

Glycerophospholipids in cell membranes and virus envelopes often affect viral entry and budding. However, the role of glycerophospholipids in membrane-associated events in viral replication in herpesvirus-infected cells has not been reported to date. In this study, we have presented data showing that cellular PE biosynthesis mediated by Pcyt2 is important for HSV-1 envelopment in the cytoplasm, as well as for viral replication and pathogenicity in vivo. This is the first report showing the importance of PE biosynthesis in herpesvirus infections. Our results showed that inhibition of Pcyt2, a key cell enzyme for PE synthesis, significantly inhibited HSV-1 replication and pathogenicity in mice. This suggested that the PE biosynthetic pathway, as well as the HSV-1 virion maturation pathway, can be a target for the development of novel anti-HSV drugs.
KEYWORDS: envelopment, herpes simplex virus, phospholipids, viral pathogenesis
ABSTRACT
Glycerophospholipids are major components of cell membranes. Phosphatidylethanolamine (PE) is a glycerophospholipid that is involved in multiple cellular processes, such as membrane fusion, the cell cycle, autophagy, and apoptosis. In this study, we investigated the role of PE biosynthesis in herpes simplex virus 1 (HSV-1) infection by knocking out the host cell gene encoding phosphate cytidylyltransferase 2, ethanolamine (Pcyt2), which is a key rate-limiting enzyme in one of the two major pathways for PE biosynthesis. Pcyt2 knockout reduced HSV-1 replication and caused an accumulation of unenveloped and partially enveloped nucleocapsids in the cytoplasm of an HSV-1-infected cell culture. A similar phenotype was observed when infected cells were treated with meclizine, which is an inhibitor of Pcyt2. In addition, treatment of HSV-1-infected mice with meclizine significantly reduced HSV-1 replication in the mouse brains and improved their survival rates. These results indicated that PE biosynthesis mediated by Pcyt2 was required for efficient HSV-1 envelopment in the cytoplasm of infected cells and for viral replication and pathogenicity in vivo. The results also identified the PE biosynthetic pathway as a possible novel target for antiviral therapy of HSV-associated diseases and raised an interesting possibility for meclizine repositioning for treatment of these diseases, since it is an over-the-counter drug that has been used for decades against nausea and vertigo in motion sickness.
IMPORTANCE Glycerophospholipids in cell membranes and virus envelopes often affect viral entry and budding. However, the role of glycerophospholipids in membrane-associated events in viral replication in herpesvirus-infected cells has not been reported to date. In this study, we have presented data showing that cellular PE biosynthesis mediated by Pcyt2 is important for HSV-1 envelopment in the cytoplasm, as well as for viral replication and pathogenicity in vivo. This is the first report showing the importance of PE biosynthesis in herpesvirus infections. Our results showed that inhibition of Pcyt2, a key cell enzyme for PE synthesis, significantly inhibited HSV-1 replication and pathogenicity in mice. This suggested that the PE biosynthetic pathway, as well as the HSV-1 virion maturation pathway, can be a target for the development of novel anti-HSV drugs.
INTRODUCTION
Interactions between viruses and host cell membranes are critical for viral infections, particularly during viral entry, when the virion crosses the host cell plasma membrane or endosomal membrane, during transmission of clusters of virions via host cell membranous vesicles, and during viral egress, when in some cases nascent progeny viruses acquire a host cell membrane by budding (1, 2). Cell membranes consist mainly of lipids, including glycerophospholipids, sphingolipids, and sterols. Therefore, lipids in cell membranes and virus envelopes often affect the entry and budding processes of various viruses (1–4). Glycerophospholipids have a glycerol backbone, a functional head group, and two fatty acids of various lengths and degrees of unsaturation and are major components of cell membranes (5). The fatty acid chains are hydrophobic but the head, which consists mainly of the phosphate group, is hydrophilic, leading to the amphipathic nature of glycerophospholipids. The glycerophospholipids are usually organized in a bilayer in membranes, with the polar hydrophilic heads sticking outward into the aqueous environment and the nonpolar hydrophobic tails pointing inward (5). Differences in the functional head group produce various species of glycerophospholipids, such as phosphatidic acid, phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylserine (PS), phosphatidylglycerol, and diphosphatidylglycerol, which are structurally and physically different (5). Data have accumulated suggesting that some glycerophospholipids in cellular and viral membranes play multiple roles in membrane-associated viral events, including viral entry and egress (3, 4). For instance, PS in viral envelopes promotes the entry of various enveloped viruses by binding directly and indirectly to PS receptors on the host cell plasma membrane, e.g., directly to members of the T cell immunoglobulin and mucin receptor (TIM) family and indirectly to members of the Tyro3-Axl-Mer (TAM) family (3, 6–9). Like PS, PE in the envelopes of some enveloped viruses binds to TIM1 and promotes TIM1-mediated viral entry (10). PS in the plasma membrane promotes assembly and egress of Ebola virus (11). In addition, either PI-4,5-bisphosphate or PI-4-phosphate is required in host cell membranes for the efficient egress of HIV, murine leukemia virus, Ebola virus, and hepatitis C virus (12–14).
Herpes simplex virus 1 (HSV-1) is a member of the Herpesviridae family and is one of the most extensively studied members of this family (15). HSV-1 causes various mucocutaneous and skin diseases in humans, including herpes labialis, genital herpes, herpetic whitlow and keratitis, and life-threatening herpes simplex encephalitis (HSE) (15). After HSV-1 entry into a host cell, replication of the viral genome and formation of nucleocapsids take place in the nucleus of HSV-1-infected cells (15). Nascent progeny virus nucleocapsids become enveloped by budding through the inner nuclear membrane (INM) into the perinuclear space between the INM and the outer nuclear membrane (ONM) (primary envelopment). The enveloped nucleocapsids subsequently fuse with the ONM to release the nucleocapsids into the cytoplasm (deenvelopment) (16, 17). The nucleocapsids acquire their final envelopes in the cytoplasm by budding through cytoplasmic membranes, such as those derived from the trans-Golgi network and/or endosomes (secondary envelopment) (17, 18). Enveloped virions are then transported to the plasma membrane through a secretory pathway (17, 18). Therefore, herpesviruses need to cross cellular membranes five times during their replication. However, there is a lack of information on the role(s) of glycerophospholipids in cellular and viral membranes in the membrane-associated events in HSV-1 replication, including viral entry, primary envelopment, deenvelopment, secondary envelopment, and release from the plasma membrane.
PE is the second most abundant glycerophospholipid in mammalian cells, constituting 15 to 25% of the total glycerophospholipids (19) and having significant roles in cellular processes such as membrane fusion, the cell cycle, autophagy, and apoptosis (20, 21). Therefore, we sought to investigate the role of PE and/or PE biosynthesis in the membrane-associated events in HSV-1 infection. PE can be produced via two major biochemical pathways, i.e., the CDP-ethanolamine Kennedy pathway at the endoplasmic reticulum and the mitochondrial PS decarboxylation pathway (20, 21). Phosphate cytidylyltransferase 2, ethanolamine (Pcyt2), is the key rate-limiting enzyme in the biosynthesis of PE from ethanolamine and diacylglycerol by the CDP-ethanolamine Kennedy pathway (21, 22). Therefore, in this study, we examined the effects of a decrease in Pcyt2 expression and of the chemical inhibition of Pcyt2 on HSV-1 replication and pathogenicity in vitro and in vivo.
RESULTS
Effect of Pcyt2 knockout on the accumulation of PE.
To examine the significance of PE biosynthesis mediated by Pcyt2 in HSV-1 infection in a HeLa cell culture, we generated HeLa/Pcyt2 knockout (KO) cells in which the Pcyt2 gene was knocked out by the CRISPR/Cas9 system (Fig. 1A). To examine off-target effects in HeLa/Pcyt2 KO cells, we also constructed HeLa/Pcyt2 KO/Flag-Pcyt2 cells, in which Flag-tagged Pcyt2 (Flag-Pcyt2) was ectopically expressed. As expected, Pcyt2 was not detectable in HeLa/Pcyt2 KO cells (Fig. 1B). HeLa/Pcyt2 KO/Flag-Pcyt2 cells expressed Flag-Pcyt2 efficiently, and the level of Flag-Pcyt2 expression in these cells was comparable to that of endogenous Pcyt2 expression in HeLa cells (Fig. 1B). The Pcyt-2 KO had no effect on the viability of HeLa cells (Fig. 1C).
FIG 1.
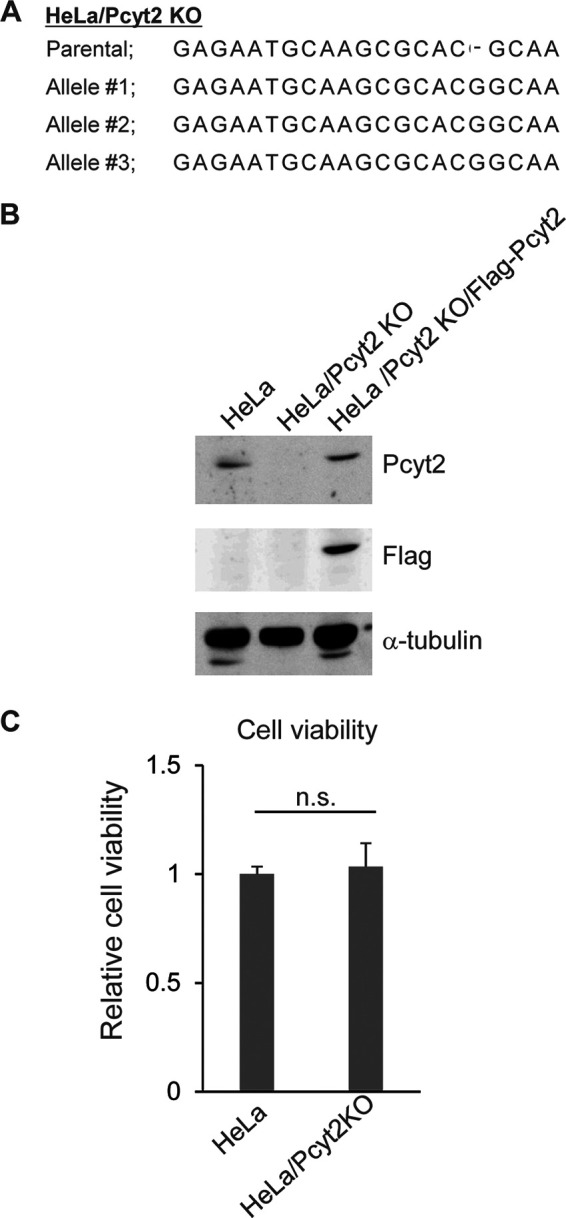
Characterization of HeLa/Pcyt2 KO cells. (A) Sequences of the Pcyt2 alleles and the parental sequence in HeLa/Pcyt2 KO cells. (B) Lysates of HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells analyzed by immunoblotting with the indicated antibodies. (C) Cell viability of HeLa and HeLa/Pcyt2 KO cells. The data are shown as the mean ± standard error of the results of 3 independent experiments and are expressed relative to the mean for HeLa cells, which was normalized to 100%. n.s., not significant by the Student's t test.
HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells were then mock infected or infected with wild-type HSV-1(F) at a multiplicity of infection (MOI) of 5. At 18 h postinfection, the infected cells were harvested and the amounts of PE and PC in the infected cells were quantitated by mass spectrometry. As shown in Fig. 2A, the amount of PE in mock-infected HeLa/Pcyt2 KO cells was smaller than that in mock-infected HeLa cells and HeLa/Pcyt2 KO/Flag-Pcyt2 cells (1.9- and 1.6-fold, respectively). Similarly, the amount of PE in HSV-1(F)-infected HeLa/Pcyt2 KO cells was smaller than that in HSV-1(F)-infected HeLa cells and HSV-1(F)-infected HeLa/Pcyt2 KO/Flag-Pcyt2 cells (1.6- and 1.4-fold, respectively) (Fig. 2A). However, these differences did not reach statistical significance. In contrast, the amount of PC in mock-infected and HSV-1(F)-infected HeLa/Pcyt2 KO cells was comparable to that in mock-infected and HSV-1(F)-infected HeLa cells and in mock-infected and HSV-1(F)-infected HeLa/Pcyt2 KO/Flag-Pcyt2 cells (Fig. 2B). These results indicated that Pcyt-2 KO specifically reduced the accumulation of PE in both mock-infected and HSV-1-infected HeLa cells.
FIG 2.
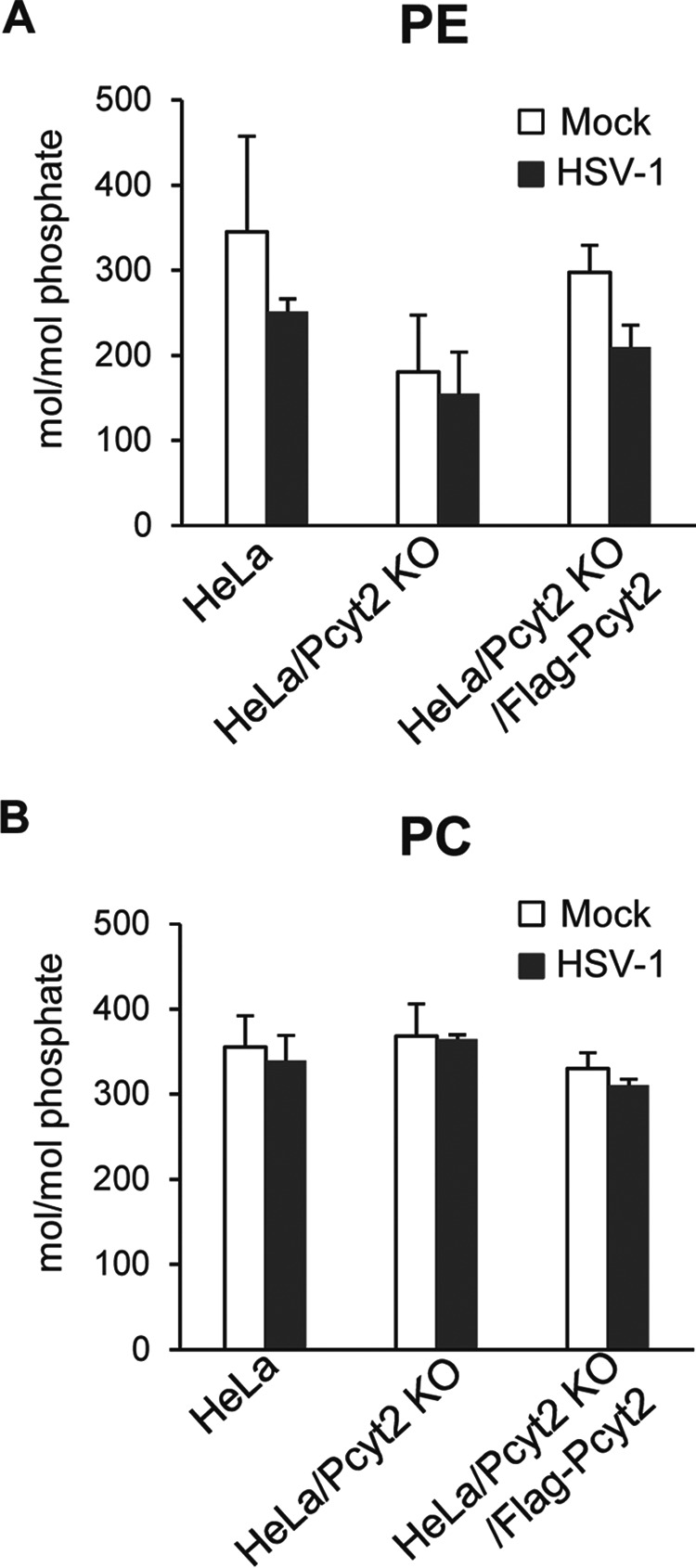
Effect of Pcyt2 on the accumulation of PE. HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells were mock infected or infected with HSV-1(F) at an MOI of 5. At 18 h postinfection, the cells were lysed and the intracellular amounts of PE (A) and PC (B) were analyzed by mass spectrometry. The data are shown as the mean ± standard error of the results of 3 independent experiments.
Effect of Pcyt2 KO on HSV-1 infection in vitro.
To investigate the effect of Pcyt2 KO on viral protein production in HSV-1-infected in cell cultures, HeLa and HeLa/Pcyt2 KO cells were mock infected or infected with HSV-1(F) at an MOI of 5, harvested 24 h postinfection, and analyzed by immunoblotting. As shown in Fig. 3, HSV-1(F)-infected HeLa/Pcyt2 KO cells accumulated viral proteins ICP4, ICP8, and glycoprotein D (gD) at levels similar to those in HeLa cells infected with HSV-1(F).
FIG 3.
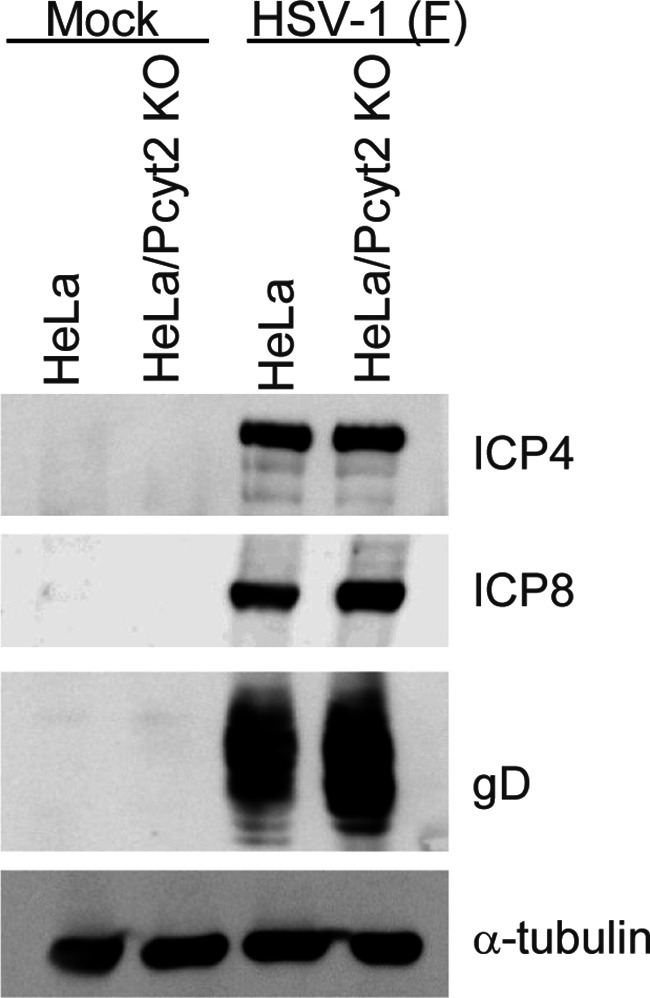
Effect of Pcyt2 on the accumulation of viral proteins. HeLa and HeLa/Pcyt2 KO cells were infected with HSV-1(F) at an MOI of 5 and harvested at 24 h postinfection. The viral proteins in these cells were then analyzed by immunoblotting with the indicated antibodies.
To investigate the effect of Pcyt2 KO on HSV-1 replication in cell cultures, HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells were infected with HSV-1(F) at an MOI of 5 or 0.01 and progeny viruses were assayed at various times postinfection. The viral growth kinetics in infected HeLa and HeLa/Pcyt2 KO cells are shown in Fig. 4A and B. HSV-1(F) growth in infected HeLa/Pcyt2 KO cells was less than that in infected HeLa cells. In particular, the progeny virus yield in HeLa/Pcyt2 KO cells infected at an MOI of 5 was 2.7-fold less at 24 h postinfection and that in HeLa/Pcyt2 KO cells infected an MOI of 0.01 was 7.9- and 5.3-fold less at 48 h and 72 h postinfection, respectively. In addition, the progeny virus yields in HSV-1-infected HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells are shown in Fig. 4C and D. The progeny virus yield in HeLa/Pcyt2 KO cells was significantly less than that in infected HeLa and HeLa/Pcyt2 KO/Flag-Pcyt2 cells. In particular, the yield was 5.1-fold less at 24 h postinfection in cells infected at an MOI of 5 (HeLa/Pcyt2 KO cells vs HeLa and HeLa/Pcyt2 KO/Flag-Pcyt2 cells) and 10- and 27-fold less at 48 h postinfection in cells infected at an MOI of 0.01 (HeLa/Pcyt2 KO cells vs HeLa or HeLa/Pcyt2 KO/Flag-Pcyt2 cells, respectively).
FIG 4.

Effect of Pcyt2 on HSV-1 growth. (A and B) HeLa and HeLa/Pcyt2 KO cells were infected with HSV-1(F) at an MOI of 5 (A) or 0.01 (B). Total virus from the cell culture supernatants and the infected cells was harvested at the indicated times postinfection and assayed on Vero cells. The data are shown as the mean ± standard error of the results of 6 (A) or 5 (B) independent experiments. *, P < 0.05 (by the unpaired Student's t test). (C and D) HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells were infected with HSV-1(F) at an MOI of 5 (C) or 0.01 (D). The total progeny virus yield from the cell culture supernatants and the infected cells at 24 h (C) and 48 h (D) postinfection was assayed on Vero cells. The data are shown as the mean ± standard error of the results of 10 (C) or 3 (D) independent experiments. *, P < 0.05 (by the Tukey test).
Taken together, these results indicated that, while Pcyt2 was not required for the accumulation of HSV-1 proteins in infected cells, it was required for efficient viral replication in cell cultures.
Effect of Pcyt2 KO on HSV-1 virion morphogenesis.
To determine the step(s) during HSV-1 replication at which Pcyt2 acts, we analyzed viral morphogenesis by quantitating the number of virus particles at different morphogenetic stages by electron microscopy of HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells infected with HSV-1(F) at an MOI of 5. As shown in Fig. 5, at 20 h postinfection, partially enveloped capsids were readily detectable in the cytoplasm of HeLa/Pcyt2 KO cells, with unenveloped and partially enveloped capsids aberrantly accumulated in the cytoplasm in these cells. In HeLa and HeLa/Pcyt2 KO/Flag-Pcyt2 cells infected with HSV-1(F), 10.9 and 11.7%, respectively, of virus particles were unenveloped or partially enveloped capsids in the cytoplasm (Fig. 5 and Table 1). However, 25.4% of the virus particles in HeLa/Pcyt2 KO cells infected with HSV-1(F) were unenveloped or partially enveloped capsids in the cytoplasm (Fig. 5 and Table 1). In addition, in HeLa and HeLa/Pcyt2 KO/Flag-Pcyt2 cells infected with HSV-1(F), 34.6 and 43.1%, respectively, of the virus particles were enveloped virions in the cytoplasm and extracellular space. However, in HeLa/Pcyt2 KO cells infected with HSV-1(F), the fraction of virus particles that were enveloped virions in the cytoplasm and extracellular space in HeLa/Pcyt2 KO cells was 23.8% (Fig. 5 and Table 1). The fractions of virus particles in the nucleus and perinuclear space in HSV-1(F)-infected HeLa (54.5%), HeLa/Pcyt2 KO (50.7%), and HeLa/Pcyt2 KO/Flag-Pcyt2 cells (45.1%) were comparable (Table 1). These results indicated that Pcyt2 KO resulted in an increase in the fraction of virus capsids in the cytoplasm and a decrease in the fraction of secondary enveloped virions in the cytoplasm and extracellular space. This phenotype has been reported to reflect a block in a process (or processes) for HSV-1 secondary envelopment (17, 23–28). Therefore, our results indicated that Pcyt2 was required for efficient HSV-1 secondary envelopment.
FIG 5.

Effect of Pcyt2 on HSV-1 morphogenesis. HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells were infected with wild-type HSV-1(F) at an MOI of 5. At 20 h postinfection, the cells were harvested, embedded, sectioned, stained, and examined by electron microscopy. The image on the lower right is a magnification of the boxed area in the image on the lower left. Arrowheads indicate partially enveloped capsids in the cytoplasm. Bars, 500 nm.
TABLE 1.
Effect of Pcyt2 KO on the distribution of virus particles in infected HeLa cells
| Cell type | % of virus particles (mean ± SE) (no. of virus particles) in morphogenetic stage of: |
Total no. of particles/cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HeLa | 50.4 ± 10.9 (635) | 4.1 ± 1.6 (52) | 10.9 ± 2.4a (138) | 6.3 ± 1.4 (79) | 28.3 ± 5.2a (357) | 1,261/13 |
| HeLa/Pcyt2 KO | 46.3 ± 5.8 (563) | 4.4 ± 2.1 (54) | 25.4 ± 4.7 (309) | 9.0 ± 1.6 (110) | 14.8 ± 3.5 (180) | 1,216/13 |
| HeLa/Pcyt2 KO/Flag-Pcyt2 | 43.2 ± 7.1 (534) | 1.9 ± 0.7 (24) | 11.7 ± 4.2a (144) | 4.3 ± 1.0 (54) | 38.8 ± 8.3a (479) | 1,235/13 |
The difference between this value and that for HeLa/Pcyt2 KO cells was statistically significant, according to the Tukey test (P < 0.05).
Subcellular localization of PE in HSV-1-infected cells.
To further investigate the linkage between Pcyt2-mediated PE biosynthesis and HSV-1 secondary envelopment, we carried out two series of experiments to analyze the subcellular localization of PE, HSV-1 envelope glycoprotein gD, and HSV-1 capsids in infected cells. In the first series of experiments, HeLa cells were mock infected or infected with HSV-1(F) at an MOI of 5, harvested at 18 h postinfection, stained using the PE-binding molecular probe duramycin (29, 30) and anti-gD antibody, and analyzed by confocal microscopy. As reported previously (31), intracellular membranous structures were detected with duramycin in mock-infected cells (Fig. 6A). In HSV-1(F)-infected cells, PE-stained structures in the cytoplasm colocalized with gD (Fig. 6A). In the second series of experiments, HeLa cells were mock infected or infected with YK601 (VenusA206K-VP26), a recombinant HSV-1(F) that expresses a small capsid protein fused to the monomeric Venus fluorescent protein (32), at an MOI of 5, harvested at 18 h postinfection, stained with duramycin, and analyzed using the Airyscan system (Zeiss), a superresolution confocal imaging system. As shown in Fig. 6B, PE-stained structures in the cytoplasm were associated with and colocalized with punctate dots of VenusA206K-VP26, which mark capsids (32). Taken together, these results suggested that PE was localized at the site of secondary envelopment in HSV-1-infected cells.
FIG 6.

Subcellular localization of PE in HSV-1-infected cells. (A) HeLa cells were mock infected or infected with wild-type HSV-1(F) at an MOI of 5. At 18 h postinfection, the cells were fixed, permeabilized, and stained with duramycin (red) or anti-gD antibody (green). Each lower image is a magnification of the boxed area in the image above it. Bars, 10 μm. (B) HeLa cells were mock infected or infected with HSV-1 YK601 (VenusA206K-VP26) at an MOI of 5. At 18 h postinfection, the cells were fixed, permeabilized, stained with duramycin, and analyzed with the Airyscan system. Each lower image is a magnification of the boxed area in the image above it. Arrowheads indicate PE-stained structures associated with capsids. Bars, 10 μm.
Effect of Pcyt2 inhibition on HSV-1 infection in vitro and in vivo.
To confirm the effect of the loss of Pcyt2 in Pcyt2 KO cells on HSV-1 infection, as described above, and to examine the role of Pcyt2 in HSV-1 infection in vivo, we used meclizine, which is a specific and direct inhibitor of Pcyt2 (33, 34). Meclizine is an antihistamine (35); therefore, we included pyrilamine, which is also an antihistamine, as a control in several experiments. First, as shown in Fig. 7A, treatment of HeLa cells with solvent, meclizine, or pyrilamine had no effect on cell viability. Second, HeLa cells were mock infected or infected with HSV-1(F) at an MOI of 5. At 6 h postinfection, the cells were treated with solvent, meclizine, or pyrilamine for a further 12 h. The infected cells were then assayed for progeny virus and analyzed by immunoblotting and electron microscopy. In agreement with the results for HeLa/Pcyt2 KO cells, treatment of HSV-1(F)-infected cells with meclizine significantly reduced the progeny virus yield, but treatment with solvent or pyrilamine did not have an effect on progeny virus yield (Fig. 7B). Of note, the decrease in virus yields in meclizine-treated HeLa cells (Fig. 7) appeared to be greater than that in HeLa/Pcyt2 KO cells (Fig. 4), suggesting that meclizine might exert additional effects such as off-target effects. Furthermore, treatment of human keratinocyte HaCaT cells with meclizine significantly reduced virus yield of wild-type HSV-1(F) without affecting cell viability (Fig. 8). Third, treatment of HSV-1(F)-infected cells with solvent, meclizine, or pyrilamine had no effect on the accumulation of ICP4, ICP8, and gD (Fig. 7C). Fourth, partially enveloped capsids were readily detectable by electron microscopy in the cytoplasm of HSV-1(F)-infected cells treated with meclizine. Meclizine treatment led to the accumulation of unenveloped and partially enveloped capsids in the cytoplasm of HSV-1-infected cells and a reduction of enveloped virions in the cytoplasm and extracellular space in these cells (Fig. 9 and Table 2). Thus, in HSV-1(F)-infected cells treated with solvent or pyrilamine, 7.0 or 4.6%, respectively, of the virus particles were unenveloped or partially enveloped capsids in the cytoplasm. However, the fraction of virus particles that were unenveloped or partially enveloped capsids in the cytoplasm of HSV-1(F)-infected cells treated with meclizine increased to 17.0% (Table 2). In contrast, in HSV-1(F)-infected cells treated with solvent or pyrilamine, 22.7 and 21.6%, respectively, of the virus particles were enveloped virions in the cytoplasm and extracellular space; however, in infected cells treated with meclizine, only 11.1% of the virus particles were enveloped virions in the cytoplasm and extracellular space (Table 2).
FIG 7.

Effect of meclizine on HSV-1 replication in HeLa cells. (A) HeLa cells were incubated in medium containing solvent, 50 μM meclizine, or 50 μM pyrilamine for 12 h, and cell viability was then measured. The data are shown as the mean ± standard error of the results of 3 independent experiments and are expressed relative to the mean for solvent-treated HeLa cells, which was normalized to 100%. n.s., not significant by the Tukey test. (B) HeLa cells were infected with HSV-1(F) at an MOI of 5. At 6 h postinfection, the cells were treated with solvent, 50 μM meclizine, or 50 μM pyrilamine for a further 12 h. The infected cells and supernatant were collected, and progeny viruses were assayed on Vero cells. The data are shown as the mean ± standard error of the results of 3 independent experiments. *, P < 0.05 (by the Tukey test). (C) HeLa cells were mock infected or infected with HSV-1(F) at an MOI of 5. At 6 h postinfection, the cells were treated with solvent, 50 μM meclizine, or 50 μM pyrilamine for a further 12 h. The cells were then harvested and analyzed by immunoblotting. The images shown are representative of 3 independent experiments.
FIG 8.
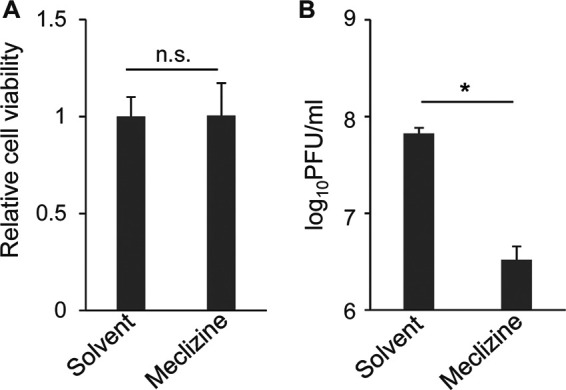
Effect of meclizine on HSV-1 replication in HaCaT cells. (A) HaCaT cells were incubated for 12 h in medium containing solvent or 50 μM meclizine, and cell viability was then measured. The data are shown as the mean ± standard error of the results of 3 independent experiments and are expressed relative to the mean for solvent-treated cells, which was normalized to 100%. n.s., not significant by the two-tailed Student's t test. (B) HaCaT cells were infected with HSV-1(F) at an MOI of 5. At 6 h postinfection, the cells were treated with solvent or 50 μM meclizine for a further 12 h. The infected cells and supernatant were collected, and progeny viruses were assayed on Vero cells. The data are shown as the mean ± standard error of the results of 3 independent experiments. *, P < 0.01 (by the two-tailed Student's t test).
FIG 9.

Effect of meclizine and pyrilamine on HSV-1 morphogenesis. HeLa cells were infected with HSV-1(F) at an MOI of 5. At 6 h postinfection, the cells were treated with solvent, 50 μM meclizine, or 50 μM pyrilamine for a further 12 h. The cells were then harvested, embedded, sectioned, stained, and examined by electron microscopy. The image on the lower right is a magnification of the boxed area in the image on the lower left. Arrowheads indicate partially enveloped capsids. The images are representative of 3 independent experiments. Bars, 500 nm.
TABLE 2.
Effect of pharmacological inhibition of Pcyt2 on the distribution of virus particles in infected HeLa cells
| Treatment | % of virus particles (mean ± SE) (no. of virus particles) in morphogenetic stage of: |
Total no. of particles/cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| Solvent | 67.6 ± 8.0 (503) | 2.7 ± 2.5 (20) | 7.0 ± 4.8a (52) | 3.2 ± 1.3 (24) | 19.5 ± 4.2a (145) | 744/12 |
| Meclizine | 70.2 ± 16.7 (565) | 1.7 ± 0.7 (14) | 17.0 ± 3.4 (137) | 4.5 ± 1.3 (36) | 6.6 ± 2.1 (53) | 805/12 |
| Pyrilamine | 72.2 ± 11.7 (608) | 1.5 ± 0.9 (13) | 4.6 ± 1.5a (39) | 2.7 ± 0.8 (23) | 18.9 ± 3.2a (159) | 842/12 |
The difference between this value and that for HeLa/Pcyt2 KO cells was statistically significant, according to the Tukey test (P < 0.05).
Finally, we carried out two series of experiments to examine the effect of meclizine on HSV-1 replication and pathogenicity in vivo. In the first series of experiments, mice were inoculated with HSV-1(F) intracranially together with solvent or meclizine (Fig. 10A). The mice were inoculated again with solvent or meclizine daily for an additional 2 days (Fig. 10A). At 3 days postinfection, brains of the infected mice were collected and virus titers were assayed (Fig. 10B). As shown in Fig. 10B, virus titers from the brains of mice treated with meclizine were significantly (3.9-fold) lower than those from mice treated with solvent. In the second series of experiments, mice were pretreated with solvent or meclizine intraperitonially (Fig. 10C). At 4 h after pretreatment, the mice were treated again intraperitonially with solvent or meclizine. Two hours later, the mice were inoculated intracranially with HSV-1(F) and with solvent or meclizine. The mice were treated intraperitonially with solvent or meclizine at 18 h postinfection and then were monitored daily (Fig. 10C). As shown in Fig. 10D, treatment of the mice with meclizine significantly improved the survival rate of the infected mice. Taken together, these results indicated that meclizine significantly inhibited HSV-1 replication and pathogenicity in infected mice, suggesting that Pcyt2 was required for efficient HSV-1 replication and pathogenicity in mice.
FIG 10.

Effects of meclizine on HSV-1 replication and pathogenicity in vivo. (A) Time course for HSV-1 infection of 3-week-old female ICR mice to analyze the intracranial viral titer after meclizine treatment. The mice were inoculated intracranially with 100 PFU HSV-1(F) together with solvent or 46 μg meclizine per mouse, and they were inoculated again with solvent or 46 μg meclizine per mouse at 1 day and 2 days postinfection. At 3 days postinfection, the brains of infected mice were collected, and virus titers were assayed on Vero cells. (B) Viral titers in the brains of mice infected with HSV-1 as described in panel A. Each data point is the virus titer in the brain of one mouse. The horizontal bars indicate the mean for each group. *, P < 0.05 (by the two-tailed Student's t test). (C) Time course for HSV-1 infection of mice to analyze mortality rates after meclizine treatment. The 3-week-old female ICR mice (n = 11 for solvent and 12 for meclizine) were pretreated intraperitonially with solvent or meclizine (200 μg/mouse). At 4 h after pretreatment, the mice were treated intraperitonially again with solvent or meclizine (200 μg/mouse). Two hours later, mice were intracranially inoculated with 100 PFU HSV-1(F) together with solvent or meclizine (46 μg/mouse). At 18 h postinfection, the mice were treated again intraperitonially with solvent or meclizine (200 μg/mouse); they were then monitored daily for 20 days. (D) Survival rates of HSV-1-infected mice described in panel C. Differences in the mortality rates of the infected mice were statistically analyzed by the log-rank test. *, P < 0.001.
DISCUSSION
In this study, we investigated the role of PE biosynthesis mediated by Pcyt2 in HSV-1 infection by knocking out the cell gene encoding Pcyt2, which is the key enzyme in one of the two major pathways for PE biosynthesis. In some studies, we used meclizine, a specific inhibitor of Pcyt2. One of the significant results of this study was that meclizine inhibited not only HSV-1 secondary envelopment and replication in cell cultures but also viral replication and pathogenicity in mice. These results indicated that the PE biosynthesis pathway, as well as the HSV-1 virion maturation pathway, could be a target for development of novel anti-HSV drugs, although many of the molecular aspects of the PE biosynthetic and HSV-1 virion maturation pathways in infected cells remain to be elucidated.
Meclizine is a first-generation piperazine H1-antihistamine that has been used as an over-the-counter drug for decades for prophylaxis against nausea and vertigo in motion sickness (35). It is notable that meclizine can be transported to the central nervous system (CNS) through the blood-brain barrier (33). Therefore, meclizine could be an interesting candidate for an alternative drug to treat HSE, since meclizine has been shown to cross the blood-brain barrier to access the CNS. Also, meclizine has been prescribed for many years, so there are adequate clinical data on the safety of this drug. However, it could be argued that it is too speculative to raise this possibility, in light of the fact that a commercially available, highly active HSV-1 inhibitor such as acyclovir does exist. Notably, once dysregulation of CNS inflammatory responses is initiated, treatment with antiherpetic drugs such as acyclovir is not sufficient to prevent fetal HSV-1 encephalitis (36–38), which results in death or severe neurological defects in a significant fraction of survivors. Therefore, it is of interest to evaluate the clinical use of meclizine for HSE patients and possibly patients with other HSV-related diseases. Furthermore, because the pathway for the secondary envelopment of HSV-1 seems to be conserved throughout the Herpesviridae viruses (17, 18, 39), meclizine may be effective for the treatment of infectious diseases caused by other herpesviruses.
The other significant result of this study was that proper host cell PE biosynthesis mediated by Pcyt2 was required for efficient HSV-1 secondary envelopment. To our best knowledge, this is the first report showing the importance of PE biosynthesis in herpesvirus infections. However, it remains uncertain at present how PE biosynthesis mediated by Pcyt2 affects efficient HSV-1 secondary envelopment in HSV-1-infected cells. It is possible that proper PE accumulation in cell membranes directly facilitates HSV-1 secondary envelopment. In general, membrane curvature is essential to initiate viral envelopment, in which infected cell membranes are deformed to wrap around nascent nucleocapsids. PE contains a polar head group with a small diameter, relative to its fatty acid chains, and this PE cone shape has been shown to form nonlamellar membrane structures, thereby modulating membrane curvature (19, 21). It has been reported that an HSV-1 heterodimeric complex consisting of the viral UL31 and UL34 proteins induces INM deformation in viral primary envelopment (16, 17). This viral complex-mediated membrane deformation in vitro is dependent on the membrane lipid composition, although the specific lipid (or lipids) required was not investigated in those studies (40–42). It has also been demonstrated that the HSV-1 envelope glycoprotein gD can deform cell membranes and that the arginine cluster in the gD cytoplasmic tail is required for efficient viral secondary envelopment in infected cells (28). Interestingly, arginine clusters in certain proteins have been shown to be involved in protein-lipid interactions (43, 44). Thus, PE in cell membranes may interact with an HSV-1 protein, such as gD, to induce membrane curvature for viral secondary envelopment.
Another possible explanation for these results may be that particular mitochondrial functions in HSV-1-infected cells could be required for efficient viral secondary envelopment. Since phosphoethanolamine is a Pcyt2 substrate, Pcyt2 inhibition results in aberrant accumulation of phosphoethanolamine. However, phosphoethanolamine is an inhibitor of mitochondrial respiration, and thus Pcyt2 inhibition downregulates mitochondrial respiration (34). It has been noted that HSV-1 secondary envelopment depends on the cellular endosomal sorting complexes required for transport III (ESCRT-III) system (45–47) and that inhibition of the activity of Vps4, an ATPase that is crucial for ESCRT-III function, causes a severe defect in HSV-1 secondary envelopment and accumulation of partially enveloped capsids in the cytoplasm (46). This was also observed in this study with Pcyt2 KO and meclizine treatment. Therefore, mitochondrial dysfunction mediated by Pcyt2 inhibition might impair VPS4 activity through an ATP shortage, thereby resulting in a defect in HSV-1 secondary envelopment. In addition, some HSV-1 proteins that act in viral secondary envelopment, such as UL7 and UL16 (23, 48, 49), have been reported to localize in mitochondria (50–53). Mitochondrial dysfunction mediated by Pcyt2 inhibition may also impair the functions of these viral proteins in viral secondary envelopment. Alternatively, there is a possibility that Pcyt2 inhibition mislocalizes PE distribution in HSV-1-infectd cells, thereby impairing viral secondary envelopment. However, this is less likely, based on the observation that Pcyt2 KO had no obvious effect on the localization of PE in HeLa cells that were mock infected or infected with wild-type HSV-1(F) (data not shown).
MATERIALS AND METHODS
Cells and viruses.
Vero, HeLa, HaCaT, and Plat-GP cells were grown as described previously (32, 54–56). Wild-type HSV-1(F) and recombinant HSV-1 YK601, encoding capsid protein VP26 fused to the VenusA206K fluorescent protein, were described previously (32).
Mass spectrometry.
HeLa cells (1 × 106) were seeded onto 60-mm plates. After 24 h, the cells were mock infected or infected with HSV-1(F) at an MOI of 5. The inocula were replaced with fresh medium at 1 h postinfection; at 18 h postinfection, the infected cells were washed with phosphate-buffered saline, scraped, collected, suspended in methanol, and stored at –20°C. Lipids were extracted from cells by the method of Bligh and Dyer (57). The extracted lipids were dried with a centrifugal evaporator, dissolved in 1:1 methanol/isopropanol, and stored at −20°C. PE and PC were quantified by liquid chromatography-electrospray ionization mass spectrometry-based lipidomics, as described previously (58).
Plasmids.
pMXs-Flag-Pcyt2-puro, a retrovirus vector that expresses Pcyt2 fused to a Flag tag, was constructed by PCR amplification of the Pcyt2 open reading frame sequence from cDNA synthesized from the total RNA of HeLa cells, as described previously (55), and cloned into pMxs-puro. Sense and antisense oligonucleotides (i.e., 5′-CACCGAGAATGCAAGCGCACG-3′ and 5′-AAACCGTGCGCTTGCATTCTC-3′) were designed for insertion into the BbsI site in the pX458 bicistronic expression vector, which expresses Cas9 and a synthetic single-guide RNA (Addgene), to produce pX459-Pcyt2. The DNA oligonucleotides were annealed and incorporated into the pX459 vector that had been linearized with the BbsI restriction enzyme.
Establishment of HeLa/Pcyt2 KO cells.
HeLa cells were transfected with pX459-Pcyt2 using Lipofectamine 2000 (Invitrogen). At 48 h posttransfection, the cells were treated with 1 μg puromycin/ml for 24 h. These cells were then diluted and seeded onto plates to make single colonies. The single colonies were picked for further analysis to identify HeLa/Pcyt2 KO cells. To determine the genotype of each allele in the HeLa/Pcyt2 KO cells, genomic DNA from these cells was amplified by PCR and sequenced. The sequenced PCR products showed that all the pcyt2 alleles were modified and carried the same sequence (Fig. 1A).
Plat-GP cells were cotransfected with pMxs-Flag-Pcyt2-puro and pMDG. At 48 h posttransfection, the transfected cell supernatants were harvested. HeLa/Pcyt2 KO cells were transduced with these supernatants and selected with 1 μg puromycin/ml. Resistant cells were designated HeLa/Pcyt2 KO/Flag-Pcyt2 cells.
Antibodies.
Mouse monoclonal antibodies against α-tubulin (DM1A; Sigma), Flag (M2; Sigma), and viral proteins gD (DL6; Santa Cruz Biotechnology), ICP8 (10A3; Millipore), and ICP4 (58S; ATCC) were used in this study. The PE-interacting agent duramycin conjugated to biotin (Molecular Targeting Technologies) was used for intracellular localization of PE.
Immunoblotting and immunofluorescence.
Immunoblotting and immunofluorescence were carried out as described previously (24, 59). For superresolution imaging, image acquisition was performed using an LSM800 microscope with Airyscan (Zeiss) and image reconstruction was carried out using ZEN2.3 software (Zeiss).
Antihistamine drug treatment.
HeLa or HaCaT cells were infected with HSV-1(F) at an MOI of 5. Infected cells were treated at 6 h postinfection with 0.05% dimethyl sulfoxide (DMSO), 50 μM meclizine dissolved in 0.05% DMSO, or 50 μM pyrilamine dissolved in 0.05% DMSO, grown for a further 12 h, and then analyzed.
Electron microscopy.
HeLa, HeLa/Pcyt2 KO, and HeLa/Pcyt2 KO/Flag-Pcyt2 cells infected with HSV-1(F) at an MOI of 5 for 20 h were examined by ultrathin-section electron microscopy as described previously (24, 60, 61). For drug treatment, HeLa cells were infected with HSV-1(F) at an MOI of 5 for 6 h, with or without treatment with the indicated drugs for a further 12 h, and analyzed. The number of virus particles at different morphogenetic stages was quantitated in randomly chosen cells.
Animal studies.
To measure virus titers in the brains of infected mice, 3-week-old female ICR mice were each inoculated intracranially with 100 PFU HSV-1(F) in a sample containing either 46 μg meclizine dissolved in 1% DMSO or 1% DMSO. At 1 day postinfection, a sample containing either meclizine (46 μg/mouse) dissolved in 1% DMSO or 1% DMSO was inoculated intracranially; this was repeated at 2 days postinfection. At 3 days postinfection, the brains of the mice were harvested and virus titers were assayed on Vero cells. In experiments to determine the survival of mice after HSV-1(F) infection, 3-week-old female ICR mice were inoculated intraperitonially with either meclizine (200 μg/mouse) dissolved in 10% Cremophor or 10% Cremophor. Four hours later, the mice were again treated intraperitonially with either meclizine (200 μg/mouse) dissolved in 10% Cremophor or 10% Cremophor. Two hours later, each mouse was inoculated intracranially with 100 PFU HSV-1(F) and either meclizine (46 μg/mouse) dissolved in 1% DMSO or 1% DMSO. At 18 h after HSV-1 infection, the mice were treated intraperitonially with either meclizine (200 μg/mouse) dissolved in 10% Cremophor or 10% Cremophor; the mice were then monitored for 20 days.
Statistical analysis.
The unpaired Student's t test was used to compare groups of two. The Tukey test was used for multiple comparisons. P values of >0.05 were considered not significant. Statistical analysis was carried out using GraphPad Prism 7 (GraphPad Software).
ACKNOWLEDGMENTS
We thank Risa Abe and Hiroshi Sagara for their excellent technical assistance.
This study was supported by Grants for Scientific Research and a Grant-in-Aid for Scientific Research (S) (grant 20H05692) from the Japan Society for the Promotion of Science, Grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology of Japan (grants 16H06433, 16H06429, 16K21723, 19H05286, and 19H05417), contract research funds from the Program of the Japan Initiative for the Global Research Network on Infectious Diseases (grant JP18fm0108006), the Research Program on Emerging and Re-emerging Infectious Diseases (grants 19fk018105h, 20wm0125002h, 20wm0225017s, and 20wm0225009h), and the Japan Program for Infectious Diseases Research and Infrastructure (grant 20wm0325005h) from the Japan Agency for Medical Research and Development, a grant from the International Joint Research Project of the Institute of Medical Science, the University of Tokyo, grants from the Takeda Science Foundation, and a grant from ONO Medical Research Foundation.
REFERENCES
- 1.Lorizate M, Krausslich HG. 2011. Role of lipids in virus replication. Cold Spring Harb Perspect Biol 3:a004820. doi: 10.1101/cshperspect.a004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan RB, Tanner L, Wenk MR. 2010. Implications for lipids during replication of enveloped viruses. Chem Phys Lipids 163:449–459. doi: 10.1016/j.chemphyslip.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ketter E, Randall G. 2019. Virus impact on lipids and membranes. Annu Rev Virol 6:319–340. doi: 10.1146/annurev-virology-092818-015748. [DOI] [PubMed] [Google Scholar]
- 4.Chukkapalli V, Heaton NS, Randall G. 2012. Lipids at the interface of virus-host interactions. Curr Opin Microbiol 15:512–518. doi: 10.1016/j.mib.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hishikawa D, Hashidate T, Shimizu T, Shindou H. 2014. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res 55:799–807. doi: 10.1194/jlr.R046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amara A, Mercer J. 2015. Viral apoptotic mimicry. Nat Rev Microbiol 13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, Bu X, Farzan M, Freeman GJ, Umetsu DT, Dekruyff RH, Choe H. 2013. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog 9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, Chen IS. 2011. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9:286–298. doi: 10.1016/j.chom.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. 2006. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol 80:10109–10116. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard AS, Zhang A, Park SJ, Farzan M, Zong M, Choe H. 2015. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc Natl Acad Sci U S A 112:14682–14687. doi: 10.1073/pnas.1508095112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adu-Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA, Gratton E, Tessier CR, Stahelin RV. 2015. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J Virol 89:9440–9453. doi: 10.1128/JVI.01087-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. 2010. Targeting of murine leukemia virus Gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J Virol 84:503–515. doi: 10.1128/JVI.01134-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishe B, Syed GH, Field SJ, Siddiqui A. 2012. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J Biol Chem 287:27637–27647. doi: 10.1074/jbc.M112.346569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed. Lippincott-Williams &Wilkins, Philadelphia, PA. [Google Scholar]
- 16.Mettenleiter TC. 2016. Vesicular nucleo-cytoplasmic transport-herpesviruses as pioneers in cell biology. Viruses 8:266. doi: 10.3390/v8100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 18.Henaff D, Radtke K, Lippe R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449. doi: 10.1111/j.1600-0854.2012.01399.x. [DOI] [PubMed] [Google Scholar]
- 19.Calzada E, Onguka O, Claypool SM. 2016. Phosphatidylethanolamine metabolism in health and disease. Int Rev Cell Mol Biol 321:29–88. doi: 10.1016/bs.ircmb.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel D, Witt SN. 2017. Ethanolamine and phosphatidylethanolamine: partners in health and disease. Oxid Med Cell Longev 2017:4829180. doi: 10.1155/2017/4829180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vance JE, Tasseva G. 2013. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Pavlovic Z, Bakovic M. 2013. Regulation of phosphatidylethanolamine homeostasis: the critical role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Int J Mol Sci 14:2529–2550. doi: 10.3390/ijms14022529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albecka A, Owen DJ, Ivanova L, Brun J, Liman R, Davies L, Ahmed MF, Colaco S, Hollinshead M, Graham SC, Crump CM. 2017. Dual function of the pUL7-pUL51 tegument protein complex in herpes simplex virus 1 infection. J Virol 91:e02196-16. doi: 10.1128/JVI.02196-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2018. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9:3379. doi: 10.1038/s41467-018-05889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oda S, Arii J, Koyanagi N, Kato A, Kawaguchi Y. 2016. The interaction between herpes simplex virus 1 tegument proteins UL51 and UL14 and its role in virion morphogenesis. J Virol 90:8754–8767. doi: 10.1128/JVI.01258-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farnsworth A, Goldsmith K, Johnson DC. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J Virol 77:8481–8494. doi: 10.1128/jvi.77.15.8481-8494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol 81:319–331. doi: 10.1128/JVI.01842-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arii J, Shindo K, Koyanagi N, Kato A, Kawaguchi Y. 2016. Multiple roles of the cytoplasmic domain of herpes simplex virus 1 envelope glycoprotein D in infected cells. J Virol 90:10170–10181. doi: 10.1128/JVI.01396-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu K, Nagy PD. 2015. RNA virus replication depends on enrichment of phosphatidylethanolamine at replication sites in subcellular membranes. Proc Natl Acad Sci U S A 112:E1782–E1791. doi: 10.1073/pnas.1418971112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwamoto K, Hayakawa T, Murate M, Makino A, Ito K, Fujisawa T, Kobayashi T. 2007. Curvature-dependent recognition of ethanolamine phospholipids by duramycin and cinnamycin. Biophys J 93:1608–1619. doi: 10.1529/biophysj.106.101584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ireland R, Schwarz B, Nardone G, Wehrly TD, Broeckling CD, Chiramel AI, Best SM, Bosio CM. 2018. Unique Francisella phosphatidylethanolamine acts as a potent anti-inflammatory lipid. J Innate Immun 10:291–305. doi: 10.1159/000489504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. 2010. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol 28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gohil VM, Zhu L, Baker CD, Cracan V, Yaseen A, Jain M, Clish CB, Brookes PS, Bakovic M, Mootha VK. 2013. Meclizine inhibits mitochondrial respiration through direct targeting of cytosolic phosphoethanolamine metabolism. J Biol Chem 288:35387–35395. doi: 10.1074/jbc.M113.489237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen B, DeJong JM. 1972. Meclizine and placebo in treating vertigo of vestibular origin: relative efficacy in a double-blind study. Arch Neurol 27:129–135. doi: 10.1001/archneur.1972.00490140033006. [DOI] [PubMed] [Google Scholar]
- 36. Reference deleted. [Google Scholar]
- 37.Whitley RJ, Gnann JW. 2002. Viral encephalitis: familiar infections and emerging pathogens. Lancet 359:507–513. doi: 10.1016/S0140-6736(02)07681-X. [DOI] [PubMed] [Google Scholar]
- 38.Lundberg P, Ramakrishna C, Brown J, Tyszka JM, Hamamura M, Hinton DR, Kovats S, Nalcioglu O, Weinberg K, Openshaw H, Cantin EM. 2008. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 82:7078–7088. doi: 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pellet PE, Roizman B. 2013. Herpesviridae, p 1802–1822. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed. Lippincott-Williams &Wilkins, Philadelphia, PA. [Google Scholar]
- 40.Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. 2015. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bigalke JM, Heldwein EE. 2015. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34:2921–2936. doi: 10.15252/embj.201592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazerik JN, Tyska MJ. 2012. Myosin-1A targets to microvilli using multiple membrane binding motifs in the tail homology 1 (TH1) domain. J Biol Chem 287:13104–13115. doi: 10.1074/jbc.M111.336313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi S, Pryciak PM. 2007. Identification of novel membrane-binding domains in multiple yeast Cdc42 effectors. Mol Biol Cell 18:4945–4956. doi: 10.1091/mbc.e07-07-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kharkwal H, Smith CG, Wilson DW. 2014. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J Virol 88:14467–14478. doi: 10.1128/JVI.02777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 81:7380–7387. doi: 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol 83:11254–11264. doi: 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roller RJ, Fetters R. 2015. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J Virol 89:3112–3122. doi: 10.1128/JVI.02799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chadha P, Sarfo A, Zhang D, Abraham T, Carmichael J, Han J, Wills JW. 2017. Domain interaction studies of herpes simplex virus 1 tegument protein UL16 reveal its interaction with mitochondria. J Virol 91:e01995-16. doi: 10.1128/JVI.01995-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. 2007. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep 8:188–193. doi: 10.1038/sj.embor.7400878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka M, Sata T, Kawaguchi Y. 2008. The product of the herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol J 5:125. doi: 10.1186/1743-422X-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murata T, Goshima F, Daikoku T, Inagaki-Ohara K, Takakuwa H, Kato K, Nishiyama Y. 2000. Mitochondrial distribution and function in herpes simplex virus-infected cells. J Gen Virol 81:401–406. doi: 10.1099/0022-1317-81-2-401. [DOI] [PubMed] [Google Scholar]
- 54.Shindo K, Kato A, Koyanagi N, Sagara H, Arii J, Kawaguchi Y. 2016. Characterization of a herpes simplex virus 1 (HSV-1) chimera in which the Us3 protein kinase gene is replaced with the HSV-2 Us3 gene. J Virol 90:457–473. doi: 10.1128/JVI.02376-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 56.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/jvi.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka Y, Shimanaka Y, Caddeo A, Kubo T, Mao Y, Kubota T, Kubota N, Yamauchi T, Mancina RM, Baselli G, Luukkonen P, Pihlajamaki J, Yki-Jarvinen H, Valenti L, Arai H, Romeo S, Kono N. 2020. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut doi: 10.1136/gutjnl-2020-320646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeshima K, Arii J, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2019. Identification of the capsid binding site in the herpes simplex virus 1 nuclear egress complex and its role in viral primary envelopment and replication. J Virol 93:e01290-19. doi: 10.1128/JVI.01290-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arii J, Takeshima K, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2019. Roles of the interhexamer contact site for hexagonal lattice formation of the herpes simplex virus 1 nuclear egress complex in viral primary envelopment and replication. J Virol 93:e00498-19. doi: 10.1128/JVI.00498-19. [DOI] [PMC free article] [PubMed] [Google Scholar]