

Abstract
Background:
The prevalence and clinical characteristics of small bowel adenocarcinomas (SBAs) in the setting of LS have not been well-studied. We characterized SBA according to DNA mismatch repair and/or microsatellite instability (MMR/MSI) and germline mutation status and compared clinical outcomes.
Methods:
A single-institution review identified 100 SBAs. Tumors were evaluated for MSI via MSIsensor and/or corresponding MMR protein expression via immunohistochemical (IHC) staining. Germline DNA was analyzed for mutations in known cancer-predisposition genes, including MMR (MLH1, MSH2, MSH6, PMS2, EPCAM). Clinical variables were correlated with MMR/MSI status.
Results:
26% (26/100; 95% CI: 18.4-35.4) of SBAs exhibited MMR deficiency (MMR-D). LS prevalence was 10% overall and 38.5% among MMR-D SBAs. Median age at SBA diagnosis was similar in non-LS MMR-D vs MMR-proficient (MMR-P) SBAs (65 vs 61, p= 0.75), but significantly younger in LS (47.5 vs 61; p=0.03). The prevalence of synchronous/metachronous cancers was 9% (6/67) in MMR-P vs 34.6% (9/26) in MMR-D SBA, with 66.7% (6/9) of these in LS (p=0.0002). In the MMR-P group, 52.2% (35/67) of patients presented with metastatic disease, compared to 23.1% (6/26) in the MMR-D group (p=0.008). In MMR-P stage I/II patients, 88.2% (15/17) recurred, compared to 18.2% (2/11) in the MMR-D group (p=0.0002).
Conclusions:
When compared to MMR-P SBA, MMR-D SBA is associated with earlier-stage disease and lower recurrence rates, similar to observations in colorectal cancer. With a 38.5% prevalence in MMR-D SBA, germline LS testing in MMR-D SBA is warranted.
Keywords: small bowel adenocarcinoma, Lynch syndrome, mismatch repair deficiency, microsatellite instability
Introduction
Small bowel adenocarcinoma (SBA) is a rare, aggressive cancer, accounting for only 0.6% of new cancer diagnoses in the United States in 2019,(1) with approximately two-thirds diagnosed at advanced stages.(1,2) Given its rarity, clinical recommendations were historically based on colorectal cancer (CRC) guidelines. However, when compared to CRC, SBA is more aggressive with inferior outcomes at all stages.(2–5) Research has recently emerged suggesting that CRC and SBA may be more physiologically and molecularly distinct than once thought,(2–4,6) with most studies focusing on tumor molecular profiling, adenoma to carcinoma transformation, and tumor histological grade.(2–4,6) Importantly, the unique genomic landscape of SBA as compared to other gastrointestinal malignancies was elegantly demonstrated in a recent study(6), but the resultant impact on clinical treatment outcomes has not been well-described. Given the increased recognition of the distinct nature of SBA, in 2020 the clinical committee of the National Comprehensive Cancer Center (NCCN) established separate guidelines for the diagnosis and management of SBA from CRC. While adjuvant treatment recommendations mimic those in CRC, there are some differences noted in the metastatic setting, such as the recommendation against anti-EGFR treatments for RAS-wildtype SBA given the lack of demonstrated benefit.(7) However, other clinical guidelines like the European Society for Medical Oncology (ESMO) have not yet pursued independent recommendations for SBA.
It has been well-established in early-stage CRC that tumors demonstrating DNA mismatch repair deficiency and/or microsatellite instability (MMR-D) have a favorable prognosis compared to mismatch repair proficient/microsatellite stable (MMR-P) tumors. (8–11) Furthermore, patients with MMR-D CRC appear to lack clinical benefit from adjuvant 5-fluorouracil (5-FU)-based chemotherapy compared to MMR-P CRC, allowing for surgical management alone for early-stage (II) disease.(8–10) Given this, and the fact that 3% of CRCs are due to LS,(12) universal screening of all CRCs for MMR-D status is currently standard-of-care.(13) While these observations in CRC have been extrapolated to SBA, there are currently limited studies assessing the prognostic value of MMR-D status in SBA,(14) and no studies directly comparing clinical treatment response to that of MMR-P SBA. Moreover, with the Food and Drug Administration (FDA) approval of pembrolizumab therapy for all advanced MMR-D solid tumors, MMR-D analysis is now being increasingly incorporated into the care of all advanced cancer patients, including those with SBA.(15)
We have previously demonstrated that MMR-D/MSI status is associated with Lynch syndrome (LS) pan-cancer.(16) LS is an autosomal-dominant cancer predisposition syndrome with up to an 80% lifetime risk of cancer development of multiple types, requiring life-long surveillance.(12,17,18) LS patients harbor germline mutations in the mismatch repair genes (MLH1, MSH2, MSH6, PMS2, EPCAM).(12,19) As SBA is a rare, but known LS-associated tumor,(16,20,21) we sought to characterize SBA according to MMR and germline mutation status and compare clinical outcomes.
Methods
Study Population
The study comprised patients diagnosed with primary SBA in which MSI and/or MMR-D assessment was ordered at Memorial Sloan Kettering Cancer Center from 2006 to 2019. This study was conducted in accordance with the recognized ethical guidelines of the U.S. Common Rule. Included patients provided written, informed consent to either the institutional review board (IRB)-approved protocol for matched tumor/normal DNA sequencing via MSK-IMPACT (ClinicalTrials.gov identifier, NCT01775072) or to an IRB-approved protocol for prospective tracking of MMR-D tumors and/or biospecimen collection. Patient electronic medical records were reviewed. While 111 unique patients were identified as having small bowel tumors, only those with pathology-confirmed primary small bowel adenocarcinoma (SBA) were included in this analysis (n=100). Of the 11 excluded cases, four patients were found to have ampullary cancers of pancreatic origin, three were neuroendocrine tumors, three were CRC metastases, and one was a gastric cancer (signet ring cell) metastasis. Of our total cohort (n=100), two patients did not have MSI or MMR-D testing as one canceled the test request and the other had inadequate tissue available for MSI analysis.
MSI analysis and Immunohistochemical Staining
Beginning in 2014 our institution adopted universal IHC for MMR proteins (and/or MSI) on all CRC and endometrial tumors, with our much smaller numbers of SBAs also routinely undergoing such analysis. Inclusion of NGS MSI assessment in the research setting began in 2016. Prior to 2014, our institution only routinely assessed MMR status in SBA diagnosed under 50 years of age. For MSK-IMPACT-sequenced tumors, MSI assessment was conducted using MSIsensor, a next-generation sequencing (NGS)-based bioinformatics platform that incorporates data from more than 1,000 microsatellite regions, reporting the percentage of unstable loci as a cumulative score.(22,23) MSIsensor scores ≥10 designate MSI-H status, scores ≥3 to <10 an indeterminate (MSI-I) status, and scores <3 microsatellite stable (MSS).(16,22,23) MSK-IMPACT is approved by the NYS Department of Health for clinical use and authorized by the FDA for clinical reporting of somatic mutations, indels, rearrangements and MSI calculated from the microsatellite regions covered by the assay.(22–26)
Immunohistochemical (IHC) staining for DNA mismatch repair (MMR) protein expression was performed using standard procedures(27) and compared to tumor MSIsensor scores to establish concordance rates. A tumor was considered IHC/MSI concordant if the MSIsensor score was ≥3 and the IHC result demonstrated lack of MMR protein expression. For the purposes of simplification, MMR-D refers to MMR deficiency demonstrated either via IHC or MSI via MSIsensor.
Germline Analysis
In patients with known LS, genetic testing reports were reviewed to confirm diagnoses. For patients that underwent MSK-IMPACT germline assessment, DNA extracted from peripheral blood was used for analysis of up to 88 known cancer-predisposition genes, including MMR genes (MLH1, MSH2, MSH6, PMS2, EPCAM). Only deletions/rearrangements involving the 3’ region of EPCAM, causative of LS, were considered.(28,29) Single-exon and PMS2 deletions that encompassed exons 13 and 14 were only included if confirmed by orthogonal method, given the presence of known pseudogenes, as per standard operating procedures. Variant calling was performed in accordance with the American College of Medical Genetics and Genomics variant classification standards.(26,30) All research-based genetic testing was confirmed with clinical-grade testing in a Clinical Laboratory Improvement Amendments (CLIA)-approved laboratory. Only patients with pathogenic or likely pathogenic variants were considered germline-positive.
Statistical Analysis
Continuous variables were compared using either a two-tailed t-test or a Mann-Whitney U-test, as appropriate. Categorical variables were compared using either a χ2 or Fisher’s exact test, depending on sample size. P-values <0.05 were considered statistically significant.
Results
Lynch syndrome prevalence
We identified that the prevalence of MMR-D was 26% (26/100) (95% CI 18.4-35.4), with most MMR-D tumors (65.4%; 17/26) demonstrating loss of expression of MLH1 and/or PMS2 proteins on IHC. 15.4% (4/26) of MMR-D tumors demonstrated loss of MSH2/MSH6 protein expression. The overall LS prevalence in our SBAs was 10% (10/100), with all LS-associated cases exhibiting MMR-D. When assessing LS prevalence in MMR-D SBA, 38.5% (10/26) (95% CI 22.4–57.5) had LS (Figure 1A). In comparison, among 826 CRC patients included in our prior study,(16) 16.6% (137) of tumors exhibited MMR-D; of these, 19% (26/137) were found to have LS(16) (Figure 1B; p=0.002). Of our 10 SBA patients with LS, the distribution of germline mutations was 50% (5/10) in MLH1, 30% (3/10) MSH2, and 20% (2/10) PMS2. (Figure 2). The prevalence of MMR-D after the adoption of routine MMR-D assessment in SBA in 2014, was 25%, equivalent to the overall cohort MMR-D prevalence.
Figure 1. Prevalence of MMR-D and LS in SBA compared to CRC.

Panel A: MMR-D and LS prevalence in small bowel adenocarcinoma (SBA). Pie chart demonstrates that 26% (26/100) of SBAs found to be mismatch repair deficient (MMR-D). Of the MMR-D cases, 38.5% (10/26) of patients had underlying Lynch syndrome (LS) as indicated by top portion of bar graph. Panel B: MMR-D and LS prevalence in colorectal cancer (CRC) from our prior study (Latham et al., JCO 2019). Pie chart demonstrates that 83.4% (689/826) of CRC found to be mismatch repair proficient (MMR-P), while 16.6% (137/826) of CRC found to be mismatch repair deficient (MMR-D). Of the MMR-D cases, 19% (26/137) of patients had underlying Lynch syndrome (LS) as indicated by top portion of bar graph. Abbreviations: MMR-P: mismatch repair proficient; MMR-D non-LS: mismatch repair deficient without underlying Lynch syndrome; LS: Lynch syndrome
Figure 2. Distribution of MMR gene mutations in MMR-D SBA.
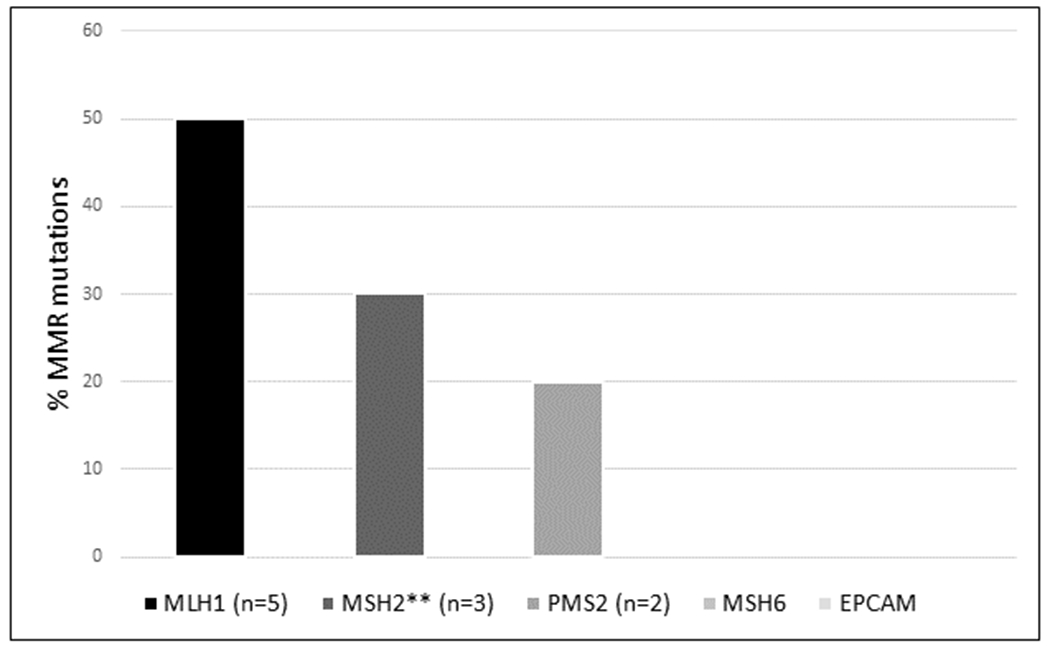
Mismatch repair genes (MMR) listed on the x-axis. Percentage of mutations per gene indicated on the y-axis. 50% (5/10) of germline mutations were found in the MLH1 gene. 30% (3/10) of germline mutations found in MSH2, and 20% (2/10) in PMS2. There were no underlying MSH6 or EPCAM germline mutations. Abbreviations: MMR: mismatch repair proficient **One patient with germline MSH2 mutation also found to harbor germline NTHL1 mutation.
Patient and Tumor Characteristics
Of the 100 SBA patients, 95% had clinical information available for review. Five were consult-only patients referred to our tertiary center without additional clinical follow-up. Two did not have MSI or IHC analysis conducted on their tumors, and there was no additional tumor available to complete these assays. As such, there were 93 patients in which demographic, clinical, and tumor pathology were available for review. Patient demographics and clinical characteristics are demonstrated in Table 1.
Table 1.
Patient Clinical Characteristics
| Characteristic | Total Cohort(N=100) | MMR-P(n=72)** | MMR-D non-LS (n=16) | MMR-D+ LS (n=10) |
|---|---|---|---|---|
| Median Age at SBA diagnosis | 60 | 60 | 65 | 47.5 |
| Male | 62%(59) | 65.7% (44) | 50% (8) | 50% (5) |
| Female | 38%(36) | 34.3% (23) | 50% (8) | 50% (5) |
| Race/Ethnicity | ||||
| Non-Hispanic White | 74.7%(70) | 73.1% (49) | 75% (12) | 80% (8) |
| Non-Hispanic Black | 10.5%(10) | 11.9% (8) | 6.3% (1) | 10% (1) |
| Asian | 6.3%(6) | 4.5% (3) | 12.5% (2) | 10% (1) |
| Hispanic | 4.2%(4) | 4.5% (3) | 6.3% (1) | 0% |
| Patient Declined to Answer | 5.3%(5) | 6% (4) | 0% | 0% |
| Positive Smoking History | 43.2%(41) | 40.3% (27) | 37.5% (6) | 60% (6) |
| Tumor Location | ||||
| Duodenum | 41% (41) | 38.9% (28) | 43.8% (7) | 50% (5) |
| Jejunum | 39% (39) | 36.1% (26) | 53.3% (9) | 30% (3) |
| Ileum | 18% (18) | 22.2% (16) | 0% | 20% (2) |
| Small Bowel, NOS | 2% (2) | 2.8% (2) | 0% | 0% |
| Tumor Differentiation on Pathology | ||||
| well-differentiated | 2% (2) | 2.8% (2) | 0% | 0% |
| moderately-differentiated | 44% (44) | 43.1% (31) | 25% (4) | 70% (7) |
| poorly differentiated | 45% (45) | 41.7% (30) | 75% (12) | 30% (3) |
| unknown | 9% (9) | 12.5% (9) | 0% | 0% |
| SBA as Index Cancer | 82% (78) | 86.6% (58) | 93.8% (15) | 50% (5) |
| Synchronous/Metachronous Tumors | 15.8% (15) | 9% (6) | 18.8% (3) | 60% (6) |
| Stage at Diagnosis | ||||
| Stage I | 4.2% (4) | 1.5% (1) | 0% | 30%(3) |
| Stage II | 25.2% (24) | 23.9% (16) | 25% (4) | 40%(4) |
| Stage III | 23.2% (22) | 19.4% (13) | 43.8% (7) | 20% (2) |
| Stage IV (metastatic) | 45.3% (43) | 52.2% (35) | 31.3% (5) | 10% (1) |
| Unknown | 2.1% (2) | 3%(2) | 0% | 0% |
| Median LN Dissected (range)^ | 11(1-77) | 10 (1-77) | 14 (2-27) | 22 (5-39) |
| Median Positive LN (range)^ | 0(0-15) | 1 (0-15) | 1 (0-7) | 0 (0-4) |
| Initial Treatment | ||||
| Surgery alone | 11.6% (11) | 6% (4) | 12.5% (2) | 50% (5) |
| Chemotherapy | 73.7% (70) | 83.6%(56) | 68.8% (10) | 20% (2) |
| Chemo+ immunotherapy | 6.3% (6) | 1.5%(1) | 12.5% (2) | 30% (3) |
| XRT alone | 1.1% (1) | 1.5% (1) | 0% | 0% |
| Declined/Palliation | 2.1% (2) | 3% (2) | 0% | 0% |
| Unknown or treated elsewhere^^ | 5.3% (5) | 4.5% (3) | 12.5% (2) | 0% |
| Recurrence in early-stage (I/II) | 17.9%(17) | 88.2%(15) | 25%(1) | 14.3%(1) |
There were 2 patients in which MMR/MSI status was unknown, but demographic, clinical staging, and tumor histology is represented as part of overall cohort
5 MMR-P/MSS tumors had limited clinical information, but some demographic, tumor pathology, TMN staging, and tumor grade are represented.
68 patients underwent surgery in total, with four patients (three in MMR-P and one in non-LS MMR-D group) at outside institution and LN yield and positivity were not available.
one MMR-D/MSI non-LS pt on RCT for pembrolizumab: unknown if in treatment or placebo group, and is therefore counted as “unknown”
While there was not a significant difference in median age of diagnosis between the MMR-proficient (MMR-P) and non-LS-associated MMR-D groups (60 and 65 years, respectively), LS-associated SBA was diagnosed in significantly younger patients, at a median age of 47.5 (p=0.03). Despite the earlier age of SBA onset in LS, 50% (5/10) had prior cancer, while the majority of MSS and sporadic MMR-D groups (86.6% and 93.8%, respectively) were diagnosed with SBA as their index cancer (p= 0.007) (Table 1).
We assessed the prevalence of synchronous and/or metachronous cancers. We considered any primary cancer detected within two months of the SBA diagnosis as synchronous, whereas subsequent primary cancers (detected >2 months post-SBA diagnosis) were metachronous, in accordance with the SEER database.(31) Among patients in the MMR-P group, 9% (6/67) had synchronous and/or metachronous cancers, compared to 34.6% (9/26) in the MMR-D group, with 66.7% (6/9) of these occurring in LS (p=0.0002) (Table 1). The majority of these tumors (60%; 9/15) were other primary gastrointestinal (GI) cancers (synchronous SBA, CRC, gastric), whereas additional endometrial and urothelial tumors were also identified among LS patients.
Of the entire SBA cohort, 41% of tumors were located in the duodenum, 39% in the jejunum, and 18% in the ileum. There was no statistically-significant difference in tumor location when compared across groups. Among LS, 50% (5/10) were located in the duodenum, compared to 38.9% (28/72) and 43.8% (7/16) in the MMR-P and MMR-D non-LS groups, respectively (p=0.77) (Figure 3).
Figure 3. Distribution of tumors by location, MMR, and LS status.
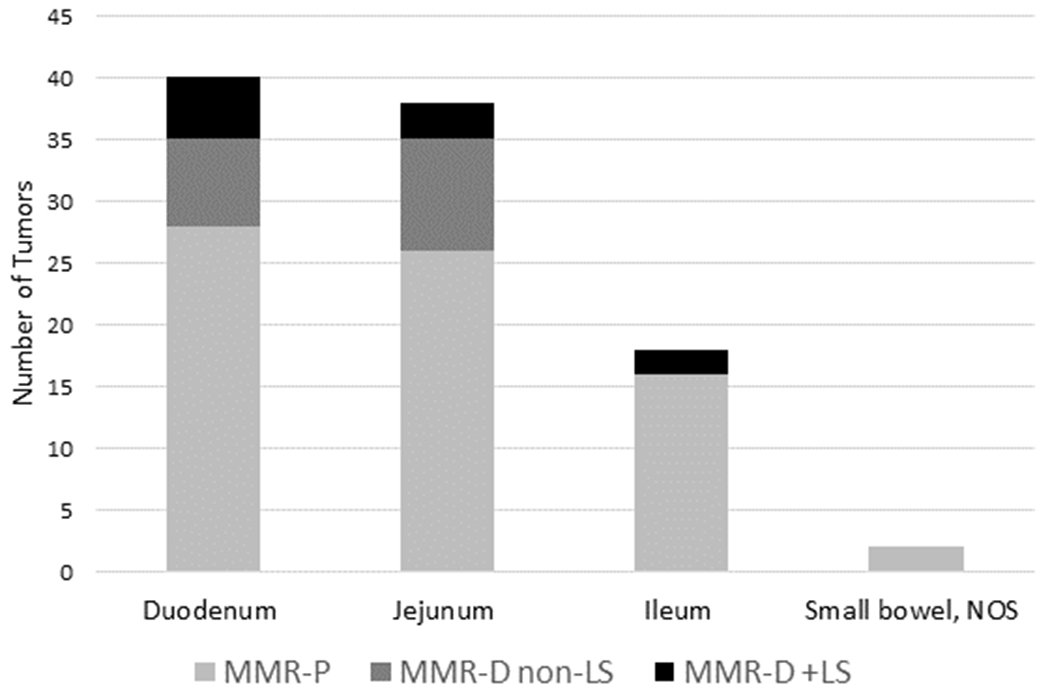
Tumor location indicated on the x-axis (duodenum, jejunum, ileum, small bowel NOS). Number of tumors per location indicated on the y-axis, with largest number of tumors located in the duodenum. Each bar sub-divided by group as indicated. Abbreviations: MMR-P: mismatch repair proficient; MMR-D non-LS: mismatch repair deficient without underlying Lynch syndrome; LS: Lynch syndrome
As lymph node (LN) positivity informs treatment decisions, we assessed LN dissection among patients that underwent surgical resection, comparing total LNs resected and LN positivity across groups (Table 1). There were four patients that underwent surgery at an outside institution and LN yield and positivity were not available. Among MMR-P tumors, median LN yield was 10 (range 1-77), with a median number of LNs positive for malignancy of one (range 0-15). There was no difference when compared to the MMR-D group overall, where median LN yield was 14 (range 2-39), with a median number of positive LSs of 0 (range 0-7) (MMR-P vs. MMR-D LN yield; p=0.07; MMR-P %LN positive vs. MMR-D %LN positive; p=0.32). Additionally, we compared LN positivity across all three groups, and found that 50%(23/46) of MMR-P cases were LN positive, compared to 58.3%(7/12) and 11.1%(1/9 ) in our non-LS MMR-D and LS-associated SBA surgical cases, respectively, but this did not reach statistical significance (p=0.066).
We also assessed tumor differentiation, as prior studies suggest that MMR-D CRCs have a propensity toward poorly differentiated histology, partially driven by the more frequent presence of medullary carcinoma subtype.(10,32) While we noted that the majority of MMR-D tumors were poorly differentiated (57.7%; 15/26), histology was not statistically different when compared to the MMR-P group overall (Table 1, Figure 4A). However, when assessing non-metastatic disease alone, there was a difference in tumor differentiation, with 60% (12/20) of MMR-D tumors being poorly differentiated compared to 30% (9/30) in the MMR-P group (p= 0.03), consistent with prior observations in CRC.(32) Figure 4B.
Figure 4. Tumor differentiation by MMR and LS status.
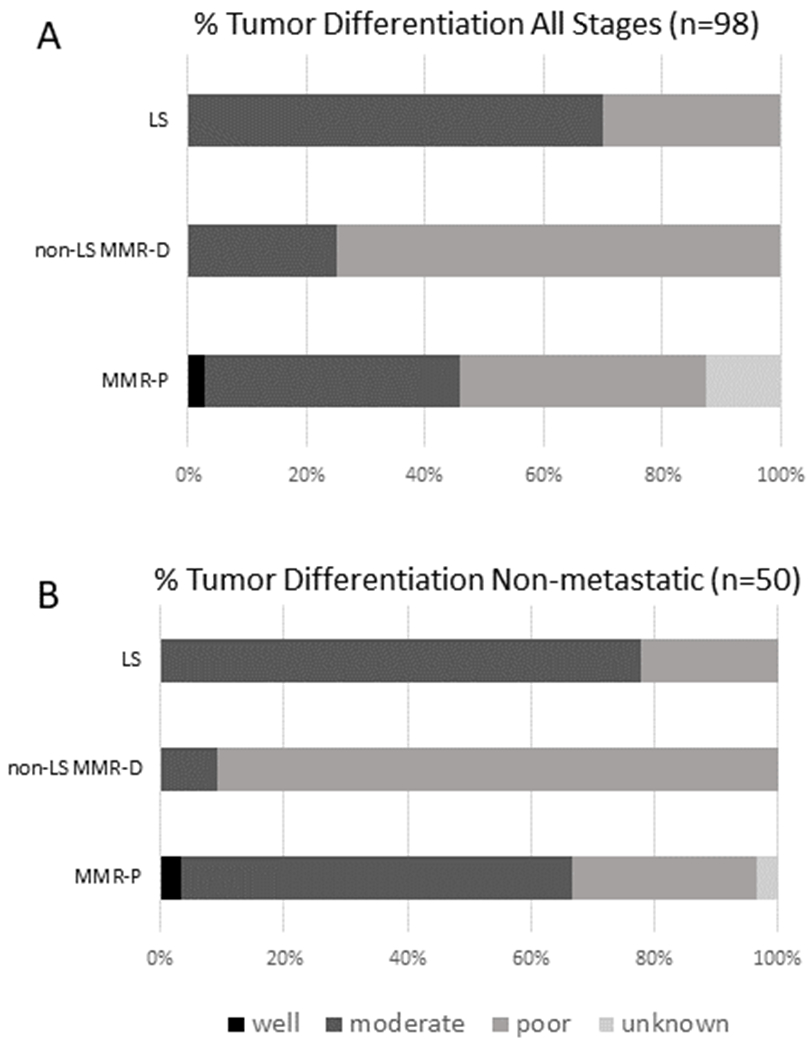
Panel A: Bar graph representing entire cohort in which MMR status available. (both early and metastatic disease). Black represents well-differentiated tumors, blue represents moderately-differentiated tumors, and yellow represents poorly-differentiated tumors. . Panel B: Bar graph representing non-metastatic cohort in which MMR status available. Black represents well-differentiated tumors, blue represents moderately-differentiated tumors, and yellow represents poorly-differentiated tumors. Figure demonstrates that while no differences noted in histology overall, when assessing non-metastatic cases, MMR-D tumors were of predominately poorly-differentiated histology. Abbreviations: MMR-P: mismatch repair proficient; MMR-D non-LS: mismatch repair deficient without underlying Lynch syndrome; LS: Lynch syndrome
Patient clinical outcomes
In the MMR-P group 52.2% (35/67) of patients presented with metastatic disease, compared to 23.1% (6/26) in the MMR-D group (p=0.008). Among early-stage(I/II) MMR-P SBA, 88.2% (15/17) recurred, compared to 18.2% (2/11) in the MMR-D group (p=0.0002). When assessing the entire cohort, there was no statistically significant difference in the proportion of patients receiving systemic chemotherapy. 85.1% (57/67) of the MMR-P group received either neoadjuvant or adjuvant chemotherapy at initial diagnosis compared to 65.4% (17/26) in the MMR-D group (p=0.068). All patients that received systemic chemotherapy for their primary SBA (n=74) underwent 5-FU-based regimens.
As MMR-D status is used in chemotherapy decision-making in stage II CRC,(8–11) we assessed chemotherapy utilization and recurrence in stage II SBA alone. While 81.3% (13/16) of stage II patients in the MMR-P group received systemic chemotherapy compared to 50% (4/8) in the MMR-D group, 84.6% (11/13) recurred, compared to 50% (2/4) in the MMR-D group; however, this did not reach statistical significance, likely due to sample size (p=0.15). Clinical characteristics, treatment, and recurrence rates of all patients with stage II SBA are demonstrated in Supplemental Table 1.
We also assessed utilization and response to immune-checkpoint blockade in MMR-D patients. Among the five patients with metastatic MMR-D SBAs receiving immunotherapy, 60% (3/5) demonstrated treatment response. One patient with a LS-associated MMR-D SBA was started on immunotherapy after progression of disease (POD) with FOLFIRINOX. While the patient demonstrated no clinical evidence of disease (NED) for 28 months, the patient developed a new primary metastatic MMR-D gastric cancer, which was the ultimate cause of death six months later. One additional patient with stage III SBA was placed on FOLFOX and demonstrated POD with development of a metachronous stage IV rectal cancer while on treatment. Immunotherapy was initiated, and that patient is NED for both malignancies. One deceased MMR-D patient only received immunotherapy right before death, as the drug had just received FDA approval.
Etiology of MMR-D
We assessed potential somatic causes of MMR-D among LS-negative SBA patients (n=16). While two patients declined additional work-up, among remaining patients (n=14), we identified a somatic driver of MMR-D status in 64.3% (9/14), with the majority of tumors (77.8% 7/9) demonstrating MLH1 promoter hypermethylation, a known somatic driver of MMR-D.(33) Table 2.
Table 2.
MSI/ IHC Concordance, Germline Status, and Somatic Drivers in MMR-D Tumors
| Age at DX | MSIsensor Score | IHC result | Tumor Location | LS-positive (Y/N) | SBA was index cancer? (Y/N) | SBA detected on surveillance? (Y/N) | Personal or Family history of SBA? (Y/N) | Gene(s) Mutated in Germline | Somatic Driver of MMR-D status |
|---|---|---|---|---|---|---|---|---|---|
| 67 | NA | PMS2 absent | jejunum | N | Y | N | N | UNK | UNK |
| 75 | 53.85 | MLH1/PMS2/MSH3 absent | jejunum | N | Y | N | N | NR | MLH1 hypermethylation |
| 56 | 48.09 | MLH1/PMS2 absent | duodenum | N | Y | N | N | NR | MLH1 hypermethylation |
| 65 | 43.42 | MLH1/PMS2 absent | duodenum | N | Y | N | N | NR | MLH1 hypermethylation |
| 70 | 43.28 | MSH2/MSH6 absent | jejunum | N | Y | N | N | NEG | hypermutated tumor, POLE exon32 pR1364Vfs*5 |
| 70 | 40.06 | MLH1/PMS2 absent | duodenum | N | Y | N | N | NEG | ultramutated tumor. Six somatic POLE mutations, encompassing exonuclease domain. Biallelic somatic MLH1 mutations |
| 61 | 36.55 | MLH1/PMS2 absent | jejunum | N | Y | N | N | NEG | MLH1 hypermethylation |
| 29 | 35.5 | MLH1/PMS2 absent | jejunum | N | Y* | N | N | APC | MLH1 hypermethylation |
| 71 | 34.9 | NORMAL | jejunum | N | N | N | N | APC I1307K | MLH1 hypermethylation |
| 41 | 27.02 | MLH1/PMS2 absent | jejunum | N | Y | N | N | LS NEG | UNK |
| 28 | 24.19 | MLH1/PMS2 absent | jejunum | N | Y | N | N | NF1 | MLH1 hypermethylation |
| 82 | 22.19 | MLH1/PMS2 absent | jejunum | N | Y | N | N | NEG | UNK |
| 66 | 20.59 | NA | duodenum | N | Y | N | N | NEG | UNK |
| 54 | 18.2 | MLH1/PMS2 absent | duodenum | N | Y | N | N | UNK | UNK |
| 65 | 10.02 | NORMAL | duodenum | N | Y | N | N | NEG | UNK |
| 57 | 8.14 | MSH2/MSH6 absent | duodenum | N | Y | N | N | NEG | UNK |
| 72 | NA | MSH2/MSH6 absent | jejunum | Y | N | N | Y | MSH2; NTHL1 | NR |
| 62 | NA | MSH2/MSH6 absent | jejunum | Y | N | N | N | MSH2 | NR |
| 62 | NA | MLH1/PMS2 absent | duodenum | Y | N | Y** | Y | MLH1 | NR |
| 54 | NA | MLH1/PMS2 absent | duodenum | Y | N | N | N | MLH1 | NR |
| 45 | 47.28 | NA | jejunum | Y | Y | N | N | MSH2 | NR |
| 44 | 31.68 | MLH1/PMS2 absent | duodenum | Y | Y* | N | N | MLH1 | NR |
| 50 | 31.29 | MLH1 focal/PMS2 absent | duodenum | Y | Y | N | N | PMS2 | NR |
| 35 | 25.65 | MLH1/PMS2 absent | duodenum | Y | Y | N | N | MLH1 | NR |
| 44 | 25.06 | NORMAL | ileum | Y | N | N | N | MLH1 | NR |
| 44 | 23.44 | PMS2 absent | ileum | Y | Y | N | N | PMS2 | NR |
Notes: hypermutated tumor defined as >50 somatic mutations. Ultramutated tumor defined as >150 somatic mutations. MSIsensor scores: MSI-High ≥10, MS-Indeterminate 3-10, MS-stable <3. POLE exonuclease domain spanning exons 9-14.
UNK: Unknown, pt lost to follow-up
Pt declined additional work-up.
LS NEG: Lynch syndrome testing negative. NR: Not Relevant. NA: Not available*synchronous colon cancer
Found on screening endoscopic ultrasound completed at outside institution for FH of pancreas cancer
Among the entire cohort, 76 patients underwent comprehensive tumor/normal paired NGS analysis (MSK-IMPACT). In those undergoing germline genetic assessment (n=38), in addition to MMR genes, pathogenic and/or likely pathogenic (P/LP) variants were identified in NF1, APC, ATM, BRCA2, MUTYH, and NTHL1, with most (55.6%; 5/9) of these among patients with MMR-P tumors (Supplemental Table 2). Of the four patients with non-LS germline mutations but MMR-D tumors, two harbored APC mutations with one being diagnostic of familial adenomatous polyposis (FAP) and the other being the APC p.I1307K Ashkenazi Jewish founder mutation not implicated in FAP.(18,34,35) Both patients’ tumors demonstrated MLH1 promoter hypermethylation. An additional patient had a known neurofibromatosis type 1 (NF1) diagnosis with an MLH1/PMS2-absent jejunal tumor. The patient received full MSK-IMPACT germline assessment and clinical LS testing, which did not reveal any additional germline findings. A second NF1 somatic mutation was found in the tumor in addition to MLH1 promoter hypermethylation.(Supplemental Table 2).
Of note, there was one LS patient with an MSI-H SBA (MSIsensor score 25.06) that had intact MMR protein expression on IHC (Table 2, Supplemental Table 2). The patient had a personal history of endometrial cancer (EC), harbored a germline MLH1 pathogenic missense variant (c.55A>T(p.Ile19Phe)), and had a family history of LS-associated cancers in her mother and maternal grandmother (CRC and EC). Her prior EC also demonstrated retained MMR protein expression, suggesting a failed screening test for LS using IHC rather than MSI in this patient in both of her LS-associated malignancies. We have previously reported that germline MMR missense mutations may lead to retained protein expression in the tumor despite demonstrating MSI status.(36)
Discussion
In contrast to CRC, where the role of MMR-D in LS screening and as a prognostic and predictive marker for clinical outcome has been well-established, the role of MMR-D in SBA has not been well-characterized, in part due to the rarity of this malignancy. Our data builds upon prior studies demonstrating that the prevalence of MMR-D in SBA may in fact be higher than in CRC (5,6), as we identified a slightly increased MMR-D prevalence among SBAs of 26% compared to the reported 15–20% of CRC (16,37,38). While one prior study characterizing the genomic landscape of SBA reported the MSI-H prevalence in SBA to be 7.6% (6), it is worth noting that only 50% of tumors included were assessed for MSI/MMR-D status, including CRC, in which only 4% were found to be MSI-H. Perhaps more importantly, as noted by Schrock et al., their cohort was enriched for metastatic cases leading to an underrepresentation of MMR-D tumors, which are more likely to be associated with early stage disease.(5,6) Interestingly, despite this, they demonstrated a nearly two-fold increase in MMR-D prevalence in SBA compared to CRC (7.6% vs 4%, respectively). Comparatively, our increase in MMR-D prevalence in SBA compared to CRC was more modest (26% vs. 19%, respectively), but it also aligns with a prior study demonstrating a 23% MMR-D prevalence among a cohort of 61 SBA patients(5), providing additional supporting evidence that MMR-D prevalence in SBA may be higher than in CRC.
Second, as in CRC, our data suggests that MMR-D status appears to predict for earlier stage disease. This observation does not reflect potential screening bias in LS-associated cases, as only one LS patient was found to have SBA during a screening procedure for family history of pancreas cancer (Table 2). 50% of LS-associated SBA’s were the patient’s index cancer and 80% of our LS cohort had no family history of SBA. Additionally, it is not currently recommended for all patients with LS to undergo surveillance for SBA given the lack of proven efficacious methods(39).
Our data also demonstrates that MMR-D SBA may in fact be associated with better disease prognosis, as we found a statistically-significant lower rate of disease recurrence, as only 8% (2/11) of our stage I/II MMR-D cancers recurred compared to 88% (15/17) of stage I/II MMR-P tumors (p=0.0002). This is analogous to CRC data, wherein especially in early-stage (II) patients, MMR-D is associated with a significantly better prognosis.(10,11,32) In fact, based on this improved prognosis and a demonstrated lack of benefit from 5FU-based chemotherapy, NCCN guidelines recommend against the use of adjuvant chemotherapy in patients with stage II MMR-D CRC, irrespective of other risk features.(13) Given that stage-for-stage SBA has worse clinical outcomes than CRC,(2–4) this observation has important implications for identifying low-risk, early-stage patients, namely MMR-D stage II SBA, that could potentially be spared chemotherapy toxicity. Notably, improved outcomes persisted despite an increased rate of synchronous and/or metachronous cancers among our MMR-D cohort, as over one-third of patients with MMR-D SBA had synchronous and/or metachronous cancers compared to less than 10% in the MMR-P group. Not surprisingly, the majority (66.7%) were in LS patients. Given our small sample size in this rare malignancy, additional large-scale studies are needed to best inform treatment decisions specific to stage II SBA.
We identified a 10% (10/100) LS prevalence in SBA overall, and a 38.5% (10/26) prevalence among MMR-D tumors. Moreover, we identified a two-fold increase in LS prevalence among MMR-D SBAs compared to that of our MMR-D CRC cases (38.5% vs. 19%, respectively).(16) Despite the relatively small sample size of LS-associated SBA, we noted a consistent trend among the distribution of germline mutations when comparing to LS-associated CRC, as 80% (8/10) of patients with LS-associated SBAs had underlying MLH1 and MSH2 germline mutations. This is not surprising, as mutations in MLH1 and MSH2 are considered to be highly penetrant.(16,40,41) We also noted that 20% (2/10) of patients with LS-associated SBAs harbored PMS2 germline mutations. Both were found to have concordant IHC and MSIsensor scores (Table 2), suggesting the PMS2 germline mutation was driving the malignancy. This is important to highlight as the penetrance of PMS2 mutations is an ongoing area of controversy in which some groups argue for revised clinical management, suggesting that PMS2 mutation carriers are not at a significantly increased risk of extra-colonic malignancies above the general population.(42–44) Additionally, while we did find additional germline mutations in other cancer-predisposition genes, as we did not perform uniform multi-gene panel testing across the entire cohort, the implications of such findings is beyond the scope of this study. Additional research is warranted.
There are several limitations to our study. First, this was a retrospective study across a single institution; however, the uniformity of clinical and genomic characterization allowed for in-depth analysis. Second, our cohort reflects that of a large referral center primarily comprised of non-Hispanic white patients. Third, as our cases of LS-associated SBA were diagnosed at a significantly younger age than in MMR-D non-LS SBA (47.5 vs 65, respectively) and prior research in rectal cancer has demonstrated that younger age may predict for higher LN yield and positivity(45), it is possible that age may be a confounder of LN yield in our LS population. However, since clinical outcomes were compared between MMR-P vs all other MMR-D SBA inclusive of LS patients, this difference in our small LS cohort did not drive the improved outcomes demonstrated. Finally, our study population size was relatively small, again reflecting the rarity of this malignancy. However, to our knowledge, this is the largest study comparing clinical outcomes and systemic treatment response of SBA patients according to MMR and germline status.
Our analysis demonstrates that among SBA, there is a significant proportion of underlying MMR-D tumors, as we found that over one-fourth of our SBA cohort were MMR-D. Given the recently updated NCCN guidelines recommending MMR-D assessment in SBA, we would suspect the true MMR-D proportion will soon be elucidated as clinicians become accustomed to incorporating this into routine management as is done in CRC.(7) Moreover, 50% of LS patients presented with SBA as their index cancer and would not have been identified as having LS had they not undergone MMR assessment. As it is standard-of-care for all CRC to have MMR-D assessment and subsequent LS testing in MMR-D CRCs, there is low likelihood of an underestimation of LS among CRC. Taken together, this seemingly suggests that MMR-D status among SBA may be more predictive of underlying LS than in CRC. As such and given the implications for patients and their at-risk relatives, MMR-D assessment in all SBAs with subsequent germline testing for LS is warranted.
Supplementary Material
Statement of Translational Relevance:
Small bowel adenocarcinoma (SBA) is a rare, aggressive cancer known to be associated with Lynch syndrome (LS). Clinical management guidelines for SBA have historically been extrapolated from those of colorectal cancer (CRC). Our study systematically characterizes SBA according to mismatch repair (MMR) and LS status.
Our data shows that one-quarter of SBAs were MMR-deficient (MMR-D) with nearly 40% of patients with MMR-D tumors having LS. Patients with MMR-D SBA were more likely to have early-stage disease and lower recurrence rates.. Assessment of all SBAs for MMR-D with recognition of better prognosis and potential futility of chemotherapy in early-stage MMR-D SBAs has immediate clinical implications. Moreover, 50% of LS patients presented with SBA as their index cancer and would not have been identified as having LS had they not undergone MMR assessment, which not only has direct implications for patients, but also their at-risk family members.
Acknowledgments
Funding sources:
This research was funded in part through the Romeo Milio Lynch Syndrome Foundation, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, the Robert and Kate Niehaus Center for Inherited Cancer Genomics, the Fieldstone Family Fund, and the NIH/NCI Cancer Center Support Grant P30 CA008748
Conflicts of Interest Reported:
J. Hechtman: Honoraria: Medscape, Consulting or Advisory Role: Navigant Consulting
Research Funding: Boehringer Ingelheim, Bayer, Eli Lilly
R. Yaeger: Research Funding: Array BioPharma, GlaxoSmithKline, Novartis
Travel, Accommodations, Expenses: Array BioPharma
N. H. Segal: Consulting or Advisory Role: Bristol-Myers Squibb, Pfizer, AstraZeneca/MedImmune, Imugene, Roche/Genentech, Amgen, Calithera Biosciences, Pieris Pharmaceuticals, Synlogic, Kyocera, Aduro Biotech, Kyn Therapeutics, Boehringer Ingelheim
Research Funding: MedImmune, Bristol-Myers Squibb, Pfizer, Genentech, Merck, Incyte
L.B. Saltz: Consulting or Advisory Role (Immediate Family Member): McNeil PPC
Research Funding: Taiho Pharmaceutical
D. Reidy-Lagunes: Honoraria: Novartis, Consulting or Advisory Role: Ipsen, Pfizer, Novartis
Research Funding: Novartis
J. Garcia Aguilar: Consulting or Advisory Role: Medtronic, Intuitive Surgical, Johnson & Johnson
A. Zehir: Honoria: Illumina
M.F. Berger: Consulting or Advisory Role: Roche
Research Funding: Illumina
Zsofia K. Stadler: Consulting or Advisory Role (Immediate Family Member): Allergan, Adverum Biotechnologies, Alimera Sciences, Fortress Biotech, Genentech/Roche, Novartis, Optos, Regeneron, Regenxbio, and Spark Therapeutics
References
- 1.Program. NCIUSCCR. SEER Cancer Stat Facts: Small Intestine Cancer. 2019.
- 2.Overman MJ, Hu CY, Kopetz S, Abbruzzese JL, Wolff RA, Chang GJ. A population-based comparison of adenocarcinoma of the large and small intestine: insights into a rare disease. Ann Surg Oncol 2012;19(5):1439–45 doi 10.1245/s10434-011-2173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedersen KS, Raghav K, Overman MJ. Small Bowel Adenocarcinoma: Etiology, Presentation, and Molecular Alterations. J Natl Compr Canc Netw 2019;17(9):1135–41 doi 10.6004/jnccn.2019.7344. [DOI] [PubMed] [Google Scholar]
- 4.Akce M, Jiang R, Zakka K, Wu C, Alese OB, Shaib WL, et al. Clinical Outcomes of Small Bowel Adenocarcinoma. Clin Colorectal Cancer 2019. doi 10.1016/j.clcc.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Aparicio T, Svrcek M, Zaanan A, Beohou E, Laforest A, Afchain P, et al. Small bowel adenocarcinoma phenotyping, a clinicobiological prognostic study. Br J Cancer 2013;109(12):3057–66 doi 10.1038/bjc.2013.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrock AB, Devoe CE, McWilliams R, Sun J, Aparicio T, Stephens PJ, et al. Genomic Profiling of Small-Bowel Adenocarcinoma. JAMA Oncol 2017;3(11):1546–53 doi 10.1001/jamaoncol.2017.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Small Bowel Adenocarcinoma, Version 1.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2019;17(9):1109–33 doi 10.6004/jnccn.2019.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertagnolli MM, Redston M, Compton CC, Niedzwiecki D, Mayer RJ, Goldberg RM, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29(23):3153–62 doi 10.1200/JCO.2010.33.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merok MA, Ahlquist T, Royrvik EC, Tufteland KF, Hektoen M, Sjo OH, et al. Microsatellite instability has a positive prognostic impact on stage II colorectal cancer after complete resection: results from a large, consecutive Norwegian series. Ann Oncol 2013;24(5):1274–82 doi 10.1093/annonc/mds614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010;28(20):3219–26 doi 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sargent DJ, Marsoni S, Thibodeau SN, Labianca R, Hamilton SR, Torri V, et al. Confirmation of deficient mismatch repair (dMMR) as a predictive marker for lack of benefit from 5-FU based chemotherapy in stage II and III colon cancer (CC): A pooled molecular reanalysis of randomized chemotherapy trials. 2008;26(15_suppl):4008- doi 10.1200/jco.2008.26.15_suppl.4008. [DOI] [Google Scholar]
- 12.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352(18):1851–60 doi 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 13.Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen YJ, Ciombor KK, et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J Natl Compr Canc Netw 2018;16(4):359–69 doi 10.6004/jnccn.2018.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brueckl WM, Heinze E, Milsmann C, Wein A, Koebnick C, Jung A, et al. Prognostic significance of microsatellite instability in curatively resected adenocarcinoma of the small intestine. Cancer Lett 2004;203(2):181–90 doi 10.1016/j.canlet.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Administration USFD. 2017 Oct 5 FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. U.S. Food & Drug Administration. Accessed 2017 Oct 5 [Google Scholar]
- 16.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol 2019;37(4):286–95 doi 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26(35):5783–8 doi 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hampel H, Peltomaki P. Hereditary colorectal cancer: risk assessment and management. Clin Genet 2000;58(2):89–97 doi 10.1034/j.1399-0004.2000.580201.x. [DOI] [PubMed] [Google Scholar]
- 19.de la Chapelle A The incidence of Lynch syndrome. Fam Cancer 2005;4(3):233–7 doi 10.1007/s10689-004-5811-3. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Bigas MA, Vasen HF, Lynch HT, Watson P, Myrhoj T, Jarvinen HJ, et al. Characteristics of small bowel carcinoma in hereditary nonpolyposis colorectal carcinoma. International Collaborative Group on HNPCC. Cancer 1998;83(2):240–4 doi . [DOI] [PubMed] [Google Scholar]
- 21.Jun SY, Lee EJ, Kim MJ, Chun SM, Bae YK, Hong SU, et al. Lynch syndrome-related small intestinal adenocarcinomas. Oncotarget 2017;8(13):21483–500 doi 10.18632/oncotarget.15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precision Oncology 2017(1):1–17 doi 10.1200/po.17.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014;30(7):1015–6 doi 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.EVALUATION OF AUTOMATIC CLASS III DESIGNATION FOR MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets). US Food and Drug Administration; 2017 2017. Report nr DEN170058. [Google Scholar]
- 25.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DT, Prasad M, Chekaluk Y, Benayed R, Sadowska J, Zehir A, et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics 2017;10(1):33 doi 10.1186/s12920-017-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T, Stadler ZK, Zhang L, Weiser MR, Basturk O, Hechtman JF, et al. Immunohistochemical null-phenotype for mismatch repair proteins in colonic carcinoma associated with concurrent MLH1 hypermethylation and MSH2 somatic mutations. Fam Cancer 2018;17(2):225–8 doi 10.1007/s10689-017-0031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ligtenberg MJ, Kuiper RP, Geurts van Kessel A, Hoogerbrugge N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam Cancer 2013;12(2):169–74 doi 10.1007/s10689-012-9591-x. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Cabornero L, Infante Sanz M, Velasco Sampedro E, Lastra Aras E, Acedo Becares A, Miner Pino C, et al. Frequency of rearrangements in Lynch syndrome cases associated with MSH2: characterization of a new deletion involving both EPCAM and the 5′ part of MSH2. Cancer Prev Res (Phila) 2011;4(10):1556–62 doi 10.1158/1940-6207.CAPR-11-0080. [DOI] [PubMed] [Google Scholar]
- 30.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405–24 doi 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruhl JL CC, Hurlbut A, Ries LAG, Adamo P, Dickie L, Schussler N Summary Stage 2018: Codes and Coding Instructions. Bethesda, MD: National Cancer Institute; 2018. [Google Scholar]
- 32.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349(3):247–57 doi 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 1998;95(12):6870–5 doi 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boursi B, Sella T, Liberman E, Shapira S, David M, Kazanov D, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49(17):3680–5 doi 10.1016/j.ejca.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 35.Prior TW, Chadwick RB, Papp AC, Arcot AN, Isa AM, Pearl DK, et al. The I1307K polymorphism of the APC gene in colorectal cancer. Gastroenterology 1999;116(1):58–63 doi 10.1016/s0016-5085(99)70229-5. [DOI] [PubMed] [Google Scholar]
- 36.Hechtman JF, Rana S, Middha S, Stadler ZK, Latham A, Benayed R, et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod Pathol 2019. doi 10.1038/s41379-019-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol 2010;28(20):3380–7 doi 10.1200/JCO.2009.27.0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J Natl Compr Canc Netw 2019;17(9):1032–41 doi 10.6004/jnccn.2019.0044. [DOI] [PubMed] [Google Scholar]
- 40.Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev 2017;26(3):404–12 doi 10.1158/1055-9965.EPI-16-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305(22):2304–10 doi 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- 42.Sjursen W, Haukanes BI, Grindedal EM, Aarset H, Stormorken A, Engebretsen LF, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. Journal of medical genetics 2010;47(9):579–85 doi 10.1136/jmg.2010.077677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moller P, Seppala TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 2018;67(7):1306–16 doi 10.1136/gutjnl-2017-314057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 2020;22(1):15–25 doi 10.1038/s41436-019-0596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer JE, Cohen SJ, Ruth KJ, Sigurdson ER, Hall MJ. Young Age Increases Risk of Lymph Node Positivity in Early-Stage Rectal Cancer. J Natl Cancer Inst 2016;108(1) doi 10.1093/jnci/djv284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.