

Abstract
Designer Receptors Exclusively Activated by Designer Drugs (DREADD) are a preclinical chemogenetic approach with clinical potential for various disorders. In vivo visualization of DREADDs has been achieved with positron emission tomography (PET) using 11C radiotracers. The objective of this study was to develop DREADD radiotracers labeled with 18F for a longer isotope half-life. A series of non-radioactive fluorinated analogs of clozapine with a wide range of in vitro binding affinities for the hM3Dq and hM4Di DREADD receptors has been synthesized for PET. Compound [18F]7b was radiolabeled via a modified 18F-deoxyfluorination protocol with a commercial ruthenium reagent. [18F]7b demonstrated encouraging PET imaging properties in a DREADD hM3Dq transgenic mouse model, whereas the radiotracer uptake in the wild type mouse brain was low. [18F]7b is a promising long-lived alternative to the DREADD radiotracers [11C]clozapine ([11C]CLZ) and [11C]deschloroclozapine ([11C]DCZ).
Keywords: DREADD, hM3Dq, hM4Di, SAR, PET, 18F, chemogenetics
Graphical Abstract
INTRODUCTION
Designer Receptors Exclusively Activated by Designer Drugs (DREADD) are a chemogenetic approach for remote manipulation of neuronal activity in freely-moving animals.1 DREADDs comprise mutated G protein-coupled receptors (GPCRs) that do not respond to their endogenous neurotransmitter, but do respond to an otherwise “inert” exogenous substance. The most widely used DREADDs are derived from the human muscarinic (hM3 and hM4) receptors and are referred to as hM3Dq and hM4Di. When expressed in the brain, these muscarinic DREADDs do not respond to their endogenous ligand (i.e. acetylcholine), but are selectively activated using sub-therapeutic doses of clozapine (CLZ)2, the DREADD pro-drug clozapine N-oxide (CNO) or other novel designer drugs.3 hM3Dq activation via its ligand elicits recruitment of Gq-proteins, activating a signaling cascade that generally leads to neuronal excitation, whereas activation of hM4Di recruits Gi/o proteins, activating a signaling cascade that leads to neuronal inhibition.
DREADD technology has significant potential for clinical translation, especially for indications that presently use invasive therapeutic approaches such as epilepsy,4 Parkinson’s disease,5 and pain management.6
One critical development needed for translation of DREADD technology to the clinic involves the necessity to visualize and monitor exogenous DREADD expression noninvasively and longitudinally in non-human primates (NHPs) or humans. Currently, this can be achieved using [11C]CLZ or [11C]DCZ and positron emission tomography (PET).2, 7–9 However, these carbon-11 labeled radiotracers have short half-lives (~20 minutes) and therefore access to these radioligands is limited to research institutions and hospitals with cyclotrons and dedicated radiochemistry infrastructure.
The objective of this study was to develop novel fluorinated DREADD ligands that would exhibit high selectivity and potency, and furthermore, that would be amenable to radiolabeling using the PET radioisotope 18F. Fluorine-18 has a half-life of ~110 minutes and can be transported from a production site to a remote PET imaging site. Accordingly, a 18F-labeled DREADD ligand would facilitate PET studies in animals and human subjects at institutions that lack cyclotron and PET radiochemistry infrastructure. In addition, strategic replacement of metabolically labile hydrogen atoms with fluorine can enhance the metabolic stability of drugs10 which would also improve their performance as in vivo actuators. This report presents the synthesis, in vitro binding affinity and structure-activity relationship of a series of fluorine-bearing analogs of clozapine as potential DREADD ligands. 18F-radiolabeling and evaluation in rodents of selected compounds within the series are also presented.
RESULTS AND DISCUSSION
Design and Synthesis
The antipsychotic drug clozapine (CLZ), exhibits high binding affinity for various receptors including the 5-HT2A, D4, M1 and adrenergic receptors (1–10 nM),11 with weaker affinity for the D2 receptor (75 – 385 nM).12 Radiolabeled [11C]CLZ was developed in 1986 as a potential serotonergic and dopaminergic PET radiotracer, but it manifested poor specific signal in monkey13 and human brain14 and was abandoned. A recent study, however, demonstrated that clozapine was a highly potent DREADD (hM3Dq) agonist (EC50 = 1.1 nM)15 and radiolabeled [11C]CLZ was re-introduced as a specific PET radioligand for DREADD imaging.2, 7, 9 Recently, [11C]-deschloroclozapine ([11C]DCZ) was developed as another promising PET tracer for DREADD imaging with similar properties to those of [11C]CLZ8.
The previous structure-activity relationship (SAR) investigation by Chen et al.15 demonstrated that CLZ and several derivatives of CLZ bind at DREADD receptor subtypes hM3Dq and hM4Di.
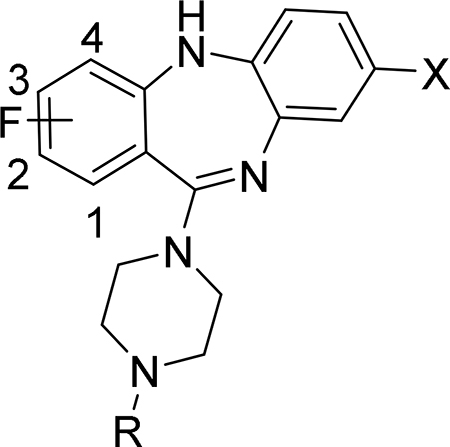
From that SAR, the N-ethyl analog of CLZ (1, Fig. 1), a highly potent DREADD agonist with excellent selectivity (hM3Dq EC50 = 1.3 nM; hM3 EC50 > 30000 nM),15 was selected as the lead for the development of fluorinated DREADD ligands. The DREADD binding affinities of 1 are comparable with those of CLZ and DCZ (Table 1). The main aim was to synthesize a series of fluorinated analogs of 1 with high binding affinity and selectivity for hM3Dq and hM4Di and to radiolabel suitable compounds with 18F.
Figure 1.

Lead 115 and projected fluorinated analogs
Table 1.
In vitro binding affinity values, molecular weight (MW) and lipophilicity (cLogD7.4) of CLZ, DCZ, 1 and fluorinated isosteres of 1. Binding affinity data are mean Ki ± SD (n = 3)
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | F position | X | Ki, nM | Structural properties1 | |||
| hM3Dq | hM4Di | MW | cLogD7.4 | |||||
| CLZ2 | Me | - | Cl | 3.5 ± 1.3 | 2.8 ± 0.7 | 327 | 2.9 | |
| DCZ8 | Me | - | H | 6.3 | 4.2 | 292 | 2.5 | |
| 115 | Et | - | Cl | 4.3 ± 2.9 | 4.3 ± 0.1 | 341 | 3.4 | |
| 7a18 | Et | 1 | Cl | 1.7 ± 0.1 | 8.7 ± 2.1 | 359 | 3.1 | |
| 7b18 | Et | 2 | Cl | 10.5 ± 1.5 | 23.5 ± 4.5 | 359 | 2.9 | |
| 7c | Et | 3 | Cl | 51.4 ± 7.5 | 51 ± 9 | 359 | 3.0 | |
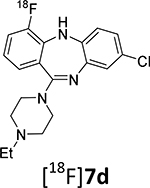
| 7d18 | Et | 4 | Cl | 1.9 ± 0.2 | 3.6 ± 0.8 | 359 | 3.1 | |
| 7e | Et | 1 | H | 4.8 ± 0.6 | 14.2 ± 3.3 | 324 | 2.3 | |
| 7f | Et | 2 | H | 33.5 ± 4.1 | 118 ± 23 | 324 | 2.1 | |
| 7g | Et | 3 | H | 101 ± 20 | 149 ± 28 | 324 | 2.2 | |
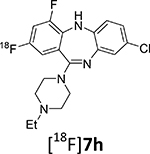
| 7h | Et | 2, 4 | Cl | 8.1 ± 0.9 | 18.1 ± 5.4 | 377 | 3.4 | |
| 7i | Et | - | F | 23.7 ± 2.5 | 12.9 ± 2.2 | 324 | 2.3 | |
Calculated using ACD/Percepta software
The synthesis of new fluorinated compounds 7a - 7i was performed using a conventional method of the synthesis of clozapine and its analogs (Scheme 1).16, 17 All intermediates of this synthesis were used in the next step without further purification. An Ullmann coupling of the fluorinated 2-amino-benzoic acids 2a - 2d and the 1-iodo-2-nitrobenzenes 3a - 3c formed the nitro-diarylamines 4a - 4i, which were then reduced to the corresponding amino-diarylamines 5a - 5i with zinc in methanol. Intramolecular amidation of 5a - 5i with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) gave the 5,10-dihydro-11H-dibenzo[b,e][1,4]diazepin-11-ones 6a - 6i. In the final step, the cyclic amides 6a - 6i were converted in high yield to the final compounds 7a - 7i by a TiCl4 - assisted reaction with N-ethyl piperazine.
Scheme 1. Synthesis of fluoro compounds 7a - 7i.

Reagents and conditions: (a) Cu, K2CO3, 1-pentanol, reflux; (b) Zn, NH4Cl, MeOH, rt; (c) EDC·HCl, CH2Cl2, rt; (d) TiCl4, 1,4-dioxane, 1-ethylpiperazine, reflux.
This publication expands upon the limited biological disclosure of compounds 7a (JHU37152), 7b (JHU37107) and 7d (JHU37160)18 to encompass the full structure-activity relationship data for the whole series, as well as the novel radiolabeling details and PET imaging properties for the critical compounds. This presents a complete thorough development of the key DREADD ligands and sets the stage for future necessary advances.
Binding assays and structure-activity relationship
The in vitro inhibition binding affinity of compounds 7a - 7i was determined using the radioligand [3H]CLZ and membrane preparations from human embryonic kidney cells (HEK293) transiently transfected with hM3Dq or hM4Di cDNAs. Non-transfected HEK293 cells showed negligible binding to [3H]CLZ when compared to HEK293 cells expressing DREADDs2, 18, hence the Ki values shown in Table 1 were determined through competition curves using [3H]CLZ.
CLZ, DCZ and lead 1 exhibited similar binding affinities (Table 1). The introduction of fluorine on the aromatic ring of 1 had a clear position-dependent effect on hM3Dq and hM4Di binding affinity. Compounds 7a, 7b and 7d with fluorine in positions 1, 2 and 4 exhibited the highest binding affinity within the series. Incorporation of fluorine at the 3-position, however, caused a sharp decrease of binding affinity (7c). Comparison of the Ki values between 7a - 7c and 7e - 7g suggested that the 8-chloro substitution causes an approximately 2 – 3 -fold increase of the binding affinity. Interestingly, the 2,4-di-fluoro compound 7h exhibited an intermediate binding affinity between the corresponding mono-fluoro compounds 7b and 7d. The analog 7i with 8-fluorine instead of 8-chlorine exhibited a reduced binding affinity.
In addition, 7a, 7b, 7d, 7e and 7h were tested in a large panel of binding and enzyme assays for their relative selectivity, potential adverse effects, and potential alternate mechanisms of action (Supplemental Info, Supplemental Tables 1 and 2). These compounds demonstrated a similar pharmacology to that of CLZ within the panel.
Selection of candidates for 18F radiolabeling
DREADDs can exhibit a very high level of expression in the mammalian brain. Via a radioligand binding assay, the DREADD density in hM3Dq-expressing mice has been measured to have a Bmax equal to 8.5 – 10 pmol/mg protein or approximately 300 – 1,000 nmol/L tissue.19 That is higher than the Bmax of most natural brain receptors such as dopaminergic (BmaxD2 = 10 – 80 nmol/L20) and serotonergic receptors (Bmax5HT2 = 20 – 40 nmol/L21). The conventional criteria for a suitable PET radiotracer is for the , where KD is the dissociation constant.22 This equation suggests that a suitable DREADD PET radiotracer should exhibit a Ki < 30 – 100 nM. These calculations agree with the experimental evidence for [11C]CLZ (Ki = 2.8 – 3.5 nM, Table 1) and [11C]DCZ (Ki = 4.2 – 6.3 nM), DREADD radiotracers with suitable PET imaging properties in animal experiments2, 7–9.
Several members (7a, 7b, 7d, 7e and 7h) of the series 7a - 7i manifest comparable or better hM3Dq binding affinity values and comparable pharmacology to those of CLZ (Table 1, Supplemental Info).
The other necessary properties for cerebral PET tracers include optimal lipophilicity (1 < logD7.4 < 4) and molecular weight MW < 500 Da23,24. The structural properties of 7a, 7b, 7d, 7e and 7h (Table 1) are in a suitable range for brain PET (MW = 320 – 377 Da; logD7.4 = 2.3 – 3.4). Accordingly, the DREADD ligands 7a, 7b, 7d, 7e and 7h are attractive candidates for radiolabeling with 18F for PET imaging of DREADD.
Unfortunately, the backbone of the fluorinated series, 11-(4-methylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine, is not readily suitable for conventional aromatic nucleophilic substitution with [18F]fluoride due to the of the lack of electron-withdrawing groups.25 In recent years, however, many methods have been developed for 18F-labeling of electron-rich aromatic compounds.25, 26 Our initial attempts to radiolabel [18F]7a and [18F]7d using one of these methods utilizing iodonium(III) ylide precursors were unsuccessful.
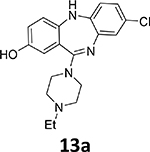
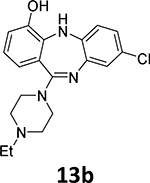
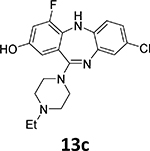
Another suitable method involves the 18F-deoxyfluorination of phenols using ruthenium π-complexes.27 Inspired by this report, we synthesized phenols 13a, 13b, and 13c as potential precursors for 18F-radiolabeling of 7b, 7d and 7h, respectively
Synthesis of precursors for 18F radiolabeling
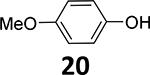
Starting from commercially available 2-amino-5-methoxybenzoic acid 8a, the methoxy arene 12a was synthesized in four steps, followed by demethylation in BBr3/CH2Cl2 solution to obtain the phenol precursor 13a (Scheme 2). The phenol precursors 13b and 13c were prepared similarly to 13a (Scheme 2).
Scheme 2.

Synthesis of phenol precursors 13a – 13c for radiolabeling. Reagents and conditions: (a) Cu, K2CO3, 1-pentanol, reflux; (b) Zn, NH4Cl, MeOH, rt; (c) EDCHCl, CH2Cl2, room temperature; (d) TiCl4, 1,4-dioxane, 1-ethylpiperazine, reflux; (e) BBr3, CH2Cl2, 0°C to room temperature.
18F-Deoxyfluorination using ruthenium reagent 15.
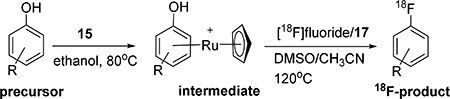
In order to efficiently convert the phenol precursors into their 18F analogs, the use of a deoxyfluorination methodology developed by Ritter and coworkers has considerable advantages.27 Unfortunately, a disadvantage to that methodology is the lack of commercial availability of the necessary CpRu(COD)Cl complex 14 (Fig. 2), coupled with the toxicity hazard involved in the synthesis of 14. It was envisioned that an analogous, commercially available (STREM Chemicals) tris(acetonitrile)cyclopentadienylruthenium(II) hexafluorophosphate, 15 (Fig. 2) would also enable the 18F-deoxyfluorination of phenols and we sought to investigate this.
Figure 2.

Structures of CpRu(COD)Cl, 14 and commercial ruthenium reagent 15.
To test this hypothesis on a model system, a stoichiometric amount of CpRu(MeCN)3PF6 complex 15 was heated with model phenol 16 in degassed ethanol. The desired organometallic complex 16a was quickly and quantitatively obtained (Scheme 3), as determined by 1H-NMR and LC-MS. Notably, the PF6 anion was also present as determined by 19F-NMR. Using similar conditions to those of Ritter’s protocol,27 this organometallic complex was then converted to the aryl fluoride 16b with the addition of cesium fluoride and N,N’-bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride, 17. Importantly, in the same reaction the ruthenium ligand 16a was decomplexed from the aryl group. Alternative aryl-cyclopentadienyl ruthenium complexes prepared via commercially-available chloro(pentamethylcyclopentadienyl)(cyclooctadiene)ruthenium(II) 18, failed to decomplex from the aryl group under these conditions (Scheme 3).
Scheme 3.

Deoxyfluorination synthesis of 1-(4-(4-[19F]fluorophenyl)piperazin-1-yl)ethan-1-one 16b.
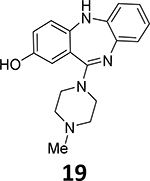
With the model system in hand, 19F-deoxyfluorination on a model closer to desired substrates, the clozapine metabolite GMC-1–116, 19 (Scheme 4), was attempted. Again, this reaction sequence also performed quite well, and demonstrated that the methodology should work well in the context of 18F radiofluorination. However, the presence of the PF6 anion had the potential to substantially reduce the radiochemical yield and specific activity. In order to remove the PF6 anion, simple treatment of the ruthenium organometallic complex (19a) with excess ion exchange resin rapidly and efficiently exchanged the PF6 anion for a chloride anion. This could be easily monitored by 19F-NMR and the disappearance of the fluorine signal after treatment with the exchange resin.
Scheme 4.

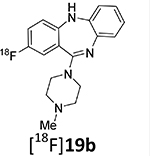
Deoxyfluorination synthesis of 8-[19F]fluoro-11-(4-methylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine, 19b.
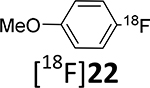
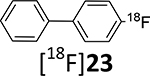
With the methodology worked out in 19F systems, the 18F radiolabeling within model systems and target substrates was examined. The radiolabeling 18F-deoxyfluorination reactions of model phenol precursors 19 - 21 with ruthenium complex 15 are summarized in Table 2.
Table 2.
18F-Deoxyfluorination reactions with commercially available ruthenium compound 15.
 | ||
|---|---|---|
| Precursor | 18F-product | Radiochemical yield, % (HPLC)a |
 |
 |
34% |
 |
 |
10.1 ± 4.5 (n = 4) |
 |
 |
18% |
 |
 |
40.2 ± 13.3 (n = 13) |
 |
 |
<0.1% |
 |
 |
~1% (n = 2) |
Non-decay-corrected radiochemical yield that was determined using reverse-phase high performance liquid chromatography (RP-HPLC) analysis of the reaction mixture.
The model radiochemical syntheses of [18F]19b, [18F]22 and [18F]23 via phenols 19 - 21 were performed using an automated radiofluorination TRACERlab module (General Electric Medical Systems). In brief, prior to the radiochemical synthesis, phenol precursors (19 - 21) and ruthenium reagent 15 were dissolved in ethanol, heated for 30 min, and evaporated to dryness under a nitrogen flow. The dry residue of intermediate phenol/15 complex was dissolved in a 1:1 DMSO/CH3CN mixture. [18F]Fluoride solution in [18O]water from a cyclotron target was loaded onto an anion-exchange solid-phase extraction cartridge. The cartridge was rinsed with methanol and [18F]fluoride was eluted with a methanol solution of N,N’-bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride (17) into the reaction vessel. The degassed solution of intermediate phenol/15 complex in DMSO/CH3CN was then added. The reaction mixture was heated at 120°C for 30 min in a sealed reaction vessel, cooled, and the radiochemical yield of the 18F-labeled products [18F]19b, [18F]22 and [18F]23 in the reaction mixture was determined by analytical RP-HPLC.
The relatively low radiochemical yield of the model 4-[18F]fluoroanisole, [18F]22, may be due to its volatility. The radiochemical yields of the other two model compounds, [18F]19b and [18F]23, were higher (18 – 34%). The radiolabeling conditions were not optimized.
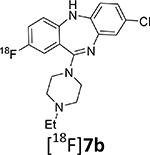
The radiosynthesis of the DREADD radiotracer [18F]7b via 18F-deoxyfluorination of phenol 13a was done under the same conditions that were used for the model radiolabeling. The radiochemical yield of [18F]7b was high as determined by RP-HPLC analysis of the reaction mixture (40.2 ± 13.3%; n=13). The final product [18F]7b was purified by semi-preparative RP-HPLC (Supplemental info) and formulated in saline containing 9% ethanol with a final radiochemical yield of 7 – 9% (non-decay corrected) and a radiochemical purity of 95%. The difference between the radiochemical yields determined by RP-HPLC analysis and final dose assay is due to a loss of radioactivity in the mechanical steps of the radiosynthesis (evaporation, fluid transfer, filtration, etc.) that is common in PET radiochemistry. Because the radiochemical yield of the final product [18F]7b was sufficient for animal experiments, it has not been optimized. The radiolabeling of [18F]7b was first described in our previous publication, but without chemistry and radiolabeling details.18
As expected from previous 18F-labeling research,28 the specific (molar) radioactivity of [18F]7b (123 ± 120 GBq/μmol (3,315 ± 3,237 mCi/μmol) (n = 10)) depends on the starting amount of [18F]fluoride (Fig. 3). The original [18F]deoxyfluorination study reported similar specific radioactivity ((~111 GBq/μmol (~3,000 mCi/μmol)) of final 18F-product.27
Figure 3.

Specific (molar) radioactivity of [18F]7b versus starting amount of [18F]fluoride.
Such specific radioactivities are sufficiently high for animal studies, but are lower than are normally observed in the authors’ laboratory using conventional nucleophilic 18F-fluorination reactions (>740 GBq/μmol (>20,000 mCi/μmol)). This difference may be due to trace amounts of carrier [19F]fluoride from the PF6− anion of the ruthenium compound 15.
Surprisingly, 18F-deoxyfluorination of phenol 13c, which is structurally similar to 13a, under the same conditions, gave a corresponding radiolabeled compound [18F]7h in low yield (~1%). One possible explanation is that 15 chelates with the fluorine atom in position 4 of the aromatic backbone. Another possible explanation might be that having another electronegative substituent on the ring decreases the rate of the 18F-fluorination reaction.
18F-Radiofluorination of phenol 13b yielded only trace amounts of corresponding [18F]7d, which may be because of the ortho-steric effect of the neighboring NH substituent. This substrate also proved to be quite difficult during the 19F-deoxyfluorination attempts.
PET imaging study in mice
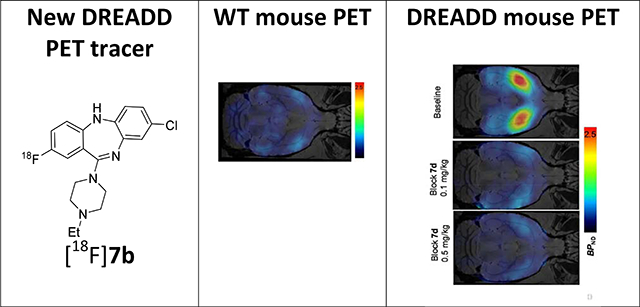
Given its favorable binding affinity profile compared to [11C]CLZ and [11C]DCZ (Table 1 and Supplemental info) and high synthetic yield, purity and specific radioactivity, radiotracer [18F]7b was selected for DREADD PET imaging experiments in animals.
In a previous study18 the use of [18F]7b for imaging of DREADDs in macaque monkeys, rats and mice expressing DREADDs via viral transduction has been validated. That preliminary strategy allowed us to identify the vector injection site. The goal of this study was to validate the use of [18F]7b as a PET radiotracer in a transgenic mouse model expressing hM3Dq receptors18 in a selected subset of neurons expressed throughout the brain and evaluate the radiotracer specificity in dose-escalation blocking experiments with a selective DREADD ligand.
Regional brain distribution of [18F]7b was determined in PET experiments in transgenic mice expressing hM3Dq whereas wild-type mice (n = 3 per group), that lacked DREADD expression, were used as a control (Figs. 4, 5). The D1-HM3Dq mice express hM3Dq in dopamine D1 receptor (D1R) positive neurons and were generated by crossing D1-Cre mice (D1-Cre, FK150 line, C57BL/6J congenic, Gensat, RRID: MMRRC_036916-UCD) and hM3Dq mice (R26-hM3Dq/mCitrine, Jackson Laboratory, stock no. 026220). Hence, only animals carrying both transgenes would express DREADDs. Accordingly, hM3Dq expression in these mice correlates with D1R expression and they express a high level of hM3Dq in the striatum, moderate and disperse levels in cortex and low levels in the cerebellum.5, 18
Figure 4.

PET-[18F]7b experiments in transgenic mice expressing hM3Dq. Time-uptake curves in baseline (panel A) and blocking studies (blocker – 7d) (panel C, blocker dose = 0.1 mg/kg; panel D, blocker dose = 0.5 mg/kg). Data: mean SUV ± SD (n=3). SUV = standardized uptake value in the mouse striatum (STR), cortex (CTX) and cerebellum (CB). Target-to-non-target ratio versus time (panel B)
Figure 5.

Panel A: Transverse PET-[18F]7b image of wild-type mouse; Panel B: Transverse PET-[18F]7b images of transgenic mice expressing hM3Dq (baseline and dose-escalation blocking); Panel C: Brain regional binding potential (BPND) values in baseline and dose-escalation blocking PET scans with [18F]7b in transgenic mice expressing hM3Dq. Abbreviation: STR = striatum; CTX = cortex; CB = cerebellum). Blocker – 7d. Statistical analysis: comparison of baseline versus blocking in the same region. *P < 0.05; **P > 0.05 (ANOVA).
PET imaging with [18F]7b in a wild-type mouse showed low accumulation of radioactivity in the brain (Fig. 5A). That result is not a surprise because of the structural and pharmacological similarity of 7b to CLZ (Table 1, Supplemental Info). Previous extensive PET imaging experiments with radiolabeled [11C]CLZ demonstrated low specific signal in normal monkey13 and human brain14 due to insufficient binding affinity at serotonergic and dopaminergic receptors.
In agreement with brain regional expression of hM3Dq in the DREADD mice,5, 18 a PET imaging study in transgenic hM3Dq mice demonstrated robust uptake of [18F]7b in the mouse brain regions with a peak at 2–10 min followed by washout (Fig. 4). The highest accumulation of [18F]7b-induced radioactivity occurred in the striatum (1.93 ± 0.33 SUV [mean ± SD of 3]) (Figs. 4A, 5). The clearance rate of [18F]7b from the cerebellum was higher than from any other regions studied. The ratios of tissue to the cerebellum increased steadily, reaching maximum values of 3.0 in the striatum and 1.8 in the cortex at 50–60 min after injection (Fig. 4B).
The [18F]7b radioactivity accumulation in the striatum and cortex in the DREADD mice was specific and mediated by hM3Dq binding as determined in a dose-escalation blocking study with 7d, a high binding affinity and selective DREADD ligand (Figs. 4C, 4D and 5B,C). As expected5, 18, the binding potential (BPND) was high in the striatum and lower in the cortex. The regional binding potential values of [18F]7b in the striatum and cortex were BPND = 1.6 and 0.6, respectively (Fig. 5C). In all blocking experiments, the binding potential values in the striatum were significantly reduced versus baseline, whereas in the cortex the blocking was significant only at the highest dose of blocker (0.5 mg/kg) (Fig. 5B,C). In the cerebellum, the blocking effect was insignificant. The imaging properties of [18F]7b are comparable to those of [11C]CLZ7, 9, 18 and [11C]DCZ (STR/CB ratio value = 2.7 in hM3Dq Tg mice8), but the longer half-life of [18F]7b affords a more practical radiotracer for PET imaging experiments.
CONCLUSION
A series of fluorinated derivatives of 8-chloro-11-(4-ethylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine with high binding affinities for DREADD (Ki = 1.7 − 149 nM) has been synthesized with the potential application for PET imaging. Five members of the series, 7a, 7b, 7d, 7e and 7h, with the highest DREADD binding affinities (Ki = 1.7 – 10.5 nM) and selectivity were chosen as candidates for radiolabeling and DREADD imaging research.
Several model [18F]fluoro-arenes were synthesized with good radiochemical yields via a modified Ritter’s [18F]deoxyfluorination protocol using a commercially available ruthenium reagent. Under the same radiolabeling conditions, DREADD radiotracer [18F]7b was prepared with sufficient radiochemical yield, specific radioactivity and purity whereas radiochemical yields of [18F]7d and [18F]7h were lower. Further radiolabeling experiments with [18F]7a, 7d, 7e and 7h are warranted.
In PET studies, [18F]7b readily entered the mouse brain and specifically and selectively labeled cerebral hM3Dq DREADD receptors in a transgenic mouse model. The binding potential (BPND) values and target-to-non-target ratios of [18F]7b in the mouse brain regions were in agreement with previously published regional brain distribution of DREADD in these animals and comparable with previous DREADD PET radiotracers [11C]CLZ and [11C]DCZ.
The suitable PET imaging properties and longer half-life of [18F]7b make it a practical alternative to [11C]CLZ and [11C]DCZ for in vivo quantification of DREADD receptors. The presented study demonstrated the suitability of [18F]7b for evaluation of non-labeled DREADD ligands in vivo. This research also shows promise towards expanding the DREADD PET imaging field with a range of new and powerful radioligands.
EXPERIMENTAL SECTION
General.
Analytical thin layer chromatography (TLC) was performed on 0.20 mm silica gel 60 plates POLYGRAM SIL G/UV254 (Machery-Nagel, Germany). The TLC spots were visualized using UV light or staining with iodine, ninhydrin, phosphomolybdic acid, or vanillin followed by heating. Column chromatography for purification of crude products was carried out on silica gel (60, Irregular, particle size 0.040–0.060 mm, VWR). All NMR spectra were recorded with either a Bruker AV 500 Ultra instrument or a Varian 400 MHz spectrometer. The chemical shifts (δ) were expressed in parts per million (ppm). 1H-NMRs were referenced to CDCl3 at δ 7.26 ppm and 13C-NMR at 77.16 ppm. Signals are reported as multiplet (m), singlet (s), doublet (d), triplet (t), quartet(q), or broad singlet (bs); and coupling constants are reported in Hertz (Hz). Purity of the final compounds were confirmed with quantitative NMR using pyrazine (purity >99%) as a standard and DMSO-d6 as solvent, the molar ratio of pyrazine/compound was 1:1 (see Supplemental data for q-NMR results).
Low-resolution mass spectra were obtained on an Agilent 1200 series LC/MS equipped with a Luna C18 (3 mm x 75 mm, 3 μm) reversed-phase column with UV detection at 220 and 254 nm. The mobile phase consisted of water containing 0.05% trifluoroacetic acid as component A and acetonitrile containing 0.025% trifluoroacetic acid as component B. A linear gradient was run as follows: 0 min 4% B; 7 min 100% B; 8 min 100% B at a flow rate of 0.8 mL/min. High-resolution mass spectra were recorded utilizing electrospray ionization (ESI) at the University of Notre Dame Mass Spectrometry facility. IUPAC chemical names of compounds were generated by ChemDraw Professional 16.0.
8-Chloro-11-(4-ethylpiperazin-1-yl)-1-fluoro-5H-dibenzo[b,e][1,4]diazepine (7a) (General procedure)
2-Amino-6-fluorobenzoic acid (2a, 2.48 g, 16 mmol) in pentanol solution (50 mL) was heated to 140°C. 4-Chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) was added to the reaction mixture, followed by K2CO3 (2.21 g, 16.00 mmol), and Cu powder (78 mg, 1.22 mmol). The reaction mixture was refluxed for 6 h. After cooling down, the precipitated solid was filtered, dissolved in water, and acidified with aqueous HCl to form a yellow solid that was dried under vacuum. This crude solid compound 2-((4-chloro-2-nitrophenyl)amino)-6-fluorobenzoic acid (4a) (4.89 g, 98.4% yield) was used for the next step without further purification.
The above crude 4a (0.5 g, 1.6 mmol) was dissolved in methanol (10 mL) and treated with NH4Cl (0.46 g, 8 mmol) and Zn (0.52 g, 8 mmol) at 5 – 10°C. The mixture was stirred at room temperature for 2 h, and then filtered to remove solids. The filtrate was concentrated under vacuum to yield a green solid 5a. This solid was dissolved in dichloromethane (20 mL) and treated with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) (0.92 g, 4.8 mmol, 3 equiv.). The reaction mixture was stirred at room temperature under argon for 12 h, which then was concentrated to obtain a pale residue. The residue was treated with TiCl4 (1M in toluene) (1.9 mL, 1.9 mmol) and 1-ethylpiperazine (0.92 g, 8 mmol) in 1,4-dioxane (50 mL) under argon. The mixture was heated to 100°C for 12 h. After cooling down, the mixture was acidified with 1M aqueous HCl, and then extracted with ethyl acetate (EtOAc). The aqueous layer was basified with 1M aqueous NaOH, and then extracted with EtOAc. The organic layer was dried with Na2SO4, and concentrated for purification on a silica gel column to produce the pure product 7a as a yellow solid (0.225 g, 39.4% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.28 – 7.23 (m, 1H), 7.06 (s, 1H), 6.79 (d, J = 8.3 Hz, 1H), 6.76 (t, J = 9.0 Hz, 1H), 6.67 (d, J = 7.9 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.95 (s, 1H), 3.59 (bs, 4H), 2.59 (bs, 1H), 2.48 (d, J = 7.0 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3): 160.6, 158.6, 156.2 (d, J = 2.1 Hz), 155.1 (d, J = 6.4 Hz), 142.4, 139.6, 132.3 (d, J = 10.6 Hz), 129.7, 126.4, 122.6, 120.3, 116.1, 112.0 (d, J = 15.8 Hz), 111.0 (d, J = 22.9 Hz), 52.86, 52.83, 52.5, 12.1. HRMS (ESI+) calculated for C19H21ClFN4 (M+H)+: 359.1456; found: 359.1435.
8-Chloro-11-(4-ethylpiperazin-1-yl)-2-fluoro-5H-dibenzo[b,e][1,4]diazepine (7b)
According to the general procedure, 2-amino-5-fluorobenzoic acid (2b, 4.96 g, 32 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 9.06 g, 32 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-5-fluorobenzoic acid was obtained (8.93 g, 90% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-5-fluorobenzoic acid (4b, 1.6 mmol) was used to produce the product 7b as a yellow solid (0.145 g, 25% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.08 (d, J = 2.3 Hz, 1H), 7.03 (td, J = 8.2, 2.9 Hz, 1H), 6.99 (dd, J = 8.7, 2.9 Hz, 1H), 6.85 (dd, J = 8.3, 2.4 Hz, 1H), 6.63 (d, J = 8.3 Hz, 1H), 4.83 (s, 1H), 3.50 (bs, 4H), 2.56 (bs, 4H), 2.50 (dd, J = 14.4, 7.3 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3): 171.3, 161.5, 159.8, 157.8, 148.6 (d, J = 2.2 Hz), 141.7, 140.5, 129.4, 127.0, 125.0 (d, J = 6.6 Hz), 123.5, 121.5 (d, J = 7.9 Hz), 120.2, 118.9 (d, J = 22.8 Hz), 116.6 (d, J = 23.5 Hz), 60.6, 52.8, 52.5, 12.1 (see Fig. S4). HRMS (ESI+) calculated for C19H21ClFN4 (M+H)+: 359.1439; found: 359.1435.
8-Chloro-11-(4-ethylpiperazin-1-yl)-3-fluoro-5H-dibenzo[b,e][1,4]diazepine (7c)
According to the general procedure, 2-amino-4-fluorobenzoic acid (2c, 4.96 g, 32 mmol) and 1-iodo-4-chloro-2-nitrobenzene (3a, 9.06 g, 32 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-4-fluorobenzoic acid (4c) was obtained. (7.86 g, 79.2% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-4-fluorobenzoic acid (4c, 1.6 mmol) was used to produce the product 7c as a yellow solid (0.165 g, 28.8% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.27 – 7.23 (m, 1H), 7.07 (d, J = 2.3 Hz, 1H), 6.84 (dd, J = 8.3, 2.4 Hz, 1H), 6.74 (td, J = 8.4, 2.4 Hz, 1H), 6.61 (d, J = 8.3 Hz, 1H), 6.57 (dd, J = 9.2, 2.4 Hz, 1H), 4.91 (s, 1H), 3.47 (bs, 4H), 2.55 (bs, 4H), 2.49 (dd, J = 14.4, 7.2 Hz, 2H), 1.13 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3): 166.0, 164.0, 162.1, 154.6 (d, J = 9.1 Hz), 141.9, 139.9, 132.1 (d, J = 9.8 Hz), 129.5, 127.0, 123.4, 120.3, 119.7 (d, J = 3.2 Hz), 110.3 (d, J = 21.7 Hz), 107.5 (d, J = 23.0 Hz), 52.8, 52.5, 47.5, 12.0. HRMS (ESI+) calculated for C19H21ClFN4 (M+H)+: 359.1456; found: 359.1435.
8-Chloro-11-(4-ethylpiperazin-1-yl)-4-fluoro-5H-dibenzo[b,e][1,4]diazepine (7d)
According to the general procedure, 2-amino-3-fluorobenzoic acid (2d, 2.48 g, 16 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-fluorobenzoic acid (4d) was obtained. (4.91 g, 98.6% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-fluorobenzoic acid (4d, 1.6 mmol) was used to produce the product 7d as a yellow solid (0.265 g, 44.9% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.15 – 7.09 (m, 1H), 7.07 (d, J = 2.3 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.95 (td, J = 8.0, 5.1 Hz, 1H), 6.85 (dd, J = 8.3, 2.4 Hz, 1H), 6.68 (d, J = 8.3 Hz, 1H), 5.34 (d, J = 3.8 Hz, 1H), 3.49 (bs, 4H), 2.55 (bs, 3H), 2.49 (q, J = 7.2 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3): 161.7, 154.5, 152.6, 141.8, 140.5 (d, J = 15.2 Hz), 140.0, 129.5, 127.1, 126.0, 125.6 (d, J = 3.7 Hz), 123.4, 123.1 (d, J = 7.5 Hz), 120.9, 117.4 (d, J = 20.0 Hz), 52.9, 52.6, 47.5, 12.1. HRMS (ESI+) calculated for C19H21ClFN4 (M+H)+: 359.1456; found: 359.1435.
11-(4-Ethylpiperazin-1-yl)-1-fluoro-5H-dibenzo[b,e][1,4]diazepine (7e)
According to the general procedure, 2-amino-6-fluorobenzoic acid (2a, 4.96 g, 32 mmol) and 1-iodo-2-nitrobenzene (3b, 7.97 g, 32 mmol) were used. The crude compound 6-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4e) was obtained (7.36 g, 83.3% yield).
The above crude compound 6-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4e, 1.6 mmol) was used to produce the product 7e as a yellow solid (0.131 g, 25.3% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.22 (dd, J = 14.5, 7.5 Hz, 1H), 7.08 (d, J = 7.8 Hz, 1H), 6.99 (t, J = 7.5 Hz, 1H), 6.85 (t, J = 7.5 Hz, 1H), 6.72 (t, J = 8.3 Hz, 2H), 6.67 (d, J = 7.9 Hz, 1H), 5.02 (s, 1H), 3.55 (s, 4H), 2.60 (s, 2H), 2.53 – 2.46 (m, 4H), 1.12 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3): 160.9, 160.6, 155.73, 155.68, 141.2, 140.8, 132.2 (d, J = 10.7 Hz), 126.7, 124.8, 123.4, 119.7, 116.2 (d, J = 2.7 Hz), 111.7 (d, J = 15.4 Hz), 110.6 (d, J = 22.9 Hz), 52.5, 52.4, 45.5, 11.4. HRMS (ESI+) calculated for C19H22FN4 (M+H)+: 325.1828; found: 325.1810.
11-(4-Ethylpiperazin-1-yl)-2-fluoro-5H-dibenzo[b,e][1,4]diazepine (7f)
According to the general procedure, 2-amino-5-fluorobenzoic acid (2b, 4.96 g, 32 mmol) and 1-iodo-2-nitrobenzene (3b, 7.97 g, 32 mmol) were used. The crude compound 5-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4f) was obtained (6.35 g, 71.9% yield).
The above crude compound 5-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4f, 1.6 mmol) was used to produce the product 7f as a yellow solid (0.144 g, 27.5% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.12 – 7.07 (m, 1H), 7.04 – 6.97 (m, 3H), 6.90 (td, J = 7.6, 1.4 Hz, 1H), 6.79 (dd, J = 9.4, 4.7 Hz, 1H), 6.74 – 6.69 (m, 1H), 4.86 (s, 1H), 3.49 (bs, 4H), 2.57 (bs, 4H), 2.50 (q, J = 7.2 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3): 160.4, 159.8, 157.9, 149.2, 141.9, 139.9, 127.5, 124.69, 124.66, 124.5 (d, J = 6.5 Hz), 121.6 (d, J = 7.9 Hz), 119.6, 119.1 (d, J = 22.9 Hz), 116.3 (d, J = 23.5 Hz), 52.6, 51.6, 46.0, 10.7. HRMS (ESI+) calculated for C19H22FN4 (M+H)+: 325.1828; found: 325.1819.
11-(4-Ethylpiperazin-1-yl)-3-fluoro-5H-dibenzo[b,e][1,4]diazepine (7g)
According to the general procedure, 2-amino-4-fluorobenzoic acid (2c, 4.96 g, 32 mmol) and 1-iodo-2-nitrobenzene (3b, 7.97 g, 32 mmol) were used. The crude compound 4-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4g) was obtained (6.68 g, 75.6% yield).
The above crude compound 4-fluoro-2-((2-nitrophenyl)amino)benzoic acid (4g, 1.6 mmol) was used to produce the product 7g as a yellow solid (0.189 g, 36.1% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.30 – 7.24 (m, 1H), 7.09 (d, J = 7.7 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.90 (t, J = 7.4 Hz, 1H), 6.73 – 6.68 (m, 2H), 6.57 (d, J = 9.2 Hz, 1H), 4.95 (s, 1H), 3.46 (bs, 4H), 2.56 (bs, 4H), 2.49 (dd, J = 14.4, 7.2 Hz, 2H), 1.13 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3): 165.9, 163.9, 161.6, 155.0 (d, J = 9.1 Hz), 141.3, 140.5, 132.1 (d, J = 9.6 Hz), 127.5, 124.8, 124.0, 119.9 (d, J = 3.2 Hz), 119.6, 110.1 (d, J = 21.7 Hz), 107.3 (d, J = 22.9 Hz), 52.9, 52.6, 47.5, 12.0. HRMS (ESI+) calculated for C19H22FN4 (M+H)+: 325.1828; found: 325.1822.
8-Chloro-11-(4-ethylpiperazin-1-yl)-2,4-difluoro-5H-dibenzo[b,e][1,4]diazepine (7h)
According to the general procedure, 2-amino-3,5-difluorobenzoic acid (2e, 2.77 g, 16 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-3,5-difluorobenzoic acid (4h) was obtained (4.62 g, 88.0% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-3,5-difluorobenzoic acid (4h, 3.2 mmol) was used to produce the product 7h as a brown solid (0.531 g, 44% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.08 (s, 1H), 6.93 (t, J = 8.5 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.82 (d, J = 8.6 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.20 (s, 1H), 3.51 (bs, 4H), 2.57 (s, 4H), 2.51 (s, 1H), 1.15 (t, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3): 160.5 (d, J = 2.0 Hz), 158.6 (d, J = 11.4 Hz), 156.6 (d, J = 11.6 Hz), 154.2 (d, J = 12.3 Hz), 152.2 (d, J = 12.1 Hz), 141.5, 139.8, 136.4 (d, J = 15.4 Hz), 129.7, 127.2, 126.6 (d, J = 7.6 Hz), 123.7, 120.9, 111.9 (dd, J = 23.2, 3.9 Hz), 106.5 – 106.1 (m), 52.8, 52.5, 47.5, 12.1. HRMS (ESI+) calculated for C19H20ClF2N4 (M+H)+: 377.1345; found: 377.1347.
11-(4-Ethylpiperazin-1-yl)-8-fluoro-5H-dibenzo[b,e][1,4]diazepine (7i)
According to the general procedure, 2-aminobenzoic acid (2f, 4.39 g, 32 mmol) and 4-fluoro-1-iodo-2-nitrobenzene (3c, 8.54 g, 32 mmol) were used. The crude compound 2-((4-fluoro-2-nitrophenyl)amino)benzoic acid (4i) was obtained. (8.54 g, 96.6% yield).
The above crude compound 2-((4-fluoro-2-nitrophenyl)amino)benzoic acid (4i, 1.6 mmol) was used to produce the product 7i as a yellow solid (0.289 g, 55.7% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.34 – 7.27 (m, 2H), 7.02 (t, J = 7.4 Hz, 1H), 6.84 (d, J = 7.7 Hz, 1H), 6.79 (d, J = 10.1 Hz, 1H), 6.67 – 6.60 (m, 1H), 6.57 (t, J = 8.2 Hz, 1H), 4.87 (s, 1H), 3.51 (bs, 4H), 2.56 (bs, 4H), 2.50 (q, J = 6.6 Hz, 2H), 1.14 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3): 162.9, 161.1, 159.2, 153.3, 142.2 (d, J = 11.2 Hz), 138.0, 132.0, 130.4, 123.6, 123.1, 120.2, 119.7 (d, J = 9.4 Hz), 113.5 (d, J = 22.8 Hz), 109.8 (d, J = 22.8 Hz), 52.9, 52.6, 47.4, 12.1. HRMS (ESI+) calculated for C19H22FN4 (M+H)+: 325.1828; found: 325.1847.
The purity of all final DREADD compounds 7a-7i was confirmed using quantitative NMR analysis as shown in the Table 4 (see Supplemental information for the spectra).
Table 4.
Chemical purities of final compounds 7a-7i obtained from quantitative NMR analysis with pyrazine as a standard (see qNMR spectra in Fig. S3)
| Compound | qNMR purity |
|---|---|
| 7a | >99% |
| 7b | >99% |
| 7c | >97% |
| 7d | >99% |
| 7e | >96% |
| 7f | >97% |
| 7g | >98% |
| 7h | >98% |
| 7i | >99% |
8-Chloro-11-(4-ethylpiperazin-1-yl)-2-methoxy-5H-dibenzo[b,e][1,4]diazepine (12a)
According to the general procedure, 2-amino-5-methoxybenzoic acid (8a, 2.68 g, 16 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-5-methoxybenzoic acid was obtained (9a, 4.18 g, 81.0% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-3,5-difluorobenzoic acid (9a, 3.2 mmol) was used to produce the product 12a as a brown solid (0.661 g, 55.7% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.06 (d, J = 2.3 Hz, 1H), 6.88 (dd, J = 8.5, 2.8 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 2.6 Hz, 1H), 6.77 (d, J = 8.6 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.74 (s, 1H), 3.75 (s, 3H), 3.52 (s, 4H), 2.55 (s, 4H), 2.50 (s, 2H), 1.14 (s, 3H). HRMS (ESI+) calculated for C20H24ClN4O (M+H)+: 371.1639; found: 371.1640.
8-Chloro-11-(4-ethylpiperazin-1-yl)-4-methoxy-5H-dibenzo[b,e][1,4]diazepine (12b)
According to the general procedure, 2-amino-3-methoxybenzoic acid (8b, 2.68 g, 16 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-methoxybenzoic acid (9c) was obtained(4.39 g, 85.0% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-methoxybenzoic acid (9b, 3.2 mmol) was used to produce the product 12b as a yellow solid (0.583 g, 49.1% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.05 (d, J = 2.4 Hz, 1H), 6.96 – 6.91 (m, 1H), 6.90 – 6.87 (m, 1H), 6.85 (dd, J = 7.7, 1.2 Hz, 1H), 6.81 (dd, J = 8.3, 2.4 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 5.76 (s, 1H), 3.88 (s, 3H), 3.50 (s, 4H), 2.54 (bs, 4H), 2.48 (q, J = 7.2 Hz, 2H), 1.12 (t, J = 7.2 Hz, 3H). HRMS (ESI+) calculated for C20H24ClN4O (M+H)+: 371.1639; found: 371.1640.
8-Chloro-11-(4-ethylpiperazin-1-yl)-4-fluoro-2-methoxy-5H-dibenzo[b,e][1,4]diazepine (12c)
According to the general procedure, 2-amino-3-fluoro-5-methoxybenzoic acid (8c, 2.96 g, 16 mmol) and 4-chloro-1-iodo-2-nitrobenzene (3a, 4.53 g, 16 mmol) were used. The crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-fluoro-5-methoxybenzoic acid (9c) was obtained (3.98 g, 73.0% yield).
The above crude compound 2-((4-chloro-2-nitrophenyl)amino)-3-fluoro-5-methoxybenzoic acid (9c, 1.6 mmol) was used to produce the product 12c as a yellow solid (170.0 mg, 27.5% yield over three steps).
1H NMR (500 MHz, CDCl3): 7.06 (s, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 11.3 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 6.61 (s, 1H), 5.13 (s, 1H), 3.74 (s, 3H), 3.52 (s, 4H), 2.56 (s, 4H), 2.51 (s, 2H), 1.14 (t, J = 7.1 Hz, 3H). HRMS (ESI+) calculated for C20H23ClFN4O (M+H)+: 389.1544; found: 389.1546.
8-Chloro-11-(4-ethylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepin-2-ol (13a)
To 8-chloro-11-(4-ethylpiperazin-1-yl)-2-methoxy-5H-dibenzo[b,e][1,4]diazepine (12a, 370.9 mg, 1.0 mmol) in CH2Cl2 (10 mL), BBr3 solution in CH2Cl2 (1M, 10 mL, 10 equiv.) was added dropwise at 0°C. The mixture was slowly warmed to room temperature and stirred overnight under argon. The reaction was quenched by pouring over ice. The crude mixture was diluted with CH2Cl2 (30 mL), neutralized with NaHCO3 solution, and then washed with H2O (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4. Subsequent filtration and evaporation of the solvent gave the crude product. This crude product was recrystallized using hexanes and CH2Cl2 (v:v=3:1) to afford the pure product 13a as a gray-yellow solid (220.0 mg, 0.62 mmol, 62% yield).
1H NMR (500 MHz, CD3OD): 6.96 (s, 1H), 6.87 – 6.84 (m, 2H), 6.84 – 6.78 (m, 2H), 6.73 (s, 1H), 3.51 (s, 4H), 2.67 (s, 4H), 2.58 (dd, J = 14.1, 7.0 Hz, 2H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): 162.8, 153.1, 145.7, 143.6, 142.2, 127.8, 126.8, 125.9, 124.2, 122.9, 121.8, 120.8, 119.5, 115.7, 52.6, 52.1, 47.1, 12.3 (see Fig. S5). HRMS (ESI+) calculated for C19H22ClN4O (M+H)+: 357.1482; found: 357.1483.
8-Chloro-11-(4-ethylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepin-4-ol (13b)
To 8-chloro-11-(4-ethylpiperazin-1-yl)-4-methoxy-5H-dibenzo[b,e][1,4]diazepine (12b, 370.9 mg, 1.0 mmol) in CH2Cl2 (10 mL), BBr3 solution in CH2Cl2 (1M, 10 mL, 10 equiv.) was added dropwise at 0°C. The mixture was slowly warmed to room temperature and stirred overnight under argon. The reaction was quenched by pouring over ice. The crude mixture was diluted with CH2Cl2 (100 mL), neutralized with NaHCO3 solution and then washed with H2O (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4. Subsequent filtration and evaporation of the solvent gave the crude product. This crude product was recrystallized using hexanes, CH2Cl2 and methanol (v:v:v = 3:3:1) to afford the pure product 13b as a yellow solid (72.0 mg, 0.2 mmol, 20% yield).
1H NMR (500 MHz, DMSO-d6): 10.03 (s, 1H), 6.89 (d, J = 8.1 Hz, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.85 – 6.83 (m, 2H), 6.82 (dd, J = 5.9, 2.4 Hz, 1H), 6.66 (d, J = 7.6 Hz, 1H), 6.43 (s, 1H), 3.33 (s, 4H), 2.43 (s, 4H), 2.37 (dd, J = 14.2, 7.1 Hz, 2H), 1.02 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): 163.2, 148.3, 142.6, 142.4, 141.2, 127.1, 125.8, 124.6, 123.0, 122.5, 122.2, 120.3, 117.3, 52.7, 52.1, 47.7, 12.5. HRMS (ESI+) calculated for C19H22ClN4O (M+H)+: 357.1482; found: 357.1483.
8-Chloro-11-(4-ethylpiperazin-1-yl)-4-fluoro-5H-dibenzo[b,e][1,4]diazepin-2-ol (13c)
To 8-chloro-11-(4-ethylpiperazin-1-yl)-4-fluoro-2-methoxy-5H-dibenzo[b,e][1,4]diazepine (12c, 194.5 mg, 0.5 mmol) in CH2Cl2 (5 mL), BBr3 solution in CH2Cl2 (1M, 5 mL, 10 equiv.) was added dropwise at 0°C. The mixture was slowly warmed to room temperature and stirred overnight under argon. The reaction was quenched by pouring over ice. The crude mixture was diluted with CH2Cl2 (20 mL), neutralized with NaHCO3 solution and then washed with H2O (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4. Subsequent filtration and evaporation of the solvent gave the crude product. This crude product was recrystallized using hexanes and CH2Cl2 (v:v = 3:1) to afford the pure product 13c as a gray-blue solid (77.0 mg, 0.21 mmol, 41% yield).
1H NMR (500 MHz, CD3OD): 6.98 (s, 1H), 6.87 (s, 2H), 6.70 (d, J = 11.3 Hz, 1H), 6.56 (s, 1H), 3.52 (s, 4H), 2.66 (s, 4H), 2.57 (q, J = 6.7 Hz, 2H), 1.18 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CD3OD): 162.3, 154.8, 153.5, 153.4, 152.9, 141.8 (d, J = 49.8 Hz), 132.0 (d, J = 16.0 Hz), 128.2, 126.1 (d, J = 3.3 Hz), 125.9, 125.0 (d, J = 6.1 Hz), 123.1, 120.7, 110.9 (d, J = 3.0 Hz), 105.4 (d, J = 22.8 Hz), 52.1, 52.0, 47.9, 10.3. HRMS (ESI+) calculated for C19H21ClFN4O (M+H)+: 375.1388; found: 375.1390.
1-(4-(4-fluorophenyl)piperazin-1-yl)ethan-1-one (16b)
To 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethan-1-one (16, 20 mg, 0.09 mmol) in ethanol (0.2 mL) in a 0.5 mL microwave vial was added tris(acetonitrile)cyclopentadientylruthenium(II) hexafluorophosphonate (15, 40 mg, 0.09 mmol, Strem catalog 44–7870) as a solid. The vial was sealed, and the solution was degassed by nitrogen bubbling for 2 minutes. The reaction was heated to 85°C by microwave irradiation, cooled, unsealed and the volume of ethanol reduced to ~0.05 mL by use of a stream of nitrogen. Then 2-chloro-1,3-bis(2,6-diisopropyl)-1H-imidazol-3-ium chloride (17, 85 mg, 0.18 mmol, 2.0 eq) was added to the vial as a solid, followed by cesium fluoride (0.13 mmol, 1.4 eq). Acetonitrile (0.2 mL) and DMSO (0.2 mL) were added. The vial was sealed, and the solution degassed by nitrogen bubbling for ~ 2 minutes. The reaction was then heated to 125°C by microwave irradiation for 2 hours, cooled, and filtered through a silica gel plug, which was then washed with 10% methanol in dichloromethane. The resulting organic solvent was removed by rotary evaporation, and the residue was purified by silica gel chromatography (0% to 100% ethyl acetate in hexanes, Isco MPLC) to give the desired product in 24% yield (5.0 mg). Both the 1H and 19F-NMR spectra matched the commercially available product (Sigma-Aldrich) and are reported below.
1H NMR (400 MHz, CDCl3): 7.03 – 6.91 (m, 2H), 6.91 – 6.79 (m, 2H), 3.73 (t, J = 5.3 Hz, 2H), 3.58 (t, J = 5.2 Hz, 2H), 3.08 – 3.00 (m, 4H), 2.10 (s, 3H). 19F NMR (376 MHz, CDCl3): −123.38 (tt, J = 8.6, 4.6 Hz). LC-MS: MW (M+H) = 223.1.
2-Fluoro-11-(4-methylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine (19b)
11-(4-methylpiperazine-1-yl)-5H-dibenzo[b,e][1,3]diazepin-8-ol (19, 0.01 g, 0.03 mmol) and tris(acetonitrile)cyclopentadientylruthenium(II) hexafluorophosphonate (15, 0.015 g, 0.03 mmol) were placed in a microwave-compatible vial with a stir bar. The vial was sealed, and ethanol (0.2 mL) was added. Nitrogen gas was then bubbled through the solution for 5 minutes. Then the vial was heated to 85°C for 30 minutes via microwave irradiation. The vial was cooled, unsealed, and the solvent volume was reduced to ~0.05 mL by evaporation with a stream of nitrogen gas. Then 2-chloro-1,3-bis(2,6-diisopropyl)-1H-imidazol-3-ium chloride (17, 0.03 g, 0.06 mmol) was added, followed by cesium fluoride (0.006 g, 0.04 mmol). DMSO (0.2 mL) and acetonitrile (0.2 mL) were added. The vial was sealed, and nitrogen gas was bubbled through the reaction mixture for 5 minutes. The vial was then heated to 125°C for 30 minutes by microwave irradiation. The reaction was cooled, and the solvent volume was reduced by rotary evaporation. The product was then purified by semi-preparative HPLC using a Phenomenex Luna C18 reverse phase (5 μm, 30 mm × 75 mm) column running at a flow rate of 45 mL/min. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and the temperature was maintained at 50°C. A gradient was run from 5% acetonitrile to 95 % acetonitrile. The eluent was removed under vacuum, and the purified product was dissolved in an ethyl acetate-aqueous saturated sodium bicarbonate mixture, extracted into ethyl acetate, and the solvent removed under vacuum to give the desired product in 20% yield (0.002g).
1H NMR (400 MHz, CDCl3): 7.34 – 7.27 (m, 2H), 7.06 – 6.97 (m, 1H), 6.83 (ddd, J = 7.9, 1.2, 0.4 Hz, 1H), 6.79 (dd, J = 10.2, 2.8 Hz, 1H), 6.66 – 6.52 (m, 2H), 4.86 (s, 1H), 3.58 – 3.40 (m, 4H), 2.58 – 2.44 (m, 4H), 2.36 (s, 3H). 19F NMR (376 MHz, CDCl3): −120.77 – −120.87 (m). LC-MS: MW (M+H) = 311.1.
Radiochemistry
11-(4-Ethylpiperazin-1-yl)-2-[18F]fluoro-5H-dibenzo[b,e][1,4]diazepine ([18F]7b)
Step 1:
Ruthenium compound 15 (8 mg) and precursor 13a (4 mg) were dissolved in 1 mL of ethanol in a 4-cc screw-top vial and residual air was removed with a flow of research purity nitrogen (15 sec). The solution was sealed and heated at 80°C for 30 min and the solvent was evaporated under nitrogen flow at 40°C. The brownish residue was dissolved in a mixture of 0.5 mL anhydrous acetonitrile and 0.5 mL anhydrous DMSO. The solution of 15/13a complex was degassed with nitrogen flow.
Step 2:
Aqueous [18F]fluoride flowing from the cyclotron target was trapped on a Chromafix 30-PS-HCO3 (ABX GmbH) anion-exchange cartridge followed by a rinse with 1 mL of methanol. The cartridge was eluted consecutively with a solution of 9 mg N,N’-bis(2,6-diisopropylphenyl)-2-chloroimidazolium chloride, 17 in 1 mL of methanol and rinsed with 0.5 mL of neat methanol into a Pyrex reaction vessel. The [18F]fluoride and 17 solution was dried under nitrogen flow and vacuum at 100–120°C. The solution of 15/13a complex from step 1 was added to the reaction vessel. The vessel was sealed and heated at 120°C for 30 min. The reaction mixture was diluted with 0.5 mL of acetonitrile and transferred to a collection vial. Water (0.5–0.7 mL) was added to the collection vial, and the mixture was injected onto a semi-preparative RP-HPLC column (10 × 250 mm, XBridge, Waters). The column was eluted with acetonitrile:water:45% aqueous trimethylamine (430:570:1) at flow rate of 10 mL/min. The radiolabeled compound [18F]7b (tR = 16.5 min; see Fig. S1) was collected in a reformulation reservoir containing 50 mL of water. The diluted aqueous solution of [18F]7b was passed through an Oasis HLB Light cartridge (SPE) (Waters, Milford, MA). The SPE was washed with 10 mL of deionized water after which the radiolabeled product was eluted with 1 mL of absolute ethanol followed by 10 mL of normal saline. The solution was passed through a sterile 0.22 μ Millex FG filter unit (MilliporeSigma, Burlington, MA) into a sterile pyrogen-free vial. [18F]7b was isolated with an average radiochemical yield of 7 – 9% (non-decay corrected) (n=10). An aliquot (0.1 mL) was assayed for radioactivity, and checked by analytical HPLC using a Luna C18 column (4.6×250 mm, 10 μ) and a mobile phase of acetonitrile:water:trifluoroacetic acid (230:770:1) at flow rate of 3 mL/min. A single radioactive peak (tR = 6.2 min) corresponding to [18F]7b was observed (see Fig. S2).
All other 18F-labeled compounds ([18F]7d, [18F]7h, [18F]19b, [18F]22, [18F]23) were prepared under similar reaction conditions to those of [18F]7b. The preparative and analytical HPLC conditions of the 18F-labeled compounds are shown in the Table 5.
Table 5. HPLC conditions.
Preparative HPLC column: XBridge, 10 × 250 mm (Waters), 10 mL/min; Analytical HPLC column: Luna C18 (10 micron) 4.6×250 mm (Phenomenex), 3 mL/min
| Compound | Prep HPLC (Rt, min) | Analytical HPLC (Rt, min) |
|---|---|---|
| [18F]19b | A (13.8) | B (7.2) |
| [18F]23 | C (11.3) | D (7.0) |
| [18F]22 | E (13.6) | F (5.8) |
| [18F]7d | G (13.2) | - |
| [18F]7h | H (21.2) | I (4.8) |
Mobile phase (mL):
acetonitrile : water : 45% aqueous trimethylamine (320:680:1)
acetonitrile : water : ammonium formate (400:600:6)
acetonitrile : water : 45% aqueous trimethylamine (500:500:1)
acetonitrile : water : ammonium formate (600:400:6)
acetonitrile : water : 45% aqueous trimethylamine (310:690:1)
acetonitrile : water : trifluoroacetic acid (140:860:1)
acetonitrile : water : 45% aqueous trimethylamine (430:570:1)
acetonitrile : water : 45% aqueous trimethylamine (440:560:1)
acetonitrile : water : trifluoroacetic acid (300:700:1)
Radioligand binding assays
Human embryonic kidney (HEK-293, ATCC) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 2 mM L-glutamine, antibiotic/antimycotic (all supplements from Gibco) and 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Inc. Flowery Branch, GA, USA) and kept in an incubator at 37 °C and 5% CO2. Cells were routinely tested for myclopasma contamination (MycoAlert® Mycoplasma Detection Kit, Lonza). Cells were seeded on 60 cm2 dishes at 4 × 106 cells/dish 24 h before transfection. The cells were transfected with 5 μg/dish of AAV packaging plasmids encoding for hM3Dq (Addgene #89149), hM4Di (Addgene #89150) or a control vector and harvested 48 h after transfection. The cells were suspended in Tris-HCl 50 mM pH 7.4 supplemented with protease inhibitor cocktail (1:100, Sigma-Aldrich, St. Louis, MO, USA). HEK-293 cells were disrupted with a Polytron homogenizer (Kinematica, Basel, Switzerland). Homogenates were centrifuged at 48,000 g (50 min, 4 °C) and washed twice in the same conditions to isolate the membrane fraction. Protein was quantified by the bicinchoninic acid method (Pierce). For competition experiments, membrane suspensions (50 μg of protein/mL) were incubated in 50 mM Tris-HCl (pH 7.4) containing 10 mM MgCl2, 2.5 nM of [3H]clozapine (3070 GBq/mmol (83 Ci/mmol), Novandi Chemistry AB, Södertälje, Sweden) and increasing concentrations of the competing drugs during 2 h at RT. Nonspecific binding was determined in the presence of 10 μM clozapine. In all cases, free and membrane-bound radioligand were separated by rapid filtration of 500 μL aliquots in a 96-well plate harvester (Brandel, Gaithersburg, MD, USA) and washed with 2 mL of ice-cold Tris-HCl buffer. Microscint-20 scintillation liquid (65 μL/well, PerkinElmer) was added to the filter plates. The plates were incubated overnight at RT and radioactivity counts were determined in a MicroBeta2 plate counter (PerkinElmer, Boston, MA, USA) with an efficiency of 41%. One-site competition curves were fitted using Prism 7 (GraphPad Software, La Jolla, CA, USA). Ki values were calculated using the Cheng–Prusoff equation.
PET imaging
All experiments and procedures complied with all relevant ethical regulations for animal testing and research, and followed NIH guidelines, and were approved by each institute’s animal care and use committees. Transgenic mice were bred at the NIDA breeding facility. Heterozygous transgenic mice expressing the enzyme cre recombinase under the control of the dopamine D1 receptor promoter (D1-Cre, FK150 line, C57BL/6J congenic, Gensat, RRID: MMRRC_036916-UCD) were crossed with heterozygous transgenic mice with cre recombinase-inducible expression of hM3Dq DREADD (R26-hM3Dq/mCitrine, Jackson Laboratory, stock no. 026220). Mice carrying both transgenes and hence expressing hM3Dq in D1 positive neurons18 were used as experimental animals, whereas littermates lacking both transgenes were used as wild-type controls. Mouse PET experiments were performed on a NanoScan PET/CT model 1225 with 0.7 mm resolution (Mediso Medical Imaging Systems, Budapest, Hungary). Three animals per group were used.
Mice were anesthetized with isoflurane and placed in a prone position on the scanner bed. The mice were injected intravenously (~100–200 μL) with [18F]7b (~13 MBq (~350 μCi)) with specific radioactivity of 81 GBq/μmol (2,200 mCi/μmol), and dynamic scanning commenced. Animals were pretreated with either vehicle or 7d blocker solution 10 min before injection of the PET radiotracer. The total acquisition time was 60 min.
In all cases, the PET data were reconstructed and corrected for dead-time and radioactive decay. All qualitative and quantitative assessments of the PET images were performed using the PMOD software environment (PMOD Technologies, Zurich Switzerland). Binding potential BPND (a relative measure of specific binding) was calculated using a reference tissue model using the cerebellum as a reference tissue. In all cases, the dynamic PET images were coregistered to MRI templates and time-activity curves were generated using manually drawn volumes of interest. BPND parametric maps were generated by pixel-based kinetic modeling using a multilinear reference tissue model29 using the cerebellum as a reference region and the start time (t*) was set to 16 min.
Supplementary Material
Highlights.
New ligands for Designer Receptors Exclusively Activated by Designer Drugs
Compounds 7a-7i are suitable candidates for DREADD PET imaging
[18F]7b is radiolabeled via a modified 18F-deoxyfluorination protocol [18F]7b specifically labels hM3Dq in PET study in a DREADD transgenic mouse model
ACKNOWLEDGMENTS
We thank Drs. Amy Newman and Kenner Rice of NIH and Drs. Tobias Ritter and Jacob Hooker of Harvard University for fruitful discussions.
This research was supported by NIH grants R21AG054802 and R33AG054802 (AGH and MGP), NIDA/NIH contract (MGP), EB024495 (MGP and AGH) and the NIH intramural research fund.
Footnotes
Declaration of interest statement
FH, JB, MGP, MM and AGH are co-inventors on a U.S. patent covering PET imaging with [18F]7b and as such are entitled to a portion of any licensing fees and royalties generated by this technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roth BL DREADDs for Neuroscientists. Neuron 2016, 89, 683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomez JL; Bonaventura J; Lesniak W; Mathews WB; Sysa-Shah P; Rodriguez LA; Ellis RJ; Richie CT; Harvey BK; Dannals RF; Pomper MG; Bonci A; Michaelides M Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017, 357, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armbruster BN; Li X; Pausch MH; Herlitze S; Roth BL Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 2007, 104, 5163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieb A; Weston M; Kullmann DM Designer receptor technology for the treatment of epilepsy. EBioMedicine 2019, 43, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alcacer C; Andreoli L; Sebastianutto I; Jakobsson J; Fieblinger T; Cenci MA Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson’s disease therapy. J Clin Invest 2017, 127, 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyer SM; Vesuna S; Ramakrishnan C; Huynh K; Young S; Berndt A; Lee SY; Gorini CJ; Deisseroth K; Delp SL Optogenetic and chemogenetic strategies for sustained inhibition of pain. Sci Rep 2016, 6, 30570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagai Y; Kikuchi E; Lerchner W; Inoue KI; Ji B; Eldridge MA; Kaneko H; Kimura Y; Oh-Nishi A; Hori Y; Kato Y; Hirabayashi T; Fujimoto A; Kumata K; Zhang MR; Aoki I; Suhara T; Higuchi M; Takada M; Richmond BJ; Minamimoto T PET imaging-guided chemogenetic silencing reveals a critical role of primate rostromedial caudate in reward evaluation. Nat Commun 2016, 7, 13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagai Y; Miyakawa N; Takuwa H; Hori Y; Oyama K; Ji B; Takahashi M; Huang XP; Slocum ST; DiBerto JF; Xiong Y; Urushihata T; Hirabayashi T; Fujimoto A; Mimura K; English JG; Liu J; Inoue KI; Kumata K; Seki C; Ono M; Shimojo M; Zhang MR; Tomita Y; Nakahara J; Suhara T; Takada M; Higuchi M; Jin J; Roth BL; Minamimoto T Deschloroclozapine, a potent and selective chemogenetic actuator enables rapid neuronal and behavioral modulations in mice and monkeys. Nat Neurosci 2020. [DOI] [PubMed] [Google Scholar]
- 9.Ji B; Kaneko H; Minamimoto T; Inoue H; Takeuchi H; Kumata K; Zhang MR; Aoki I; Seki C; Ono M; Tokunaga M; Tsukamoto S; Tanabe K; Shin RM; Minamihisamatsu T; Kito S; Richmond BJ; Suhara T; Higuchi M Multimodal Imaging for DREADD-Expressing Neurons in Living Brain and Their Application to Implantation of iPSC-Derived Neural Progenitors. J Neurosci 2016, 36, 11544–11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller K; Faeh C; Diederich F Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 11.Meltzer HY An overview of the mechanism of action of clozapine. J Clin Psychiatry 1994, 55 Suppl B, 47–52. [PubMed] [Google Scholar]
- 12.Seeman P Clozapine, a fast-off-D2 antipsychotic. ACS Chem Neurosci 2014, 5, 24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartvig P; Eckernas SA; Lindstrom L; Ekblom B; Bondesson U; Lundqvist H; Halldin C; Nagren K; Langstrom B Receptor binding of N-(methyl-11C) clozapine in the brain of rhesus monkey studied by positron emission tomography (PET). Psychopharmacology (Berl) 1986, 89, 248–52. [DOI] [PubMed] [Google Scholar]
- 14.Lundberg T; Lindstrom LH; Hartvig P; Eckernas SA; Ekblom B; Lundqvist H; Fasth KJ; Gullberg P; Langstrom B Striatal and frontal cortex binding of 11-C-labelled clozapine visualized by positron emission tomography (PET) in drug-free schizophrenics and healthy volunteers. Psychopharmacology (Berl) 1989, 99, 8–12. [DOI] [PubMed] [Google Scholar]
- 15.Chen X; Choo H; Huang X-P; Yang X; Stone O; Roth BL; Jin J The First Structure–Activity Relationship Studies for Designer Receptors Exclusively Activated by Designer Drugs. ACS Chemical Neuroscience 2015, 6, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao Y; DeBoer P; Meier E; Wikstrom H Synthesis and pharmacological evaluation of triflate-substituted analogues of clozapine: identification of a novel atypical neuroleptic. J Med Chem 1997, 40, 4146–53. [DOI] [PubMed] [Google Scholar]
- 17.Sasikumar TK; Burnett DA; Zhang H; Smith-Torhan A; Fawzi A; Lachowicz JE Hydrazides of clozapine: a new class of D1 dopamine receptor subtype selective antagonists. Bioorg Med Chem Lett 2006, 16, 4543–7. [DOI] [PubMed] [Google Scholar]
- 18.Bonaventura J; Eldridge MAG; Hu F; Gomez JL; Sanchez-Soto M; Abramyan AM; Lam S; Boehm MA; Ruiz C; Farrell MR; Moreno A; Galal Faress IM; Andersen N; Lin JY; Moaddel R; Morris PJ; Shi L; Sibley DR; Mahler SV; Nabavi S; Pomper MG; Bonci A; Horti AG; Richmond BJ; Michaelides M High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat Commun 2019, 10, 4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander GM; Rogan SC; Abbas AI; Armbruster BN; Pei Y; Allen JA; Nonneman RJ; Hartmann J; Moy SS; Nicolelis MA; McNamara JO; Roth BL Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 2009, 63, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuwabara H; McCaul ME; Wand GS; Earley CJ; Allen RP; Weerts EM; Dannals RF; Wong DF Dissociative changes in the Bmax and KD of dopamine D2/D3 receptors with aging observed in functional subdivisions of the striatum: a revisit with an improved data analysis method. J Nucl Med 2012, 53, 805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang K; Huang XF; Wang Q; Deng C Decreased density of serotonin 2A receptors in the superior temporal gyrus in schizophrenia--a postmortem study. Prog Neuropsychopharmacol Biol Psychiatry 2009, 33, 867–71. [DOI] [PubMed] [Google Scholar]
- 22.Eckelman WC; Reba RC; Gibson RE Receptor-binding radiotracers: A class of potential radiopharmaceuticals. J Nucl Med 1979, 20, 350–357. [PubMed] [Google Scholar]
- 23.Waterhouse RN Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol 2003, 5, 376–89. [DOI] [PubMed] [Google Scholar]
- 24.Horti AG; Raymont V; Terry GE PET imaging of endocannabinoid system. In PET and SPECT of Neurobiological Systems, Dierckx R; Otte A; De Vries, E. F.; Van Waarde A, Eds. Springer: Berlin-Heidelberg, 2014; pp 251–319. [Google Scholar]
- 25.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for Positron Emission Tomography: Recent Advances in (11) C-, (18) F-, (13) N-, and (15) O-Labeling Reactions. Angew Chem Int Ed Engl 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preshlock S; Tredwell M; Gouverneur V (18)F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem Rev 2016, 116, 719–66. [DOI] [PubMed] [Google Scholar]
- 27.Neumann CN; Hooker JM; Ritter T Concerted nucleophilic aromatic substitution with (19)F(−) and (18)F(−). Nature 2016, 534, 369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horti AG; Villemagne VL The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Current Pharmaceutical Design 2006, 12, 3877–3900. [DOI] [PubMed] [Google Scholar]
- 29.Ichise M; Ballinger JR; Golan H; Vines D; Luong A; Tsai S; Kung HF Noninvasive quantification of dopamine D2 receptors with iodine-123-IBF SPECT. J Nucl Med 1996, 37, 513–20. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.