

Abstract
Background:
Vitamin K antagonists (VKAs), such as warfarin, have remained the cornerstone of oral anticoagulation therapy in the prevention and treatment of thromboembolism for over half a century. They function by impairing the biosynthesis of vitamin K-dependent (VKD) clotting factors through the inhibition of vitamin K epoxide reductase (VKOR). The challenge of VKAs therapy is their narrow therapeutic index and highly variable dosing requirements, which are partially due to the genetic variations of VKOR.
Objectives:
The goal of this study was to search for an improved VKA that is tolerant to the genetic variations of its target enzyme.
Methods:
A series of vitamin K derivatives with benzyl and related side-chain substitutions at the 3-position of 1,4-naphthoquinone were synthesized. The role of these compounds in VKD carboxylation was evaluated by mammalian cell-based assays and conventional in vitro activity assays.
Results:
Our results showed that replacing the phytyl side-chain with a methylene cyclooctatetraene (COT) moiety at the 3-position of vitamin K1 converted it from a substrate to an inhibitor for VKD carboxylation. Strikingly, this COT-vitamin K derivative displayed a similar inhibition potency in warfarin-resistant VKOR mutations whose warfarin resistance varied over 400-fold. Further characterization of COT-vitamin K for the inhibition of VKD carboxylation suggested that this compound targets multiple enzymes in the vitamin K redox cycle. Importantly, the anticoagulation effect of COT-vitamin K can be rescued with high doses of vitamin K1.
Conclusion:
We discovered a vitamin K analogue that functions as a VKA, and is tolerant to genetic variations in the target enzyme.
Keywords: Coagulation factors carboxylation, Cyclooctatetraene, Oral anticoagulant, Vitamin K antagonist, Vitamin K epoxide reductase
Introduction
Thromboembolic events, resulting from atrial fibrillation (AF) and venous thromboembolism (VTE), are common complications that have high mortality and morbidity[1]. It is estimated that AF currently affects over 33 million individuals worldwide[2]. According to the America Heart Association, VTE (including deep vein thrombosis and pulmonary embolism) is the third leading vascular diagnosis after heart attacks and strokes, affecting between 300,000 to 600,000 Americans each year. Additionally, patients with active cancer are at an increased risk of arterial and venous thromboembolism[3]. Anticoagulant therapy has proven to be an effective strategy for the prevention and treatment of thromboembolism[4]. For over half a century, vitamin K antagonists (VKAs), such as warfarin, have remained the cornerstone of oral anticoagulation therapy[5].
Warfarin, approved by the US Food and Drug Administration (FDA) in 1954, is the most commonly prescribed oral anticoagulant worldwide[6, 7]. In 2017, more than 15 million prescriptions for warfarin were written in the United States alone[8]. Warfarin functions by impairing the biosynthesis of active vitamin K-dependent (VKD) clotting factors through the inhibition of vitamin K epoxide reductase (VKOR), a key enzyme for the redox cycling of vitamin K to support posttranslational carboxylation of clotting factors. Since the discovery of the gene encoding VKOR[9, 10], the mechanism of action and pharmacogenomics of warfarin have been extensively studied. Single-nucleotide polymorphisms in the VKOR gene have been identified as one of the key factors that are strongly associated with large warfarin dose variation phenotypes in humans[11, 12]. A cell-based functional study suggested that warfarin resistance among naturally occurring VKOR mutations varied over 100-fold[13]. Clinical observations indicate, that to achieve the desired anticoagulation efficacy, the interpatient variability of warfarin therapeutic dosages can have a 10- to 20-fold difference[14]. To assist physicians in estimating the appropriate warfarin dosage in anticoagulation therapy, several pharmacogenetic-based dosing algorithms using VKOR genotypes have been proposed[11, 15, 16]. It has been shown that approximately 30% of patients receiving warfarin would benefit from VKOR pharmacogenetics at the beginning of their warfarin therapy[17]. VKOR pharmacogenetics is thought to be so clinically useful that the US FDA revised its warfarin product labeling to reflect the contribution of VKOR genotypes in warfarin dose selection[18].
Despite the effectiveness, widespread use, and genotype-guided dosing, warfarin therapy requires routine monitoring of the International Normalized Ratio (INR) to ensure an optimal warfarin efficacy while limiting the risk of bleeding[6, 19]. This has been the main cost and obstacle in warfarin therapy. Over the past decade, several novel (or non-VKA) oral anticoagulants (NOACs) that directly targeting thrombin (dabigatran) or factor Xa (apixaban, edoxaban, and rivaroxaban) have been advocated as warfarin replacements. These NOACs do not require laboratory monitoring and offer a number of additional attractive advantages over warfarin, including: fixed dosing, predictable pharmacokinetics, and fewer food–drug interactions[20, 21]. Nevertheless, compared to warfarin, these NOACs are expensive, have limited antidotes available[22], have higher medication risks[23, 24], and have poorer patient adherence due to lack of monitoring[25]. Additionally, the NOACs often require an assessment of kidney functions and their efficacies have not been extensively tested in patients with severe renal dysfunction[23, 26]. These situations lead physicians to choose VKAs for anticoagulation control[27].
In addition to warfarin, acenocoumarol and phenprocoumon (4-hydroxycoumarin derivatives like warfarin) and fluindione (a derivative of 1,3-indandione, Figure 1A) are also widely prescribed VKAs for anticoagulation therapy[27, 28]. Warfarin is the most commonly prescribed anticoagulant worldwide, whereas other VKAs are commonly used in many European countries[29]. In general, the use of these VKAs is challenging because of their narrow therapeutic index and high variability in dosage requirements that are effected by many factors, including genetic variations of VKOR. In this study, we explored the possibility of converting vitamin K from a substrate to an inhibitor for VKD carboxylation. Vitamin K is a family of 2-methyl-1,4-naphthoquinone derivatives, which include the naturally occurring phylloquinone (vitamin K1) and menaquinones (vitamin K2), and the synthetic menadione (vitamin K3)[30]. Menaquinones differ from phylloquinone in that the side chain at the 3-position comprises a number of repeating prenyl units rather than the semi-saturated phytyl chain. The menaquinones are named according to the number of prenyl units contained within the side chain (i.e. MK-n). Since the core structure of vitamin K is distinct from that of the current clinically used VKAs, we reasoned that, as an inhibitor, vitamin K analogues should have different inhibition and binding characteristics to the target enzyme. Our results show that substituting the phytyl side-chain of vitamin K1 with a methylene cyclooctatetraene (COT) moiety resulted in a vitamin K analogue that functions as an inhibitor for VKD carboxylation. Importantly, the inhibition potency of the COT-vitamin K derivative was tolerant to genetic variations of VKOR.
Figure 1. Evaluation of vitamin K derivatives for supporting VKD carboxylation in HEK293 cells.
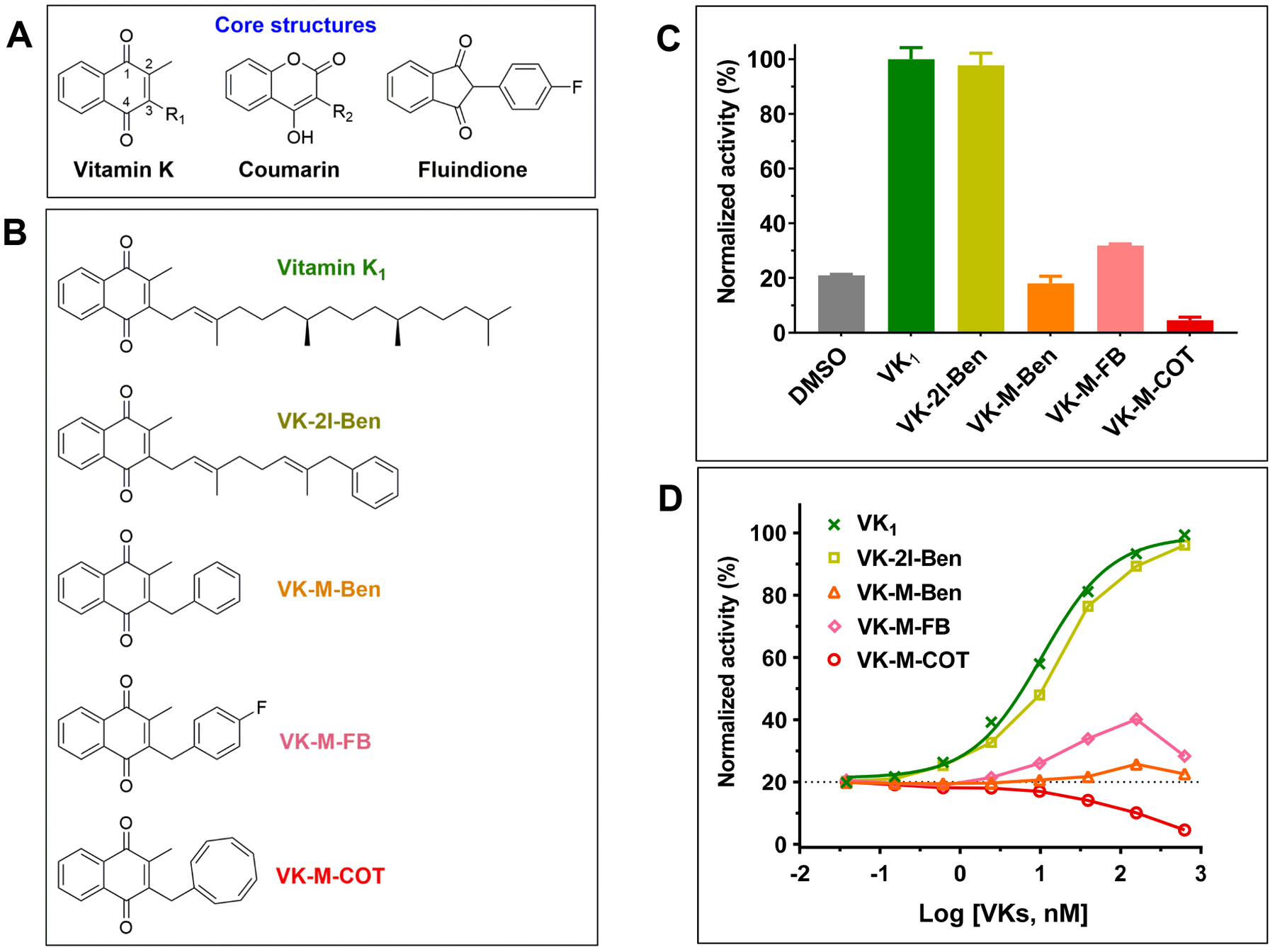
(A) Core structures of vitamin K and clinically used vitamin K antagonists. (B) Chemical structures of vitamin K derivatives used in this study. (C) Cell-based VKD carboxylation activity of different vitamin K derivatives in FIXgla-PC/HEK293 reporter cells. The reporter cells were cultured in cell culture medium containing 1 μM vitamin K derivative as the substrate for 24 hours. Carboxylation activity was evaluated by determining the carboxylation efficiency of the reporter-protein FIXgla-PC using ELISA. The carboxylation activity of vitamin K1 (VK1) as the substrate was normalized to 100%. (D) Concentration titration of vitamin K derivatives in FIXgla-PC/HEK293 reporter cells. The reporter cells were cultured with increasing concentrations of vitamin K derivatives for 24 hours. Reporter-protein FIXgla-PC carboxylation was determined and normalized as described in the legend of Figure 1C.
Materials and Methods
Reagents and cell lines –
Warfarin, vitamin K1, vitamin K1 epoxide (KO), menadione, menaquinone-4 (MK-4), and MK-4 epoxide were obtained from Sigma-Aldrich (St. Louis, MO). Vitamin K derivatives (novel and known) were synthesized based on previously described methods[31–36]. Synthetic procedures, full characterization and copies of 1H and 13C NMR spectra can be found in Supporting Information. Xfect transfection reagent was from Clontech Laboratories, Inc. (Mountain View, CA). Mouse anti-carboxylated factor IX gla domain (FIXgla) monoclonal antibody was from Green Mountain Antibodies (Burlington, VT). Horseradish peroxidase conjugated sheep anti-human protein C (PC) was from Affinity Biologicals, Inc. (Ancaster, ON, Canada). Cell counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. (Rockville, MD). ABTS (2,2’-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) peroxidase substrate kit for enzyme-linked immunosorbent assay (ELISA) was from KPL Inc. (Gaithersburg, MD).
Human embryonic kidney 293 (HEK293) cells were from ATCC (Manassas, VA). HEK293 cells stably expressing the reporter-protein FIXgla-PC (protein C with its Gla domain exchanged with that of factor IX) (FIXgla-PC/HEK293) were obtained, as previously described[37]. FIXgla-PC/HEK293 cells with their endogenous VKOR and VKOR-like enzyme (VKORL) knocked out (double gene knockout, DGKO) were obtained by TALENs-mediated genome editing[38]. FIXgla-PC/HEK293 cells with their endogenous gamma-glutamyl carboxylase (GGCX) or UbiA prenyltransferase domain-containing protein 1 (UBIAD1) gene knockout were obtained by CRISPR-Cas9-mediated genome editing[39].
DNA Manipulations and plasmids construction –
The cDNA encoding Metridia luciferase (used as an internal control for transfection efficiency) was cloned into one of the multi-cloning sites of the mammalian dual expression vector pBudCE4.1 (Life Technologies Corp., San Diego, CA). The resulting vector pBudCE4.1-Met.Luc was used as the cloning and expression vector for expressing VKOR and its variants. Naturally occurring VKOR mutations were created by QuickChange site-directed mutagenesis. The nucleotide sequences of all the constructs were verified by DNA sequencing at Eton Bioscience Inc. (Research Triangle Park, NC).
Evaluation of vitamin K derivatives for VKD carboxylation in HEK293 cells –
A cell-based activity assay was used to evaluate the vitamin K derivatives as a substrate or inhibitor for VKD carboxylation in FIXgla-PC/HEK293 cells, as previously described[37]. To assess the ability of the vitamin K derivatives as a substrate for VKD carboxylation, FIXgla-PC/HEK293 cells were cultured with a fixed concentration or with increasing concentrations of the vitamin K derivative in cell culture medium. To evaluate the efficiency of vitamin K derivatives as an inhibitor for VKD carboxylation, we cultured FIXgla-PC/HEK293 cells with increasing concentrations of the vitamin K derivative or warfarin (positive control) in cell culture medium containing 5 μM KO. After 24 hours incubation, the carboxylation efficiency of the reporter-protein FIXgla-PC was determined directly from the cell culture medium by ELISA[37].
Vitamin K reductase or KO reductase activity assay –
The effect of vitamin K derivatives as inhibitors of vitamin K reductase or KO reductase activity was evaluated in FIXgla-PC/HEK293 reporter cells with either the endogenous VKOR/VKORL (DGKO) or GGCX knocked out, as previously described[40]. Enzymatic activity was evaluated using ELISA-based reporter-protein carboxylation and conventional HPLC-based vitamin K conversion assays. For the ELISA-based KO reductase activity assay, FIXgla-PC/HEK293 cells were cultured with a fixed concentration or with increasing concentrations of the vitamin K derivative in cell culture medium containing 5 μM KO. After 24 hours incubation, the efficiency of reporter-protein carboxylation was determined in the cell culture medium by ELISA, as previously described[37]. ELISA-based vitamin K reductase activity was performed using DGKO reporter cells with vitamin K1 as the substrate. For the conventional HPLC-based KO reductase activity assay, GGCX-knockout FIXgla-PC/HEK293 cells were cultured with a fixed concentration or with increasing concentrations of the vitamin K derivative in cell culture medium containing 5 μM KO. K vitamins were extracted from the cells and the conversion of KO to vitamin K1 was determined by reverse-phase HPLC[40]. Vitamin K reductase activity was evaluated using DGKO reporter cells with vitamin K1 as the substrate. Since reduced vitamin K1 (KH2), an intermediate product in VKD carboxylation, is unstable and difficult to accurately quantify from cells directly[41], we coupled the vitamin K reduction and epoxidation reactions to quantitate the final stable product, KO. Details of differentially determining the vitamin K cycle enzyme activity were summarized in Supplemental Table 1.
Vitamin K epoxidation activity assay –
The in vitro vitamin K epoxidation activity of GGCX was determined by the quantitation of KO formed during the carboxylation of FLEEL in the presence of FIX’s propeptide, as previously described[42]. GGCX enriched microsomes from insect cells (Sf9) were used in this assay. K vitamins were extracted from the reaction mixture and quantitated by an HPLC assay.
Effect of VKOR genetic variations on the inhibition potency of VKAs –
The effect of VKOR mutations on the inhibition potency of VKAs were performed in DGKO cells, as previously described[38]. Briefly, the plasmid DNA of pBudCE4.1-Met.Luc containing the cDNA of wild-type or mutant VKOR was transiently expressed in the DGKO reporter cells. Transfected cells were cultured with complete medium containing 5 μM KO and increasing concentrations of VKAs. Reporter-protein carboxylation was determined by ELISA after 24 hours incubation. The VKAs inhibition potency was evaluated by determining the half-maximal inhibition concentration (IC50) of the VKAs using GraphPad Prism 8 software.
UBIAD1-dependent carboxylation activity of vitamin K derivatives –
To determine the UBIAD1-dependent carboxylation activity of the vitamin K derivatives, UBIAD1-knockout FIXgla-PC/HEK293 cells were cultured with cell culture medium containing increasing concentrations of vitamin K derivatives for 24 hours. The efficiency of reporter-protein carboxylation was determined by ELISA. To confirm that the vitamin K derivatives unable to support VKD carboxylation in the UBIAD1-knockout reporter-cells was due to the absence of the UBIAD1 gene, the UBIAD1 cDNA was re-introduced back into these reporter-cells by transient expression and the reporter-protein carboxylation in the transfected cells was determined. To confirm that the vitamin K analogue supported VKD carboxylation as a result of the conversion of the vitamin K derivatives to MK-4 by UBIAD1, HEK293 or UBIAD1-knockout, HEK293 reporter cells were cultured with the vitamin K derivatives and the production of MK-4 within these cells were determined using an HPLC assay[40].
Results
The 3-position side-chain of vitamin K plays an essential role in VKD carboxylation
Current clinically used VKAs are derivatives of 4-hydroxycoumarin and 1,3-indandione[43] (Figure 1A). The inhibition potency of these drugs to their target enzyme VKOR varies 10- to 200-fold depending on the genetic variations of VKOR[13, 44, 45]. To search for improved VKAs with less inhibition variabilities, we synthesized a series of vitamin K analogues (2-methyl-1,4-naphthoquinone derivatives) with different side-chains at the 3-position (Figure 1B). Modification of the 3-position side-chain has been shown to significantly affect vitamin K activity in supporting VKD carboxylation[46], and has successfully converted vitamin K to drugs with varying pharmacological functions[32, 47, 48]. In this study, we explored the possibility of using these types of vitamin K analogues as inhibitors for VKD carboxylation. Since the newly designed vitamin K analogues were different from the 4-hydroxycoumarin and 1,3-indandione in terms of containing a different core structure (Figure 1A), we expected that, as an inhibitor, the vitamin K analogues might also have a different mechanism of action from the current clinically used VKAs.
First, we determined the capability of these compounds as a substrate to support VKD carboxylation in HEK293 cells using our cell-based assay. Results in Figure 1C showed that side-chain substitutions at the 3-position of vitamin K differentially affected VKD carboxylation activity. For example, introduction of a phenyl group to the ω-position of the 3-position side-chain (VK-2I-Ben) only had a minor effect on carboxylation activity. However, removal of the isoprenyl unit from VK-2I-Ben (VK-M-Ben) abolished carboxylation activity. On the other hand, introducing a fluorine atom at the 4-position of the phenyl group (VK-M-FB) recovered ~20% carboxylation activity. Strikingly, the methylene COT derived vitamin K (VK-M-COT) derivative displayed activity lower than the background activity (DMSO) (Figure 1C). We have shown previously that this background activity is a result of residual K vitamins in the fetal bovine serum supplemented in the cell culture medium[40]. Therefore, these results indicate that VK-M-COT could be an inhibitor for VKD carboxylation.
To better understand the effect of these vitamin K derivatives on VKD carboxylation, we performed a serial-concentration titration by incubating FIXgla-PC/HEK293 reporter cells with increasing concentrations of different vitamin K analogues in cell culture medium. Results in Figure 1D show that, consistent with the above results, VK-2I-Ben has a similar dose-response curve in the stimulation of VKD carboxylation as vitamin K1. However, VK-M-COT displays an inhibitory dose-response curve, suggesting that VKD carboxylation activity decreases when the concentration of VK-M-COT increases (Figure 1D). Together, these results suggest that VK-M-COT functions as an inhibitor for VKD carboxylation.
The COT-vitamin K analogue targets multiple enzymes in the vitamin K cycle
To further confirm that COT-vitamin K was indeed an inhibitor for VKD carboxylation, we determined the inhibition potency of VK-M-COT on VKD carboxylation in HEK293 cells. We cultured FIXgla-PC/HEK293 cells with 5 μM KO and with increasing concentrations of VK-M-COT or warfarin (positive control), and determined the carboxylation efficiency of reporter-protein FIXgla-PC. Our results showed that VK-M-COT had a dose-response inhibition curve similar to warfarin for FIXgla-PC carboxylation (Figure 2A), although the inhibition potency of VK-M-COT was ~20-fold lower than warfarin (IC50, 249 nM vs. 12.5 nM) (Figure 2B). This result supported the above observation that VK-M-COT functions as an inhibitor in VKD carboxylation.
Figure 2. Characterization of VK-M-COT as an inhibitor for VKD carboxylation in HEK293 reporter cells.
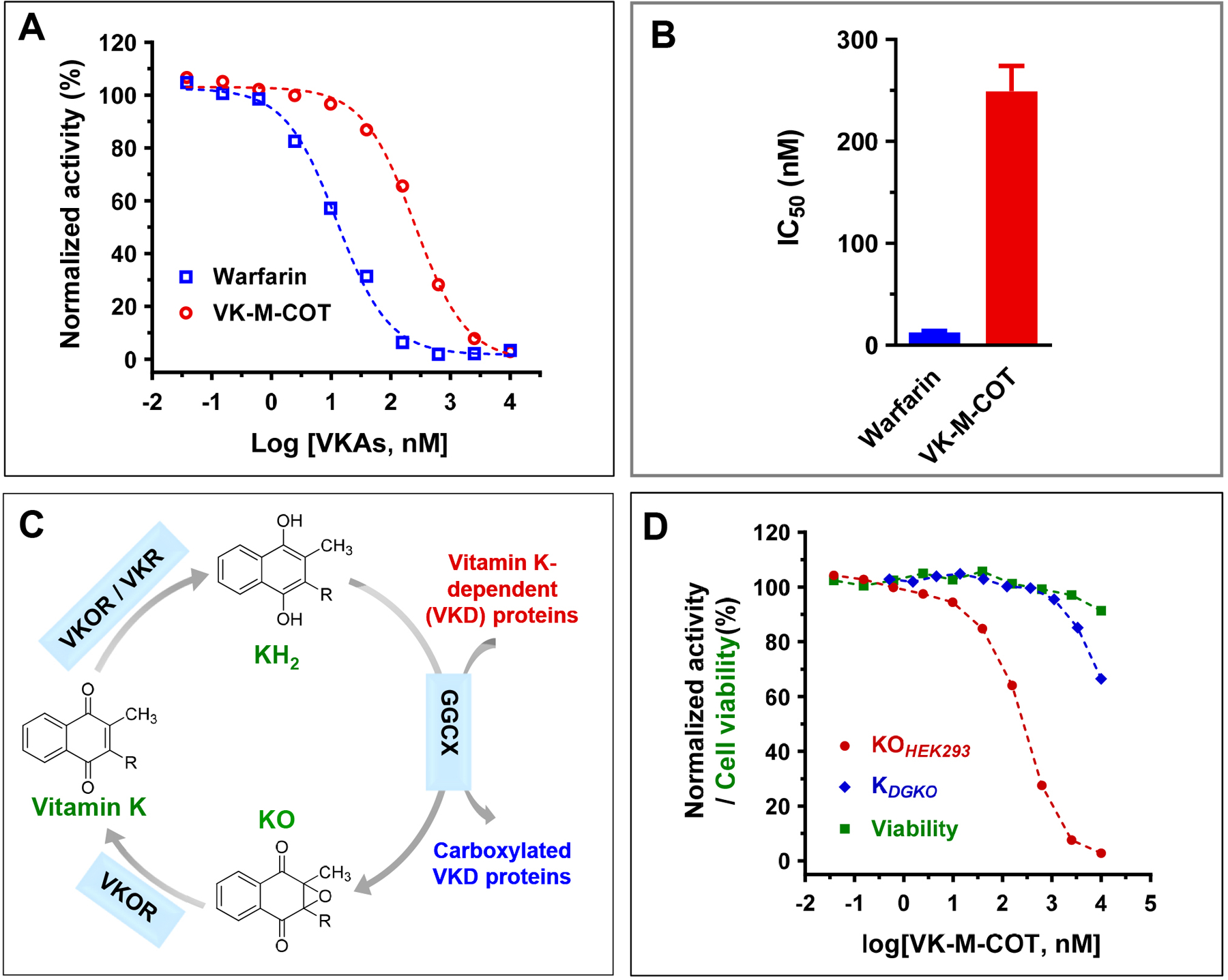
(A) Inhibition of VKD carboxylation by VK-M-COT (ο) and warfarin (◻) in HEK293 cells. FIXgla-PC/HEK293 cells were cultured with increasing concentrations of VK-M-COT or warfarin in cell culture medium containing 5 μM KO for the cell-based activity assay, as described in the legend of Figure 1C. (B) Inhibition potency of warfarin and VK-M-COT for VKD carboxylation. Half-maximal inhibition concentrations (IC50) of VK-M-COT and warfarin on VKD carboxylation were determined from Figure 2A using GraphPad Prism 8. (C) The vitamin K redox cycle. When VKD proteins are carboxylated by GGCX, the cofactor reduced vitamin K (KH2) is oxidized to vitamin K epoxide (KO), which needs to be reduced back to KH2 by a two-step reduction of KO to vitamin K and vitamin K to KH2. (D) Inhibition of VKD carboxylation by VK-M-COT in HEK293 and DGKO reporter cells. FIXgla-PC/HEK293 or DGKO reporter cells were cultured with increasing concentrations of VK-M-COT in cell culture medium containing 5 μM KO (• KOHEK293) or vitamin K1 (♦ KDGKO), respectively, for the cell-based activity assay and cell viability assays (■ viability).
In the vitamin K cycle, when the VKD protein is carboxylated by GGCX, the reduced form of vitamin K (KH2) is oxidized to KO, which must be reduced back to vitamin K and then to KH2 by a two-step reduction process (Figure 2C). To clarify whether VK-M-COT inhibits the reduction of KO to vitamin K, vitamin K to KH2 or the epoxidation of KH2 to KO, we used our recently established cell-based activity assays[40] and the conventional in vitro activity assay[42] (Supplemental Table 1). First, we examined the inhibition of reporter-protein carboxylation by VK-M-COT in FIXgla-PC/HEK293 cells using KO as the substrate and in DGKO cells using vitamin K1 as the substrate. To exclude the effect of cytotoxicity in our cell-based assay, we also determined cell viability in the corresponding concentrations of VK-M-COT. Results in Figure 2D show that VK-M-COT inhibited reporter-protein carboxylation when either KO or vitamin K1 were used as substrate, and these inhibitions were not due to the cytotoxicity of VK-M-COT.
Next, we determined KO and vitamin K reductase activities in HEK293 cells by directly measuring K vitamins within the cells using a conventional reversed-phase HPLC assay[40]. GGCX-knockout or DGKO reporter cells were cultured with VK-M-COT together with KO or vitamin K1 as described above, and K vitamins were extracted from the treated cells for the HPLC assay. Since reduced vitamin K (KH2) is unstable and difficult to accurately quantify from cells directly[41], we coupled the vitamin K reduction and epoxidation reactions to measure the final stable product KO (Figure 2C). Our results show that VK-M-COT significantly decreased both the reduction of KO (Figure 3A) and vitamin K (Figure 3B), which are consistent with the results from our cell-based carboxylation activity assays (Figure 2D).
Figure 3. Characterization of VK-M-COT as an inhibitor in the vitamin K redox cycle.
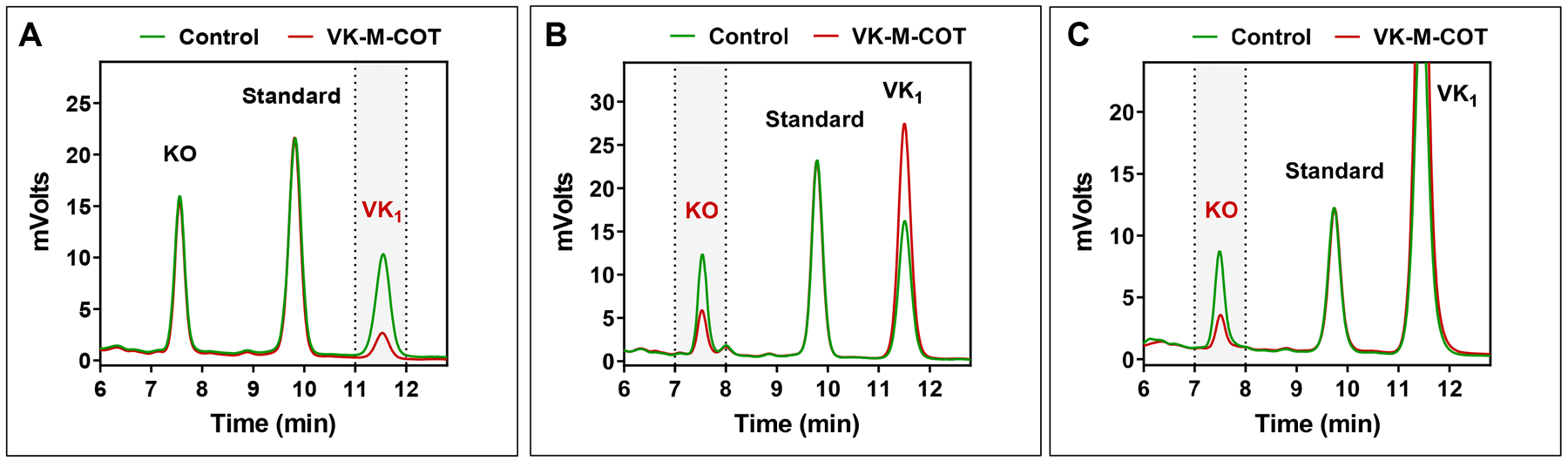
(A) Inhibition of KO to vitamin K1 reduction by VK-M-COT in HEK293 cells. GGCX-knockout HEK293 cells were cultured with cell culture medium containing 10 μM KO in the absence or presence of 10 μM VK-M-COT for 6 hours. DMSO (solvent used for dissolving VK-M-COT) treated cells were used as the control. K vitamins were extracted from the treated cells for the conventional HPLC assay. KO reduction activity is evaluated by the production of vitamin K1 (VK1) (labeled in red and marked with vertical dash lines) in the chromatogram. Standard: vitamin E acetate. (B) Inhibition of vitamin K reduction by VK-M-COT in HEK293 cells. DGKO cells were cultured with 10 μM vitamin K1 in the absence or presence of 10 μM VK-M-COT for 6 hours. DMSO treated cells were used as the control. K vitamins were extracted from the treated cells for the conventional HPLC assay. Vitamin K reduction activity was evaluated by the production of KO (labeled in red and marked with vertical dash lines) in the chromatogram. (C) Inhibition of vitamin K epoxidation by VK-M-COT. GGCX enriched microsomes were cultured with FLEEL, FIX-propeptide, and fresh prepared KH2 in the absence or presence of 100 μM VK-M-COT. K vitamins were extracted from the reaction mixture for the conventional HPLC assay. Vitamin K epoxidation activity was evaluated by the production of KO (labeled in red and marked with vertical dash lines) in the chromatogram.
It should be noted that the above vitamin K reductase activity is a coupled activity of vitamin K reduction and epoxidation. Therefore, the inhibition of either vitamin K reductase, GGCX, or both of the enzymes will affect the coupled enzymatic activity assay. Since the identity of vitamin K reductase is unknown and the product (KH2) of the enzyme is unstable[41], we determined the effect of VK-M-COT on GGCX activity using a conventional in vitro activity assay[42]. Our result showed that VK-M-COT inhibits GGCX activity for vitamin K epoxidation (Figure 3C). Together, these results suggested that, unlike warfarin that only targets VKOR[37], VK-M-COT targets multiple enzymes in the vitamin K cycle.
The anticoagulation effect of COT-vitamin K is tolerant to VKOR’s genetic variations
Our previous study shows that all current clinically used VKAs are susceptible to genetic variations of VKOR[13]. As VKOR is also the target of VK-M-COT (Figure 2D and 3A) and the core structure of VK-M-COT (2-methyl-1,4-naphthoquinone) is different from that of clinically used VKAs (Figure 1A), we assumed that VK-M-COT could have a different binding site in the target enzyme, and thus, have different sensitivities to genetic variations of VKOR. To test this hypothesis, we examined the inhibition potency of VK-M-COT among the most warfarin-resistant VKOR mutations (A26P, F55A, W59R, L128R, and Y139F) located at different regions of the VKOR molecule. Residue A26 is located within the first transmembrane domain (TMD1). Residues F55 and W59 are located in the loop region between TMD1 and TMD2, while residues L128 and Y139 are located within the last TMD near VKOR’s active site. Consistent with previous studies[13, 49, 50], our results showed that compared to wild-type VKOR, the inhibition curves of these VKOR mutations by warfarin dramatically shifted toward higher warfarin concentrations, suggesting that these mutations are resistant to warfarin inhibition (Figure 4A). The IC50 values of warfarin to these mutations increased 60-fold (A26P) to 417-fold (F55A) (Table I). However, the inhibition potency of VK-M-COT to these warfarin-resistant VKOR mutations was similar to that of wild-type VKOR (IC50 variation is less than 3-fold) (Figure 4B and Table I). These results suggested that the anticoagulation efficiency of VK-M-COT is tolerant to VKOR genetic variations.
Figure 4. Effect of VKOR mutations on the inhibition potency of warfarin and VK-M-COT.
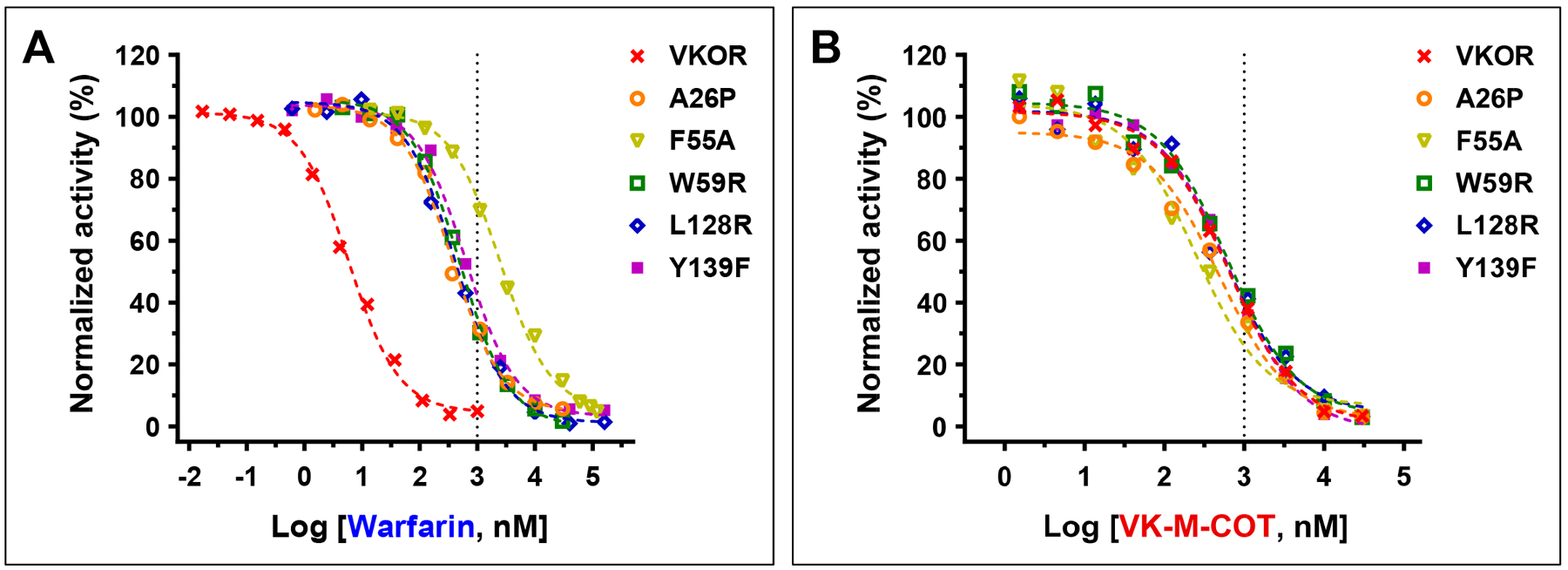
VKOR mutations were transiently expressed in DGKO reporter cells. Transfected cells were cultured with 5 μM KO with increasing concentrations of warfarin (A) or VK-M-COT (B) for 24 hours. The efficiency of reporter-protein FIXgla-PC carboxylation was determined as described in the legend of Figure 1C.
Table I.
Inhibition potency of warfarin and VK-M-COT for VKOR mutations
| VKOR | IC50 ± standard deviation (nM) | |
|---|---|---|
| VK-M-COT | Warfarin | |
| Wild type | 616 ± 74 | 5.9 ± 0.6 |
| A26P | 487 ± 75 | 360 ± 33 |
| F55A | 249 ± 66 | 2460 ± 235 |
| W59R | 615 ± 101 | 498 ± 37 |
| L128R | 575 ± 132 | 416 ± 49 |
| Y139F | 658 ± 54 | 633 ± 67 |
A ketone group at the 3-position side-chain of the vitamin K analogues recovered their VKD carboxylation activity
It has been shown that a benzoyl substitution at the 3-position of menadione markedly influenced its redox properties through increased delocalization of π-electrons induced by the ketone group[31]. To explore this effect on VKD carboxylation, we introduced keto functionality into the core of the vitamin K derivatives, which either significantly decreased VKD carboxylation activity (VK-M-Ben and VK-M-FB) or functioned as an inhibitor for VKD carboxylation (VK-M-COT) (Figure 5A). We then examined the ability of these compounds as a substrate to support VKD carboxylation in HEK293 cells. Our results showed that, compared with the parent compounds (Figure 1), introducing a ketone group at the 3-position significantly increased VKD carboxylation activity (Figure 5B and 5C). For example, the ketone group increased the carboxylation activity of VK-M-Ben and VK-M-FB from 3% and 11% (Figure 1C) to 41% and 69% (Figure 5B), respectively. Interestingly, the introduction of a ketone group converted the COT-vitamin K derivative from an inhibitor (VK-M-COT) (Figure 2) to a substrate (VK-K-COT) for VKD carboxylation.
Figure 5. Effect of a ketone group at the 3-positon side-chain of the vitamin K derivatives on VKD carboxylation.
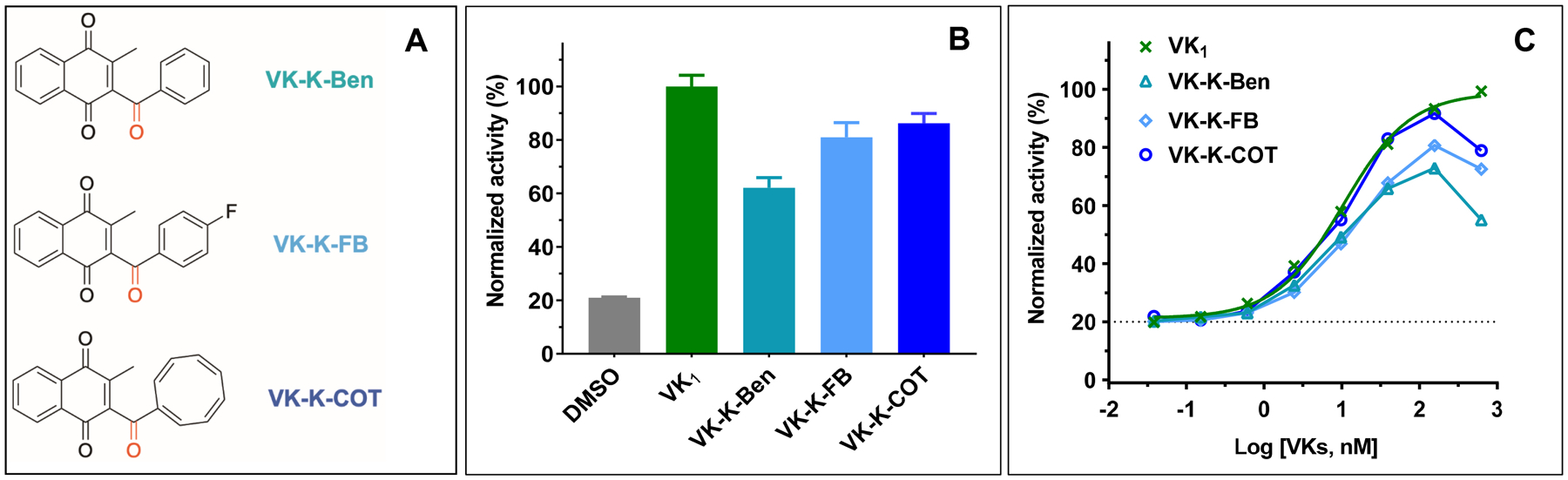
(A) Chemical structures of the ketone derived vitamin K analogues. (B) Cell-based VKD carboxylation activity of the ketone derived vitamin K analogues in FIXgla-PC/HEK293 reporter cells. Carboxylation activity was determined as described in the legend of Figure 1C. (C) Concentration titration of the ketone derived vitamin K analogues in FIXgla-PC/HEK293 reporter cells as described in the legend of Figure 1D.
VKD carboxylation activity of VK-K-COT is dependent on UBIAD1
Previous studies showed that in tissues, dietary phylloquinone is converted to menadione by the cleavage of the 3-position side-chain; then, menadione is converted to MK-4 by the prenyltransferase UBIAD1[51–53] (Figure 6A). In vitro studies showed that both vitamin K1 and MK-4, but not menadione, can efficiently support VKD carboxylation[54, 55]. To clarify whether VK-K-COT is directly involved in VKD carboxylation or is converted to MK-4 by UBIAD1 to support VKD carboxylation, we determined the VKD carboxylation activity of VK-K-COT in UBIAD1-knockout FIXgla-PC/HEK293 reporter cells. Results in Figure 6B show that both vitamin K1 and MK-4 can efficiently support VKD carboxylation in UBIAD1-knockout cells. However, menadione was unable to support reporter-protein carboxylation in these cells. This result suggests that vitamin K1 and MK-4, but not menadione, can directly function as cofactors for VKD carboxylation, which is consistent with previous in vitro and cell-based studies[39, 54, 55]. It is noteworthy that, unlike in HEK293 cells (Figure 5C), VK-K-COT was unable to support VKD carboxylation in UBIAD1-knockout reporter cells (Figure 6B), suggesting that the VKD carboxylation activity of VK-K-COT is dependent on UBIAD1.
Figure 6. Characterization of UBIAD1-dependent VKD carboxylation of COT derived vitamin K analogue in HEK293 cells.
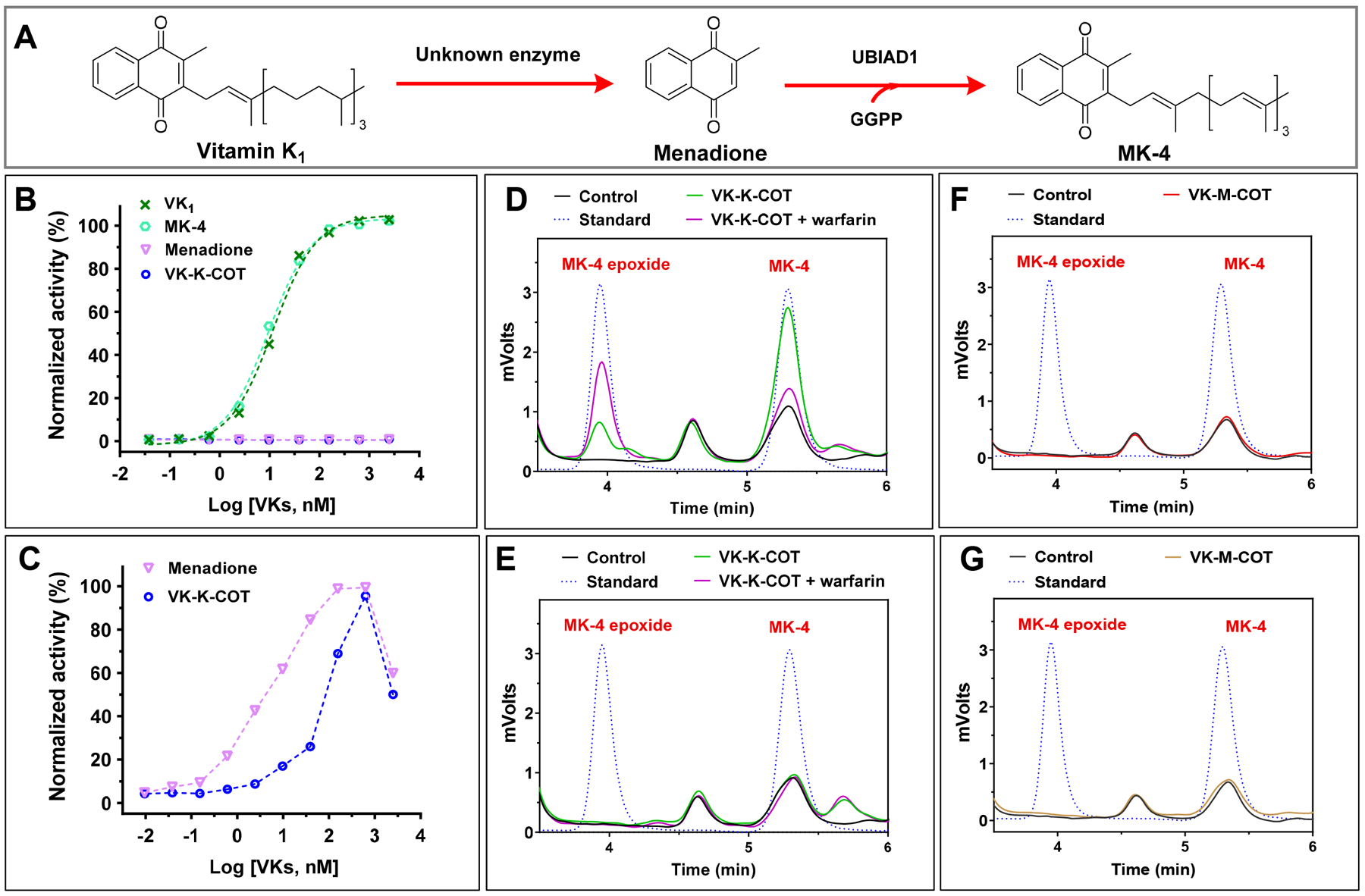
(A) Schematic diagram of the conversion of vitamin K1 to MK-4 mediated by UBIAD1. (B) VKD carboxylation in UBIAD1-knockout HEK293 reporter cells using vitamin K1 (VK), MK-4, menadione, or VK-K-COT as the substrate. UBIAD1-knockout reporter cells were cultured with increasing concentrations of the vitamin K analogues, and the efficiency of the reporter-protein FIXgla-PC carboxylation was determined as described in the legend of Figure 1D (C) Contribution of UBIAD1 to VKD carboxylation using menadione and VK-K-COT as the substrate. UBIAD1 was transiently expressed in UBIAD1-knockout reporter cells and the transfected cells were cultured with increasing concentrations of menadione or VK-K-COT. Carboxylation efficiency of reporter-protein FIXgla-PC was determined as described in the legend of Figure 1C. (D) (E) Conversion of VK-K-COT to MK-4 in HEK293 (D) and UBIAD1-deficent cells (E). FIXgla-PC/HEK293 cells (D) or UBIAD1-deficent HEK293 reporter cells (E) were cultured with 10 μM VK-K-COT in the absence or presence of 5 μM warfarin for 6 hours. DMSO treated cells were used as the control. K vitamins were extracted from the treated cells for the conventional HPLC assay. Pure MK-4 and MK-4 epoxide were used as standards to define the retention of these compounds in the chromatogram. (F) (G) Determination of the conversion of VK-M-COT to MK-4 in HEK293 (F) and UBIAD1-deficent cells (G) as described in the legend of Figure 6 D and E.
To further confirm the role of UBIAD1 in VK-K-COT supported VKD carboxylation, we introduced UBIAD1 back into the UBIAD1-knockout cells by transient expression. Our results showed that in the presence of UBIAD1, both menadione and VK-K-COT regained VKD carboxylation activity (Figure 6C), supporting the notion that the VKD carboxylation activity of VK-K-COT is dependent upon UBIAD1. We assumed that this UBIAD1-dependent carboxylation activity of VK-K-COT results from the conversion of VK-K-COT to MK-4. To test this hypothesis, we determined the production of MK-4 in wild-type and UBIAD1-knockout FIXgla-PC/HEK293 cells when these cells were cultured with VK-K-COT. Our results showed that, a significant amount of MK-4 and small amounts of MK-4 epoxide were observed in wild-type HEK293 cells where UBIAD1 was present (Figure 6D). Additionally, MK-4 epoxide accumulated in cells when VKOR was inactivated by warfarin. This result is consistent with the observation that KO accumulated in plasma when rats were treated with warfarin or in patients with VKA anticoagulation therapy[56, 57]. However, neither MK-4 nor MK-4 epoxide was observed in UBIAD1-deficent cells under the same conditions (Figure 6E). Together, these results support the hypothesis that VK-K-COT cannot directly function as a cofactor for VKD carboxylation, rather it needs to be converted to MK-4 by UBIAD1 in order to be active in the vitamin K cycle.
It is worth noting that no MK-4 production was observed from VK-M-COT, the counterpart of VK-K-COT which functions as an inhibitor of VKD carboxylation, by the UBIAD1-dependet metabolic pathway (Figure 6F and 6G). This suggests that the ketone group, at the 3-position side-chain, makes the side-chain of the vitamin K analogues more susceptible for cleavage and conversion to MK-4 as a substrate for VKD carboxylation.
The anticoagulation effect of VK-K-COT can be reversed by vitamin K1
One of the challenges of anticoagulation therapy is reversing the anticoagulation effect to reduce bleeding risks[58]. Vitamin K1 administration is an effective strategy for reversing excessive anticoagulation in patients receiving VKA therapy[59–61]. It is believed that the antidotal effect of vitamin K1 is due to the existence of warfarin-resistant vitamin K reductase that directly reduces vitamin K1 to KH2 to support VKD carboxylation in the presence of warfarin[37, 62, 63]. Since VK-M-COT targets multiple enzymes in the VKD carboxylation process, we explored whether the anticoagulation effect of VK-M-COT could be reversed using high concentrations of vitamin K1. We cultured FIXgla-PC/HEK293 reporter-cells with a fixed concentration of warfarin or VK-M-COT to inactive VKD carboxylation, and then using increasing concentrations of vitamin K1 to reverse their anticoagulation effect. Results in Figure 7 show that the inhibition of VKD carboxylation by both warfarin and VK-M-COT can be reversed equally with higher concentrations of vitamin K1.
Figure 7. Reversal of the anticoagulation effect of VK-M-COT and warfarin by vitamin K1.
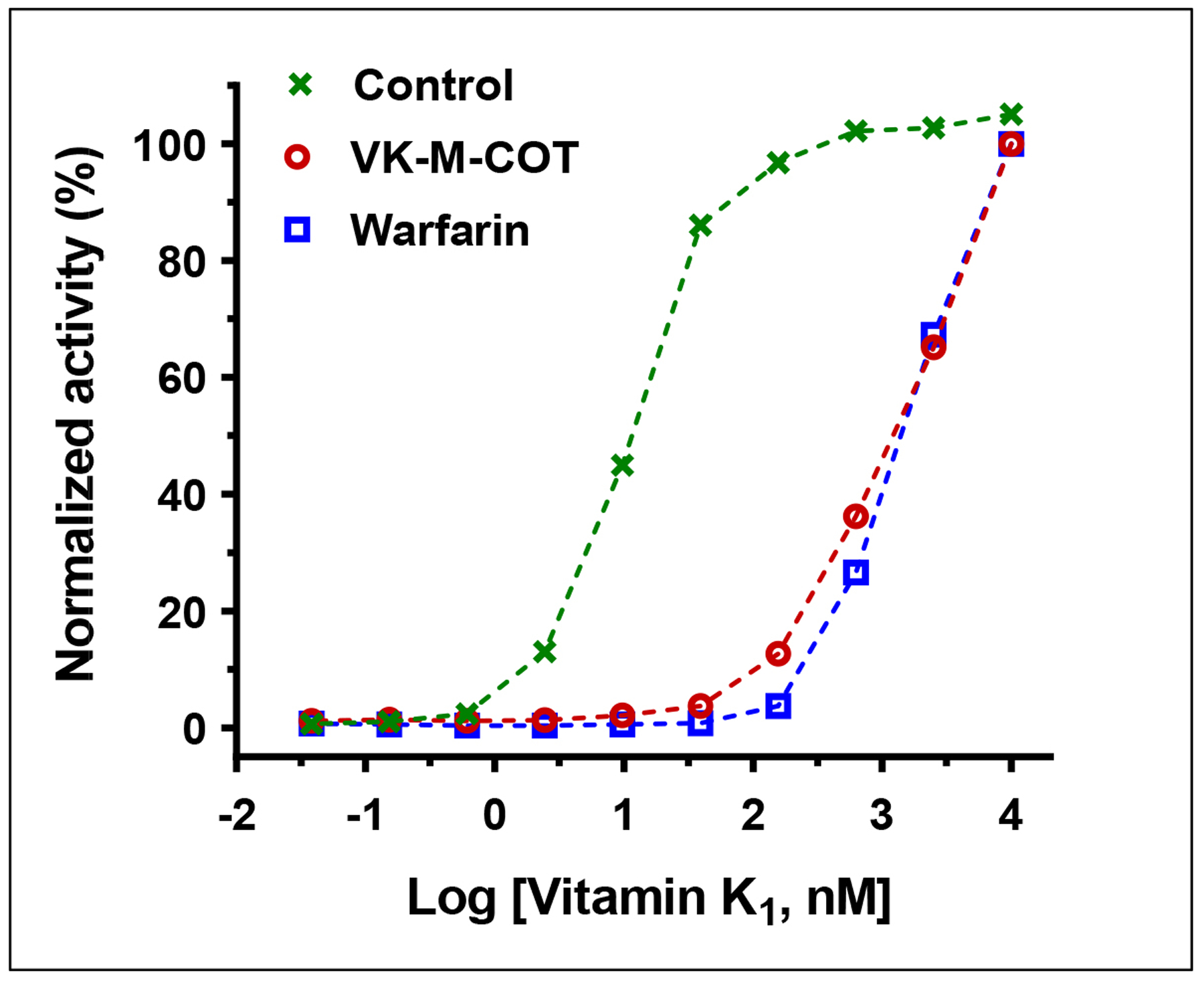
FIXgla-PC/HEK293 reporter cells were cultured in cell culture medium containing 3 μM VK-M-COT, 3 μM warfarin or DMSO (control) and increasing concentrations of vitamin K1 for 24 hours. The carboxylation efficiency of the reporter-protein FIXgla-PC was determined as described in the legend of Figure 1D.
Discussion
Vitamin K antagonists have been the mainstay of anticoagulation therapy for the past 60 years. Since the discovery of the first anticoagulant, dicoumarol, from spoiled sweet clover[64], numerous coumarin derivatives have been chemically synthesized or isolated from natural sources for anticoagulation tests[27, 65, 66]. Currently, three coumarin derivatives (warfarin, acenocoumarol, and phenprocoumon) have been clinically used for anticoagulation therapy. These compounds have different half-lives, with long-acting coumarins appearing to provide a more stable anticoagulation effect[5]. Nevertheless, coumarin-derived VKAs have a narrow therapeutic range and require regular coagulation monitoring and dose adjustments to keep the anticoagulation intensity within the therapeutic range. This is partially due to the genetic variations of their target enzyme VKOR.
One of our long-term goals has been to search for an improved VKA that is not susceptible to the genetic variations of VKOR with the hope that a fixed dose of anticoagulant would achieve the desired anticoagulation effect, despite the genetic backgrounds of patients. Recently, we have shown that replacing the phenyl group of warfarin with COT significantly decreased the variation of warfarin resistance among naturally occurring VKOR mutations[34]. However, the resistance of VKOR mutations to COT-warfarin still had an over 25-fold difference. Additionally, COT-warfarin and warfarin displayed a similar pattern of resistance to all naturally occurring VKOR mutations. This could be due to the fact that their core structure, 4-hydroxycoumarin (Figure 1A), binds to the same site in VKOR. Therefore, it seemed logical that compounds with different core structures (i.e. other than coumarin) may be associated with a different binding site in the target enzyme, and thus have a different mechanism of action.
As a continuation to our previous work, we explored the possibility of derivatizing vitamin K as an inhibitor for VKD carboxylation. Vitamin K comprises a group of structurally similar compounds that share a 2-methyl-1,4-naphthoquinone core structure, and are distinctly different from the coumarins (Figure 1A), but all differ in the side-chain at the 3-position[30, 46]. Lowenthal et al have shown that replacing the 2-methyl group of phylloquinone with a chlorine atom resulted in an analogue (Chloro-K) that was a potent antagonist of vitamin K[67]. Because of its distinct structure and alternative mechanism of action from the coumarin anticoagulants[68], Chloro-K appears to be an effective anticoagulant in warfarin-resistant rats[69].
In this study, we focused on derivatization of the 3-postion side-chain of vitamin K. Our results showed that substitution of the 3-position side-chain significantly affected its activity as a substrate for VKD carboxylation (Figure 1C). Interestingly, replacement of the phytyl side-chain with methylene COT converted the vitamin K core from a substrate to an inhibitor for VKD carboxylation (Figure 2). Characterization of methylene COT-vitamin K with the most warfarin-resistant VKOR mutations showed that methylene COT-vitamin K displayed a similar inhibition potency to wild-type VKOR and all the warfarin-resistant VKOR mutations, although these mutations affected the inhibition potency of warfarin for VKD carboxylation up to 400-fold (Table 1). These results suggested that, different from warfarin, a fixed dose of methylene COT-vitamin K should achieve a similar anticoagulation effect, despite genetic variations of VKOR.
Bleeding complications are the most concerning adverse event with the use of any anticoagulant. Therefore, a specific reversal agent is desired when anticoagulant therapy needs to be reversed or neutralized[70]. Unlike warfarin, methylene COT-vitamin K appears to target multiple enzymes in the VKD carboxylation pathway (Figure 2D and Figure 3). We reasoned that a COT-vitamin K analogue that possesses vitamin K activity could be an ideal specific reversal agent for methylene COT-vitamin K. As mentioned, a previous study suggested that a ketone group at 3-position can alternate redox properties of the vitamin K core[31]. Therefore, we introduced a ketone group to the vitamin K derivatives used in this study. Our results showed that the ketone addition converted the COT-vitamin K derivative from an inhibitor (VK-M-COT) to a substrate (VK-K-COT) for VKD carboxylation (Figure 2 and 5). Further characterization of VK-K-COT supported VKD carboxylation, suggesting that VK-K-COT is not directly involved in vitamin K redox cycling, rather, its VKD carboxylation activity resulted from the conversion of VK-K-COT to MK-4 mediated by UBIAD1 (Figure 6). Nevertheless, our results showed that vitamin K1 can efficiently reverse the anticoagulation effect of COT-vitamin K as in warfarin (Figure 7).
It should be noted that, as an analogue of vitamin K, VK-M-COT appears to target multiple enzymes in the vitamin K cycle (Figure 3). Therefore, genetic variations of other enzymes in the cycle, like GGCX, may also affect the anticoagulation efficacy of VK-M-COT. Future studies will explore how naturally occurring GGCX mutations affect the inhibition potency of VK-M-COT for VKD carboxylation. GGCX mutations mainly cause the combined vitamin K-dependent clotting factors deficiency (VKCFD)[71], and this symptom can be ameliorated by the administration of vitamin K1. As GGCX mutations do not appear to affect the efficiency of vitamin K therapy, we do not expect GGCX mutations to have a significant effect on the inhibition potency of the vitamin K analogue, VK-M-COT, on VKD carboxylation. Additionally, since the inhibition potency of VK-M-COT to VKD carboxylation was ~20-fold less than that of warfarin (Figure 2A and 2B), the other objective is to improve the binding affinity of VK-M-COT to the target enzymes by undertaking a structure activity relationship study, without compromising the resistance tolerance to genetic variations in vitamin K cycle enzymes.
Supplementary Material
Essentials.
Warfarin therapy has been the mainstay of treatment for thromboembolism
The efficacy of warfarin therapy relies on the genetic variation of its target VKOR
A cyclooctatetraene (COT) derived vitamin K analogue can function as an anticoagulant
The anticoagulation efficacy of COT-vitamin K is independent on VKOR’s genetic variation
Acknowledgments
This work was supported by grant HL131690 from the National Institutes of Health, USA (to J.K.T. and D.W.S.), the University of Queensland (UQ) and the CSIRO (Melbourne). C.M.W. gratefully acknowledges the Australian Research Council for a Future Fellowship Award (grant number FT110100851).
Footnotes
Conflict-of-Interest Disclosure G. P. S. and C. M. W. have formal and informal commercial relationships with companies developing and supplying cubane intermediates. All other authors declare no competing financial interests.
References
- 1.Sogaard KK, Schmidt M, Pedersen L, Horvath-Puho E, Sorensen HT. 30-year mortality after venous thromboembolism: a population-based cohort study. Circulation. 2014; 130: 829–36. 10.1161/CIRCULATIONAHA.114.009107. [DOI] [PubMed] [Google Scholar]
- 2.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr., Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014; 129: 837–47. 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosarla RC, Vaduganathan M, Qamar A, Moslehi J, Piazza G, Giugliano RP. Anticoagulation Strategies in Patients With Cancer: JACC Review Topic of the Week. J Am Coll Cardiol. 2019; 73: 1336–49. 10.1016/j.jacc.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makaryus JN, Halperin JL, Lau JF. Oral anticoagulants in the management of venous thromboembolism. Nat Rev Cardiol. 2013; 10: 397–409. 10.1038/nrcardio.2013.73. [DOI] [PubMed] [Google Scholar]
- 5.Zirlik A, Bode C. Vitamin K antagonists: relative strengths and weaknesses vs. direct oral anticoagulants for stroke prevention in patients with atrial fibrillation. J Thromb Thrombolysis. 2017; 43: 365–79. 10.1007/s11239-016-1446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moualla H, Garcia D. Vitamin K Antagonists - Current Concepts and Challenges. Thrombosis Research. 2011; 128: 210–5. 10.1016/j.thromres.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Pirmohamed M Warfarin: almost 60 years old and still causing problems. Br J Clin Pharmacol. 2006; 62: 509–11. 10.1111/j.1365-2125.2006.02806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warfarin - Drug Usage Statistics. ClinCalc. 2020; https://clincalc.com/DrugStats/Drugs/Warfarin. [Google Scholar]
- 9.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, Muller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004; 427: 537–41. 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 10.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004; 427: 541–4. 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 11.Tavares LC, Marcatto LR, Santos P. Genotype-guided warfarin therapy: current status. Pharmacogenomics. 2018; 19: 667–85. 10.2217/pgs-2017-0207. [DOI] [PubMed] [Google Scholar]
- 12.D’Andrea G, D’Ambrosio RL, Di Perna P, Chetta M, Santacroce R, Brancaccio V, Grandone E, Margaglione M. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005; 105: 645–9. 10.1182/blood-2004-06-2111. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Jin DY, Stafford DW, Tie JK. Evaluation of oral anticoagulants with vitamin K epoxide reductase in its native milieu. Blood. 2018; 132: 1974–84. 10.1182/blood-2018-05-846592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson JA. Warfarin pharmacogenetics: a rising tide for its clinical value. Circulation. 2012; 125: 1964–6. 10.1161/CIRCULATIONAHA.112.100628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arwood MJ, Deng J, Drozda K, Pugach O, Nutescu EA, Schmidt S, Duarte JD, Cavallari LH. Anticoagulation endpoints with clinical implementation of warfarin pharmacogenetic dosing in a real-world setting: A proposal for a new pharmacogenetic dosing approach. Clin Pharmacol Ther. 2017; 101: 675–83. 10.1002/cpt.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos PC, Marcatto LR, Duarte NE, Gadi Soares RA, Cassaro Strunz CM, Scanavacca M, Krieger JE, Pereira AC. Development of a pharmacogenetic-based warfarin dosing algorithm and its performance in Brazilian patients: highlighting the importance of population-specific calibration. Pharmacogenomics. 2015; 16: 865–76. 10.2217/pgs.15.48. [DOI] [PubMed] [Google Scholar]
- 17.Lund K, Gaffney D, Spooner R, Etherington AM, Tansey P, Tait RC. Polymorphisms in VKORC1 have more impact than CYP2C9 polymorphisms on early warfarin International Normalized Ratio control and bleeding rates. Br J Haematol. 2012; 158: 256–61. 10.1111/j.1365-2141.2012.09150.x. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JA, Cavallari LH. Warfarin pharmacogenetics. Trends Cardiovasc Med. 2015; 25: 33–41. 10.1016/j.tcm.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Lopez JA, Sterne JAC, Thom HHZ, Higgins JPT, Hingorani AD, Okoli GN, Davies PA, Bodalia PN, Bryden PA, Welton NJ, Hollingworth W, Caldwell DM, Savovic J, Dias S, Salisbury C, Eaton D, Stephens-Boal A, Sofat R. Oral anticoagulants for prevention of stroke in atrial fibrillation: systematic review, network meta-analysis, and cost effectiveness analysis. BMJ. 2017; 359: j5058. 10.1136/bmj.j5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almutairi AR, Zhou L, Gellad WF, Lee JK, Slack MK, Martin JR, Lo-Ciganic WH. Effectiveness and Safety of Non-vitamin K Antagonist Oral Anticoagulants for Atrial Fibrillation and Venous Thromboembolism: A Systematic Review and Meta-analyses. Clin Ther. 2017; 39: 1456–78 e36. 10.1016/j.clinthera.2017.05.358. [DOI] [PubMed] [Google Scholar]
- 21.Ng SS, Lai NM, Nathisuwan S, Jahan NK, Dilokthornsakul P, Kongpakwattana K, Hollingworth W, Chaiyakunapruk N. Comparative efficacy and safety of warfarin care bundles and novel oral anticoagulants in patients with atrial fibrillation: a systematic review and network meta-analysis. Sci Rep. 2020; 10: 662. 10.1038/s41598-019-57370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mujer MTP, Rai MP, Atti V, Dimaandal IL, Chan AS, Shrotriya S, Gundabolu K, Dhakal P. An Update on the Reversal of Non-Vitamin K Antagonist Oral Anticoagulants. Adv Hematol. 2020; 2020: 7636104. 10.1155/2020/7636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fanaroff AC, Ohman EM. Non-Vitamin K Antagonist Oral Anticoagulants in the Treatment of Atrial Fibrillation. Annu Rev Med. 2019; 70: 61–75. 10.1146/annurev-med-042617-092334. [DOI] [PubMed] [Google Scholar]
- 24.Czuprynska J, Patel JP, Arya R. Current challenges and future prospects in oral anticoagulant therapy. Br J Haematol. 2017; 178: 838–51. 10.1111/bjh.14714. [DOI] [PubMed] [Google Scholar]
- 25.Yeh CH, Hogg K, Weitz JI. Overview of the new oral anticoagulants: opportunities and challenges. Arterioscler Thromb Vasc Biol. 2015; 35: 1056–65. 10.1161/ATVBAHA.115.303397. [DOI] [PubMed] [Google Scholar]
- 26.Hokusai VTEI, Buller HR, Decousus H, Grosso MA, Mercuri M, Middeldorp S, Prins MH, Raskob GE, Schellong SM, Schwocho L, Segers A, Shi M, Verhamme P, Wells P. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013; 369: 1406–15. 10.1056/NEJMoa1306638. [DOI] [PubMed] [Google Scholar]
- 27.Kasperkiewicz K, Ponczek MB, Owczarek J, Guga P, Budzisz E. Antagonists of Vitamin K-Popular Coumarin Drugs and New Synthetic and Natural Coumarin Derivatives. Molecules. 2020; 25. 10.3390/molecules25061465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Heuzey JY, Ammentorp B, Darius H, De Caterina R, Schilling RJ, Schmitt J, Zamorano JL, Kirchhof P. Differences among western European countries in anticoagulation management of atrial fibrillation. Data from the PREFER IN AF registry. Thrombosis and haemostasis. 2014; 111: 833–41. 10.1160/TH13-12-1007. [DOI] [PubMed] [Google Scholar]
- 29.Mekaj YH, Mekaj AY, Duci SB, Miftari EI. New oral anticoagulants: their advantages and disadvantages compared with vitamin K antagonists in the prevention and treatment of patients with thromboembolic events. Ther Clin Risk Manag. 2015; 11: 967–77. 10.2147/TCRM.S84210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shearer MJ, Newman P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J Lipid Res. 2014; 55: 345–62. 10.1194/jlr.R045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidorov P, Desta I, Chesse M, Horvath D, Marcou G, Varnek A, Davioud-Charvet E, Elhabiri M. Redox Polypharmacology as an Emerging Strategy to Combat Malarial Parasites. Chemmedchem. 2016; 11: 1339–51. 10.1002/cmdc.201600009. [DOI] [PubMed] [Google Scholar]
- 32.Kimura K, Hirota Y, Kuwahara S, Takeuchi A, Tode C, Wada A, Osakabe N, Suhara Y. Synthesis of Novel Synthetic Vitamin K Analogues Prepared by Introduction of a Heteroatom and a Phenyl Group That Induce Highly Selective Neuronal Differentiation of Neuronal Progenitor Cells. J Med Chem. 2017; 60: 2591–6. 10.1021/acs.jmedchem.6b01717. [DOI] [PubMed] [Google Scholar]
- 33.Xing H, Houston SD, Chen X, Jin DY, Savage GP, Tie JK, Williams CM. Determining the necessity of phenyl ring pi-character in warfarin. Bioorg Med Chem Lett. 2019; 29: 1954–6. 10.1016/j.bmcl.2019.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing H, Houston SD, Chen X, Ghassabian S, Fahrenhorst-Jones T, Kuo A, Murray CP, Conn KA, Jaeschke KN, Jin DY, Pasay C, Bernhardt PV, Burns JM, Tsanaktsidis J, Savage GP, Boyle GM, De Voss JJ, McCarthy J, Walter GH, Burne THJ, Smith MT, Tie JK, Williams CM. Cyclooctatetraene: A Bioactive Cubane Paradigm Complement. Chemistry. 2019; 25: 2729–34. 10.1002/chem.201806277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houston SD, Fahrenhorst-Jones T, Xing H, Chalmers BA, Sykes ML, Stok JE, Farfan Soto C, Burns JM, Bernhardt PV, De Voss JJ, Boyle GM, Smith MT, Tsanaktsidis J, Savage GP, Avery VM, Williams CM. The cubane paradigm in bioactive molecule discovery: further scope, limitations and the cyclooctatetraene complement. Org Biomol Chem. 2019; 17: 6790–8. 10.1039/c9ob01238a. [DOI] [PubMed] [Google Scholar]
- 36.Chalmers BA, Xing H, Houston S, Clark C, Ghassabian S, Kuo A, Cao B, Reitsma A, Murray CE, Stok JE, Boyle GM, Pierce CJ, Littler SW, Winkler DA, Bernhardt PV, Pasay C, De Voss JJ, McCarthy J, Parsons PG, Walter GH, Smith MT, Cooper HM, Nilsson SK, Tsanaktsidis J, Savage GP, Williams CM. Validating Eaton’s Hypothesis: Cubane as a Benzene Bioisostere. Angew Chem Int Ed Engl. 2016; 55: 3580–5. 10.1002/anie.201510675. [DOI] [PubMed] [Google Scholar]
- 37.Tie JK, Jin DY, Straight DL, Stafford DW. Functional study of the vitamin K cycle in mammalian cells. Blood. 2011; 117: 2967–74. 10.1182/blood-2010-08-304303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tie JK, Jin DY, Tie K, Stafford DW. Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J Thromb Haemost. 2013; 11: 1556–64. 10.1111/jth.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tie JK, Carneiro JD, Jin DY, Martinhago CD, Vermeer C, Stafford DW. Characterization of vitamin K-dependent carboxylase mutations that cause bleeding and nonbleeding disorders. Blood. 2016; 127: 1847–55. 10.1182/blood-2015-10-677633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Li C, Jin D, Ingram B, Hao Z, Bai X, Stafford DW, Hu K, Tie JK. A cell-based high-throughput screen identifies drugs that cause bleeding disorders by off-targeting the vitamin K cycle. Blood. 2020; 136: 898–908. 10.1182/blood.2019004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rishavy MA, Hallgren KW, Wilson LA, Usubalieva A, Runge KW, Berkner KL. The Vitamin K Oxidoreductase Is a Multimer That Efficiently Reduces Vitamin K Epoxide to Hydroquinone to Allow Vitamin K-dependent Protein Carboxylation. Journal of Biological Chemistry. 2013; 288: 31556–66. 10.1074/jbc.M113.497297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tie JK, Zheng MY, Pope RM, Straight DL, Stafford DW. Identification of the N-linked glycosylation sites of vitamin K-dependent carboxylase and effect of glycosylation on carboxylase function. Biochemistry. 2006; 45: 14755–63. 10.1021/bi0618518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Formiller M, Cohon MS. Coumarin and indandione anticoagulants. Potentiators and antagonists. Am J Hosp Pharm. 1969; 26: 574–82. [PubMed] [Google Scholar]
- 44.Hodroge A, Matagrin B, Moreau C, Fourel I, Hammed A, Benoit E, Lattard V. VKORC1 mutations detected in patients resistant to vitamin K antagonists are not all associated with a resistant VKOR activity. J Thromb Haemost. 2012; 10: 2535–43. 10.1111/jth.12019. [DOI] [PubMed] [Google Scholar]
- 45.Czogalla KJ, Biswas A, Wendeln AC, Westhofen P, Muller CR, Watzka M, Oldenburg J. Human VKORC1 mutations cause variable degrees of 4-hydroxycoumarin resistance and affect putative warfarin binding interfaces. Blood. 2013; 122: 2743–50. 10.1182/blood-2013-05-501692. [DOI] [PubMed] [Google Scholar]
- 46.Vermeer C, van ‘t Hoofd C, Knapen MHJ, Xanthoulea S. Synthesis of 2-methyl-1,4-naphthoquinones with higher gamma-glutamyl carboxylase activity than MK-4 both in vitro and in vivo. Bioorg Med Chem Lett. 2017; 27: 208–11. 10.1016/j.bmcl.2016.11.073. [DOI] [PubMed] [Google Scholar]
- 47.Hirota Y, Suhara Y. New Aspects of Vitamin K Research with Synthetic Ligands: Transcriptional Activity via SXR and Neural Differentiation Activity. Int J Mol Sci. 2019; 20. 10.3390/ijms20123006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou CJ, Inks ES, Josey BJ. Vitamin K: a structural basis for the design of novel neuroprotective agents? Future Med Chem. 2013; 5: 857–60. 10.4155/fmc.13.70. [DOI] [PubMed] [Google Scholar]
- 49.Shen G, Cui W, Zhang H, Zhou F, Huang W, Liu Q, Yang Y, Li S, Bowman GR, Sadler JE, Gross ML, Li W. Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat Struct Mol Biol. 2017; 24: 69–76. 10.1038/nsmb.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Czogalla KJ, Biswas A, Honing K, Hornung V, Liphardt K, Watzka M, Oldenburg J. Warfarin and vitamin K compete for binding to Phe55 in human VKOR. Nat Struct Mol Biol. 2017; 24: 77–85. 10.1038/nsmb.3338. [DOI] [PubMed] [Google Scholar]
- 51.Hirota Y, Tsugawa N, Nakagawa K, Suhara Y, Tanaka K, Uchino Y, Takeuchi A, Sawada N, Kamao M, Wada A, Okitsu T, Okano T. Menadione (vitamin K3) is a catabolic product of oral phylloquinone (vitamin K1) in the intestine and a circulating precursor of tissue menaquinone-4 (vitamin K2) in rats. J Biol Chem. 2013; 288: 33071–80. 10.1074/jbc.M113.477356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y, Okano T. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010; 468: 117–21. 10.1038/nature09464. [DOI] [PubMed] [Google Scholar]
- 53.Shearer MJ, Okano T. Key Pathways and Regulators of Vitamin K Function and Intermediary Metabolism. Annu Rev Nutr. 2018; 38: 127–51. 10.1146/annurev-nutr-082117-051741. [DOI] [PubMed] [Google Scholar]
- 54.Buitenhuis HC, Soute BA, Vermeer C. Comparison of the vitamins K1, K2 and K3 as cofactors for the hepatic vitamin K-dependent carboxylase. Biochim Biophys Acta. 1990; 1034: 170–5. 10.1016/0304-4165(90)90072-5. [DOI] [PubMed] [Google Scholar]
- 55.Houser RM, Searcey MT, Gardner EJ, Scheinbuks J, Subba Rao GN, Jones JP, Hall AL. Nature of the vitamin K-dependent CO2 fixation in microsomal membranes. Fed Proc. 1978; 37: 2610–4. [PubMed] [Google Scholar]
- 56.Shearer MJ, McBurney A, Breckenridge AM, Barkhan P. Effect of warfarin on the metabolism of phylloquinone (vitamin K1):dose-response relationships in man. Clin Sci Mol Med. 1977; 52: 621–30. 10.1042/cs0520621. [DOI] [PubMed] [Google Scholar]
- 57.Bell RG, Matschiner JT. Warfarin and the inhibition of vitamin K activity by an oxide metabolite. Nature. 1972; 237: 32–3. 10.1038/237032a0. [DOI] [PubMed] [Google Scholar]
- 58.Kuramatsu JB, Sembill JA, Huttner HB. Reversal of oral anticoagulation in patients with acute intracerebral hemorrhage. Crit Care. 2019; 23: 206. 10.1186/s13054-019-2492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wentzien TH, O’Reilly RA, Kearns PJ. Prospective evaluation of anticoagulant reversal with oral vitamin K1 while continuing warfarin therapy unchanged. Chest. 1998; 114: 1546–50. 10.1378/chest.114.6.1546. [DOI] [PubMed] [Google Scholar]
- 60.Dezee KJ, Shimeall WT, Douglas KM, Shumway NM, O’Malley PG. Treatment of excessive anticoagulation with phytonadione (vitamin K): a meta-analysis. Arch Intern Med. 2006; 166: 391–7. 10.1001/.391. [DOI] [PubMed] [Google Scholar]
- 61.Buecking B, Eschbach D, Bliemel C, Oberkircher L, Struewer J, Ruchholtz S, Sachs UJ. Effectiveness of vitamin K in anticoagulation reversal for hip fracture surgery--a prospective observational study. Thromb Res. 2014; 133: 42–7. 10.1016/j.thromres.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 62.Wallin R, Martin LF. Vitamin K-dependent carboxylation and vitamin K metabolism in liver. Effects of warfarin. J Clin Invest. 1985; 76: 1879–84. 10.1172/JCI112182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van der Meer J, Hemker HC, Loeliger EA. Pharmacological aspects of vitamin K1. A clinical and experimental study in man. Thromb Diath Haemorrh Suppl. 1968; 29: 1–96. [PubMed] [Google Scholar]
- 64.Stahmann MA, Huebner CF, Link KP. Studies on the hemorrhagic sweet clover disease V. Identification and synthesis of the hemorrhagic agent. Journal of Biological Chemistry. 1941; 138: 513–27. [Google Scholar]
- 65.Link KP. The discovery of dicumarol and its sequels. Circulation. 1959; 19: 97–107. 10.1161/01.cir.19.1.97. [DOI] [PubMed] [Google Scholar]
- 66.Lei L, Xue YB, Liu Z, Peng SS, He Y, Zhang Y, Fang R, Wang JP, Luo ZW, Yao GM, Zhang JW, Zhang G, Song HP, Zhang YH. Coumarin derivatives from Ainsliaea fragrans and their anticoagulant activity. Sci Rep-Uk. 2015; 5. ARTN 13544 10.1038/srep13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lowenthal J, Macfarlane JA, McDonald KM. The inhibition of the antidotal activity of vitamin K1 against coumarin anticoagulant drugs by its chloro analogue. Experientia. 1960; 16: 428–9. 10.1007/BF02178853. [DOI] [PubMed] [Google Scholar]
- 68.Cheung A, Suttie JW. Synthesis of menaquinone-2 derivatives as substrates for the liver microsomal vitamin K-dependent carboxylase. Biofactors. 1988; 1: 61–5. [PubMed] [Google Scholar]
- 69.Suttie JW. Anticoagulant-resistant rats: possible control by the use of the chloro analog of vitamin K 1. Science. 1973; 180: 741–3. 10.1126/science.180.4087.741. [DOI] [PubMed] [Google Scholar]
- 70.Smythe MA, Trujillo T, Fanikos J. Reversal agents for use with direct and indirect anticoagulants. Am J Health Syst Pharm. 2016; 73: S27–48. 10.2146/ajhp150959. [DOI] [PubMed] [Google Scholar]
- 71.Napolitano M, Mariani G, Lapecorella M. Hereditary combined deficiency of the vitamin K-dependent clotting factors. Orphanet J Rare Dis. 2010; 5: 21. 10.1186/1750-1172-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.