

Herpes simplex virus 1 (HSV-1) is the causative agent of several pathologies ranging in severity from the common cold sore to life-threatening encephalitic infection. A critical step during productive HSV-1 infection is the cleavage and packaging of replicated, concatemeric viral DNA into preformed capsids. A key knowledge gap is how the capsid engages the replicated viral genome and the subsequent packaging of a unit-length HSV genome. Here, biochemical and structural studies focused on the unique portal vertex of wild-type HSV and packaging mutants provide insights into the mechanism of HSV genome packaging. The significance of our research is in identifying the portal proteins pUL6 and pUL17 as key viral factors for engaging the terminase complex with the capsid and the subsequent cleavage, packaging, and stable incorporation of the viral genome in the HSV-1 capsid.
KEYWORDS: HSV-1, pUL6 portal protein, pUL17, CVSC, genome packaging, cryo-electron microscopy, cryoEM
ABSTRACT
The packaging of DNA into preformed capsids is a critical step during herpesvirus infection. For herpes simplex virus, this process requires the products of seven viral genes: the terminase proteins pUL15, pUL28, and pUL33; the capsid vertex-specific component (CVSC) proteins pUL17 and pUL25; and the portal proteins pUL6 and pUL32. The pUL6 portal dodecamer is anchored at one vertex of the capsid by interactions with the adjacent triplexes as well as helical density attributed to the pUL17 and pUL25 subunits of the CVSC. To define the roles and structures of the CVSC proteins in virus assembly and DNA packaging, we isolated a number of recombinant viruses expressing pUL25, pUL17, and pUL36 fused with green or red fluorescent proteins as well as viruses with specific deletions in the CVSC genes. Biochemical and structural studies of these mutants demonstrated that (i) four of the helices in the CVSC helix bundle can be attributed to two copies each of pUL36 and pUL25, (ii) pUL17 and pUL6 are required for capsid binding of the terminase complex in the nucleus, (iii) pUL17 is important for determining the site of the first cleavage reaction generating replicated genomes with termini derived from the long-arm component of the herpes simplex virus 1 (HSV-1) genome, (iv) pUL36 serves no direct role in cleavage/packaging, (v) cleavage and stable packaging of the viral genome involve an ordered interaction of the terminase complex and pUL25 with pUL17 at the portal vertex, and (vi) packaging of the viral genome results in a dramatic displacement of the portal.
IMPORTANCE Herpes simplex virus 1 (HSV-1) is the causative agent of several pathologies ranging in severity from the common cold sore to life-threatening encephalitic infection. A critical step during productive HSV-1 infection is the cleavage and packaging of replicated, concatemeric viral DNA into preformed capsids. A key knowledge gap is how the capsid engages the replicated viral genome and the subsequent packaging of a unit-length HSV genome. Here, biochemical and structural studies focused on the unique portal vertex of wild-type HSV and packaging mutants provide insights into the mechanism of HSV genome packaging. The significance of our research is in identifying the portal proteins pUL6 and pUL17 as key viral factors for engaging the terminase complex with the capsid and the subsequent cleavage, packaging, and stable incorporation of the viral genome in the HSV-1 capsid.
INTRODUCTION
The herpes simplex virus 1 (HSV-1) capsid is an elaborate and multifunctional assembly comprising hundreds of copies of the major capsid protein, pUL19, organized with icosahedral geometry as pentamers (pentons) at all but one of the vertices and hexamers (hexons) in between, while heterotrimeric “triplex” molecules bind at external sites of local 3-fold symmetry between these capsomers. The 12th vertex is occupied by a portal complex that is essential for the packaging and cleavage of replicated viral DNA into the preformed capsid. This process requires the products of seven viral genes in addition to the capsid itself, and most of these gene products have been assigned functions based on genetic and biochemical studies. The portal itself is a dodecamer of the pUL6 protein that forms a channel through which viral DNA enters and exits the capsid and is also proposed to nucleate capsid formation (1–7). The pUL15, pUL28, and pUL33 proteins comprise the viral terminase complex that functions to package and cleave the viral genome (8, 9) while the pUL17 and pUL25 proteins form part of the complex termed the capsid vertex-specific component (CVSC) (10–12) that is required for both DNA cleavage and stable packaging of DNA-filled capsids (13). The UL32 protein is the least well characterized of the seven but may play a role in localizing capsids to sites of DNA packaging (14). In this report, we focus on the critical functions of the CVSC molecule at the portal vertex and its organization and interactions with other members of the DNA packaging machinery.
The CVSC is essential for several steps in virus infection including (i) microtubule-mediated trafficking of the capsid to the nucleus, (ii) capsid interaction with nuclear pores and release of the viral genome into the nucleus, (iii) DNA packaging, and (iv) egress of the mature DNA-filled capsid (1, 15–18). Structural studies of the CVSC complex have focused on the icosahedrally symmetric penton vertices where the pUL17 and pUL25 subunits form a star-shaped density that extends from the top of each penton to the adjacent triplexes and hexons. Although originally considered to be a component of the tegument, the CVSC consists of proteins possessing both capsid and tegument properties that bridge the two component architectures together. A third essential component of the CVSC around pentons is pUL36 (19, 20), a large viral protein that may recruit tegument to the mature capsid (10, 21). Recent high-resolution reconstructions of capsids determined from cryo-electron microscopy (cryoEM) images of HSV-1 and pseudorabies virus (PRV) virions revealed a bundle of 5 helices on the capsid exterior bridging two triplexes and extending toward the adjacent penton (20, 22), and a similar helical bundle has also been observed in the Kaposi’s sarcoma-associated herpesvirus (KSHV) capsid (23). The bundle is most likely composed of helices from pUL17, pUL25, and pUL36, although the stoichiometry remains unclear (19, 20).
The unique portal vertex was not observed in these cryoEM reconstructions since icosahedral symmetry was imposed to maximize resolution and contrast. Consequently, the presence of the CVSC at the portal was inferred only by the requirement of its subunits for packaging, cleavage, and retention of the genome. However, several recent studies of virion capsids were successful in visualizing the portal vertex by discriminating it from the other 11 penton vertices to allow correct nonicosahedral alignment of the cryoEM images and applying only 5-fold symmetry or no symmetry around the portal axis (6, 7). The first such structure at 8-Å resolution revealed that the pUL6 portal dodecamer is anchored to the capsid by interactions with the five peripentonal triplexes and density corresponding to pUL17. The 5-helix bundle of the CVSC was clearly visible, confirming that CVSC subunits are also bound around the portal vertex where they participate in the packaging, cleavage, and retention of the viral genome (6, 7). However, neither capsid structure could definitively identify the proteins making up the density at this vertex. A more recent cryoEM structure of the virion capsid from Kaposi’s sarcoma-associated herpesvirus (KSHV), a gammaherpesvirus, suggested that the triplex molecule nearest the portal was rotated ∼120° compared to those at the equivalent position around pentons (5), a finding confirmed in HSV-2 (24), suggesting that the CVSC binding site around vertices differs not only by the occupancy of a penton or portal but perhaps also by the underlying triplex orientation. This difference may not only affect the binding affinity of CVSC subunits but also govern the functional role they play.
HSV-infected cells contain four types of nucleocapsids. The first closed capsid structure is the procapsid that is fragile and difficult to isolate but is the precursor for the other three stable capsid types (A-, B-, and C-capsids) that accumulate in the nucleus (13, 25). These stable capsids can be distinguished morphologically in electron micrographs and are separable on sucrose density gradients. They differ primarily in the material present inside the capsid cavity: C-capsids contain the viral genome while A-capsids lack both DNA and protein and B-capsids contain VP22a, the cleaved form of the scaffolding protein. Replication of viral DNA results in the formation of head-to-tail concatemers that are first cleaved to generate the L-end of the viral genome, and once a genome-sized DNA molecule is packaged, a second cleavage generates the S-end of the genome (26–28). The DNA is packaged into procapsids to generate C-capsids, which are the only capsid form that exits the nucleus (13, 25). Empty A-capsids are thought to result from abortive attempts at DNA encapsidation, while B-capsids never entered the packaging pathway (15, 16).
Viruses lacking each of the CVSC components have been examined biochemically and structurally to probe the interactions between them and the underlying capsid surface and their functions in DNA packaging. Each of the pUL17-, pUL25-, and pUL36-null viruses shows a unique phenotype. DNA cleavage requires pUL17, and only nuclear B-capsids and uncleaved viral DNA are found in pUL17-null-infected cells (29). The same phenotype is found with any of the terminase (pUL15, pUL28, and pUL33) null viruses or with the portal (pUL6)- and pUL32-null viruses (3, 30–34). Capsid-bound pUL25 is not required for DNA cleavage but functions to stabilize capsids after DNA is packaged; pUL25-null virus yields abundant levels of both nuclear A- and B-capsids and no C-capsids (15, 16). pUL36 is not required for either cleavage or packaging of the viral genome; A- and C-capsids are present in the nucleus, and DNA-filled C-capsids accumulate in the cytoplasm with the pUL36-null virus (17). The encapsidation of HSV-1 DNA is mediated by the terminase complex, which is involved with the recognition of packaging signals present in the replicated viral genomes and the association with procapsids via an interaction at the capsid portal vertex. Terminase initiates packaging by cleaving the viral DNA at a specific sequence (pac2) present between adjacent genomes within concatemers and following packaging of a unit length genome cleaves at a second packaging signal (pac1). The three terminase proteins pUL15, pUL28, and pUL33 are detected in nuclear A- and B-capsids but not in nuclear C-capsids, suggesting that they are lost once DNA is packaged. The structure of the portal vertex determined from purified virions (5, 6) revealed density of the portal dodecamer as well as of a unique 5-fold symmetric assembly. The pUL25 C-terminal domains that bind pentons at the pentonal vertices are instead proposed to be positioned differently at the portal, giving rise to a small tail-like assembly on the exterior of the portal vertex in a position where they could act to prevent release of the viral genome from the capsid.
The focus of the current study is on determining the functions of the CVSCs and their interactions with the portal vertex and with the terminase complex in cleavage and retention of the packaged viral genome. Our results demonstrate that pUL17 anchors the terminase complex to the capsid and is important for determining the site of the first cleavage reaction, and after packaging is complete, pUL17 interacts with pUL25 at the portal to retain the viral genome.
RESULTS
Isolation and characterization of HSV-1 mutants expressing CVSC fusion proteins.
Higher-resolution reconstructions of virion capsids have revealed that the CVSC density includes a 5-helix bundle bridging two triplexes and extending toward the penton (20). Sections through this bundle revealed four long helices and a fifth shorter helix, which were tentatively assigned as domains from each of pUL17, pUL25, and pUL36 (20). Recent studies have proposed that two copies of pUL25 each contribute one long helix to the bundle (22, 35), but the proposed origin of the five helices is based only on predictions from cryoEM structures and protein sequences. Here, we test the stoichiometry experimentally by tagging the pUL25 and pUL36 with bulk labels for cryoEM visualization. We are particularly interested in the contribution of pUL36, which, unlike pUL25, has no structural data confirming its participation in the CVSC molecule or its interactions with the capsid.
We previously reported on the structure of a pUL25-green fluorescent protein (GFP) fusion mutant, vFH422, with GFP inserted between amino acids 50 and 51 of the pUL25 protein (16). We have now developed two additional HSV-1 mutants: vFH561, in which the 717-bp GFP gene is fused to the C terminus of the UL36 open reading frame, and vFH679, which encodes both the pUL25-GFP fusion as before and pUL36-red fluorescent protein (RFP) with RFP appended to the C terminus (Table 1). The new fusion proteins were introduced into the viral genome through genetic manipulation of an HSV-1 (KOS) genome maintained in a recombinant bacterial artificial chromosome (BAC). The BACs were transfected into Vero cells, and the recovered virus was plaque purified and titrated on Vero cells. Both mutant viruses produced high-titer stocks on Vero cells (Table 1). In one-step growth analyses, vFH561 and vFH679 showed reduced virus propagation on Vero cells compared to wild-type KOS with titers down approximately 0.6 and 0.3 logs, respectively (Fig. 1). Vero cells were infected with either KOS, vFH561, or vFH679, and virions were isolated by sucrose gradient centrifugation from the medium of the infected cells. The virion band was collected with a syringe by side puncture of the gradient, virions were pelleted, and their protein compositions were determined on stained gels and by Western blotting (Fig. 2). With the exception of pUL36, there was no difference in the protein profiles for KOS, vFH561, or vFH679 virions by SDS-PAGE and imperial blue staining. The UL36 fusion proteins for vFH561 and vFH679 migrated slightly more slowly than the KOS wild-type (wt) UL36 protein due to the fusion of 25-kDa GFP or RFP domains added to the C terminus of pUL36 (Fig. 2). Western blot analysis confirmed that these were indeed UL36 fusion proteins, as identical blots were probed with either a pUL36, pUL25, GFP, or RFP antibody (Fig. 2). The pUL36-GFP and pUL36-RFP fusion proteins were detected with both the anti-pUL36 antibody and either anti-GFP (vFH561) or anti-RFP (vFH679) antibodies, respectively. Virions isolated from cells infected with the pUL25-GFP insertion mutant, vFH422 (16), were also included to compare with the same pUL25-GFP fusion protein expressed from the vFH679 double mutant virus. A protein approximately 25 kDa larger than wild-type pUL25 was detected with both the anti-pUL25 and anti-GFP antibodies. Taken together, these results demonstrate that virus replication was not significantly altered by either the expression of pUL36-GFP fusion proteins alone or the expression of both pUL25-GFP and pUL36-RFP fusion proteins together.
TABLE 1.
Recombinant HSV used in this studya
| Virus | Protein tags and gene deletions | Titer on Vero cells (PFU/ml) | Titer on complementing cell line (PFU/ml) | Reference(s) for virus and/or complementing cells: |
|---|---|---|---|---|
| vFH422 | pUL25-GFP | 2 × 109 | ND | Virus: Cockrell et al. (16) |
| vFH561 | pUL36-GFP | 6 × 109 | ND | Virus: this study |
| vFH679 | pUL25-GFP, pUL36-RFP | 3 × 109 | ND | Virus: this study |
| vFH479 | UL6-null | <104 | 5.9 × 108 on 31 cells | Virus: this study; cells: Lamberti and Weller (3) |
| Hr81-1 | UL15-null | <104 | 5.2 × 109 on C2 cells | Virus + cells: Yu et al. (57) |
| vFH632 | UL17-null | <104 | 2 × 108 on G5 cells | Virus: this study; cells: Desai et al. (75) |
| vFH439 | UL25-null | <104 | 4.5 × 109 on 8-1 cells | Virus: Cockrell et al. (16); cells: McNab et al. (58) |
| GCB | UL28-null | <104 | 7 × 108 on CV28 cells | Virus: Tengelsen et al. (33); cells: Yang and Baines (85) |
| hr64 | UL32-null | <104 | 2.8 × 109 on 158 cells | Virus + cells: Lamberti and Weller (14) |
| ΔUL33 | UL33-null | <104 | 2 × 109 on D4 cells | Virus: Cunningham and Davison (34); cells: Salmon et al. (56) |
| ARΔUL36 | UL36-null | <104 | 1.1 × 109 on HAUL36-1 cells | Virus + cells: Roberts et al. (21) |
| vFH693 | UL17i | <104 | 2 × 109 on G5 cells | Virus: this study; cells: Desai et al. (75) |
Virus stocks were titered on Vero cells or complementing cells expressing the indicated HSV protein. ND, not done.
FIG 1.
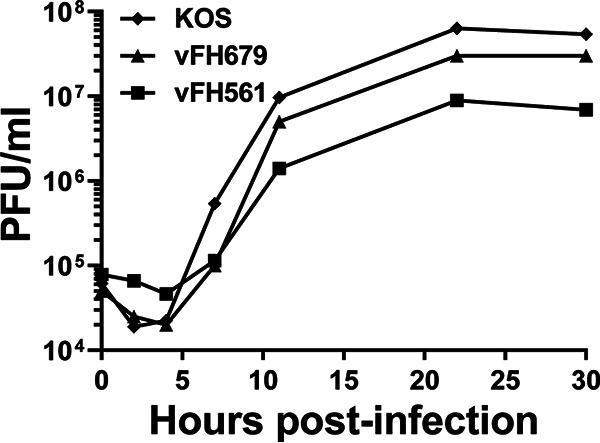
Single-step growth of viruses expressing pUL25 and pUL36 fusion proteins. Vero cells were infected with KOS or viruses expressing UL36-GFP (vFH561) or both UL36-RFP and UL25-GFP (vFH679) at an MOI of 5 at 4°C for 1 h and incubated at 37°C until the indicated time points. Cells were lysed by multiple freeze-thaws, and viral progeny were quantified by plaque assay on Vero cells. The results shown are averages from duplicate experiments.
FIG 2.
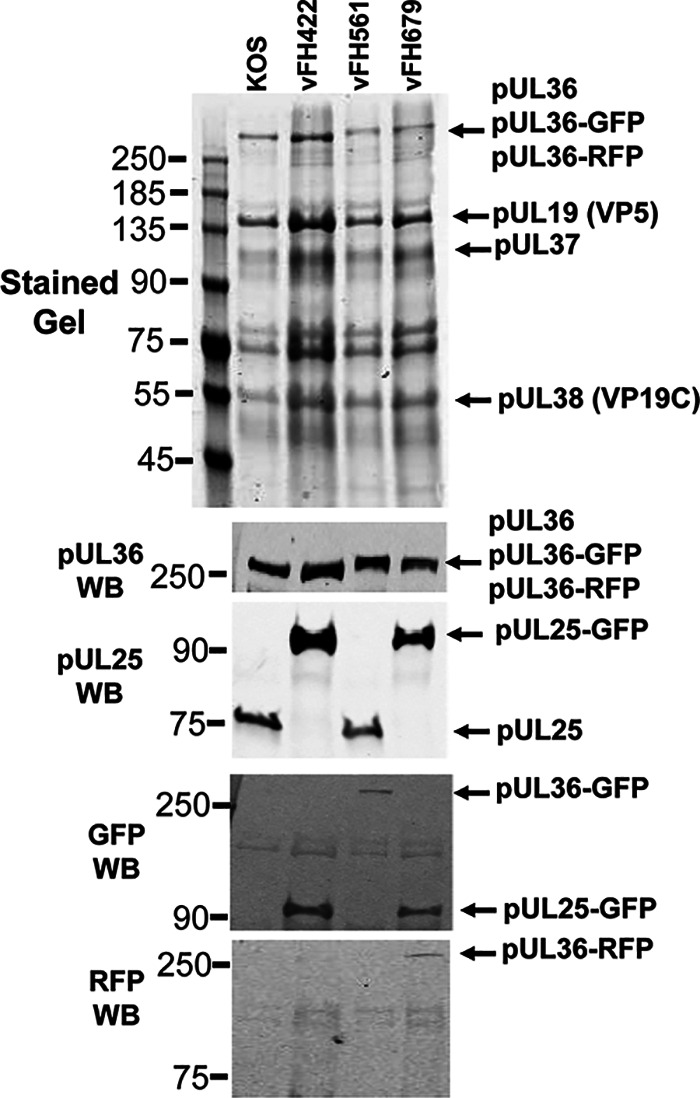
Detection of pUL25 and pUL36 fusion proteins in virions. Virions isolated from Vero cells infected with KOS or the pUL25-GFP (vFH422), pUL36-GFP (vFH561), or pUL25-GFP/pUL36-RFP (vFH679) viruses were analyzed by SDS-PAGE and immunoblotting. (Top) Vero cells infected with the indicated viruses at an MOI of 5 PFU per cell were harvested at 18 h postinfection, and virions were isolated from the medium on 20% to 60% sucrose gradients. Equivalent amounts of virions were separated by SDS-PAGE, and the proteins were detected by Coomassie blue staining. The positions of the HSV virion proteins are indicated on the right. (Bottom) Immunoblot analysis of virions with pUL36, pUL25, GFP, or RFP antibodies. Molecular mass standards are given in kilodaltons. WB, Western blot.
CVSC stoichiometry revealed by cryoEM structures of GFP- and RFP-tagged subunits.
The fusion protein-expressing mutant viruses vFH561 (pUL36-GFP) and vFH679 (pUL25-GFP and pUL36-RFP) (Table 1) were imaged by cryoEM. Capsid structures were calculated with icosahedral symmetry imposed to locate the tagged residues so that the sources of the helices in the triplex-bridging helical bundle density of CVSC could be determined. Surface views from corresponding areas of these maps, as well as from our previous wild-type map (20) and from our existing mutant vFH422 (pUL25-GFP) (10, 16), are shown in Fig. 3 together with the locations of the fusion proteins within the viral protein sequences. The additional density of the labels is evident in the density maps of the mutants, although in all cases the quality of the tag density is lower than that of the capsid due to mobility. Nonetheless, the UL25-GFP mutant shows clear evidence for two tags, consistent with the recent proposition that two copies of pUL25 are present in the CVSC molecule (23, 35). It should be noted that we previously interpreted a reconstruction of this same mutant as representing one copy of the label in two relatively stable positions 180° apart as the label region is diffuse due to flexibility (10).
FIG 3.

Composition of the CVSC helix bundle probed by cryoEM of fusion protein-labeled capsids. (A) The locations of insertions into the pUL25 and pUL36 sequences are indicated. Note that GFP-labeled pUL36 was used in the single mutant vFH561, but the double mutant vFH679 used RFP-labeled pUL36. (B) Views of the CVSC molecule in capsid reconstructions from virions isolated from Vero cells infected by KOS and each of the three mutants. Additional density in the mutant maps is colored green for the pUL25 label and red for the pUL36 label. Both of the single-mutant maps reveal two extra density regions, indicating that the two copies of the labeled protein, pUL25 or pUL36, are present in each case. The double mutant appears to show the crowding of four labels in close proximity. (C) A model of CVSC organization incorporating two copies each of pUL25 and pUL36 and one copy of pUL17, accommodating the observation that the helix bundle in the CVSC molecule comprises 5 helical rods (20). The fusion labels are colored green (pUL25) and red (pUL36) and indicate the approximate locations of corresponding density in panel B and the positions on the proteins they label.
The single-label vFH561 pUL36-GFP mutant similarly shows two distinct but well-separated density labels connected to the CVSC density, indicating that the C-terminal-most region of pUL36 is also present in two copies per CVSC molecule. The C terminus of pUL36 is predicted to fold as alpha-helix and could thus account for two of the helices in the CVSC helix bundle. The connectivity of pUL36 tags suggests that they connect to two helices directly beneath those tagged by the pUL25 label. This organization would account for the relative separation of the pUL36-GFP tags, and the constraints of the surrounding capsid structures, especially the neighboring hexons (excised from Fig. 3 for clarity), may explain the small relative stagger along the helix bundle. The double-label vFH679 mutant shows the features of the two single-label maps combined, although crowding of the GFP and RFP labels likely prevents the appearance of a simple superposition. Nonetheless, we believe this map to be consistent with the four labels representing two copies of the pUL25 tag and two copies of the pUL36 tag.
The locations of the tagged residues in the CVSC density are indicated in a schematic that updates our understanding of CVSC organization (Fig. 3C). As each of the four tags is associated with a different helix in the CVSC bundle, we assign the remaining helix to pUL17 in the absence of evidence that any other protein is involved. In this model, the two C-terminal helices of pUL36 are intimate partners with pUL25 in making up the bulk of the helix bundle, and they interface between pUL25 and the underlying pUL17, explaining why the absence of either pUL25 or pUL36 abolishes the bundle from density maps (19, 20).
Capsid-terminase binding requires CVSC subunit pUL17, portal pUL6, and terminase subunit pUL28.
We have shown that pUL25 is anchored to the capsid at penton vertices through N-terminal interactions with pUL17 and that the C-terminal domain contacts the penton directly (20). However, the organization of pUL25 and the other CVSC subunits around the portal vertex is likely to be different. A recent structure of the HSV-1 virion portal vertex suggested that 10 copies of the C-terminal domain of pUL25 were located on the exterior surface of the portal (6). These pUL25 subunits are arranged putatively as two tiers of star-shaped density above the portal (35), supporting the genetic and biochemical studies demonstrating pUL25’s essential role in retaining the viral genome inside capsids (5, 6). However, since the terminase complex is responsible for packaging and cleavage of the viral genome, a question arises as to the order in which terminase and CVSC subunits associate with the portal. To address this, we purified intranuclear capsids from Vero cells infected with wild-type (wt) KOS virus and isolated A-, B-, and C-capsids for immunoblot analysis to detect the presence of the three terminase proteins (Fig. 4). The 81-kDa pUL15, 85-kDa pUL28, and 14-kDa pUL33 were detected in gradient fractions containing A- and B-capsids, but only pUL33 was found on the C-capsid fraction but at significantly reduced levels compared to A- and B-capsids (Fig. 4, bottom). These findings confirm that the two larger terminase proteins do not remain associated with the capsid after the viral genome is packaged (36).
FIG 4.
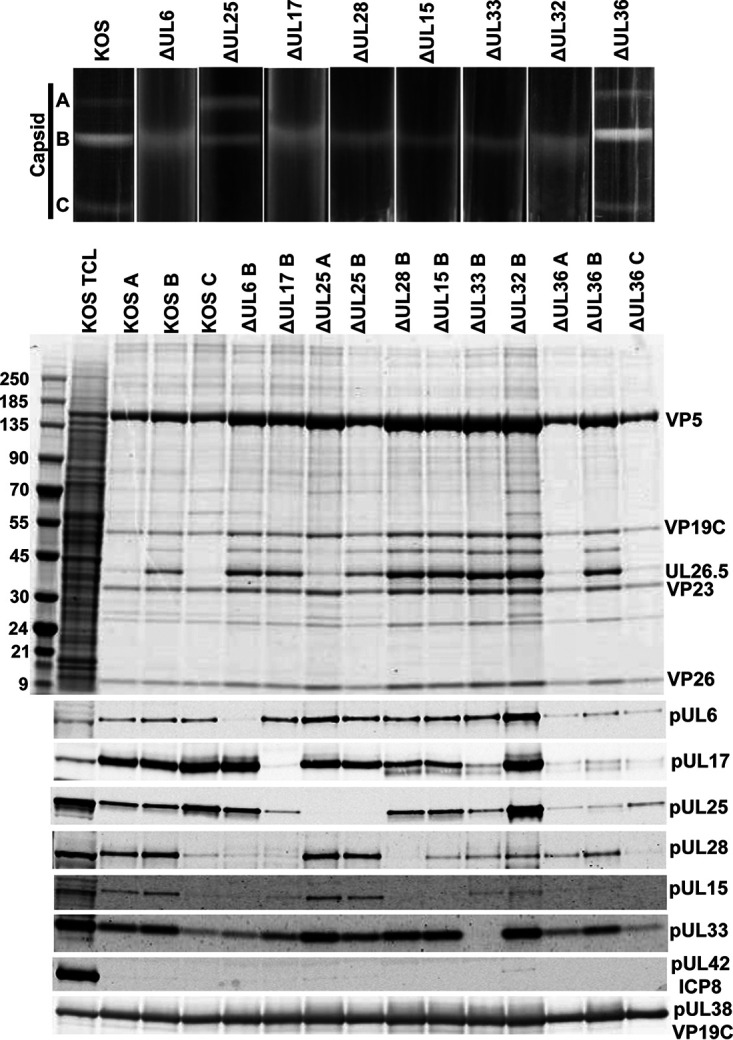
Analysis of capsid-bound packaging proteins. (Top) Rate-velocity sedimentation of capsids. Cells were infected with KOS or the indicated packaging mutants. Vero cells were infected with 10 PFU per cell for 18 h and nuclear lysates were layered onto 20% to 60% sucrose gradients and centrifuged at 24,000 rpm for 1 h. The positions of A-, B-, and C-capsid bands are indicated. (Center) Coomassie blue-stained gel of capsids (A, B, or C) isolated from KOS and the indicated packaging mutants. Positions of the major capsid protein (VP5), triplex proteins (VP19C and VP23), and scaffold protein (UL26.5) are indicated; KOS TCL, total cell lysate. (Bottom) Western blots of the capsid fractions probed with antibodies for individual packaging proteins. ICP8 (DNA replication protein) and pUL38 (capsid protein) serve as negative and positive controls, respectively. Molecular mass standards (kDa).
We expected that pUL25 would not be required for terminase-capsid interactions since it is not required for cleavage of the replicated genome. Western blot analyses of the nuclear A- and B-capsids isolated from cells infected with the UL25-null virus showed that pUL15, pUL28, and pUL33 were present (Fig. 4, bottom). Similar results were found with the UL36-null virus, although the amount of pUL17, pUL25, and pUL15 on all capsid forms was smaller (Fig. 4, bottom). The decrease in pUL17 and pUL25 capsid association can be attributed to pUL36’s role in stable binding of the CVSC proteins to the penton vertices (19). The decrease in pUL15 on A-capsids is likely due to the small amount of A-capsids found with the pUL36-null virus (Fig. 4, top and center).
Genetic studies have demonstrated that pUL17 is required for cleavage and packaging of the viral genome (29) while cryoEM structures locate pUL17 at the exterior surface of the peripentonal triplexes where it anchors pUL25 and pUL36 to the capsid (20, 23). pUL17 binds similarly to triplexes around the portal vertex, where it could also serve to anchor the terminase to the capsid (5, 6). To determine the role of pUL17 in the interaction of the terminase proteins with capsids, we isolated nuclear B-capsids from Vero cells infected with the UL17-null virus (Fig. 4, top and center). Western blot analyses of the pUL17-null B-capsids did not detect the pUL15 or pUL28 terminase subunit, while the small terminase protein pUL33 was found (Fig. 4, bottom). Since pUL17 is required for pUL25 binding, it was not surprising to find that pUL17-null B-capsids contained only a low level of pUL25 compared to KOS B-capsids. These results indicate that the interaction of pUL15 and pUL28 with capsids was dependent on the presence of pUL17 and that the observed defects in DNA packaging with the UL17-null virus are likely due to absence of the terminase.
Since the viral genome enters the capsid through the portal, we expected the portal to be required for capsid binding of the terminase subunits, but this has not previously been confirmed experimentally. Similar to what was observed with the B-capsids from the UL17-null virus, the pUL6-null B-capsids did not contain the pUL15 or pUL28 terminase subunit, while the small terminase protein pUL33 and the CVSC proteins pUL17 and pUL25 were detected on these capsids (Fig. 4, bottom). Thus, both the pUL6 portal and pUL17 are required for binding of the pUL15 and pUL28 terminase subunits to capsids.
We next examined the effect on the binding of terminase to capsids by deleting each of the three terminase subunits and pUL32. Western blot analyses of nuclear capsids isolated from cells infected in turn with each of the UL15-, UL28-, UL33-, and UL32-null viruses are shown in Fig. 4 (bottom). All of the mutants produce only B-capsids. The UL28 deletion also resulted in the loss of pUL15 from the capsid, but not pUL33, while capsids isolated from the UL15-null virus retained both pUL28 and pUL33. The B-capsids from the UL33-null virus contained pUL28 and pUL15. In contrast, the pUL32-null B-capsid contained all three terminase proteins but with reduced amounts of pUL15 and pUL28 compared to KOS or pUL25-null capsids. These results demonstrate that pUL28 interaction with capsid (presumably only at the portal vertex) is critical for capsid binding of pUL15, consistent with previous studies showing that pUL15 interacts with pUL28 (8, 32). Finally, to demonstrate that the proteins found to associate with capsids were the result of specific capsid interactions, the presence of pUL38 (capsid triplex protein, VP19C) and ICP8 (DNA replication protein) on the capsids was also determined by Western blot analysis (Fig. 4, bottom). As expected, pUL38 was found on all capsids while ICP8 was absent.
Taken together, these results demonstrate that the terminase requires only the CVSC subunit pUL17 and the portal to bind capsids and that pUL28 is required to bind the pUL15 terminase subunit, suggesting that the capsid-terminase interface involves direct interaction between pUL17, pUL6, and pUL28. Further, pUL32 is revealed to have an important if unclear role in terminase binding that was previously unknown.
CVSC subunits required for DNA cleavage/packaging.
The mechanism for replication of the HSV genome is poorly understood, but replication is thought to occur via a rolling circle mechanism that generates DNA concatemers from which unit-length genomes are subsequently cleaved (Fig. 5A). Pulsed-field gel electrophoresis (PFGE) of HSV-infected cells yields two virus-specific bands: one that migrates as linear genome-length (152-kbp) double-stranded DNA (dsDNA) (Fig. 5A) and a second that remains at the origin of the gel together with high-molecular-weight cellular DNA (26–28). The viral DNA trapped at the origin consists of short concatemers of less than 3 genome lengths that are highly branched and so retained in the well. Pulse labeling experiments demonstrated that this well DNA is the precursor to the 152-kbp genome (26). Terminase initiates packaging by cleaving at a specific sequence (termed “a”) present between adjacent genomes within concatemers and subsequently provides force for genome insertion through the hydrolysis of ATP (Fig. 5C). Packaging is terminated by a second cleavage event at the next similarly orientated a sequence. Repeated a sequences are located at the termini of both the UL and US components and at the junction between the two components (Fig. 5B) (37–41). The cis-acting signals for DNA cleavage have been mapped to specific domains termed pac1 and pac2 that are located within the UB and UC regions of the a sequence, respectively (Fig. 5B) (42–46). Cleavage of the dsDNA occurs at a defined distance from both the pac1 and pac2 elements (44), making a site-specific cut within DR1 (42). However, it is important to note that although DR1 contains the site of cleavage, this specific sequence is not required, only the defined distance from either pac element (44).
FIG 5.

HSV genome and packaging motifs and processing of virus DNA. (A) Diagram of the 152-kbp HSV-1 genome showing the locations of the BamHI K, Q, and S fragments. (B) Structure of the a sequence elements. (C) The location of the pac1 and pac2 sequences within the UC and UB regions and the sequence of the genome ends, resulting from cleavage within the DR1 element of adjacent a sequences. (D) EtBr-stained PFGE of DNA from mock, KOS, UL15-null, and UL17i virus-infected Vero (V) cells or UL15-null and UL1i-infected complementing (C) cells. (E and F) Well DNA isolated following PFGE (E) or total infected cell DNA isolated from Vero (V) or complementing (C) cells (F) was digested with BamHI followed by Southern blot analysis with 32P-labeled BamHI K (E) or the BamHI Q (F) fragments shown in panel A. The different sizes of the BamHI S fragment are due to the presence of one or more copies of the a sequence present at the UL terminus.
Replication of the viral genome produces concatemers where only the UL terminus is exposed (26–28, 47). DNA packaging initiates at the UL terminus and completes at the US terminus, and in vitro uncoating assays have demonstrated that the US terminus exits the capsid first (48). Following this model, it is thought that the initial cleavage of the concatemer is directed by pac2, resulting in a truncated DR1 element of 18 bp with a 3′ G nucleotide extension within the terminal a sequence at the UL end of the genome (Fig. 5C). Cleavage directed by the pac1 site results in a final truncated DR1 element with a 3′ C nucleotide overhang within the terminal a sequence at the US end of the genome (Fig. 5C) (49, 50). The freed genome end is packaged, and the terminase components subsequently disassociate from the viral capsid, possibly to act in additional rounds of cleavage and packaging (51–53). Stable capsids containing a complete viral genome represent the C-capsids that can egress from the nucleus and assemble into mature virions (54, 55).
To address what viral proteins are required for the initial cleavage event, DNA trapped at the origin of the pulsed-field gel was isolated from cells infected with either wt KOS or one of seven cleavage/packaging mutant viruses. PFGE of total cell DNA from wt HSV (KOS)-infected cells contained both 152-kbp viral genome and well-associated DNA (Fig. 5D). In contrast, DNA from Vero cells infected with any of the seven cleavage/packaging mutants contained only well DNA. However, when the mutants were grown on a complementing cell line, both the 152-kbp viral genome and well DNA were found. One example of a packaging mutant (UL15-null virus, ΔUL15) is shown in Fig. 5D. The 152-kbp HSV genome was found only when the ΔUL15 virus was grown on a UL15-complementing cell line (a second, high-molecular-weight band is due to cell DNA). This same PFGE pattern is repeated for null mutants of the other viral cleavage/packaging genes (data not shown). These results are consistent with previously published studies demonstrating the absence of DNA cleavage with mutants lacking expression of pUL6, pUL15, pUL17, pUL25, pUL28, pUL32, or pUL33 (3, 14, 16, 17, 29, 33, 56, 57). The well DNA from wild type and UL15-, UL25-, and UL17-null mutants was isolated from infected Vero cells or the appropriate complementing cell line and digested with BamHI, and the presence of the viral terminal UL and US ends was detected by Southern blot analysis (Fig. 5E). The blot was probed with the joint-spanning BamHI K fragment which detects both the UL (BamHI S fragment) and US (BamHI Q fragment) ends of the genome along with the joint region BamHI K fragment (Fig. 5E). The UL end fragments were clearly present in the KOS and UL25-null virus well DNA for both the Vero cell samples and the UL25-null DNA from the UL25 complementing cells. In contrast, the UL end fragments were found only with the well DNA from the complementing cells for the UL15- and UL17-null viruses. This same result was found with the well DNA from the other packaging mutants, UL6, UL28, UL32, and UL33 (data not shown). As shown previously (26), the US end fragment was not found with the well DNA for KOS or any of the null viruses (Fig. 5F). This was confirmed by probing the blot with the BamHI Q fragment that detects only the US end (data not shown). These results demonstrate that the first cleavage of the replicated HSV genome to generate the terminal UL end fragment requires portal pUL6, CVSC subunit pUL17, the three terminase subunits pUL15, pUL28, and pUL33, and pUL32, but not the CVSC subunit pUL25, as we have shown previously (15, 16, 58).
UL17 insertion mutation.
In order to identify regions of UL17 protein that are important for viral replication and for DNA cleavage, the UL17 open reading frame (ORF) was randomly disrupted by transposon-mediated insertion of a 15-bp fragment that resulted in a 5-amino-acid insertion in the UL17 open reading frame. Fourteen clones with unique DNA insertions in the coding region of the UL17 gene were mapped by restriction digestion and DNA sequencing. The resulting amino acid insertions were found to be scattered across the UL17 ORF, and the ability of the UL17 transposon mutants to support virus replication was tested using a genetic complementation assay (see Materials and Methods). Although the majority of the mutants were able to complement replication of a UL17-null virus, several insertion mutations failed to complement, and these insertions were introduced into the viral genome through the genetic manipulation of an HSV-1 (KOS) genome maintained in a recombinant bacterial artificial chromosome (BAC). One of the replication-defective viruses, UL17i, contained a 5-amino-acid insertion (CGRNA) between amino acids 161 and 162 of pUL17. The UL17i virus failed to replicate on Vero cells, producing only B-capsids, but did show cleavage of the well DNA that was isolated following PFGE (Fig. 5E). The BamHI end fragment found in UL17i well DNA appears to be from the US end of the genome based on the detection of a fragment similar in size to the BamHI Q fragment (Fig. 5E). To confirm that the UL17i mutant was generating a US end fragment, total infected cell DNA was isolated from Vero cells infected with either KOS, UL25-null, UL17-null, or UL17i mutant or from the appropriate complementing cells infected with the UL25-null, UL17-null, or UL17i mutant. The viral DNA was digested with BamHI, and the presence of the viral terminal US end fragment was determined by probing the blot with the 32P-labeled BamHI Q fragment (Fig. 5F). A fragment the same size as the KOS BamHI Q fragment was present with DNA from complementing cells infected with the UL25-null and UL17-null viruses but not Vero cells. However, this was not the case with the UL17i mutant, where both Vero and the UL17 complementing cells contained the BamHI Q fragment. To determine if the UL17i Q fragment was the result of cleavage at the same site as wild-type virus, we cloned the Q fragment from viral DNA isolated from Vero cells infected with KOS or the UL17i mutant. Sequencing of both clones demonstrated that the US end of the genome for the UL17i genome was identical to the KOS US-end sequence shown in Fig. 5C. From these results, we conclude that the cleavage machinery (terminase: pUL15, pUL28, and pUL33) is intact in the pUL17i mutant. The aberrant cleavage suggests that pUL17 is important for determining the site of the first cleavage reaction through its interaction either with the portal or with the terminase, or both. These results suggest that the pUL17i mutant is directing the portal/terminase complex to initiate an aberrant first cleavage reaction compared to wild-type-infected cells where the initial cleavage generates a free UL end (Fig. 5E and F). The presence of the US end of the viral genome does not lead to genome packaging, as demonstrated by the presence of only B-capsids and the absence of linear genome-length (152-kbp) DNA in Vero cells infected with the pUL17i mutant (Fig. 5D). The absence of packaging suggests that the packaging reaction is directional and initiates only from the L-end of the genome.
Structural characterization of the portal vertex.
To complement our biochemical data and to demonstrate the dependencies of the CVSC subunits on each other for assembly at the capsid portal vertex, we used cryoEM to determine capsid structures of mutants lacking each of the CVSC subunits, pUL17, pUL25, and pUL36, in turn. The locations and shapes of the CVSC subunits around penton vertices have been established previously by high-resolution cryoEM structures of native virion capsids as well as of mutants with bulk labeling of the CVSC subunits, as described above (Fig. 3). We now use this knowledge of CVSC density organization around the penton as the basis for understanding the nature of density distributed around the portal vertex of wild-type virion capsids. Our new capsid reconstructions, together with the virion capsid map of McElwee et al. (6), were calculated with just 5-fold symmetry imposed around the portal vertex since it is known that the CVSC organization follows such symmetry in contrast to the 12-fold symmetry of the portal. The quality of density offers clear interpretation of subunit organization and allows comparisons between portal and penton vertices and between portal vertices of the different mutants (Fig. 6).
FIG 6.

Structural consequences of CVSC subunit deletions at the portal vertex. Reconstructions from cryoEM images of wt virion capsids, UL17-null nuclear B-capsids, UL25-null nuclear A-capsids, and UL36-null nuclear C-capsids. (A) Grayscale-coded central sections through the portal axis with the portal and opposing penton vertices marked. Protein is dark. The occupancy of CVSC subunit pUL17 is indicated by black arrows (100%), white arrows (low), and white arrowheads (absent). Scale bar is 250 Å. (B) Enlarged view of the corresponding region around the portal vertex from each density map section, as labeled. The external tips of the proximal hexon are aligned consistently (white dashed line at right) while the location of the interior-most part of the portal complex is displaced between empty and full capsids by 30 Å (white dashed line at left). Scale bar is 100 Å. (C) Schematic representation of the sections in panel B identifying density with proteins and viral DNA, as indicated in the legend, including tentative assignments (“?”) and density of unknown source. (D) Portions of the schematics for full and empty capsids indicating differences in the regions of contact between the portal and capsid. One contact (arrow) appears to be made only when the capsid is filled, while another (arrowhead) involves a reorganization or addition of density (arrow). (E) Sections displaced from the map centers reveal density from two of the long helices (orange arrowhead) in the CVSC helical bundle that is apparent only when all three subunits, pUL17, pUL25, and pUL36, are present (virion map at left). If any of the three subunits is absent, the helical bundle is also absent (e.g., pUL36-null at right—white arrowhead). Scale bar is 250 Å.
Central sections through the four density maps reveal several well-understood and expected differences between the capsid structures (Fig. 6A). The virion capsid is fully packaged with DNA and has strong density corresponding to the pUL17 subunits around both penton vertices and the portal vertex (black arrows in Fig. 6A), indicating 100% occupancy of these sites (5, 6). The portal vertex includes density interior to the capsid that is ascribed to the dodecameric portal complex, as well as external density of the CVSC pUL17 subunit and density around and above the portal that is of uncertain origin. A thin shaft of density along the axis of the portal vertex is presumed to be the last-packaged end of the viral dsDNA. The UL36-null capsid map calculated from C-capsids also contains viral DNA, while the UL25-null map is of empty A-capsids since pUL25 is required to retain packaged DNA, and the UL17-null map is of B-capsids including remnants of the scaffold core that were not expelled by DNA packaging (58).
Differences in the capsid structures reveal several key features that distinguish CVSC organization around the portal and penton vertices. While the virion capsid map shows 100% occupancy by pUL17 (Fig. 6A, black arrows), the UL17-null map shows 0% occupancy (Fig. 6A, white arrowheads), as expected, and indeed no element of CVSC density is apparent, consistent with our observation that pUL17 is essential for the other CVSC subunits, pUL25 and pUL36, to bind to the capsid (20). Interestingly, the UL25-null and UL36-null maps both show differential occupancy of pUL17—the portal vertex is fully or almost fully occupied (Fig. 6A, black arrows) while the penton vertex shows only faint density (Fig. 6A, white arrows). This is the first demonstration of differences in pUL17 distribution between portal and penton vertices during capsid assembly, and indicates that pUL17 has greater affinity for the portal vertex where it may play an essential role in DNA packaging (15, 16). We also observe that pUL36 is not required for packaging or cleavage of the viral genome since the UL36-null capsid contains DNA. Although the pUL25 subunit is not clearly represented in the UL36-null map, it must be bound to the portal vertex since it is required for retention of the packaged DNA inside the capsid. Thus, pUL25 also appears to be differentially bound to the capsid during assembly. Taken together, the cryoEM structures of the null mutant and virion capsids reveal critical roles for pUL17 and pUL25 at the portal vertex, and a sequential assembly of density that includes just the portal complex (UL17-null), the portal complex with pUL17 bound (UL25-null), and additional density external to the portal that may be pUL25 and/or other proteins (UL36-null).
As well as differences between the penton and portal vertices, we also observed that the portal is shifted radially between the empty and the DNA-filled capsid maps. Alignment of the portal vertex density (Fig. 6B) reveals a 30-Å displacement of the portal complex between the capsids that are empty of DNA (UL17-null and UL25-null) and those that have successfully packaged and retained DNA (virion and UL36-null). A similar observation was reported recently from comparing native B-capsids to virion capsids of herpes simplex virus 2 (HSV-2) (24). Our assignment of density at the portal vertex is modeled in Fig. 6C, corresponding to the density regions of Fig. 6B. Portal contacts with the adjacent hexons and triplexes appear to be modified as well as the conformation of the external tips (Fig. 6D). This transition is presumably reversible as the portal in the UL25-null capsid, which has packaged and released DNA, is at the same internal location as that of the UL17-null capsid that has not entered the packaging reaction. The outward displacement of the portal results from DNA packaging and may represent a stage in a sequential packaging pathway that offers a conformation with lower affinity for the terminase complex but to which pUL25 can bind as part of the plug mechanism to prevent uncontrolled DNA release (24).
Maps of the two DNA-filled capsids, virion and UL36-null, show additional density regions above the portal that are not present in the UL17-null and UL25-null maps. One of these is continuous with the external tips of the portal, and the other is located above the portal but appears less well ordered. Since copies of the C-terminal domain of pUL25 are located both to the side and above the pentons, we speculate that this additional density at the portal has a similar source, at least in part. Additionally, the presence of a helical bundle at the portal vertex of the virion (Fig. 6E) confirms the presence of UL36 at this special vertex, even if most of the protein is not visible in the map. If any of the three subunits are absent, the helical bundle is also absent as seen with the UL36-null map (Fig. 6E).
DISCUSSION
Herpesviruses and most double-stranded DNA bacteriophages (bacterial viruses) package their genomes into a preformed capsid using an ATP-driven terminase complex that binds the DNA and the capsid portal. In the case of herpes simplex virus, the terminase complex consists of a large terminase protein and two accessory terminase subunits (8, 9) and interacts with the portal protein and the capsid vertex-specific component. This assembly is rather more elaborate than that of the related bacteriophages, involving an additional terminase subunit and the CVSC molecule that is unique to herpesviruses, and so direct inferences from better-studied phages are limited. As a starting point for this study, we sought to confirm the organization of the CVSC molecule arrayed around penton vertices where high symmetry can be exploited for greater structural confidence. Previous studies, including our own, have produced conflicting data on the CVSC composition, but by visualizing structures with subunits labeled in turn, we demonstrated here the essential correctness of a proposed model—an asymmetric dimer of the pUL25 subunit that is anchored to the capsid by one copy of pUL17 (35). In addition, we demonstrated that two copies of pUL36 also contribute a C-terminal-most helix each to the 5-helix bundle core of the CVSC complex, confirming a model where the CVSC intimately involves pUL36 (19, 20). Indeed, this helical bundle core is observed only in the presence of all three subunits, pUL17, pUL25, and pUL36, supporting our contention that all are involved in CVSC. However, the question arises whether the CVSC molecules at the portal vertex are identical in composition and conformation to those around pentons, since contacts previously observed between pUL25 and the penton subunit, VP5, are obviously absent at the portal. Although we cannot detail the CVSC subunit folds at the portal vertex, the solid organizational knowledge gained is vital for understanding the interaction between the capsid and the terminase complex, as discussed below.
The structure of the HSV capsid portal vertex has been explored in recent cryo-electron microscopy (cryoEM) reconstructions of the virion capsid by successfully discriminating it from the other penton-containing vertices (6, 7). This structure revealed that the pUL6 portal dodecamer is anchored to the capsid by interactions with the peripentonal triplexes as well as density corresponding to a set of helices attributed to the CVSC proteins. Here, we applied biochemical and structural methods to examine the functions of the CVSC components and their interaction with the portal vertex and with the terminase complex in cleavage and retention of the packaged viral genome. We found that the packaging process involves a sequential assembly of CVSC components that are able to anchor the terminase complex to the capsid to effect DNA packaging and subsequently retain the viral genome inside the capsid.
Genetic and biochemical studies have demonstrated that pUL17 is required for cleavage and packaging of the viral genome while cryoEM structures locate pUL17 in the penton triplexes where it anchors pUL25 and pUL36 to the capsid. The recent HSV capsid portal structure indicated that pUL17 binds to a similar position at the portal vertex where it could also serve to anchor the terminase to the capsid (5–7). Our studies demonstrate that the interaction of the pUL15 and pUL28 terminase subunits with capsids and the initial cleavage of the replicated viral genome are both dependent on the presence of pUL6 (portal) and pUL17. In the absence of pUL17, defects are observed in DNA packaging, and our data from the UL6- and UL17-null viruses suggest that these are due to the absence of the terminase complex at the portal. Furthermore, in the absence of pUL25 or pUL36, pUL17 is still able to bind around the portal vertex and does so in preference to binding around penton vertices. Thus, we propose that pUL17 plays critical roles at the portal vertex for both cleavage and stable packaging of the viral genome. This multistep process involves an ordered interaction of the terminase complex with pUL17 and the pUL6 portal that leads to the cleavage and packaging of a complete HSV genome followed by release of the terminase and binding of pUL25 to pUL17 that serves to stabilize capsids after DNA is packaged. During this process, the portal is displaced outwardly by 30 Å possibly in response to the packaged DNA and in doing so may signal completion by modifying the pUL17 orientation to effect the disengagement of terminase and favor binding by pUL25.
We complemented the biochemical studies by examining the organization and composition of the portal vertex in capsids isolated from cells infected with viruses that fail to express one or more of the genes required for DNA packaging. These studies demonstrated that the structure and composition of the portal vertex for capsids isolated from cells infected with a UL17-null virus are dramatically altered compared to virion capsids, and in particular, the CVSC density is absent at all vertices, confirming the roles of pUL17 as the anchor for CVSC subunits and the terminase complex to the capsid. A corresponding reconstruction of the UL25-null capsid also reveals absence of CVSC around the penton vertices, as observed previously, but density consistent with pUL17 is present at the portal vertex. This differential binding of pUL17 suggests that the penton and portal binding sites are structurally distinct. Further, our studies of DNA packaging suggest that pUL17 alone of the CVSC subunits engages the terminase and enables the DNA to be packaged, although the lack of pUL25 prevents subsequent retention of the DNA inside the capsid. The third mutant, UL36-null, both packages and retains the viral genome, indicating that pUL17 and pUL25 are sufficient and that pUL36 has no role in this process. However, we again observe differential binding of pUL25 between the penton and portal vertices, further suggesting site-dependent structural differences affecting the CVSC conformation.
Where in the infected cell pUL36 is added to the capsid is controversial. It is not required for packaging of the HSV genome nor for nuclear egress of DNA-containing capsids (17), suggesting that pUL36 is not required in the nucleus for capsid assembly. However, both full-length and truncated forms of pUL36 have been observed in the nucleus and bound to nuclear C-capsids (59–62). Our data demonstrate that pUL36 is not needed for DNA packaging or retention, functions that require the other CVSC subunits pUL17 and pUL25, and further that the CVSC helical bundle motif that depends on pUL36 is also not required to carry out these functions. This is particularly surprising for the DNA-retaining role of pUL25 since pUL36 and pUL25 appear to form the bulk of the helical bundle. If pUL36 acts postassembly to recruit tegument to the C-capsid, its attachment to the capsid would appear to be necessary only after nuclear export, and it seems that in forming the helical bundle, it triggers a conformational change in pUL25 at all the vertices. This conformational change would be expected to stabilize the CVSC through the many intimate contacts made between pUL25 and pUL36, as well as with the underlying pUL17, and perhaps serves several purposes, including robustly docking pUL36 to the capsid for tegument recruitment and locking pUL25 into its DNA retention role. The latter function of pUL36 may explain the observation that a C-terminally truncated form of pUL36 binds to nuclear C-capsids (62). The C terminus of pUL36 is predicted to fold as an alpha-helix and likely forms two of the helices in the CVSC helix bundle (Fig. 3). Clearly, our results have outlined an area for future studies and may be significant for development of antiviral drugs that interfere with the structural evolution of the CVSC.
A recent atomic structure of the HSV terminase complex revealed a hexameric ring of the terminase complex with each of the six subunits consisting of one copy each of the HSV terminase proteins pUL15, pUL28, and pUL33 (9). Modeling indicated that pUL15 interacts with viral genome through its ATPase domain (N-terminal region of pUL15) and that the hydrolysis of ATP provides the chemical force and physical motion to translocate the viral genome into the capsid. The pUL28 and pUL33 terminase subunits do not appear to interact directly with the viral genome but serve to ensure correct assembly of the complex. The pUL15 ATPase domains are proposed to be arrayed around the interior of the ring, forming a central channel through which the DNA is translocated by sequential binding and ATP hydrolysis. The final step in the packaging reaction is triggered when a viral genome equivalent is packaged, resulting in the rearrangement of the terminase complex such that the nuclease domain of pUL15 (C-terminal region) is exposed to the viral genome, which results in the final cleavage directed by the pac1 site in the terminal repeat and stable packaging of the DNA. However, although determining the structure of the terminase complex is a major step in understanding the mechanism of DNA packaging, there are several additional viral proteins that are also essential for this process, including the portal protein and the CVSC proteins.
Our data demonstrate that the initial cleavage of the replicated viral genome to generate a free UL terminus requires not only a functional terminase complex consisting of pUL15, pUL28, and pUL33 but also the portal protein, pUL6, and one of the CVSC subunits, pUL17. An additional viral protein, pUL32, is also required, but absent evidence of any direct interaction with the capsid or any of the other six packaging proteins, its role may be indirect by localizing capsids to sites of DNA packaging (14). While we have shown that the portal protein and pUL17 both appear to be important for the interaction of the cleavage/packaging machinery with the viral genome for specific cleavage at a pac2 site (Fig. 5), the question arises of whether pUL6 or pUL17 interacts with the viral genome while the other binds terminase or whether they act in concert. The UL6 portal protein binds viral genomes very early after replication, according to a study of HSV-1 DNA labeled with a clickable nucleotide analog (EdC) that allows for covalent attachment of biotin to the labeled nucleotides and the subsequent purification of viral genomes and the viral and cellular proteins bound to the DNA (63, 64). Indeed, pUL6 was one of the most abundant proteins associated with viral replication forks, although capsid proteins and both the UL17 and UL25 CVSC proteins were also found to interact with the replicating viral genomes. Here, we show that pUL17 is essential for terminase binding to the portal vertex, and our UL17i mutant reveals a unique phenotype of making only B-capsids although the replicated viral genome is cleaved, generating what appears to be an aberrant free US end of the genome (Fig. 5E and F). These results suggest that the pUL17i mutant is directing the portal/terminase complex to initiate an aberrant first cleavage reaction on the replicated viral genome compared to wild-type-infected cells where the initial cleavage generates a free UL end (Fig. 5A). The presence of the US end of the viral genome does not lead to genome packaging with the pUL17i mutant. Taken together, the studies with the UL17i mutant strongly suggest that the initial cleavage of the replicated viral genome to generate the L-end of the HSV genome is directed by a specific interaction of pUL17 with the portal and terminase proteins.
The potential importance of the herpesvirus DNA cleavage/packaging pathway as a therapeutic target has been appreciated since 1998 when the first of several different classes of small molecules were reported to inhibit the replication of human cytomegalovirus (HCMV) and HSV by targeting proteins in this pathway (65–69), The current studies were designed to take advantage of the novel approaches that we have employed to study the structure and function of HSV capsid and DNA packaging proteins (8, 10, 15, 20). Our premise is that herpesvirus DNA is incorporated into preassembled capsids through a ring-shaped portal present at a unique capsid vertex. The HSV-1 CVSC is a minor but essential capsid vertex component that is required for the cleavage and encapsidation of the HSV genome. Based on current studies, we propose that DNA packaging is initiated by a complex consisting of the dodecameric portal (pUL6), the CVSC subunit pUL17, and the 3-subunit terminase and that association of this complex with replicated viral DNA is followed by cleavage at the pac2 site to generate the UL end of the genome. The terminase proteins (pUL15, pUL28, and pUL33) act as part of an ATP-dependent pump that drives DNA into the procapsid and cuts the concatemeric DNA at the pac1 site to generate the US end of the genome and a DNA-filled capsid. The final capsid completion step involves the release of terminase from the portal vertex and binding of pUL25 to the capsid, anchored by pUL17, to cap the portal, yielding a stable DNA-containing capsid. The cleavage/packaging and capsid completion reactions can be viewed as separate steps in the assembly of a stable DNA-containing capsid.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney cells (Vero; CCL-81 from the American Type Culture Collection, Manassas, VA) and UL25-transformed 8-1 cells were propagated as previously described (32). HSV wild-type KOS and the pUL25-GFP-expressing virus, vFH422, were previously described (29, 36). The pUL36-GFP (vFH561), UL6-null (vFH479), UL17-null (vFH632), and UL17i insertion (vFH639) mutant viruses were generated by recombination of a KOS genome contained in a bacterial artificial chromosome (BAC) as previously described (29, 42–44). The pUL25-GFP/pUL36-RFP (vFH679) virus was generated by recombination of a vFH422 (pUL25-GFP virus) genome contained in a BAC. The primers used to amplify template sequences from plasmids pEPKan-in, pEPGFP-in, or pEFRFP-in are listed in Table 2. The HSV-1-null viruses used in this study are listed in Table 1 along with the references for each virus and the complementing cell lines used to grow these viruses.
TABLE 2.
Primers used for generation of recombinant HSV-BACsa
| BAC | Mutation | Primer sequence |
|---|---|---|
| KOS BAC | UL6 ORF deletion + insertion of RNA Pol II promoter for UL5 gene | 5’ CTGTCCGTCTAGCTGGCGCTCCCCGCCGGCCGCCGCCATGGCTGGCGGCGGACGCTTCGGTAGTGAGCTTGTCGGTCCCTTAGCGCCTTTATATTGGGTGCCTAGCCGGTCGACCGCCCTTAGGGATAACAGGGTAATCGATTT 3’ |
| 5’ GGCGATGGCCTGCTTGAGGATGGTGGCGGCCGACCCCTCCATAGGGCGGTCGACCGGCTAGGCACCCAATATAAAGGCGCTAAGGGACCGACAAGCTCACTACCGAAGCGTCCGCCGCCAGCCGCCAGTGGTACAACCAATTAACC 3’ | ||
| KOS BAC | UL17 ORF deletion | 5’ CCGCATCGCCTACACACCCACACCCACCCCCGAACCCGGGCGCCTTCCCCCGGCTAGGGATAACAGGGTAATCGATTT 3’ |
| 5’ GAGTGGGTGGGCGAGGTGGCCGGGGGAAGGCGCCCGGGTTCGGGGGTGGGTGTGGCCAGTGTTACAACCAATTAACCA 3’ | ||
| KOS BAC | UL36 Cterm-GFP | 5’ CGTGCTGACCAGCCTACATCACGTGCGCATGTTACTGGGCGTGAGCAAGGGCGAGGAGCTG 3’ |
| 5’ CGGTTCGAAACTTAACAACAAAATAATCGAGCGCGTCTACTTGTACAGCTCGTCCATGCCG 3’ | ||
| bFH422 | UL25-GFP and UL36-RFP | 5’ CGTGCTGACCAGCCTACATCACGTGCGCATGTTACTGGGCGCCTCCTCCGAGGACGTCATC 3’ |
| 5’ CGGTTCGAAACTTAACACACAAAATAATCGAGCGCGTCTACAAGGCGCCGGTGGAGTGGCG 3’ | ||
| KOS BAC | UL17i | 5’ GACGAGCTCGTCCCCCCCAACACGCGCTACGCGTGCGGCCGCAACGCGGCCGACAGCACGCGCATCTAGGGATAACAGGGTAATCGATTT 3’ |
| 5’CCGACAGACGCGCATGATGCGCGTGCTGTCGGCCGCGTTGCGGCCGCACGCGTAGCGCGTGTTGGGGCCAGTGTTACAACCAATTAACCA 3’ |
Abbreviations and symbols: Pol, polymerase; Cterm, C terminal; underlining, HSV sequence; italic, Pol II promoter sequences; bold, template sequence (pEP-Kan-S, pEP-GFP-in, pEP-RFP-in).
Western blotting.
Protein samples were separated on a 4% to 12% SDS-polyacrylamide gel, and proteins were transferred to nitrocellulose. The nitrocellulose was washed twice in Tris-buffered saline (TBS) and incubated overnight in Rockland near-infrared blocking buffer (catalog number MB-070-003; Rockland Immunochemicals, Inc., Gilbertsville, PA). The primary antibodies used (dilutions in parentheses) include UL25 mouse monoclonal antibody A11E4 (1:5,000) (10), UL38 (VP23) rabbit polyclonal antibody NC2 (1:5,000) (70), UL6 mouse monoclonal antibody 1C9 (1:1,000) (4), UL17 chicken polyclonal antibody (1:25,000) (71), UL15 rabbit polyclonal antibody NT-UL15 (1:1,000) (56), UL28 rabbit polyclonal antibody GST-UL28 (1:1,000) (31), UL33 rabbit polyclonal antibody (1:500) (72), UL32 rabbit polyclonal antibody (1:1,000) (73), and UL36 rabbit polyclonal antibody (1:5,000) (59). The diluted antibodies were reacted with the blocked nitrocellulose for 2 h at room temperature, washed five times in TBS with 0.5% Tween 20, and incubated with IRDye 800-conjugated secondary antibodies, goat anti-mouse (pUL6 and pUL25), goat anti-rabbit (GFP, pUL15, pUL28, pUL33, and pUL38), or donkey anti-rabbit (pUL17) from Rockland Immunochemicals diluted 1:15,000 in Rockland near-infrared blocking buffer with 0.1% Tween 20. The blots were washed and scanned using an Odyssey system (Li-Cor, Lincoln, NE).
Capsid purification.
Vero cells (1.5 × 108) were infected overnight (18 h at 37°C) at a multiplicity of infection (MOI) of 5 PFU/cell. Infected cells were harvested, rinsed with phosphate-buffered saline (PBS), resuspended in 20 mM Tris (pH 7.5) plus protease inhibitors (Roche), adjusted to 1% Triton X-100, and incubated for 30 min on ice. The resulting nuclei were harvested by low-speed centrifugation, resuspended in 10 ml TNE (500 mM NaCl, 10 mM Tris, 1 mM EDTA, pH 7.5), and then sonicated to lyse the nuclei. The nuclear lysate was adjusted to 20 mM MgCl2 and incubated with DNase I (100 μg/ml) at room temperature for 30 min. The lysate was then cleared by low-speed centrifugation, and the resulting supernatant was layered on top of a 5-ml cushion of 35% sucrose (SW28 rotor; 23,000 rpm for 1 h). The pellets were suspended in 3 ml TNE and adjusted to 1 mM dithiothreitol, and capsids were separated by centrifugation on 20 to 50% sucrose (in TNE) gradients (SW41 rotor at 24,000 rpm for 1 h). The positions of A-, B-, or C-capsids were observed as light-scattering bands with A-capsids being found highest (least dense) on the gradient and C-capsids being found lowest (dense fractions) on the gradients. The different capsid fractions can also be identified based on the presence or absence of the scaffold protein, VP22a, since only B-capsids contain the scaffold protein. The fractions were collected using a Beckman fraction recovery system (Beckman catalog number 34890). The apparatus has a mechanism to puncture the bottoms of the tubes, and fractions are collected from the bottom to top. Fractions (0.75 ml) were collected, and protein was precipitated by adding an equal volume of 16% trichloroacetic acid. Pellets were resuspended in 35 μl of 2× PAGE loading buffer (Invitrogen) supplemented with 0.4 M Tris base. Gradient fractions were run on a 5 to 12% SDS-PAGE gel, and the gels were either stained with Imperial Blue (Pierce) to visualize capsid proteins or analyzed by immunoblotting.
Pulsed-field gel electrophoresis.
PFGE was performed on a Bio-Rad (Melville, NY) contour-clamped homogeneous electric field (CHEF) mapper. One T75 flask of Vero cells or 8-1 cells or the appropriate complementing cell line (Table 1) was infected at an MOI of 5 PFU per cell, and at 18 h postinfection the medium was removed; the cells were washed with phosphate-buffered saline (PBS), scraped into PBS, and pelleted. The cell pellet was resuspended in approximately 300 ml of 55°C 1.0% low-melting-temperature agarose (Bio-Rad) in PBS (without CaCl2 and MgCl2) and cast in three blocks of approximately 100 μl each in casting blocks provided by Bio-Rad. The blocks were removed from the mold and stored at 4°C in 50 mM EDTA (pH 8.0). Prior to electrophoresis, the blocks were incubated for 20 to 24 h at 37°C in 1.0% laurylsarcosine-0.4 M EDTA (pH 9.0)-protease K (1 mg/ml) at 37°C. Blocks were then washed five times for 15 min each in 50 mM Tris-HCl (pH 7)-1 mM EDTA (TE) at 45°C. The plugs were sealed into the wells of a 1% agarose gel made in 0.53 TBE (TBE is 0.089 M Tris [pH 8.0], 0.089 M boric acid, and 0.002 M EDTA [pH 8.0]). The gels were run at 6 V/cm (200 V) for 18 h at 14°C; the angle was 120° with a pulse time of 50 to 90 s with 0 ramping factor. The plug containing the well-associated DNA was removed from the gel and stored at 4°C in 0.5 M EDTA, pH 8. The gel was then stained with ethidium bromide at 0.5 mg/ml for 1 h at room temperature and photographed.
Southern blot analyses of total infected cell DNA.
A T175 mm flask of Vero cells or the appropriate complementing cell line (Table 1) was infected with virus at an MOI of 5 PFU per cell. At 18 h postinfection, the medium was removed and the cells were washed in 1× PBS, scraped off the plate, and pelleted. The cells were lysed, and viral DNA was prepared as previously described (74). The final DNA was digested with BamHI to assess cleavage of viral DNA. The DNA was separated by agarose gel electrophoresis, transferred to a nylon membrane, and hybridized as previously described (74). Southern blots were scanned with a Storm 840 PhosphorImager, and specific bands were quantified with ImageQuant software.
Southern blot analyses of PFGE gel plugs.
The plugs containing well-associated DNA were digested with BamHI for 24 to 36 h at 37°C. Plugs were washed five times for 15 min each in TAE (40 mM Tris acetate-2 mM EDTA) at room temperature. The plugs were sealed into the wells of a 0.8% agarose gel made in TAE, electrophoresed, transferred to a nylon membrane, and hybridized as previously described. Southern blots were scanned with a GE Amersham Typhoon PhosphorImager, and specific bands were quantified with ImageQuant software.
Transposon mutagenesis.
The transposition reaction was done following the Thermo Scientific mutation generation system (MGS). The MGS is an in vitro method for random insertion of a 1.2-kbp transposon (MuA transposase) that includes a kanamycin resistance gene. The mutagenesis is accomplished by introduction of a transposon that contains the NotI site; the majority of the transposon is then removed by restriction digestion with NotI. Religation results in a 15-bp insertion, which retains the unique NotI site. Plasmid pFH649 contains the UL17 gene expressed from the CMV IE promoter and was used as the template for the transposition as previously described (16). Following transposition, the plasmid was transformed into bacteria and plated on selective agar containing kanamycin and ampicillin. DNA extracted from individual colonies was analyzed using a restriction enzyme (NotI) to map the site of the transposition. Plasmids with insertions that mapped within the UL17 open reading frame (ORF) were digested with NotI to remove transposon sequences, religated, and transformed into bacteria. The mutants were sequenced to confirm the residues and determine positions of insertion into the UL17 gene as a result of the transposition insertion.
Complementation assays.
Vero cells (1.0 × 105 cells/well in a 24-well plate) were transfected with 0.5 μg of plasmid DNA and 1 μl of Lipofectamine 2000 (Invitrogen) diluted in 100 μl of serum-free medium. The DNA-lipid complexes were allowed to form at room temperature for 20 min, added to the cells, and incubated overnight at 37°C. The next day, the cells were infected with UL17-null virus (vFH632) at an MOI of 5 PFU/cell. After 90 min at 37°C, the medium was removed and unabsorbed virus was inactivated by washing the cells three times with citrate buffer (pH 3). The cultures were incubated for an additional 18 h, after which the cells were scraped from the plate and rinsed with PBS and the cells were frozen and used to determine virus titers on G5 cells (75) and Vero cells.
Cryo-electron microscopy and image reconstruction of capsids.
CryoEM images of nuclear or virion capsids isolated from Vero cells were collected as previously described (20). Briefly, 3.5 μl of sample was pipetted onto a freshly glow-discharged Quantifoil R2/1 EM grid (Quantifoil, Jena, Germany) and plunge-frozen into a liquefied mixture of ethane and propane (60:40 mix) (76) using a Thermo Fisher Scientific Vitrobot Mark III instrument (95% humidity; 4.5 to 8 s of blotting). Grids were imaged in Thermo Fisher Scientific (Hillsboro, Oregon) Polara or Krios microscopes operating at an accelerating voltage of 300 kV and under automated control of the Thermo Fisher Scientific EPU software package. Conditions of data acquisition and image reconstruction are summarized in Table 3. For the GFP- and RFP-mutant particles, virion-embedded capsid images were picked manually using the x3dpreprocess software (77), and density maps were determined with AUTO3DEM (78) imposing full icosahedral symmetry. For the portal reconstruction of the 3 null mutants (UL17-null, UL25-null, and UL36-null), an approach similar to that of reference 6 was used. Briefly, particles were picked with the automatic option of e2boxer.py from the EMAN2 software package (79) and then classified and reconstructed with the RELION package (80) imposing icosahedral symmetry. The resulting star file of particle orientations was subjected to symmetry expansion over the 60 quasiequivalent positions of the asymmetric unit. A circular mask, created using the bmask software of the BSOFT package (81), was then used to run a focused classification imposing C5 symmetry and using a “T” value of 20 in order to isolate the portal vertex in each particle image. For all data sets, the defocus of each micrograph was estimated with CTFFIND3 (82) and reconstruction resolutions were estimated by the Fourier shell correlation (FSC) dropping to 0.143, according to the gold standard procedure followed (83). Density maps were rendered for analysis with UCSF Chimera (84).
TABLE 3.
Details of cryoEM data collection and analysis
| Sample/capsid | Microscope | Camera | Magnification (nominal) | Pixel sizeb (Å) | Count of micrographs | Count of particles | Resolution (Å) | Symmetry imposed | EMDB ID |
|---|---|---|---|---|---|---|---|---|---|
| Wild type,a virion capsid | Krios | Falcon2 | ×75,000 | 2.2 | 50,000 | 25,637 | 6.9 | Icosahedral | 6386 |
| UL25-GFP, virion capsid | Polara | Falcon3 | ×78,000 | 2.7 | 4,574 | 4,924 | 9.8 | Icosahedral | 22592 |
| UL36-GFP, virion capsid | Polara | Falcon3 | ×78,000 | 2.7 | 762 | 1,172 | 14 | Icosahedral | 22593 |
| UL25-GFP/UL36-RFP, virion capsid | Polara | Falcon3 | ×78,000 | 2.7 | 2,570 | 2,047 | 11 | Icosahedral | 22594 |
| UL17-null, B-capsid | Polara | Falcon3 | ×93,000 | 2.3 | 3,000 | 1,785 | 9.0 | C5 | 22595 |
| UL25-null, A-capsid | Krios | Falcon3 | ×59,000 | 2.8 | 16,055 | 19,834 | 12 | C5 | 22603 |
| UL36-null, C-capsid | Krios | Falcon3 | ×59,000 | 2.8 | 6,643 | 1,593 | 13 | C5 | 22609 |
The wild-type virion capsid map was previously described in the work of Huet et al. (20).
The effective pixel size results from reducing images in size by a 2-fold binning operation.
Data availability.
CryoEM density maps have been deposited in the EM Data Bank (https://www.ebi.ac.uk/pdbe/emdb/index.html/) with accession numbers EMD-22592 through EMD-22595, EMD-22603, and EMD-22609 (Table 3).
ACKNOWLEDGMENTS
We thank Sandy Weller, Frazer Rixon, Prashant Desai, Joel Baines, and Peter O’Hare for providing viruses, complementing cell lines, and antibodies used in these studies and Alexander Makhov for technical assistance with electron microscopy.
This work was supported in part by National Institutes of Health grants AI089803 and AI132967 (J.F.C. and F.L.H.) and by the Office of the Director, National Institutes of Health, under award numbers S10 OD019995 and S10 OD025009 (J.F.C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Huffman JB, Daniel GR, Falck-Pedersen E, Huet A, Smith GA, Conway JF, Homa FL. 2017. The C terminus of the herpes simplex virus UL25 protein is required for release of viral genomes from capsids bound to nuclear pores. J Virol 91:e00641-17. doi: 10.1128/JVI.00641-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huffman JB, Newcomb WW, Brown JC, Homa FL. 2008. Amino acids 143 to 150 of the herpes simplex virus type 1 scaffold protein are required for the formation of portal-containing capsids. J Virol 82:6778–6781. doi: 10.1128/JVI.00473-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamberti C, Weller SK. 1996. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology 226:403–407. doi: 10.1006/viro.1996.0668. [DOI] [PubMed] [Google Scholar]
- 4.Newcomb WW, Thomsen DR, Homa FL, Brown JC. 2003. Assembly of the herpes simplex virus capsid: identification of soluble scaffold-portal complexes and their role in formation of portal-containing capsids. J Virol 77:9862–9871. doi: 10.1128/jvi.77.18.9862-9871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong D, Dai X, Jih J, Liu YT, Bi GQ, Sun R, Zhou ZH. 2019. DNA-Packing portal and capsid-associated tegument complexes in the tumor herpesvirus KSHV. Cell 178:1329–1343.e12. doi: 10.1016/j.cell.2019.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McElwee M, Vijayakrishnan S, Rixon F, Bhella D. 2018. Structure of the herpes simplex virus portal-vertex. PLoS Biol 16:e2006191. doi: 10.1371/journal.pbio.2006191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu YT, Jih J, Dai X, Bi GQ, Zhou ZH. 2019. Cryo-EM structures of herpes simplex virus type 1 portal vertex and packaged genome. Nature 570:257–261. doi: 10.1038/s41586-019-1248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heming JD, Huffman JB, Jones LM, Homa FL. 2014. Isolation and characterization of the herpes simplex virus 1 terminase complex. J Virol 88:225–236. doi: 10.1128/JVI.02632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Yang P, Wang N, Chen Z, Su D, Zhou ZH, Rao Z, Wang X. 2020. Architecture of the herpesvirus genome-packaging complex and implications for DNA translocation. Protein Cell 11:339–351. doi: 10.1007/s13238-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conway JF, Cockrell SK, Copeland AM, Newcomb WW, Brown JC, Homa FL. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J Mol Biol 397:575–586. doi: 10.1016/j.jmb.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol Cell 26:479–489. doi: 10.1016/j.molcel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heming JD, Conway JF, Homa FL. 2017. Herpesvirus capsid assembly and DNA packaging. Adv Anat Embryol Cell Biol 223:119–142. doi: 10.1007/978-3-319-53168-7_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J Virol 72:2463–2473. doi: 10.1128/JVI.72.3.2463-2473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. 2011. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J Virol 85:4875–4887. doi: 10.1128/JVI.00242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cockrell SK, Sanchez ME, Erazo A, Homa FL. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J Virol 83:47–57. doi: 10.1128/JVI.01889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/jvi.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan WH, Roberts AP, McElwee M, Bhella D, Rixon FJ, Lauder R. 2015. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol 89:1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huet A, Makhov AM, Huffman JB, Vos M, Homa FL, Conway JF. 2016. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat Struct Mol Biol 23:531–539. doi: 10.1038/nsmb.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts AP, Abaitua F, O’Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83:105–116. doi: 10.1128/JVI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai X, Zhou ZH. 2018. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360:eaao7298. doi: 10.1126/science.aao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai X, Gong D, Wu TT, Sun R, Zhou ZH. 2014. Organization of capsid-associated tegument components in Kaposi’s sarcoma-associated herpesvirus. J Virol 88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang N, Chen W, Zhu L, Zhu D, Feng R, Wang J, Zhu B, Zhang X, Chen X, Liu X, Yan R, Ni D, Zhou GG, Liu H, Rao Z, Wang X. 2020. Structures of the portal vertex reveal essential protein-protein interactions for herpesvirus assembly and maturation. Protein Cell 11:366–373. doi: 10.1007/s13238-020-00711-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Homa FL, Brown JC. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol 7:107–122. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Deshmane SL, Raengsakulrach B, Berson JF, Fraser NW. 1995. The replicating intermediates of herpes simplex virus type 1 DNA are relatively short. J Neurovirol 1:165–176. doi: 10.3109/13550289509113963. [DOI] [PubMed] [Google Scholar]
- 27.Severini A, Morgan AR, Tovell DR, Tyrrell DL. 1994. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology 200:428–435. doi: 10.1006/viro.1994.1206. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Efstathiou S, Simmons A. 1994. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: implications for viral DNA amplification strategies. Virology 202:530–539. doi: 10.1006/viro.1994.1375. [DOI] [PubMed] [Google Scholar]
- 29.Salmon B, Cunningham C, Davison AJ, Harris WJ, Baines JD. 1998. The herpes simplex virus type 1 U(L)17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J Virol 72:3779–3788. doi: 10.1128/JVI.72.5.3779-3788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Kobaisi MF, Rixon FJ, McDougall I, Preston VG. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380–388. doi: 10.1016/0042-6822(91)90043-B. [DOI] [PubMed] [Google Scholar]
- 31.Beard PM, Taus NS, Baines JD. 2002. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J Virol 76:4785–4791. doi: 10.1128/jvi.76.10.4785-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson JG, Yang K, Baines JD, Homa FL. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J Virol 80:12312–12323. doi: 10.1128/JVI.01766-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tengelsen LA, Pederson NE, Shaver PR, Wathen MW, Homa FL. 1993. Herpes simplex virus type 1 DNA cleavage and encapsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J Virol 67:3470–3480. doi: 10.1128/JVI.67.6.3470-3480.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunningham C, Davison AJ. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116–124. doi: 10.1006/viro.1993.1572. [DOI] [PubMed] [Google Scholar]
- 35.Liu YT, Jiang J, Bohannon KP, Dai X, Gant Luxton GW, Hui WH, Bi GQ, Smith GA, Zhou ZH. 2017. A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J Gen Virol 98:2837–2849. doi: 10.1099/jgv.0.000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu D, Weller SK. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J Virol 72:7428–7439. doi: 10.1128/JVI.72.9.7428-7439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Locker H, Frenkel N. 1979. BamI, KpnI, and SalI restriction enzyme maps of the DNAs of herpes simplex virus strains Justin and F: occurrence of heterogeneities in defined regions of the viral DNA. J Virol 32:429–441. doi: 10.1128/JVI.32.2.429-441.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roizman B. 1979. The organization of the herpes simplex virus genomes. Annu Rev Genet 13:25–57. doi: 10.1146/annurev.ge.13.120179.000325. [DOI] [PubMed] [Google Scholar]
- 39.Roizman B. 1979. The structure and isomerization of herpes simplex virus genomes. Cell 16:481–494. doi: 10.1016/0092-8674(79)90023-0. [DOI] [PubMed] [Google Scholar]
- 40.Wadsworth S, Jacob RJ, Roizman B. 1975. Anatomy of herpes simplex virus DNA. II. Size, composition, and arrangement of inverted terminal repetitions. J Virol 15:1487–1497. doi: 10.1128/JVI.15.6.1487-1497.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner MJ, Summers WC. 1978. Structure of the joint region and the termini of the DNA of herpes simplex virus type 1. J Virol 27:374–387. doi: 10.1128/JVI.27.2.374-387.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deiss LP, Chou J, Frenkel N. 1986. Functional domains within the a sequence involved in the cleavage-packaging of herpes simplex virus DNA. J Virol 59:605–618. doi: 10.1128/JVI.59.3.605-618.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nasseri M, Mocarski ES. 1988. The cleavage recognition signal is contained within sequences surrounding an a-a junction in herpes simplex virus DNA. Virology 167:25–30. doi: 10.1016/0042-6822(88)90050-5. [DOI] [PubMed] [Google Scholar]
- 44.Varmuza SL, Smiley JR. 1985. Signals for site-specific cleavage of HSV DNA: maturation involves two separate cleavage events at sites distal to the recognition sequences. Cell 41:793–802. doi: 10.1016/S0092-8674(85)80060-X. [DOI] [PubMed] [Google Scholar]
- 45.Mocarski ES, Deiss LP, Frenkel N. 1985. Nucleotide sequence and structural features of a novel US-a junction present in a defective herpes simplex virus genome. J Virol 55:140–146. doi: 10.1128/JVI.55.1.140-146.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown J, McVoy MA, Homa FL. 2002. Packaging DNA into herpesvirus capsids, p 111–155. In Bogner AHE (ed), Structure-function relationships of human pathogenic viruses. Kluwer Academic/Plenum Publishers, New York, NY. [Google Scholar]
- 47.Martinez R, Sarisky RT, Weber PC, Weller SK. 1996. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol 70:2075–2085. doi: 10.1128/JVI.70.4.2075-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Newcomb WW, Cockrell SK, Homa FL, Brown JC. 2009. Polarized DNA ejection from the herpesvirus capsid. J Mol Biol 392:885–894. doi: 10.1016/j.jmb.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baines JD, Weller SK. 2005. Cleavage and packaging of herpes simplex virus 1 DNA, herpesvirus assembly, p 135–150. In Catalano CE (ed), Viral genome packaging machines: genetics, structure, and mechanism. Kluwer Academic/Plenum Publishers, New York, NY. [Google Scholar]
- 50.Mocarski ES, Roizman B. 1982. Structure and role of the herpes simplex virus DNA termini in inversion, circularization and generation of virion DNA. Cell 31:89–97. doi: 10.1016/0092-8674(82)90408-1. [DOI] [PubMed] [Google Scholar]
- 51.Beard PM, Duffy C, Baines JD. 2004. Quantification of the DNA cleavage and packaging proteins U(L)15 and U(L)28 in A and B capsids of herpes simplex virus type 1. J Virol 78:1367–1374. doi: 10.1128/jvi.78.3.1367-1374.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheaffer AK, Newcomb WW, Gao M, Yu D, Weller SK, Brown JC, Tenney DJ. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J Virol 75:687–698. doi: 10.1128/JVI.75.2.687-698.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taus NS, Baines JD. 1998. Herpes simplex virus 1 DNA cleavage/packaging: the UL28 gene encodes a minor component of B capsids. Virology 252:443–449. doi: 10.1006/viro.1998.9475. [DOI] [PubMed] [Google Scholar]
- 54.Gibson W, Roizman B. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol 10:1044–1052. doi: 10.1128/JVI.10.5.1044-1052.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perdue ML, Cohen JC, Randall CC, O’Callaghan DJ. 1976. Biochemical studies of the maturation of herpesvirus nucleocapsid species. Virology 74:194–208. doi: 10.1016/0042-6822(76)90141-0. [DOI] [PubMed] [Google Scholar]
- 56.Salmon B, Nalwanga D, Fan Y, Baines JD. 1999. Proteolytic cleavage of the amino terminus of the U(L)15 gene product of herpes simplex virus type 1 is coupled with maturation of viral DNA into unit-length genomes. J Virol 73:8338–8348. doi: 10.1128/JVI.73.10.8338-8348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu D, Sheaffer AK, Tenney DJ, Weller SK. 1997. Characterization of ICP6::lacZ insertion mutants of the UL15 gene of herpes simplex virus type 1 reveals the translation of two proteins. J Virol 71:2656–2665. doi: 10.1128/JVI.71.4.2656-2665.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol 72:1060–1070. doi: 10.1128/JVI.72.2.1060-1070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abaitua F, O’Hare P. 2008. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J Virol 82:5234–5244. doi: 10.1128/JVI.02497-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bucks MA, O’Regan KJ, Murphy MA, Wills JW, Courtney RJ. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316–324. doi: 10.1016/j.virol.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Henaff D, Remillard-Labrosse G, Loret S, Lippe R. 2013. Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J Virol 87:4895–4906. doi: 10.1128/JVI.03292-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol 86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dembowski JA, Dremel SE, DeLuca NA. 2017. Replication-coupled recruitment of viral and cellular factors to herpes simplex virus type 1 replication forks for the maintenance and expression of viral genomes. PLoS Pathog 13:e1006166. doi: 10.1371/journal.ppat.1006166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dembowski JA, DeLuca NA. 2015. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog 11:e1004939. doi: 10.1371/journal.ppat.1004939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, Graeper S, Klenk HD, Ruebsamen-Waigmann H, Hallenberger S. 2001. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol 75:9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol 72:4721–4728. doi: 10.1128/JVI.72.6.4721-4728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newcomb WW, Brown JC. 2002. Inhibition of herpes simplex virus replication by WAY-150138: assembly of capsids depleted of the portal and terminase proteins involved in DNA encapsidation. J Virol 76:10084–10088. doi: 10.1128/jvi.76.19.10084-10088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O’Hara B, Bloom JD, Johann SV. 2000. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J Virol 74:9054–9061. doi: 10.1128/jvi.74.19.9054-9061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zacny VL, Gershburg E, Davis MG, Biron KK, Pagano JS. 1999. Inhibition of Epstein-Barr virus replication by a benzimidazole L-riboside: novel antiviral mechanism of 5,6-dichloro-2-(isopropylamino)-1-beta-L-ribofuranosyl-1H-benzimidazole. J Virol 73:7271–7277. doi: 10.1128/JVI.73.9.7271-7277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen GH, Ponce de Leon M, Diggelmann H, Lawrence WC, Vernon SK, Eisenberg RJ. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J Virol 34:521–531. doi: 10.1128/JVI.34.2.521-531.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wills E, Scholtes L, Baines JD. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J Virol 80:10894–10899. doi: 10.1128/JVI.01364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reynolds AE, Fan Y, Baines JD. 2000. Characterization of the U(L)33 gene product of herpes simplex virus 1. Virology 266:310–318. doi: 10.1006/viro.1999.0090. [DOI] [PubMed] [Google Scholar]
- 73.Albright BS, Kosinski A, Szczepaniak R, Cook EA, Stow ND, Conway JF, Weller SK. 2015. The putative herpes simplex virus 1 chaperone protein UL32 modulates disulfide bond formation during infection. J Virol 89:443–453. doi: 10.1128/JVI.01913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Homa FL, Otal TM, Glorioso JC, Levine M. 1986. Transcriptional control signals of a herpes simplex virus type 1 late (gamma 2) gene lie within bases −34 to +124 relative to the 5′ terminus of the mRNA. Mol Cell Biol 6:3652–3666. doi: 10.1128/mcb.6.11.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Desai P, DeLuca NA, Glorioso JC, Person S. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J Virol 67:1357–1364. doi: 10.1128/JVI.67.3.1357-1364.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tivol WF, Briegel A, Jensen GJ. 2008. An improved cryogen for plunge freezing. Microsc Microanal 14:375–379. doi: 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conway JF, Steven AC. 1999. Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J Struct Biol 128:106–118. doi: 10.1006/jsbi.1999.4168. [DOI] [PubMed] [Google Scholar]
- 78.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM–an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 80.Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 82.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 83.Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, Jiang W, Ludtke SJ, Medalia O, Penczek PA, Rosenthal PB, Rossmann MG, Schmid MF, Schroder GF, Steven AC, Stokes DL, Westbrook JD, Wriggers W, Yang H, Young J, Berman HM, Chiu W, Kleywegt GJ, Lawson CL. 2012. Outcome of the first electron microscopy validation task force meeting. Structure 20:205–214. doi: 10.1016/j.str.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 85.Yang K, Baines JD. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J Virol 80:5733–5739. doi: 10.1128/JVI.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
CryoEM density maps have been deposited in the EM Data Bank (https://www.ebi.ac.uk/pdbe/emdb/index.html/) with accession numbers EMD-22592 through EMD-22595, EMD-22603, and EMD-22609 (Table 3).