

Abstract
DNA methyltransferase inhibitors (DNMTIs) like 5-Azacytidine (5-Aza) are the only disease-modifying drugs approved for the treatment of higher-risk myelodysplastic syndromes (MDS), however less than 50% of patients respond, and there are no predictors of response with clinical utility. Somatic mutations in the DNA methylation regulating gene tet-methylcytosine dioxygenase 2 (TET2) are associated with response to DNMTIs, however the mechanisms responsible for this association remain unknown. Using bisulfite padlock probes, mRNA sequencing, and hydroxymethylcytosine pull-down sequencing at several time points throughout 5-Aza treatment we show that TET2 loss particularly influences DNA methylation (5mC) and hydroxymethylation (5hmC) patterns at erythroid gene enhancers and is associated with down-regulation of erythroid gene expression in the human erythroleukemia cell line TF-1. 5-Aza disproportionately induces expression of these down regulated genes in TET2KO cells and this effect is related to dynamic 5mC changes at erythroid gene enhancers after 5-Aza exposure. We identified differences in remethylation kinetics after 5-Aza exposure for several types of genomic regulatory elements, with distal enhancers exhibiting longer-lasting 5mC changes than other regions. This work highlights the role of 5mC and 5hmC dynamics at distal enhancers in regulating the expression of differentiation-associated gene signatures, and sheds light on how 5-Aza may be more effective in patients harboring TET2 mutations.
Graphical Abstract
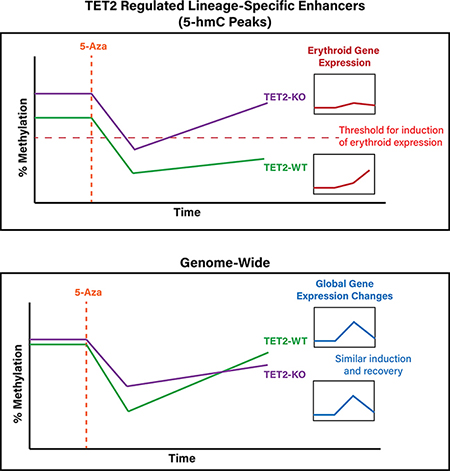
Introduction
DNA methyltransferase inhibitors (DNMTIs) such as 5-Azacytidine (5-Aza) are the only class of drugs approved for the treatment of higher-risk myelodysplastic syndromes (MDS), and are increasingly used in other cancers as monotherapy (1–3), and in combination with other epigenetic drugs (4–6). DNMTIs are cytosine analogs which are thought to act primarily through incorporation into DNA followed by covalent trapping and subsequent proteasomal degradation of DNA methyltransferase enzymes (DNMTs), leading to global hypomethylation of the genome during subsequent cell divisions (7). Despite wide usage of these drugs, overall response rates are poor, with less than 50% of MDS patients experiencing a clinical benefit, while the mechanisms which sensitize patients to these drugs remain elusive (8,9). Previous work in our laboratory and others identified TET2 mutation as a biomarker of response in MDS patients (10,11), where mutant patients were almost twice as likely to respond as their non-mutant counterparts, however the mechanisms behind this relationship remain unknown.
TET2, one of the three mammalian members of the TET family of Fe(II) and α-ketoglutarate-dependent dioxygenases (12–14), is one of the most frequently mutated genes in MDS. TET enzymes catalyze the successive conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine, together termed oxidized methylcytosines (oxi-mC) (15,16). TET proteins mediate the only known pathways for demethylation of 5mC in DNA, primarily through replication-dependent dilution of oxi-mC bases which are not recognized as substrates for symmetrical methylation by the DNMT1-UHRF1 complex (17); a minor pathway may involve the further oxidation of 5hmC into 5fC and 5caC, which are recognized and removed by thymine-DNA glycosylase and replaced with unmethylated cytosine through base excision repair (18,19). TET proteins serve diverse roles (20) but are best-known for their role in maintaining low methylation levels at transcriptional enhancers in hematopoietic cells and other cell types (21,22), a finding corroborated by observations that patients with somatic TET2 mutations in hematopoietic cells exhibit hypermethylation at enhancer elements (23,24). Methylation at enhancer elements is dynamic during cellular differentiation (25,26), and in all cases studied, TET proteins have an important role in proper lineage specification, often through their ability to be recruited by transcription factors that control the expression of lineage-specific genes (27,28).
There is ongoing debate regarding the precise mechanisms responsible for patient responses to DNMTIs, but at least in the context of TET2 mutant cases, the response does not appear to be driven by changes in the clonal burden of tumor, but rather via epigenetic induction of differentiation, a phenotype also observed in cell line models (29–33). Notably, both responding and non-responding patients exhibit similar reductions in DNA methylation (5mC) immediately following DNMTI treatment (34), however only patients who eventually developed a response exhibited sustained reductions in DNA methylation several weeks after cessation of treatment and only after several cycles (months) of therapy (29,35), suggesting that the rate of remethylation in these patients may be a significant factor determining response.
To test the hypothesis that the baseline level of 5mC as well as the kinetics of 5mC changes during 5-Aza treatment might be relevant for the increased response rates in TET2-mutant MDS patients, we identified specific 5mC changes and the accompanying biological pathways affected during 5-Aza treatment. To accomplish this, we employed CRISPR/Cas9-mediated inactivation of TET2 and massively parallel sequencing approaches to study 5mC, 5hmC and gene expression dynamics during 5-Aza exposure and recovery in a TF-1 erythroleukemia cell line model. We identified an altered sensitivity and length of recovery after 5-Aza exposure as well as differences in demethylation and remethylation rate in TET2-deficient (TET2KO) cells. We showed that downregulation of erythroid differentiation-related gene expression signatures in TET2KO can be partially corrected via 5-Aza treatment, and we show how this relates to 5hmC and 5mC changes at erythroid transcriptional enhancers. Our findings highlight the subtle relationship between 5mC deregulation and differentiation defects in TET2KO cells and provide insight as to why TET2 mutated patients may disproportionately benefit from DNMTI therapy.
Materials and Methods
TET2 gene editing and cell culture
The human GM-CSF cytokine dependent erythroleukemia cell line TF-1 (ATCC CRL-2003) was purchased from ATCC. Cells were confirmed to be negative for Mycoplasma prior to use via a PCR-based mycoplasma detection kit. Cells were maintained in medium supplemented with 10% fetal bovine serum (Omega Scientific) and 1% RPMI 1640 penicillin/ streptomycin/ L-glutamine. Cells were maintained at a density between 5×104 and 5×105 viable cells/mL and passaged every 2–3 days. Cells were passaged less than 15 times. Cell viability was assessed by Trypan Blue exclusion. CRISPR single-guide RNAs (sgRNA) were designed with CHOPCHOP (chopchop.cbu.uib.no) and cloned into px458 vector (Addgene, 48138) following the protocol in Ran et al (36,37). We followed the approach of Bauer et al. using 2 CRISPR sgRNAs to delete the catalytic residues of TET2 bounded between His1550 and Asp1981 following the design in Figure 1A (38). Biallelic deletion of this region was not achieved, however biallelic inactivation was achieved through monoallelic deletion of ~His1550 – Asp1981 and monoallelic frameshift-inducing deletion mutations near His1550 (actual deletions shown in Figure 1A for each allele of each clone). 2ug of px458 vectors with incorporated sgRNAs (1ug vector from each sgRNA) or unmodified px458 (for wild-type controls) were nucleofected into parental TF-1 cells with the Lonza Nucleofector Kit V following the manufacturer’s protocol. Nucleofected cells were allowed to grow undisturbed for 24 hours before sorting live GFP-positive cells using a Bio-Rad S3 Cell Sorter. Bulk GFP-positive sorted cell populations were allowed to grow undisturbed for 3 days before isolating single-cell clones by limiting dilution into 96-well plates. Single-cell clones were screened for inactivating out-of-frame indels/deletions by PCR followed by Sanger sequencing, and later by immunoblotting for TET2 protein and 5-hydroxymethylcytosine. CRISPR sgRNA sequences are as shown in Figure 1A.
Figure 1. CRISPR/Cas9 mediated loss of TET2 alters growth and hydroxymethylation patterns in TF1.

A) Paired CRISPR sgRNA design targeted key catalytic residues of TET2. Two independently derived TF1 clones with bi-allelic inactivating deletions were characterized. B) Western blot using C-terminal anti-TET2 antibody confirms loss of full-length TET2 protein. C) Growth curves for TF1 clones display differences in normal growth patterns for TET2 knockout. D) 5-hmC pull down and sequencing (HMCP) peak heatmaps and mean normalized counts profile for differentially hydroxymethylated regions (DHMR). E) Differential 5hmC peak enrichment volcano plot TET2KO vs. WT. F) Number of DHMR (TET2KO v. WT) observed vs. expected overlapping regulatory chromatin states in reference CD34+ HSC epigenome (from reference epigenome E051 of Roadmap Epigenomics Consortium)
Western blotting
10 million cells were lysed directly in 500uL 2x Laemmli sample buffer (Bio-Rad) freshly prepared with 2.5% B-mercaptoethanol. Lysates were sonicated at 75% amplitude for 15 seconds to shear DNA and then boiled at 95°C for 5 minutes. 20uL of whole cell lysates were separated by 7.5% Tris-Glycine SDS-PAGE and proteins were transferred onto PVDF membranes. Immunodetection of proteins was carried out in TBST (20mM Tris-HCl, 500mM NaCl, 0.05% Tween-20, pH 7.5) supplemented with 5% non-fat milk and C-terminal anti-TET2 (CST-18950), anti-DNMT1 (Abcam ab92314), anti-Lamin-B1 (Proteintech 12987–1-AP), or anti-β-actin (Biolegend poly6221), followed by incubation with HRP-conjugated secondary antibodies.
L-ascorbic acid (L-AA) treatment and 5hmC DNA immunoblotting
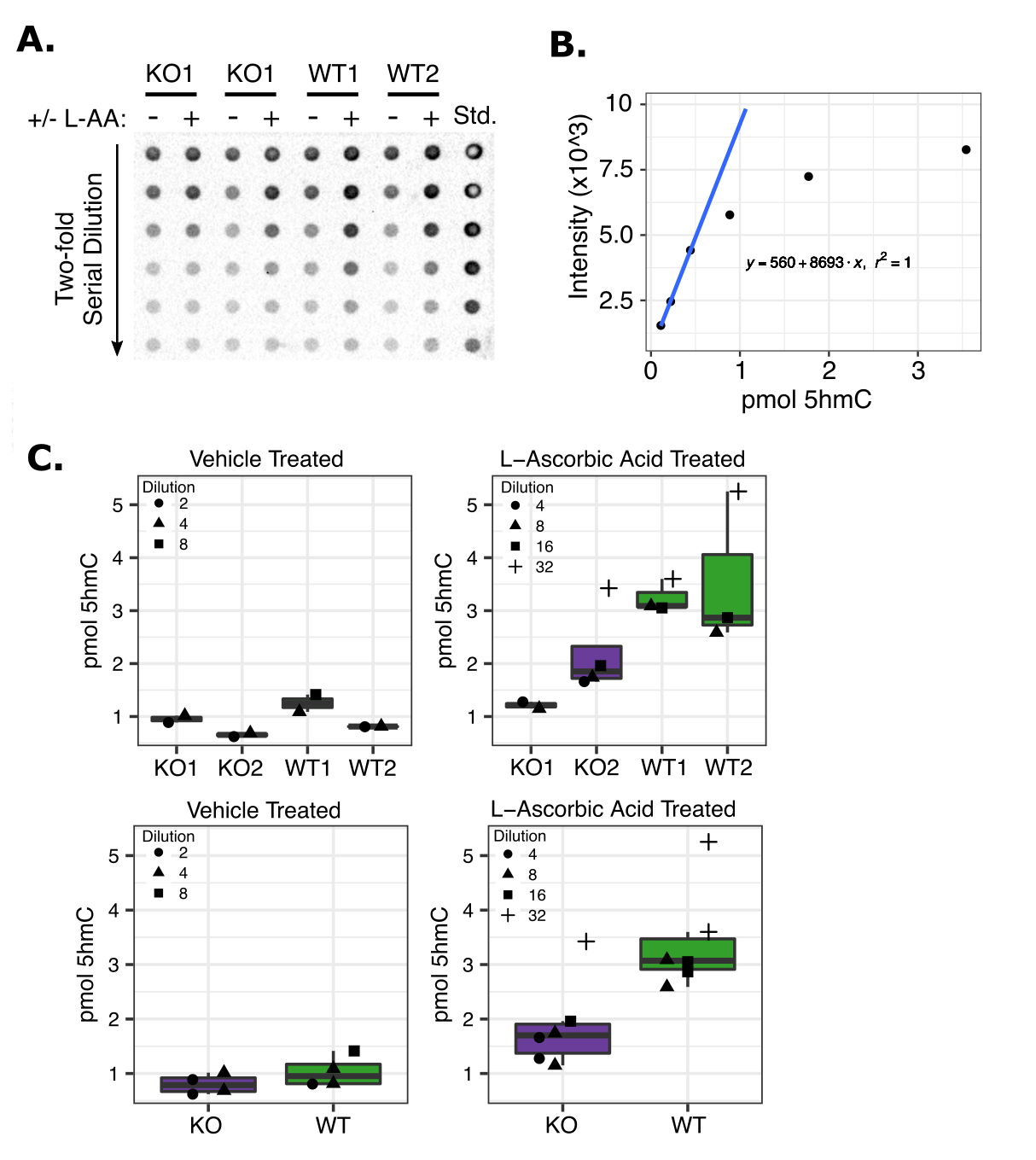
The L-AA dose was titrated to a level that minimized cell death 24hrs after exposure. Cells were treated with a minimally cytotoxic dose of 100uM L-AA and genomic DNA was extracted 24hrs after exposure for dot blot analysis. 5-hydroxymethylcytosine DNA immunoblotting was carried out as described previously (39). DNA was denatured and spotted in 2-fold serial dilutions on a nitrocellulose membrane in a Bio-Dot apparatus. A synthetic oligonucleotide containing a known quantity of 5hmC was used as standard. Immunodetection of 5hmC was carried out using anti-5hmC antibody at 1:5000 dilution (Active Motif) followed by incubation with 1:10,000 dilution of HRP-conjugated secondary antibody and visualization with West-Q Pico Dura ECL Solution (Gen-DEPOT). 5hmC dot blot signal was quantified using ImageJ. A standard curve was constructed as in Ko et al. and the linear portion of the curve was used to calculate the moles of 5hmC per sample shown in Figure S1 (40).
Drug treatment and viability assays
Viability of cultures that were treated with varying concentrations and dosing schedules of 5-Aza were assessed via CellTiter Blue viability assay following manufacturers protocol. Cells were seeded at 2,000 cells per well in 96-well culture plates with final volume of 100uL culture media 24 hours prior to addition of first drug dose. Viability was assessed 24hrs after each final treatment (e.g. at 48,72, or 96hrs for the 3 treatment schedules tested). IC50 curves were modeled using the four-parameter log-logistic regression model using the R package ‘drc’. Differences in IC50 curves between TET2-WT and TET2-KO cell lines were tested via one-way ANOVA (R ‘anova’ function). After dose-optimization, the final experiment from which genomic analyses were obtained was performed as follows: TF1 cell lines were seeded at a density of 100,000 cells/mL on Day 0 and allowed to equilibrate overnight. Freshly prepared 5-Aza or dimethyl sulfoxide vehicle was added to cultures every 24hrs for a total of three doses of 200nM starting on Day 1. Media was replaced on Day 2 and Day 4, and every 2–3 days following until Day 12. For flow-cytometry follow-up experiments the cells were grown until Day 20.
Induction of erythroid differentiation of TF-1 cells
Erythroid differentiation induction was carried out as described (41). TF1 cells growing in GM-CSF containing medium were spun down and washed three times in fresh media lacking GM-CSF before being seeded at a density of 200,000 cells/mL in 20mL media lacking GM-CSF. After 24hrs of culture in these cytokine poor conditions, recombinant human erythropoietin (Epogen, Epoetin alfa, Amgen) was added directly to cultures to a final concentration of 2U/mL. Fresh media with 2U/mL erythropoietin was added four days later and flow cytometry assessment of differentiation was performed 8 days after the first erythropoietin addition.
Flow cytometry
FACS sorting of GFP-positive px458 transfected cells was performed using the Bio-Rad S3 Cell Sorter. During 5-Aza treatment and erythroid differentiation experiments, cells were stained with PE-Cy5 conjugated anti-CD235AB (Biolegend cat. # 306605) to measure glycophorin-A expression as a surrogate for erythroid differentiation by flow cytometry on an Attune NxT Acoustic Focusing Flow Cytometer. Data were analyzed in R 3.4 and the Bioconductor ‘FlowCore’ package.
Bisulfite Padlock Probe library construction and sequencing analysis
The Bisulfite Padlock Probes used in this study were designed to selectively capture the methylation status of CpGs within genomic regions of known regulatory potential including promoters for NCBI RefSeq genes, CCCTC-binding factor binding sites, DNase I hypersensitive regions, regions that are differentially methylated during stem cell differentiation, and regions differentially methylated in cancer (42). In the present study, we obtained sufficient coverage for roughly 621,000 CpGs. BSPP libraries were constructed as previously described, and sequenced using a standard 150bp paired-end sequencing protocol on the Illumina NovaSeq (42). Sequencing reads were trimmed for low quality bases and adapter content using TrimGalore v0.4.0 and cutadapt v.1.18 before alignment to a 3-letter (bisulfite converted) version of the hg19 reference genome as previously described (43). For alignment, trimmed reads were encoded such that all cytosines converted to thymines and for the reverse complement strand, all guanines to adenines, so that the reads could align to the three-letter genome. Reads were then mapped separately to both the Watson and Crick strands of the bisulfite-converted hg19 genome using BWA mem v.0.7.12 (options: ‘-B2 -c1000’). Alignment with mapping quality scores below 5 were discarded and for reads which mapped to more than one position, and only the highest quality mapping was kept for each read. Overlapping paired-end reads were clipped with BamUtil clipOverlap function. Encoded read sequences were replaced by the original read sequences in the final BAM alignment files which were then used for extracting methylation levels for CpGs.
Statistical analyses of BSPP methylation data
Differential methylation analysis between different time points and conditions was performed using the R package ‘DSS’ version 2.26.0. Differentially methylated CpGs were determined using the ‘callDML’ function of DSS with a posterior probability threshold of 0.90 or greater that the absolute methylation difference was greater than 20% (e.g. 90% chance the difference is 20% or greater). 5mC time-profile clustering was performed as follows: First ‘dynamic CpGs’ were identified as CpGs with differential methylation between any of the three time points (Day 0 v. 4; Day 0 v. 12; Day 4 v. 12). This yielded a set of ‘dynamic CpGs’ for each genotype (KO and WT) which had some amount of overlap but were largely unique to each genotype. Next, the mean methylation value of replicates for each dynamic CpG locus was converted to a Z-score across the three time points (Days 0, 4, 12) for each genotype. We then combined the Z-scaled methylation values for both KO and WT into a single matrix and performed hierarchical clustering with Euclidean distance and Ward’s agglomeration method. To determine the optimal number of clusters for downstream analysis we examined the methylation time profiles for a range of K’s, and settled on the K=4 cluster solution because it captured the major patterns of remethylation and statistically produced the most compact clustering solution as indicated by the mean cluster silhouette width statistic.
RNAseq library preparation and analysis
Total RNA was isolated from cell lines using the Machery-Nagel Nucleospin RNA kit according to manufacturer instructions. RNA sequencing libraries were constructed using the Illumina TruSeq stranded mRNA kit and sequenced on an Illumina NovaSeq by NovoGene Inc. Fastq files were trimmed to remove polyA signals and adapter content using cutadapt and quality checked using FastQC. Reads were aligned to the hg19 genome using STAR v2.6.1d and GENCODE v19. Reads mapping to repetitive elements were removed. Transcript counts were summarized using featureCounts v1.6.2 and differential gene expression was calculated using the R package ‘DESeq2’. The threshold for determining differential expression was an adjusted p-value (FDR corrected) < 0.10. Normalized TPM values were calculated from DESeq2 normalized counts and transcript lengths from GENCODE v19. Gene set enrichment analysis (GSEA) was performed using the GSEA pre-ranked algorithm using the DESeq2 differential expression Wald statistic as the ranking metric. GSEA was carried out in R using the ‘clusterProfiler’ package (44)
5-hydroxymethylcytosine pull-down (HMCP) sequencing with reference exogenous genome spike-in normalization
We employed the HMCP with reference exogenous genome (ChIP-Rx) method of Orlando et al. for quantitative normalization of HMCP signal according to an exogenous spike-in genome (45). As exogenous control we used the DNA isolated from phage T4gt, which carries mutations in both -agt and -bgt glycosyltransferases and thus the vast majority of cytosines are only hydroxymethylated without glycosylation (46). HMCP was carried out using the 5-hydroxymethylcytosine pull-down assay (Cambridge Epigenetix) according to manufacturer’s instructions with slight modifications. Exogenous phage T4gt DNA was spiked into the TF1 cell DNA at a 1:50,000 ratio before shearing with a Covaris E220 to an average length of 150 bp. Sheared DNA samples were end-repaired and adapter ligated with barcoded Illumina TruSeq adapters. Libraries were denatured and a new copy strand was synthesized. DNA fragments containing 5hmC were pulled down using a two-step process where 5hmC were first glycosylated with T4BGT (Thermofisher) using UDP-6-azide-glucose, followed by labeling with PEG (polyethylene glycol)-biotin via copper-free CLICK chemistry. The biotin-containing DNA was then pulled down and purified using streptavidin magnetic beads. Finally, purified libraries were PCR amplified with Kapa real-time library amplification kit and sequenced using single end 50bp protocol on the Illumina NovaSeq.
Sequencing reads from HMCP were aligned to a combined genome of hg19 and the T4 phage genome (GenBank: AF158101.6) using a 2-step mapping procedure where raw reads were first mapped with BWA mem v0.7.15 (options: -M -t 8); and then any unaligned reads were trimmed for low quality base calls and adapter content with Trim-galore v0.4.3 (options: –paired –length 35 –stringency 3 –three_prime_clip_R1 1 –three_prime_clip_R2 1) before realignment using BWA-mem with more stringent mapping parameters (options: -M -t 8 -B 6). Reads aligning to ENCODE Blacklisted regions or random chromosomes, as well as any reads which mapped to both the T4 and hg19 genomes were removed. Normalization was performed based on a scaling factor for each sample which was calculated as in Orlando et al. with the modification that T4 phage genomic reads were calculated instead of Drosophila genomic reads. 5hmC peak calling was performed using MACS2 v.2.1.1 (options: callpeak -t IPbam -c INPUTbam -f BAMPE -n Name -g hs -q 0.01 --keep-dup all --nomodel --broad) and differential peak enrichment was calculated using MEDIPS v.1.34.0 and edgeR v3.24.3 (MEDIPS.meth function with options: p.adj=TRUE, diff.method=‘edgeR’, CNV=FALSE, MeDIP=FALSE, minRowSum=1, quantile=FALSE, TMM=FALSE) on the normalized read counts within peaks.
Statistical Analyses
All statistical analyses were performed in R v.3.4.4.
Data Sharing Statement
All high-throughput sequencing data from experiments in this study is publicly available under the NCBI Gene Expression Omnibus accession number GSE148120.
Results
Targeting the TET2 catalytic domain with CRISPR/Cas9 abolishes TET2 expression and function in TF-1 cells.
We used paired CRISPR Cas9 guide-RNAs (gRNA) to target key catalytic residues of the TET2 dioxygenase domain in TF-1 cells with the goal of deleting a ~1.2kb region of exon 11 (Figure 1A). To control for any potential off-target DNA damage we used separate gRNA sequences for each replicate (Figure 1A). After isolating single-cell colonies and PCR screening potential TET2KO clones, we identified one clone from each pair of CRISPR gRNAs that had bi-allelic inactivation of the TET2 catalytic domain. Notably, after PCR screening ~100 clones per gRNA pair, we did not observe any clones with a bi-allelic 1.2 kb deletion, likely due to the statistical improbability of this occurring in a single cell as mentioned in (47). We validated that full-length TET2 protein was not expressed in either of our TET2KO TF-1 clones using western blot (Figure 1B). We observed some clone-specific differences in normal growth patterns as might be expected from the initial clonal variability of the bulk cell population (Figure 1C). To confirm that the catalytic function of TET2 had been impacted we performed 5hmC DNA immunoblot, however we did not have the sensitivity to detect significant differences in 5hmC content at baseline (Figure S1), most likely because TET3 and TET1 are still expressed in the cells.
In order to enhance the 5hmC signal in our clones we took advantage of the fact that TET protein function can be enhanced by increased L-ascorbic acid (L-AA) concentrations (48,49). L-AA treatment increased 5hmC content >2 fold in TET2 WT clones, while TET2KO clones had a more modest ~1.2-fold increase. Finally, to more sensitively detect changes in 5hmC and to determine which genomic regions were affected, we performed 5-hmC pull-down followed by next-generation sequencing (HMCP-seq). The majority of 5hmC enriched regions (~51,000) were shared between TET2KO and WT cells. Of the 2,381 differentially hydroxymethylated regions (DHMR) shown in Figure 1D (unadjusted p<0.05), 1,861/2,831 (78%) had decreased 5hmC in TET2KO. Of the most statistically significant DHMRs (FDR < 0.1) 319/328 (97%) had decreased 5hmC signal in TET2KO (Figure 1E). Notably, the DHMR peaks were enriched for regions defined as actively transcribed, active enhancers, or active promoters in a reference hematopoietic stem cell epigenome (Figure 1F). Together these results indicate that TET2 is probably not be the primary regulator of 5hmC in TF-1 cells at baseline, but TET2 loss of function causes reduced 5hmC signal at regions important for gene regulation and impairs 5hmC deposition upon L-AA stimulation, which is in agreement with the known functions of TET2 (21–23,50).
TET2KO and WT TF-1 exhibit differential responses and remethylation kinetics after 5-Aza exposure.
To investigate the effects of 5-Aza we first determined the minimal dose that inhibited DNMTs while maintaining high cellular viability to avoid 5mC differences as a consequence of clonal population shifts versus true 5mC changes caused by 5-Aza. We observed that multiple lower intensity treatments of 5-Aza led to prolonged downregulation of DNMT1, while maintaining high cellular viability (Figure S2), and determined an optimal dosing regimen of 200nM 5-Aza every 24 hours for 3 days. With this regimen we did not observe a substantial difference in sensitivity to 5-Aza by TET2KO TF-1 cells as measured by IC50 (Figure 2A). Acute 5mC changes induced by 5-Aza are known to correlate with the population doubling time (PDT) in cancer cell lines (51), therefore we monitored the PDT after 5-Aza exposure as a surrogate measure of 5mC recovery. Notably, TET2KO clones had a significantly lengthened PDT compared with WT clones, however the length of time to recovery of normal PDT length was similar between the two (~12 days) (Figure 2B).
Figure 2. Differential response to 5-Aza exposure in TET2KO and WT TF-1.

A) IC50 curve for 3x once-daily dose of 5-Aza. B) Population doubling time changes following once-daily 5-Aza or vehicle exposure for 3 consecutive days. Population Doubling time calculated as (T2 – T1) x (ln(2) / ln(D2 / D1)), where T=time, D=cell density. C) Schematic of 5-Aza exposure experiment and data generated at each time point. D) Hexbin density scatter plot (each density bin represents 1 to 20 CpGs as indicated in legend) and bar plot showing baseline DNA methylation differences between TET2KO vs. WT TF-1 prior to 5-Aza exposure (only differentially methylated CpGs shown, FDR<0.05 and difference >20%). E) Global patterns of 5mC for (left) all CpGs covered and (middle) “Dynamic” CpGs that were significantly changed during treatment (posterior probability of >20% difference at FDR < 0.1 for comparisons shown in schematic at right). F) Venn diagram showing amount of overlap for Dynamic CpGs in TET2 KO vs. WT. G) Time profiles for dynamic CpGs reveal differences in remethylation rate for TET2KO vs. WT dynamic CpGs. P-values correspond to Wilcoxon signed rank test for each time point. Note: E (middle), F, and G are all visualizing the same set of “Dynamic CpGs” which were identified via differential methylation comparisons indicated in schematic E (right) using the ‘DSS’ R package (methods).
Next we used BSPP and RNAseq to measure 5mC gene expression dynamics during treatment and recovery from 5-Aza treatment at the time points indicated (Figure 2C). Surprisingly, prior to 5-Aza treatment, there were almost an equal number of hyper and hypo-methylated CpGs when comparing TET2WT and KO (12,310 hyper- and 10,246 hypo-methylated) (Figure 2D). As expected, after 5-Aza exposure there was a global decrease in methylation for both genotypes. We next defined a set of CpGs termed “dynamic CpGs” for each genotype which had the largest differences in methylation between treatment time points (posterior probability >0.90 that 5mC difference was >20%) to focus on the most notable 5mC changes during treatment. Of the 621,000 CpGs covered in all samples in our dataset, only 9,787 met the dynamic CpG criteria, and the mean change in methylation for these CpGs was 31% in WT and 27% in KO (Figure 2E). Only 1,098 dynamic CpGs were shared between genotypes, while the majority were unique (WT 4,998; KO 3,691) (Figure 2F). Finally, when examining the global remethylation kinetics, TET2KO dynamic CpGs were less demethylated and remethylated at a lower rate than WT dynamic CpGs (Figure 2G).
Dynamic CpG loci exhibit different patterns and proportions of remethylation kinetics.
To investigate different patterns of remethylation kinetics we sought to categorize CpGs based on the speed and extent with which they recovered their initial methylation value. Rather than define arbitrary thresholds to classify categories, we instead employed unsupervised hierarchical clustering of methylation values z-scored across time for each CpG locus in each genotype to define groups of loci with similar time-profiles in an unbiased manner. We examined a range of potential time-profile clustering solutions where we cut the clustering tree to produce different numbers of clusters (K) where K ranged from 2 through 9, with a goal of finding the minimum number of clusters required to describe the important patterns. We then calculated the mean silhouette width for each potential clustering solution, where larger silhouette width represents superior clustering solutions (52). We observed a peak in the mean silhouette width for K=4 clusters of CpG time-profiles, indicating that 4 clusters captured the majority of the variability in time profiles without over-fitting the results (Figure S3). The four remethylation clusters included three groups of CpGs that were significantly demethylated, with varying speeds and degree of methylation recovery (rapid, intermediate, and slow recovery), as well as one group of CpGs which actually gained methylation after 5-Aza exposure and largely recovered their initial methylation levels by day 12 (Figure 3A–B). We explored how well these four clusters reflected differences in the degree of recovery of methylation patterns by a statistic we termed ‘recovery ratio’ (Figure 3D) defined for each CpG as the change in methylation from day 0 to day 4 divided by the change in methylation from day 4 to day 12. Interestingly, we observed that the clusters could be perfectly distinguished by their recovery ratio. We examined the quantity and relative proportion of dynamic CpGs belonging to each cluster and observed that TET2KO dynamic CpGs had a smaller proportion of CpGs assigned to the rapid-recovery cluster (KO, n=1,467 (30.6%); WT, n=2,546 (41.7%); Fisher’s exact p < 0.01) and a much larger proportion of CpGs assigned to the slow-recovery cluster (KO, n=1,111 (23.2%); WT, n=804 (13.2%); Fisher’s exact p < 0.01) (Figure 3C), in agreement with our previous finding that TET2KO dynamic CpGs displayed slower global remethylation kinetics. Finally, to correlate these categories of dynamic CpGs with their regulatory functions, we determined the overlap of dynamic CpGs with regulatory chromatin states defined in a reference CD34+ hematopoietic stem cell epigenome (Figure 3E) (53). In both genotypes we observed striking enrichment for dynamic CpGs located in regions of active transcription as well as in active enhancers. Notably, there was a trend for actively transcribed regions as well as gene-body enhancers to be more rapidly remethylated (enrichment: rapid > intermediate > slow recovery), while this trend was reversed for non-gene-body enhancers and transcribed regions at gene 3’ or 5’ (slow > intermediate > rapid recovery). The one region where the trends were not identical between genotypes was for distal enhancers, where the highest enrichment was in the slow-recovery cluster for TET2KO, while for WT the highest enrichment was for intermediate-recovery CpGs, however these differences were not significant (p = 0.23).
Figure 3. Hierarchical clustering of time scaled CpG methylation values resulted in four major clusters characterized by differences in remethylation rates.

A) Time profile of dynamic CpG loci for each cluster. Each colored line represents a single CpG locus; black solid line represents mean of all loci in cluster; dashed black lines represent mean +/− 1 standard deviation; boxplots show summary statistics for WT and TET2KO. B) Heatmap of dynamic CpG mean methylation values for each genotype stratified by whether the locus was a shared dynamic CpG or unique to either genotype. C) Proportion of dynamic CpGs belonging to each cluster; p value corresponds to chi-square test; * represents p-value < 0.01 for fisher’s exact test for difference in proportions of CpGs within the indicated cluster between genotypes. D) (top) schematic of the ‘recovery ratio’ which is plotted in histogram below, colored by cluster. Clusters were perfectly separated based on the recovery ratio statistic. E) Odds ratio (+/− standard error) of enrichment for dynamic CpGs within regulatory chromatin states defined in a reference normal CD34+ HSPC epigenome (Roadmap Epigenomics, E051).
Dynamic gene expression changes during 5-Aza exposure reveal alterations in differentiation-associated gene signatures.
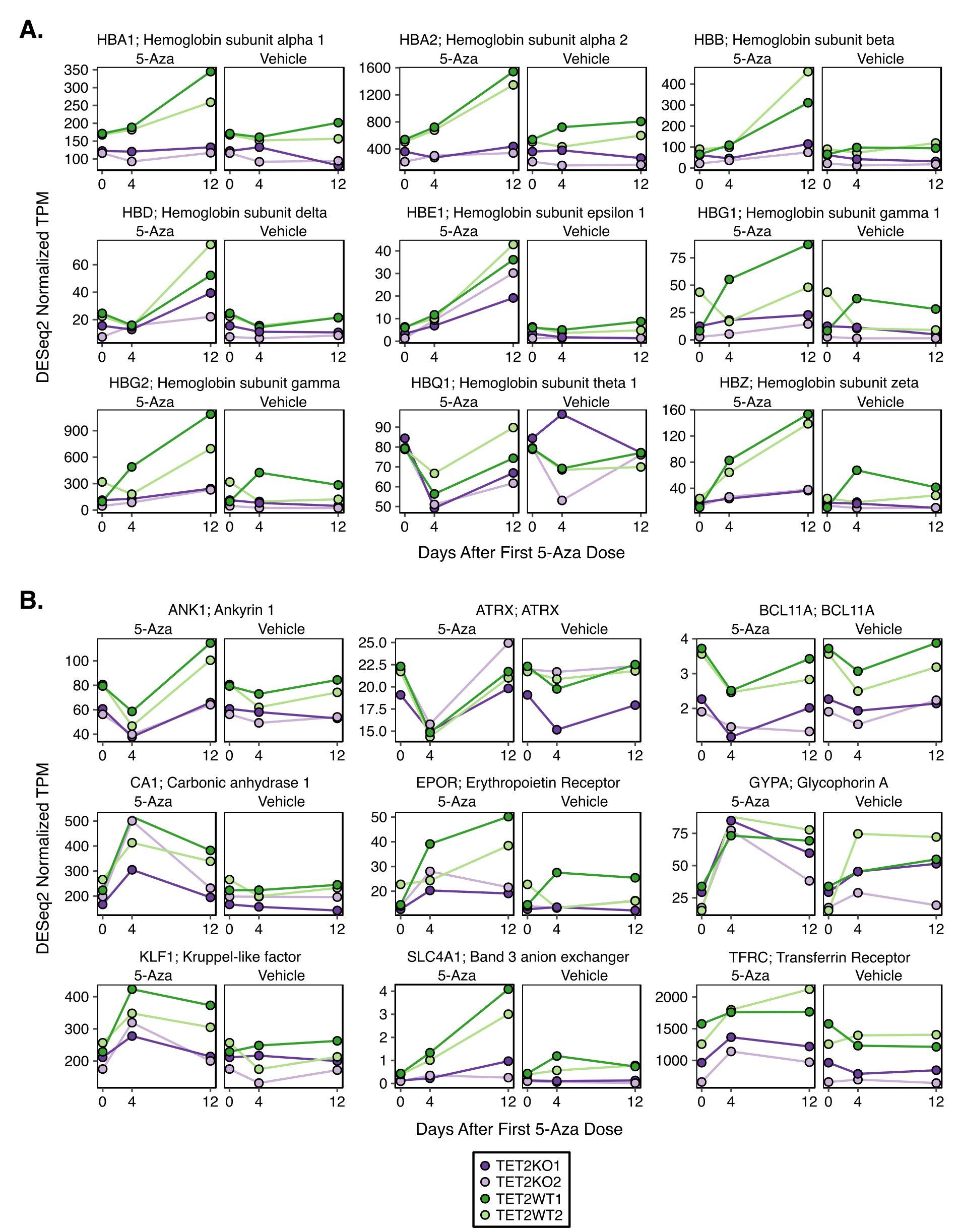
To investigate differences in gene expression pathways affected by 5-Aza exposure, we performed RNAseq on days 0, 4, and 12, as indicated in Figure 2C. Differential expression analysis identified 414 down- and 404 upregulated genes between TET2KO and WT at baseline (Day 0, pre-treatment). Gene-set enrichment analysis (GSEA)(54) identified a significant enrichment of ‘hallmark heme metabolism’ (downregulated in KO relative to WT) and ‘hallmark epithelial mesenchymal transition (EMT)’ (upregulated in KO relative to WT) gene sets (Figure 4A,C–D), among others. Next we determined differentially expressed genes (DEGs) between 5-Aza and vehicle treated conditions on days 4 (24hrs after final 5-Aza) and 12 (8 days after final dose). On day 4 we observed widespread transcriptional changes, with 2,912 genes differentially expressed in TET2KO and 2,518 in WT, with the majority of genes exhibiting the same directional change in both conditions (shared: 1,011 down, 854 up; KO unique: 432 down, 634 up; WT unique: 370 down, 270 up) (Figure 4B). By Day 12 the widespread transcriptional changes induced by 5-Aza were almost completely normalized, with only 25 genes significantly differentially expressed in either genotype compared with vehicle (shared: 8 up; KO unique: 5 up; WT unique: 9 up, 3 down). As might be expected of a DNA damaging agent like 5-Aza, gene sets involved in cell cycle and growth/metabolism were downregulated, while DNA damage response, inflammation and apoptosis gene sets were upregulated on Day 4 for both WT and KO (Figure 4E–F). By day 12 many of the same gene sets were still enriched, indicating that although many genes were no longer ‘significantly’ differentially expressed, there were still large numbers of small-magnitude changes that had not fully recovered. Notably, the heme metabolism gene set was the most significantly upregulated gene set in both genotypes on day 4, and in the case of WT cells, its enrichment grew more significant by day 12. We observed that 5-Aza treated TETKO cells showed greater fold induction of heme metabolism genes on day 4, however this induction was temporary and rapidly approached pre-treatment levels by day 12 in TET2KO cells, whereas in WT the fold change increased further from day 4 to day 12 (Figure 4G–H, Supplementary Figure S4). Notably, expression of heme metabolism genes was greater in TET2WT than KO at all time points, although the difference was smallest on day 4 (Figure 4I).
Figure 4. Gene expression changes after 5-Aza treatment reveal differences in erythroid gene signatures.

A) Volcano plot of differentially expressed genes between TET2KO and WT at baseline. B) Genes with significant differential expression after 5-Aza exposure in TET2KO and WT. C) Gene Set Enrichment Analysis (GSEA) for baseline gene expression differences (KO vs. WT). D) GSEA plot for the most significantly enriched gene set in (C): Heme metabolism genes. E-F) GSEA results for comparisons of mock vs. 5-Aza treated TF1 on Day 4 and Day 12 after treatment. Only gene sets with q-value < 0.05 shown. G) Heatmap displaying genes in the Hallmark Heme Metabolism gene set which were differentially expressed between TET2 KO and WT TF1 at any time point (top) and genes associated with terminal erythroid differentiation. H) Expression log2(fold-change) (5-Aza vs. Veh. treated) of genes in the ‘Hallmark Heme Metabolism’ gene set by genotype I) Expression log2(fold-change) (TET2KO vs. TET2WT) of genes in the ‘Hallmark Heme Metabolism’ gene set by treatment across all time points. Note: data plotted are from all genes within the gene set, not only the significantly different genes. While the log2(fold-change) (5-Aza v Veh.) is greater on day 4 for TET2KO, the raw expression is greater in TET2WT at all time points and in both treatments.
Gene expression differences in TET2KO partially explained by remethylation kinetics at 5hmC DHMRs.
After observing significant changes in erythroid differentiation-associated gene expression (heme metabolism genes), we next examined our integrated genomic datasets at the key erythroid differentiation marker gene, erythrocyte anion exchanger, band 3 (SLC4A1), which was sufficiently covered in all of our datasets. In a genome browser track of the gene (Figure 5A) we observed a DHMR within the enhancer/promoter region of the gene. Notably, there were a number of dynamic CpGs directly overlapping this enhancer region, and these CpGs had opposing patterns of remethylation kinetics in TET2KO and WT, where WT lost methylation and did not recover, while the KO rapidly recovered its original methylation (Figure 5B). When we correlated the methylation of this locus with expression patterns it was clear that 5-Aza treatment was able to induce expression of this gene in WT but to a much lesser extent in KO (Figure 5C), highlighting the importance of enhancer 5mC kinetics in controlling erythroid gene signatures. To determine if the differences in remethylation kinetics observed in the SLC4A1 enhancer may be a global phenomenon, we identified CpGs overlapping DHMR peaks on a global level (Figure 5D) and observed marked hypermethylation and more rapid remethylation kinetics in TET2KO cells compared with WT.
Figure 5. TET2 regulated enhancers are hypermethylated, more rapidly remethylated, and impact erythroid gene expression.
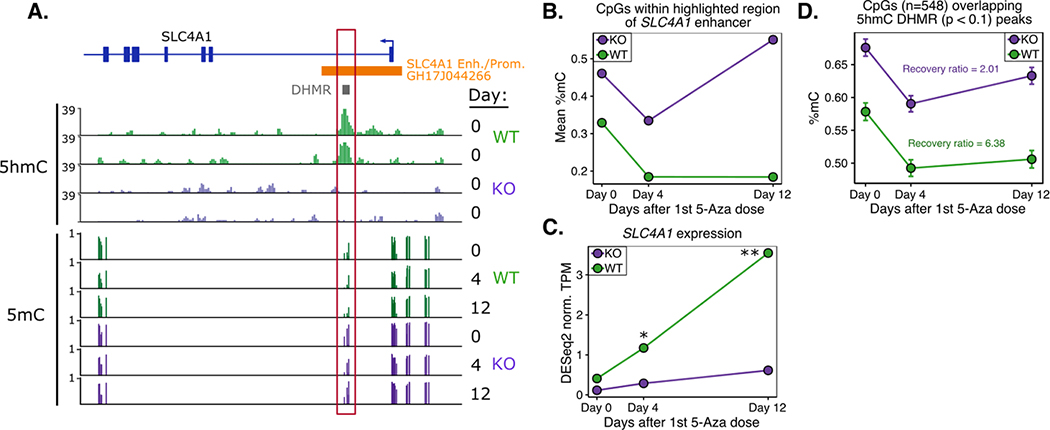
A) Genome browser track highlighting 5mC and 5hmC differences centered in the SLC4A1 enhancer/promoter (Enh./Prom.) region. Enhancer annotation was derived from GeneHancer Regulatory Interactions (double elite) table on UCSC genome browser. DHMR = Differentially Hydroxymethylated Region (as determined by differential peak enrichment analysis of HMCP-Seq data). B) Mean % methylation during 5-Aza treatment for CpGs within highlighted box in (A). C) Normalized expression of SLC4A1 during 5-Aza treatment; “*” = FDR < 0.1; “**” = FDR < 0.01. D) As shown for SLC4A1, global methylation kinetics for CpGs located within DHMR peaks show more rapid remethylation kinetics after 5-Aza in TET2KO cells.
TET2KO cells are capable of EPO-induced differentiation and 5-Aza enhances differentiation capacity.
Next we measured Glycophorin-A (GYPA) cell surface expression by flow-cytometry as a surrogate for differentiation along the erythroid lineage during 5-Aza exposure and after erythropoietin (EPO) induced erythroid differentiation (Figure 6). TET2WT cells had higher GYPA expression at baseline, and 5-Aza exposure substantially increased expression in both genotypes (Figure 6A). We also observed a trend for increased GYPA expression over time in the WT vehicle treated condition, potentially due to spontaneous differentiation or due to vehicle exposure, however TET2KO cells did not display any significant increases in GYPA expression under the same conditions. In agreement with our previous RNAseq results, the KO cells displayed peak GYPA expression on day 4, while the TET2WT cells steadily increased over time, even through day 20. When we calculated the fold-change in GYPA expression for 5-Aza vs. vehicle treated conditions, TET2KO had significantly greater relative GYPA induction compared with WT (Figure 6B). Finally, we took advantage of the ability of TF-1 cells to differentiate along the erythroid lineage when exposed to erythropoietin to assess how TET2 loss and 5-Aza exposure would impact differentiation (Figure 6C). There were wide variations in the relative amounts of differentiation among clones from both genotypes, but in general, all clones retained erythroid differentiation capacity. Notably, even after an 8-day recovery period (days 4–12), 5-Aza treated cells retained an increased capacity for differentiation compared to vehicle treated cells as can be seen through relative increases in GYPA expression and reddening of the cell pellets which is indicative of higher hemoglobin expression (Figure 6C–D).
Figure 6. Expression of the erythroid cell surface protein Glycophorin-A (GYPA) is differentially induced in KO v. WT after 5-Aza but not during terminal erythroid differentiation.

A) Percentage of GYPA-positive cells over time after 5-Aza or vehicle exposure as measured by flow cytometry. Drug/vehicle was added on Days 1–3 as in previous experiments. B) Fold-change in cell-surface GYPA expression over time after 5-Aza vs. vehicle exposure. A) and B): Each point represents the mean of 2 biological replicates and 3 independent experiments, error bars represent standard error of the mean. C) Erythroid differentiation induction by recombinant erythropoietin (EPO) starting 8 days after 5-Aza or vehicle removal and ending 8 days later, measured by GYPA flow cytometry. D) Red pigmentation of cell pellets on Day 20 (Day 8 after EPO addition) is indicative of the amount of hemoglobin expression induced.
Discussion
DNA methyltransferase inhibitors such as azacitidine (5-Aza) are increasingly used in treatments for a variety of cancers, yet the mechanisms underpinning therapeutic response to these agents remain elusive. Previous work has shown that somatic TET2 mutations present in MDS patient cells are associated with response to DNMTIs. Here we examine mechanisms underlying this association using high-throughput sequencing approaches in genetically engineered TF-1 erythroleukemia cell lines. We characterize the dynamics of 5mC, 5hmC, and gene expression changes after 5-Aza treatment in TET2 knockout and control TF-1 cells. We observed key differences in pharmacological responses to 5-Aza, as well as molecular responses in the form of differential effects on 5mC and 5hmC dynamics in genes involved in erythroid differentiation. Our results highlight the impact of TET2 loss on the 5mC kinetics of erythroid differentiation-associated genes and their enhancers, and how 5-Aza can target these regions to normalize gene expression levels and boost differentiation capacity.
We observed key differences in the pharmacological and epigenetic responses to 5-Aza between TET2KO vs WT TF-1. Notably, both genotypes had similar dose-response curves for viability, but KO cells had significantly lengthened PDT after 5-Aza. We also observed that TET2KO cells had globally reduced demethylation and degree of recovery. The reduced extent of demethylation in KO is likely related to the lengthened PDT, as passive demethylation by 5-Aza is dependent on DNA replication and cell division. Because there were likely fewer cell divisions in TET2KO after 5-Aza, we expect reduced demethylation extent. Interestingly we did observe upregulation of gene sets involved in DNA damage response (hallmark p53 pathway, hallmark UV response down) in TET2KO at baseline. Several recent studies have shown that TET2 and 5hmC play essential roles in genome stability, and deficiencies of TET enzymes and 5hmC lead to defects in chromosome segregation and DNA damage responses (55–57). Together, these data are consistent with TET2KO cells having impaired DNA damage responses which make them less resilient to the added DNA damage induced by 5-Aza exposure, thus more profound stalling of cell cycle and less passive DNA demethylation occurs. This mechanism may also help explain why 5-Aza treatment significantly decreased peripheral blood chimerism after competitive bone marrow transplantation in Tet2-null but not WT cells in mouse studies (10).
A notable finding of our study was the characterization of dynamic changes in 5mC and gene expression after Aza exposure. While we observed relatively rapid recovery of transcriptional changes induced by 5-Aza (less than 1% of day 4 DEGs present on day 12), many of the DNA methylation changes observed on day 4 were not fully recovered by day 12. This finding reinforces the role of 5-Aza as a differentiating agent as recent work has shown that 5mC alterations, especially at bivalent promoters, may impact the later differentiation ability of stem cells even in the absence of gene expression changes (58). We determined there were four major patterns of remethylation and observed differences in the proportion of loci falling in each pattern, where TET2KO exhibited a significantly higher proportion of slow-remethylating and significantly lower proportion of rapid-remethylating CpGs. We observed enrichment for regions of active gene-transcription and active enhancers when calculating the overlap of CpGs from each dynamic cluster in relation to reference HSC chromatin states. Actively transcribed regions and gene-body enhancers were highly enriched for rapid-recovery CpGs with less enrichment for slow-recovery CpGs, while active enhancers located outside of gene bodies were highly enriched for intermediate and slow-recovery remethylation CpGs with less enrichment for rapid-recovery CpGs. This finding suggests that the long-term 5mC changes induced by 5-Aza occur somewhat selectively at non-genic enhancers while minimally impacting other, more rapidly recovered regulatory loci.
A second key finding of our study was the influence of TET2 loss on erythroid differentiation-associated gene expression programs, and the ability of 5-Aza exposure to transiently normalize expression in KO cells. The most differentially enriched gene expression signatures between TET2KO and WT TF-1 at baseline were those involved in erythroblast differentiation(54,59) (downregulated in KO), and epithelial to mesenchymal transition(59) (upregulated in KO). The combination and direction of these two gene sets in particular are interesting, as both are involved in opposing directions of differentiation, and in both cases KO cells are enriched for the undifferentiated state. At least some of the discrepancy in erythroid gene expression between genotypes is likely due to loss of 5hmC at differentiation-associated enhancers in KO condition which is also associated with differences in remethylation kinetics as we showed for the erythrocyte anion exchanger, Band 3 (SLC4A1). This result is in agreement with several studies demonstrating the importance of TET2 and 5hmC in erythroid differentiation (60–62) and further highlights the importance of 5mC dynamics in this process. Additionally, while we expected some degree of spontaneous erythroid differentiation to occur during the culture of TF-1 cells based on previous work (41,63,64), we noted that this only occurred for an appreciable fraction of WT vehicle-treated cells while virtually none occurred in vehicle-treated KO cells, highlighting the relative resistance of KO cells towards differentiation. However, both KO and WT cells retained erythroid differentiation capacity when exposed to the strong differentiation stimulus of erythropoietin, indicating that KO cells do not exhibit a complete differentiation block. All of these results suggest that 5mC and 5hmC changes induced by TET2 loss shift TF-1 cells towards a more undifferentiated state and make them relatively resistant towards differentiation when compared to wild-type. The 5mC and 5hmC changes in KO appear to convey greater potential for influence by 5-Aza as shown by the greater fold induction of erythroid differentiation genes in KO vs WT, however these changes are transient. Our results are in accord with studies demonstrating that TET2 loss can impair differentiation of stem cells (22,58,65) as well as studies showing that reduction of TET2-inhibiting 2-hydroxyglutarate can improve erythroid differentiation (41,64).
Our findings also have potential clinical implications. We observed more rapid rebound methylation for TET2-regulated CpG loci following cessation of 5-Aza in KO compared with WT cells (Figure 5D). This could imply that treatment regimens with more sustained dosing schedules may more effectively suppress aberrant rebound methylation in TET2 mutated patients. In support of this concept, a recent clinical trial evaluating the efficacy of oral azacitidine with 21-day extended dosing intervals reported just one patient who achieved a complete response with extended dosing after a previous treatment failure with traditional 7-day injectable regimen, and this patient had TET2 mutated disease (66). Clearly this relationship will require further study.
A limitation of this study is that it included only one type of leukemia cell line. We chose TF-1 cells as our model for several reasons. TF-1 cells lack most MDS-associated genetic mutations and display important characteristics of a MDS-like stem cell including the capacity for differentiation which is a useful measure of 5-Aza efficacy. Future projects will explore the generalizability of our findings in other cell lines and in vivo systems.
In conclusion, we demonstrate that TET2 inactivation impairs erythroid gene expression and spontaneous differentiation of erythroleukemia cells, and that this phenotype is associated with 5hmC and 5mC dynamics at erythroid gene enhancers. We show that this TET2KO phenotype can be partially and transiently corrected by 5-azacytidine exposure. Overall our study provides insights into the mechanisms underlying the relationship between TET2 mutation and response to 5-Aza. How this occurs in MDS patients achieving a clinical response to 5-Aza merits further study.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications.
TET2 loss in erythroleukemia cells induces hypermethylation and impaired expression of erythroid differentiation genes which can be specifically counteracted by 5-Azacytidine, providing a potential mechanism for the increased efficacy of 5-Aza in TET2-mutant MDS patients.
Acknowledgements
We would like to thank Michael Love and Pablo Tamayo for their advice on pathway enrichment analysis. We would also like to thank Frank Furnari and Renate Pilz for the fruitful discussions regarding the conception and design of several experiments in this study. We kindly thank Peter Weigele (NEB) for providing the original phage T4gt which was used in HMCP experiments. This work was supported by the Evans Foundation (B. Reilly, R. Bejar) and a pre-doctoral fellowship from the American Foundation for Pharmaceutical Education (B. Reilly).
Footnotes
Competing Interests:
RB declares employment by Aptose Biosciences Inc., as well as consultancy for AbbVie Inc., Astex Pharmaceuticals Inc., Forty Seven Inc. (Gilead Sciences Inc.), Keros Therapeutics Inc., Takeda Pharmaceutical Company Ltd., and Celgene Corporation Inc. (Bristol-Meyers Squibb). RB receives research funding from Takeda Pharmaceutical Company Ltd. and Celgene Corporation Inc.
Materials and Correspondence:
All requests and correspondence should be made to Rafael Bejar.
References
- 1.Issa J-PJ, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II Study of Low-Dose Decitabine in Patients With Chronic Myelogenous Leukemia Resistant to Imatinib Mesylate. J Clin Oncol. 2005;23:3948–56. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, With Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients With Newly Diagnosed Acute Myeloid Leukemia. J Clin Oncol. 2012;30:2670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stahl M, DeVeaux M, Montesinos P, Itzykson R, Ritchie EK, Sekeres MA, et al. Hypomethylating agents in relapsed and refractory AML: outcomes and their predictors in a large international patient cohort. Blood Adv. American Society of Hematology; 2018;2:923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azad NS, el-Khoueiry A, Yin J, Oberg AL, Flynn P, Adkins D, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat: a phase 2 consortium/stand Up 2 cancer study. Oncotarget. 2017;8:35326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connolly RM, Li H, Jankowitz RC, Zhang Z, Rudek MA, Jeter SC, et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin Cancer Res. 2017;23:2691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non-Small Cell Lung Cancer. Cancer Discov. 2011;1:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Issa JPJ, Kantarjian HM. Targeting DNA methylation. Clin. Cancer Res. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Manero G Demethylating agents in myeloid malignancies. Curr. Opin. Oncol. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenaux P, Ades L. Review of azacitidine trials in Intermediate-2-and High-risk myelodysplastic syndromes. Leuk Res. 2009; [DOI] [PubMed] [Google Scholar]
- 10.Bejar R, Lord A, Stevenson K, Bar-Natan M, Pérez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. American Society of Hematology; 2014;124:2705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. Macmillan Publishers Limited; 2011;25:1147–52. [DOI] [PubMed] [Google Scholar]
- 12.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. American Association for the Advancement of Science; 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. Macmillan Publishers Limited; 2014;28:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. American Association for the Advancement of Science; 2011;333:1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011;333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lio C-WJ, Yuita H, Rao A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood. American Society of Hematology; 2019;134:1487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011;333:1303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiti A, Drohat AC. Thymine DNA Glycosylase Can Rapidly Excise 5-Formylcytosine and 5-Carboxylcytosine. J Biol Chem. 2011;286:35334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.López-Moyado IF, Tsagaratou A, Yuita H, Seo H, Delatte B, Heinz S, et al. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci. National Academy of Sciences; 2019;116:16933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasmussen KD, Jia G, Johansen J V, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29:910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell. 2014;56:286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamazaki J, Jelinek J, Lu Y, Cesaroni M, Madzo J, Neumann F, et al. TET2 Mutations Affect Non-CpG Island DNA Methylation at Enhancers and Transcription Factor-Binding Sites in Chronic Myelomonocytic Leukemia. Cancer Res. 2015;75:2833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reilly B, Tanaka TN, Diep D, Yeerna H, Tamayo P, Zhang K, et al. DNA methylation identifies genetically and prognostically distinct subtypes of myelodysplastic syndromes. Blood Adv. American Society of Hematology; 2019;3:2845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rönnerblad M, Andersson R, Olofsson T, Douagi I, Karimi M, Lehmann S, et al. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood. American Society of Hematology; 2014;123:79–89. [DOI] [PubMed] [Google Scholar]
- 27.Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L, et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev. Cold Spring Harbor Laboratory Press; 2014;28:652–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lio C-WJ, Shukla V, Samaniego-Castruita D, González-Avalos E, Chakraborty A, Yue X, et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol [Internet]. Science Immunology; 2019. [cited 2020 Apr 16];4. Available from: https://immunology.sciencemag.org/content/4/34/eaau7523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M, et al. Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat Commun. Nature Research; 2016;7:10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unnikrishnan A, Papaemmanuil E, Beck D, Wong JWH, Campbell PJ, Pimanda Correspondence JE. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017;20:572–85. [DOI] [PubMed] [Google Scholar]
- 31.Yan X, Ehnert S, Culmes M, Bachmann A, Seeliger C, Schyschka L, et al. 5-azacytidine improves the osteogenic differentiation potential of aged human adipose-derived mesenchymal stem cells by DNA demethylation. PloS One. 2014;9:e90846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manzoni EFM, Pennarossa G, deEguileor M, Tettamanti G, Gandolfi F, Brevini TAL. 5-azacytidine affects TET2 and histone transcription and reshapes morphology of human skin fibroblasts. Sci Rep. 2016;6:37017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sajadian SO, Ehnert S, Vakilian H, Koutsouraki E, Damm G, Seehofer D, et al. Induction of active demethylation and 5hmC formation by 5-azacytidine is TET2 dependent and suggests new treatment strategies against hepatocellular carcinoma. Clin Epigenetics. 2015;7:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch JS, Petti AA, Miller CA, Fronick CC, O’Laughlin M, Fulton RS, et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N Engl J Med. Massachusetts Medical Society; 2016;375:2023–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28:605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. Nature Publishing Group; 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. Oxford University Press; 2016;44:W272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bauer DE, Canver MC, Orkin SH. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. J Vis Exp JoVE. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee M, Li J, Liang Y, Ma G, Zhang J, He L, et al. Engineered Split-TET2 Enzyme for Inducible Epigenetic Remodeling. J Am Chem Soc. American Chemical Society; 2017;139:4659–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Losman J-A, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. NIH Public Access; 2013;339:1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diep D, Plongthongkum N, Gore A, Fung H-L, Shoemaker R, Zhang K. Library-free methylation sequencing with bisulfite padlock probes. Nat Methods. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2012;9:270–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo S, Diep D, Plongthongkum N, Fung H-L, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet. Nature Research; 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics J Integr Biol. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, et al. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014; [DOI] [PubMed] [Google Scholar]
- 46.Weigele P, Raleigh EA. Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses . Chem Rev. American Chemical Society; 2016;116:12655–87. [DOI] [PubMed] [Google Scholar]
- 47.Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 2014;289:21312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. Nature Publishing Group; 2017;549:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. Cold Spring Harbor Laboratory Press; 2016;30:733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell. 2014;26:577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rousseeuw PJ. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math. 1987; [Google Scholar]
- 53.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang YW, Wang Z, Xie W, Cai Y, Xia L, Easwaran H, et al. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Mol Cell. 2017;65:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L-L, Lin H-P, Zhou W-J, He C-X, Zhang Z-Y, Cheng Z-L, et al. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep. 2018;25:1485–1500.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kafer GR, Li X, Horii T, Suetake I, Tajima S, Hatada I, et al. 5-Hydroxymethylcytosine Marks Sites of DNA Damage and Promotes Genome Stability. Cell Rep. 2016;14:1283–92. [DOI] [PubMed] [Google Scholar]
- 58.Verma N, Pan H, Doré LC, Shukla A, Li QV., Pelham-Webb B, et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat Genet. Nature Publishing Group; 2018;50:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H, Wang Y, Qu X, Li J, Hale J, Huang Y, et al. Distinct roles for TET family proteins in regulating human erythropoiesis. Blood. 2017;129:2002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qu X, Zhang S, Wang S, Wang Y, Li W, Huang Y, et al. TET2 deficiency leads to stem cell factor-dependent clonal expansion of dysfunctional erythroid progenitors. Blood. 2018;132:2406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, et al. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol. 1989;140:323–34. [DOI] [PubMed] [Google Scholar]
- 64.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. American Association for the Advancement of Science; 2013;340:622–6. [DOI] [PubMed] [Google Scholar]
- 65.Madzo J, Liu H, Rodriguez A, Vasanthakumar A, Sundaravel S, Caces DBD, et al. Hydroxymethylation at Gene Regulatory Regions Directs Stem/Early Progenitor Cell Commitment during Erythropoiesis. Cell Rep. 2014;6:231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Savona MR, Kolibaba K, Conkling P, Kingsley EC, Becerra C, Morris JC, et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am J Hematol. 2018;93:1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.