkin WI. 2012. Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J Virol 86:2729–2738. doi: 10.1128/JVI.05591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deigan KE, Li TW, Mathews DH, Weeks KM. 2009. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A 106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huynen M, Gutell R, Konings D. 1997. Assessing the reliability of RNA folding using statistical mechanics. J Mol Biol 267:1104–1112. doi: 10.1006/jmbi.1997.0889. [DOI] [PubMed] [Google Scholar]
- 30.Firth AE, Wills NM, Gesteland RF, Atkins JF. 2011. Stimulation of stop codon readthrough: frequent presence of an extended 3′ RNA structural element. Nucleic Acids Res 39:6679–6691. doi: 10.1093/nar/gkr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niesters HGM, Strauss JH. 1990. Mutagenesis of the Conserved 51-Nucleotide Region of Sindbis Virus. J Virol 64:1639–1647. doi: 10.1128/JVI.64.4.1639-1647.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones JE, Long KM, Whitmore AC, Sanders W, Thurlow LR, Brown JA, Morrison CR, Vincent H, Peck KM, Browning C, Moorman N, Lim JK, Heise MT. 2017. Disruption of the opal stop codon attenuates chikungunya virus-induced arthritis and pathology. mBio 8:e01456-17. doi: 10.1128/mBio.01456-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyde JL, Chen R, Trobaugh DW, Diamond MS, Weaver SC, Klimstra WB, Wilusz J. 2015. The 5′ and 3′ ends of alphavirus RNAs: non-coding is not nonfunctional. Virus Res 206:99–107. doi: 10.1016/j.virusres.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morley VJ, Noval MG, Chen R, Weaver SC, Vignuzzi M, Stapleford KA, Turner PE. 2018. Chikungunya virus evolution following a large 3′UTR deletion results in host-specific molecular changes in protein-coding regions. Virus Evol 4:vey012. doi: 10.1093/ve/vey012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filomatori CV, Bardossy ES, Merwaiss F, Suzuki Y, Henrion A, Saleh M-C, Alvarez DE. 2019. RNA recombination at Chikungunya virus 3′UTR as an evolutionary mechanism that provides adaptability. PLoS Pathog 15:e1007706. doi: 10.1371/journal.ppat.1007706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kendall C, Khalid H, Müller M, Banda DH, Kohl A, Merits A, Stonehouse NJ, Tuplin A. 2019. Structural and phenotypic analysis of Chikungunya virus RNA replication elements. Nucleic Acids Res 47:9296–9312. doi: 10.1093/nar/gkz640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynaud JM, Kim DY, Atasheva S, Rasalouskaya A, White JP, Diamond MS, Weaver SC, Frolova EI, Frolov I. 2015. IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLoS Pathog 11:e1004863. doi: 10.1371/journal.ppat.1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, Esteban M. 2006. Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev 20:87–100. doi: 10.1101/gad.357006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toribio R, Díaz-López I, Boskovic J, Ventoso I. 2016. An RNA trapping mechanism in Alphavirus mRNA promotes ribosome stalling and translation initiation. Nucleic Acids Res 44:4368–4380. doi: 10.1093/nar/gkw172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Moreno M, Sanz MA, Carrasco L. 2016. A viral mRNA motif at the 3′-untranslated region that confers translatability in a cell-specific manner: implications for virus evolution. Sci Rep 6:19217. doi: 10.1038/srep19217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villordo SM, Carballeda JM, Filomatori CV, Gamarnik AV. 2016. RNA structure duplications and flavivirus host adaptation. Trends Microbiol 24:270–283. doi: 10.1016/j.tim.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chapman EG, Costantino DA, Rabe JL, Moon SL, Wilusz J, Nix JC, Kieft JS. 2014. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 344:307–310. doi: 10.1126/science.1250897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapman EG, Moon SL, Wilusz J, Kieft JS. 2014. RNA structures that resist degradation by Xrn1 produce a pathogenic dengue virus RNA. Elife 3:e01892. doi: 10.7554/eLife.01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villordo SM, Filomatori CV, Sánchez-Vargas I, Blair CD, Gamarnik AV. 2015. Dengue virus RNA structure specialization facilitates host adaptation. PLoS Pathog 11:e1004604. doi: 10.1371/journal.ppat.1004604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Filomatori CV, Carballeda JM, Villordo SM, Aguirre S, Pallarés HM, Maestre AM, Sánchez-Vargas I, Blair CD, Fabri C, Morales MA, Fernandez-Sesma A, Gamarnik AV. 2017. Dengue virus genomic variation associated with mosquito adaptation defines the pattern of viral noncoding RNAs and fitness in human cells. PLoS Pathog 13:e1006265. doi: 10.1371/journal.ppat.1006265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider A de B, Ochsenreiter R, Hostager R, Hofacker IL, Janies D, Wolfinger MT. 2019. Updated phylogeny of chikungunya virus suggests lineage-specific RNA architecture. Viruses 11:798. doi: 10.3390/v11090798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Filomatori CV, Merwaiss F, Bardossy ES, Alvarez DE. 2020. Impact of alphavirus 3′UTR plasticity on mosquito transmission. Semin Cell Dev Biol 11:S1084-9521(19)30193-4. doi: 10.1016/j.semcdb.2020.07.006. [DOI] [PubMed] [Google Scholar]
- 48.Boerneke MA, Ehrhardt JE, Weeks KM. 2019. Physical and functional analysis of viral RNA genomes by SHAPE. Annu Rev Virol 6:93–117. doi: 10.1146/annurev-virology-092917-043315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li P, Wei Y, Mei M, Tang L, Sun L, Huang W, Zhou J, Zou C, Zhang S, Qin C-F, Jiang T, Dai J, Tan X, Zhang QC. 2018. Integrative analysis of Zika virus genome RNA structure reveals critical determinants of viral infectivity. Cell Host Microbe 24:875–886.e5. doi: 10.1016/j.chom.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Villordo SM, Gamarnik AV. 2009. Genome cyclization as strategy for flavivirus RNA replication. Virus Res 139:230–239. doi: 10.1016/j.virusres.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Borba L, Villordo SM, Iglesias NG, Filomatori CV, Gebhard LG, Gamarnik AV. 2015. Overlapping local and long-range RNA-RNA interactions modulate dengue virus genome cyclization and replication. J Virol 89:3430–3437. doi: 10.1128/JVI.02677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L, Gamarnik AV. 2006. A 5’ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev 20:2238–2249. doi: 10.1101/gad.1444206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smola MJ, Weeks KM. 2018. In-cell RNA structure probing with SHAPE-MaP. Nat Protoc 13:1181–1195. doi: 10.1038/nprot.2018.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Firth AE. 2014. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res 42:12425–12439. doi: 10.1093/nar/gku981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sükösd Z, Swenson MS, Kjems J, Heitsch CE. 2013. Evaluating the accuracy of SHAPE-directed RNA secondary structure predictions. Nucleic Acids Res 41:2807–2816. doi: 10.1093/nar/gks1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eddy SR. 2014. Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu Rev Biophys 43:433–456. doi: 10.1146/annurev-biophys-051013-022950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levitt NH, Ramsburg HH, Hasty SE, Repik PM, Cole FE, Lupton HW. 1986. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 4:157–162. doi: 10.1016/0264-410x(86)90003-4. [DOI] [PubMed] [Google Scholar]
- 58.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT. 2011. A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease. Am J Pathol 178:32–40. doi: 10.1016/j.ajpath.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Viruses and materials used in this study will be provided upon request. Nucleotide sequences for the CHIKV 3′UTR variant infectious clones have been deposited in GenBank under accession numbers MT228631, MT228632, and MT228633. SHAPE-MaP data are available online in SNRNASM format (https://docs.google.com/spreadsheets/d/1OrnU4lmvytfHhv-nh47PHyUxMdiHc-hDe0OS0r4o9dA/edit?usp=sharing). Figure S1 and Table S1 are available online in a single PDF document (https://drive.google.com/file/d/1ZlrFGYxsrUeRE0QcUB_rWPirpLki4C6F/view?usp=sharing).