

Abstract
Alcohol elicits a neuroimmune response in the brain contributing to the development and maintenance of alcohol use disorder (AUD). While pro-inflammatory mediators initiate and drive the neuroimmune response, anti-inflammatory mediators provide an important homeostatic mechanism to limit inflammation and prevent pathological damage. However, our understanding of the role of anti-inflammatory signaling on neuronal physiology in critical addiction-related brain regions and pathological alcohol-dependence induced behaviors is limited, precluding our ability to identify promising therapeutic targets. Here, we hypothesized that chronic alcohol exposure compromises anti-inflammatory signaling in the central amygdala, a brain region implicated in anxiety and addiction, consequently perpetuating a pro-inflammatory state driving aberrant neuronal activity underlying pathological behaviors. We found that alcohol dependence alters the global brain immune landscape increasing IL-10 producing microglia and T-regulatory cells but decreasing local amygdala IL-10 levels. Amygdala IL-10 overexpression decreases anxiety-like behaviors, suggesting its local role in regulating amygdala-mediated behaviors. Mechanistically, amygdala IL-10 signaling through PI3K and p38 MAPK modulates GABA transmission directly at presynaptic terminals and indirectly through alterations in spontaneous firing. Alcohol dependence-induces neuroadaptations in IL-10 signaling leading to an overall IL-10-induced decrease in GABA transmission, which normalizes dependence-induced elevated amygdala GABA transmission. Notably, amygdala IL-10 overexpression abolishes escalation of alcohol intake, a diagnostic criterion of AUD, in dependent mice. This highlights the importance of amygdala IL-10 signaling in modulating neuronal activity and underlying anxiety-like behavior and aberrant alcohol intake, providing a new framework for therapeutic intervention.
Keywords: IL-10, amygdala, alcohol, GABA transmission, anxiety, alcohol drinking
Introduction
Repeated alcohol exposure elicits neuroimmune responses in the brain contributing to the transition and maintenance of alcohol dependence, in part through persistent changes in neuronal activity in critical addiction-related brain regions. Understanding the alcohol-induced neuroimmune response is important for the development of new therapeutic strategies for alcohol use disorder (AUD), which are currently very limited. While pro-inflammatory mediators initiate and drive the neuroimmune response, anti-inflammatory mediators provide an important homeostatic mechanism to limit inflammation and prevent pathological damage. Indeed, several anti-inflammatory drugs including minocycline, topiramate, and pioglitazone have shown efficacy in decreasing alcohol drinking in preclinical models, highlighting the potential use of anti-inflammatories for AUD (Mayfield, Ferguson, and Harris 2013). However, our understanding of the role of anti-inflammatory processes in the neurobiological and behavioral adaptions induced by chronic alcohol exposure is limited.
Interleukin-10 (IL-10), a major anti-inflammatory cytokine, and its cognate IL-10 receptor (IL-10R) are expressed in the brain (Kwilasz et al. 2015; Fickenscher et al. 2002) and have been implicated in AUD. Human genetic studies have identified an IL10 gene polymorphism associated with alcoholism (Marcos et al. 2008), and humans with an AUD have reduced IL-10R levels in the amygdala and cortex (Ponomarev et al. 2012). Limited preclinical studies have also shown that alcohol alters IL-10 levels (Roberto, Patel, and Bajo 2017), and IL-10 infusion in the basolateral amygdala decreases binge drinking (Marshall et al. 2017). Despite the growing evidence for a role of IL-10 in regulation of alcohol-related behaviors, a mechanistic understanding of IL-10 signaling and its interaction with alcohol in critical addiction-related regions is lacking. Additionally, it is unknown whether IL-10 plays a role in regulating alcohol dependence-induced behaviors such as escalated alcohol drinking.
The amygdala is important for valence and emotional processing and comprises several nuclei with discrete connectivity within and across nuclei (Janak and Tye 2015). This includes the predominantly glutamatergic basolateral amygdala (BLA) subdivided into the lateral amygdala (LA) and basal amygdala (BA) along with the predominantly GABAergic central nucleus of the amygdala (CeA) largely subdivided into the lateral and medial subdivisions (Duvarci and Pare 2014)). The lateral CeA sends GABAergic to the medial CeA in cell type and projection-specific manner to gate behavior (Ciocchi et al. 2010; Haubensak et al. 2010; Li et al. 2013; Tye et al. 2011). The CeA is regarded as the major output nucleus of the amygdala complex, wherein dysregulated GABA transmission has been critically implicated in alcohol dependence-induced behaviors (Roberto, Gilpin, and Siggins 2012). Acute and chronic alcohol increase CeA GABAergic transmission in rodent and non-human primate models (Roberto et al. 2003; Roberto et al. 2004; Augier et al. 2018; Jimenez et al. 2019), suggesting a conserved mechanism of alcohol’s action and the particular vulnerability of limbic brain regions to the effects of alcohol. Modulation of GABA transmission in this region alters alcohol consumption in preclinical rodent models of alcohol dependence (Roberto et al. 2010; Hyytia and Koob 1995; Gilpin and Roberto 2012). A defining feature of alcohol dependence is escalated alcohol intake and loss of control over alcohol use, which contributes to neurodegeneration, neuronal dysfunction and other comorbid pathologies. Moreover, the CeA also mediates negative affective states such as fear and anxiety, which can provide motivational drive for consumption of alcohol (Fadok et al. 2018). Therefore, the CeA is a critical addiction-related brain region, wherein neuroimmune signaling could have a major impact on alcohol dependence-induced behaviors.
In this study, we hypothesized that chronic alcohol exposure leads to global alterations in the brain immune cell landscape and that regional changes in neuroimmune signaling regulate local neuronal activity contributing to alcohol dependence-induced behaviors. Specifically, we tested the hypothesis that IL-10 signaling can modulate GABAergic transmission in the CeA, and that alcohol dependence induced neuroadaptations in IL-10 signaling lead to aberrant GABAergic transmission in this region underlying escalated alcohol intake. Our results demonstrate a specific cellular immune response to alcohol dependence in the brain and a critical role of IL-10 signaling in regulating neuronal activity and amygdala mediated behaviors including anxiety and dependence-induced escalation of alcohol intake. These findings highlight the important role of IL-10 signaling in the alcohol-induced neuroimmune response contributing to the escalation of alcohol intake, a hallmark feature of AUD that may be tempered by enhancing IL-10 signaling.
Methods
Animals
Male C57BL/6J and IL10tm1Flv (stock no: 008379) mice were obtained from The Jackson Laboratory (ME) and group-housed in a temperature and humidity-controlled vivarium on a 12 hour reversed light/dark cycle (lights turn off at 8AM) with food and water available ad libitum. A total of 240 mice (naïve: n=117, average weight = 31.3 ± 0.4 g; non-dependent: n=54, average weight = 31.0 ± 0.3 g; dependent: n=69, average weight = 29.6 ± 0.3 g) of a minimum age of 10 weeks were used for experiments. Mice underwent either one of two different alcohol treatment paradigms – two bottle choice-chronic intermittent ethanol vapor exposure (2BC-CIE) or CIE only (detailed below). CIE only exposure was used for multiparameter flow-cytometry analysis, while all other experiments used the CIE-2BC paradigm. All protocols involving the use of experimental animals in this study were approved by The Scripps Research Institute (TSRI) Institutional Animal Care and Use Committee and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Chronic-intermittent ethanol two-bottle choice paradigm
To induce alcohol dependence, we used the two bottle choice - chronic intermittent ethanol vapor exposure (2BC-CIE) paradigm, as previously described (Patel et al. 2019). This method consistently produces alcohol dependence in C57BL/6J mice (Becker and Lopez 2004; Bajo et al. 2016; Huitron-Resendiz et al. 2018), as exhibited by escalated alcohol intake, anxiety-like behavior and reward deficits. Briefly, mice were exposed to limited access alcohol (15% w/v) and water - two bottle choice (2BC) sessions followed by either chronic intermittent alcohol (CIE) exposure in vapor chambers (La Jolla Alcohol Research, La Jolla, CA), to induce alcohol dependence (dependent mice) or control air exposure (non-dependent control mice) in identical chambers. Naïve mice were not exposed to any alcohol either by drinking or vapor exposure.
To establish baseline drinking in the alcohol exposed mice, we performed 2BC testing 5 days per week for 4 consecutive weeks. Thirty minutes before lights off, mice were single housed and given two-hour access to two drinking tubes containing either 15% w/v alcohol or water. Following this baseline phase, mice were divided into two balanced groups with equal alcohol and water consumption (Fig. 1). Dependent and non-dependent groups were exposed to CIE vapor and air, respectively. Mice in the dependent group were i.p. injected with 1.75 g/kg alcohol + 68.1 mg/kg pyrazole (alcohol dehydrogenase inhibitor) and placed in vapor chambers for 4 days (16 hours vapor on, 8 hours off) (Becker and Lopez 2004). Naïve and non-dependent mice were injected with 68.1 mg/kg pyrazole in saline. After pyrazole injection, naïve mice were placed back in their home cages, while non-dependent mice were transferred into air chambers for the same intermittent period as the dependent group. The vapor/air exposure was followed by 72 hours of abstinence and 5 days of 2BC testing. This regimen was repeated for a total of 3-4 full rounds. On the third or fourth day of vapor exposure tail blood was collected to determine blood ethanol levels (BELs). Alcohol drip rates in the vapor chambers were altered such that BELs progressively increased over the vapor rounds to a final target of 200-250 mg/dL. Before euthanasia, dependent mice were exposed to a single alcohol vapor exposure (16 hours) (Fig. 1A), and tail blood was collected to determine terminal BELs. For flow cytometry, a separate cohort of mice (N = 13) were only exposed to consecutive weeks of vapor or air to generate the dependent and non-dependent groups, respectively, and 2BC testing was omitted. To determine the impact of CeA IL-10 overexpression on dependence-induced drinking, a cohort of mice (N = 32) was injected with control or IL-10 virus as described below in ‘Viral injections in the brain’ 4 weeks prior to starting the 2BC-CIE paradigm. Amygdala injection sites were confirmed following behavioral testing (Suppl. Fig. 1), and 6 mice were excluded due to missed injections or loss during vapor exposure.
Figure 1. Two bottle choice - chronic intermittent ethanol (2BC-CIE) vapor exposure paradigm.
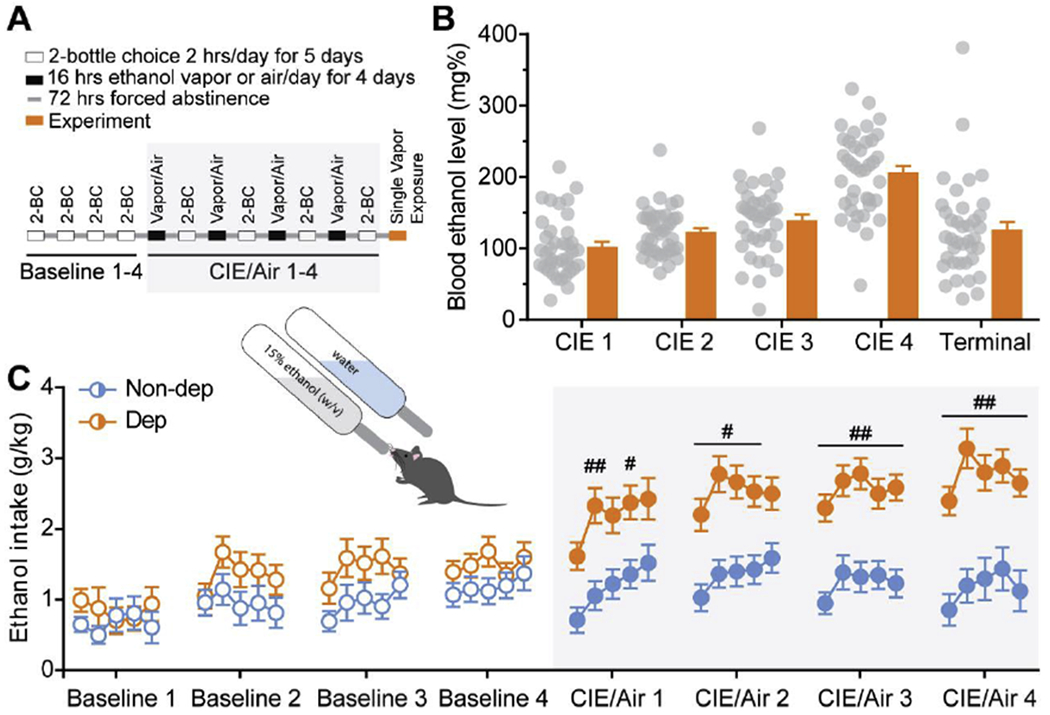
A. Schematic of the timeline for 2BC-CIE paradigm to generate non-dependent and dependent mice. B. Average blood ethanol levels achieved during weeks of CIE vapor exposure in dependent mice. C. Average ethanol intake during 2BC sessions during baseline weeks (open circles) and following air or CIE vapor exposure weeks (greyed box; filled circles) for non-dependent (blue) and dependent (orange) mice, respectively. #, p<0.05; ##, p<0.01 by two-way ANOVA; n=33-49.
In situ hybridization and confocal microscopy
Mice were anesthetized using isoflurane and perfused transcardially with cold phosphate-buffered saline (PBS) and Z-fix (Fisher Scientific, Waltham, MA). Following dissection, brains were immersion fixed in Z-fix for 24 h at 4°C, cryoprotected in 30% sucrose in PBS for 24-48 h at 4° C, flash frozen in pre-chilled isopentane on dry ice and stored at −80° C. Brains were then sliced into 20 μm thick sections, mounted on SuperFrost Plus slides (Fisher Scientific, 1255015), and stored at −80°C until use. In situ hybridization was performed using RNAscope fluorescent multiplex kit (ACD, 320850) as previously described (Wolfe et al. 2019). Briefly, a target retrieval pretreatment protocol was performed as outlined in the RNAscope manual (ACD, doc.no. 320535) by submerging slides in target retrieval buffer (ACD, 322000) at 95-98°C for 10 minutes, immediately washing in distilled water, followed by dehydration in 100% ethanol (storage at −80°C if required), and lastly Protease IV was applied to slides for 20 min at 40°C. Next, the RNAScope Fluorescent Multiplex Reagent Kit User Manual (ACD, doc.no. 320293) was followed and slides were mounted with DAPI-containing Vectashield (Fisher Scientific, NC9029229). The probes used from ACD Biotechne were as follows: negative control (320751), Il-10ra (517731-C1), Gfap (313211-C2), Itgam (311491-C2), and Rbfox3 (313311-C2). Images were acquired using a Zeiss LSM 780 laser scanning confocal microscope (40X oil immersion, 1024x1024, tile scanning of CeA at approximately bregma −1.46mm, 5-μm z-stacks). All microscope settings were kept the same within experiments during image acquisition. Quantification was performed using Fiji (Schindelin et al. 2012) by identifying nuclei based on DAPI staining in the medial subdivision of CeA and measuring corresponding fluorescence intensity within nuclei for each probe after background (mean intensity of the negative control) subtraction. The percent of positive nuclei for co-labeled cells was calculated, and in instances of multiple treatment groups normalized to the control/na’ive group to show relative values. Analysis and statistics were performed using R programming (Team 2018). All analyses were performed on raw images. Outliers detected by Grubb’s test were excluded.
Western blot analysis
Western blot analyses for brain regional changes in protein were conducted in mice as previously described in rats (Pahng et al. 2017). Mice were anesthetized with 3-5% isoflurane, decapitated, and brains were rapidly removed, snap-frozen in isopentane, and stored at −80°C. Twelve to twenty-four hours before dissection, brains were moved to −20°C. Brains were then mounted and sliced using a cryostat at −12°C, amygdala brain punches (0.5 mm thick) were obtained using a 16 gauge needle according to the mouse brain atlas (Franklin 2008). Brain punches were homogenized by sonication in a lysis buffer (in (mM): 320 sucrose, 5 HEPES, 1 EGTA, 1 EDTA, 1% SDS, protease inhibitor cocktail (diluted 1:100), and phosphatase inhibitor cocktails II and III (diluted 1:100); Sigma, St. Louis, MO, USA). Tissue homogenates were heated at 95°C for 5 minutes and the total protein concentration was measured using a detergent-compatible Lowry method (Bio-Rad, Hercules, CA, USA). The samples were aliquoted and stored at −80°C until use. Protein samples (20 μg) were separated by SDS-polyacrylamide gel electrophoresis on 12% acrylamide gels using a Tris/Tricine/SDS buffer system (Bio-Rad). The gels were electrophoretically transferred to polyvinylidene difluoride membranes (GE Healthcare, Piscataway, NJ, USA). Membranes were blocked for 1 h in 5% non-fat milk at room temperature and incubated overnight in 2.5% non-fat milk with primary antibody at 4°C. The primary antibody was mouse IL-10Rα (1:2500; R&D Systems; Cat # AF-474-NA). Membranes were washed and incubated with species-specific peroxidase-conjugated secondary antibodies (1:10,000-1:20,000; Bio-Rad) for 1 h at room temperature. Membranes were washed and incubated in a chemiluminescent reagent (Immobilon Crescendo Western HRP Substrate, Millipore Corporation, Billerica, MA, USA) and exposed to film. Following film development, membranes were stripped for 30 m at room temperature (Restore; Thermo Scientific) and reprobed for β-tubulin (1:1,000,000; Santa Cruz Biotechnology; Cat # 05-797R) levels. The immunoreactivity of the bands was detected using densitometry (Image J 1.45S; Bethesda, MD). To normalize the data across the blots, the densitometric values were expressed as a percentage of the mean of the naive controls for each gel. Individual mouse IL-10 Rα levels were normalized to individual β-tubulin protein levels to generate IL-10 Rα: β-tubulin ratio values for statistical comparison.
Multiplex assay of cytokine/chemokine levels
PBS perfused brains were flash-frozen in isopentane, and 1 mm diameter CeA punches were collected and sonicated in lysis buffer containing 0.1 M phosphate buffer saline, 1 % triton x-100, 1x Halt Protease and Phosphatase Inhibitor Cocktail (#78441; Thermo Scientific, Waltham, MA). Samples were run as doubles and were tested using the MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel kit (#MCYTOMAG-70K-PX32; MilliporeSigma, Burlington, MA) and Luminex MAGPIX® System (MilliporeSigma, Burlington, MA) according to the manufacturer’s instructions. Cytokine levels determined by the assay in each sample (pg/ml) were normalized to its total loading protein concentration (pg/mg) determined with DC™ Protein Assay (Bio-Rad, Hercules, CA). For statistical analyses and presentation, we used cytokine values expressed as a percentage of the mean of the controls for each cytokine and total protein normalized cytokine values were used for correlation analysis.
To measure the levels of viral-mediated IL-10 overexpression, we used a similar protocol with the following modifications. Tissue samples were homogenized using Bio-Plex Cell Lysis Kit (171304011, Bio-Rad, Hercules, CA) following the manufacturer’s protocol. Bio-Plex Pro Mouse Cytokine IL-10 (171G5009M, BioRad) was used, and data were acquired by Bio-Plex MAGPIX Multiplex Reader (Bio-Rad). IL-10 levels (pg/ml) were normalized against the loading protein concentrations, and IL-10 concentrations are presented as pg/mg of total proteins normalized to control.
Brain immune cell isolation and multiparameter flow cytometry analysis
Male IL10tm1Flv (IL-10 reporter) mice underwent air or CIE exposure (no 2BC testing) to generate non-dependent and dependent mice, respectively, for flow cytometry analysis. Single cell suspensions from brains were generated by mechanical disruption using a glass homogenizer. Briefly, whole brains were isolated and immediately transferred to a 7 ml glass tissue grinder (Corning Cat# 7727-07) in 5 ml of RPMI (HyClone Cat# SH30027.02). Brains were homogenized until the solution became a slurry (approximately 6-10 dounces). Homogenized brain suspensions were then passed through a 70uM strainer into a 50ml conical tube containing percoll solution (1 ml 10X PBS, 9 ml Percoll (GE Health Care Life Sciences Cat# 17-0891-02), 10 mL RPMI). Homogenized brain suspension and percoll solution was mixed thoroughly and centrifuged at 1500 RPM for 10 m. After centrifugation, myelin debris layer was aspirated and disposed using sterile disposable pipettes. 50mL conical tubes containing cells were topped off with RPMI to a total volume of 50 ml and centrifuged again at 1500 RPM for 10 m. Supernatant was aspirated off and cell pellet were resuspended in 2 ml of RPMI. Cells were counted using a hemocytometer.
Mouse brain cells were stained for extracellular markers in PBS with 1% bovine serum for 30 m, washed, fixed, permeabilized and stained for intracellular marker using the FoxP3 permeabilization buffer kit (Tonbo Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions. Cells were incubated for 10 m with Fcblock (Biolegend, San Diego, CA) before extracellular and intracellular staining. Flow cytometry analyses were performed on a four-laser Aurora (Cytek, Fremont, CA), and data was analyzed using FlowJo (Becton, Dickinson and Company, Franklin Lakes, NJ). Data were graphed and statistical analyses performed using Graphpad Prism (San Diego, CA). A list of antibody clones and vendors used for these experiments can be found in Suppl. Table 1. Single immune-cell suspensions of brains from dependent and non-dependent animals were compared using the t-Distributed Stochastic Neighbor Embedding (t-SNE) dimensionality reduction algorithm, which provides a two-dimensional visualization of multiparametric single-cell data. t-SNE maps patterns in multi-dimensional data, such as mutli-parameter flow cytometry of immune cells, by detecting clusters based on similarity of specific cell surface or intracellular marker expression(Anchang et al. 2016; Lin et al. 2015; van Unen et al. 2017; Wu and Wu 2010).
Slice preparation and electrophysiological recordings
Mice (n=62) were anesthetized with 3-5% isoflurane, decapitated, and the brains were quickly removed and placed in ice-cold oxygenated (95% O2 and 5% CO2) high-sucrose cutting solution containing (in mM): 206 sucrose, 2.5 KCl, 2.5 CaCl2, 7 MgCl2, 1.2 NaH2PO4, 26 NaHCO3, 5 glucose, 5 HEPES; pH 7.3. Coronal slices (300 μM) containing the CeA were sliced using a 1200S vibratome (Leica Microsystems, Buffalo Grove, IL) and incubated in artificial cerebrospinal fluid (ACSF) containing (in mM): 130 NaCl, 3.5 KCl, 1.25 NaH2PO4, 1.5 MgSO4, 2 CaCl2, 24 NaHCO3, 10 glucose; pH 7.3 at 37°C for 30 m and then at room temperature for at least 30 m before use (Patel et al. 2019; Bajo et al. 2019).
Neurons in the medial subdivision of the CeA that were visualized with infrared differential interference contrast (IR-DIC) optics. Whole-cell voltage-clamp and cell-attached recordings were obtained using voltage-clamp gap-free acquisition mode, digitized at 20kHz sampling frequency, and filtered at 10 kHz using Multiclamp 700B amplifier, Digidata 1440A and pClamp 10 software (Molecular Devices, Sunnyvale, CA). Glass pipettes were pulled to a resistance of 3-6 MΩ and filled with an internal solution containing (in mM): 135 KCl, 5 EGTA, 2 MgCl2, 10 HEPES, 2 Mg-ATP, 0.2 Na-GTP; 290-300 mOsms; pH 7.2-7.3 for whole-cell experiments. ACSF was used as the internal solution for cell-attached recordings. GABAA-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) were pharmacologically isolated with glutamatergic transmission blockers (20μM 6,7-dinitroquinoxaline-2,3-dione, DNQX and 30 μM DL-2-amino-5-phosphonovalerate, AP-5; Tocris Bioscience (Ellisville, MI)) and a GABAB receptor blocker (1 μM CGP55845A; Tocris Bioscience), and tetrodotoxin [0.5 μM, TTX; Sigma Aldrich (St. Louis, MO)] was added to isolate GABAA-mediated miniature inhibitory postsynaptic currents (mIPSCs). CeA neurons were voltage-clamped at −60 mV. Series resistance was not compensated, and cells with a series resistance > 20 MΩ or with a > 20% change during the recording, monitored with 10 mV pulses, or with a holding current ≥ 100 pA were excluded. Drugs were freshly dissolved in ACSF by adding a known concentration of stock solutions and were bath, gravity perfused. Recombinant mouse IL-10 and alcohol were purchased from Peprotech (Rocky Hill, NJ) and Remet (La Mirada, CA). IL-10 serum levels are typically in the 10-200 pg/ml range (Suryanarayanan et al. 2016; Meadows et al. 2017). However, to achieve sufficient perfusion of IL-10, it is necessary to increase drug concentrations. Based on previous electrophysiological studies (Suryanarayanan et al. 2016), we chose a concentration of 50 ng/ml because it achieves a maximal effect on GABAergic transmission. This also allowed for comparison with previous reports (Roberto et al. 2003; Nenov et al. 2019).
Frequency, amplitude, and kinetics of s/mIPSCs and spontaneous firing were analyzed using Mini Analysis (Synaptosoft Inc., Fort Lee, NJ) and Clampfit 10.7 (Molecular Devices), respectively, and all events were visually confirmed. Data were binned into three intervals to obtain average sIPSC characteristics. The maximum value during minute 6 through 15 of drug application was used for analysis and changes greater or equal to 15% compared to baseline control were considered as an increase or decrease. All statistical tests were two-tailed and significance was accepted for p < 0.05. One-sample t-test were used to test for significance against the null hypothesis set to 100, chi squared tests were used to determine changes in the distribution of responses across groups, and one-way ANOVA was used to compare the magnitude of effects across groups. Data were analyzed and graphed using Graphpad Prism 6.01 (San Diego, CA). Data are presented as mean ± standard error (SEM), and n and N represents the sample number of cells and mice, respectively. Brain slices from each mouse were used for different experiments to increase biological diversity in each experimental group.
Viral injections in the brain
We overexpressed IL-10 in the amygdala using a control (pLenti-CMV-copGFP-2A-Puro) and IL-10 expressing virus (pLenti-CMV-IL-10-copGFP-2A-Puro; Applied Biological Materials Inc., Richmond, BC, Canada). Briefly, mice were anesthetized with 2-5% isoflurane and secured in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). Viruses were bilaterally injected into the amygdala (AP: −1.09, DV: 4.5, ML: 3.0 – from dura) at 100 nL/min for a total volume of 1μL using custom 28 gauge injectors (Plastics One Inc., Roanoke, VA) connected to a microfluidic syringe pump (Harvard Apparatus) and 10 μL Hamilton syringes. After viral injection, injectors were kept in place for an additional 10 mins before slowly removing. Mice remained group-housed throughout the experiment and were allowed to recover for 4 weeks prior to undergoing behavioral testing. After experimental testing, brains were collected (as described above for in situ hybridization) and used for histological verification of injections sites. Briefly, slides were blocked [10% normal donkey serum (NDS), 0.5% triton x100 in PBS] at room temperature, incubated with primary antibody for rabbit anti-copepod GFP (copGFP/ppluGFP2) (ThermoFisher, PA5-22688) in antibody diluent (2%NDS, 0.1% triton x100 in PBS) overnight at 4° C, washed in PBS containing 0.2% triton x100 (PBS-T) three times for 10 m, incubated in secondary antibody Alexa Fluor 488 donkey anit-rabbit (Jackson ImmunoResearch, 711-545-152) for 2 h at room temperature and washed in PBS-T again before mounting in vectashield+DAPI (Fisher Scientific, NC9029229). All mice with injection sites in the amygdala (including the central amygdala and basolateral amygdala) were included in behavioral analysis (Suppl. Fig. 1).
Behavioral testing
Elevated plus maze test:
Mice were placed in a center of an elevated plus maze apparatus (containing two open arms (5 by 30 cm) and two enclosed arms (5 by 30 cm) connected by a central area (5 by 5 cm) positioned 30 cm above the floor) facing an open arm and videotaped for 5 minutes without the experimenter in the room. The percentage of open arm entries, open arm time, the number of times mice crossed the midline of the open arm (i.e. went towards the open ends), and total arm entries were recorded. An arm entry was defined as all four limbs of mouse on the arm.
Novelty-suppressed feeding test:
After 24 hours of food deprivation, mice were placed in a novel, open arena (50 cm X 50 cm) (center lux of 400) with a piece of food in the center, and the latency to feed was measured. Immediately after mice began to feed in the open arena, they were moved to their homecage (red light) and the same piece of food was placed in the center, and the latency to feed was again measured.
Digging behavior:
Mice were placed in the center of a standard mouse cage filled 5 cm deep with wood-chip bedding in red light for 3 minutes. Activity of mice including the latency to first digging bout, number of digging bouts, and total time digging was recorded. All behavioral testing apparati were thoroughly cleaned with 70% ethanol between mice, and behavioral testing began approximately two hours into the start of the dark cycle. Tests were separated by 2-3 days and occurred in the following order elevated plus maze test, novelty-suppressed feeding test, and digging behavior. All data were hand scored either live or from video recordings, and experimenters were blinded to conditions during testing and scoring. Two cohorts of mice (N = 16 and 23) were separately run by multiple experimenters. Data showed similar trends in both cohorts and were combined. CeA punches (1mm) from the first cohort were used to confirm viral mediated overexpression using a multiplex assay described above in ‘Multiplex assay of cytokine/chemokine levels’. Amygdala injections sites were confirmed in the second cohort (Suppl. Fig. 1)
Results
Chronic alcohol exposure alters the brain immune cell landscape.
Alcohol elicits a neuroimmune response in the brain; therefore, we hypothesized that alcohol dependence would lead to global alterations in the composition of brain immune cells. We used IL-10 reporter mice and chronic intermittent ethanol or air exposure to generate ethanol dependent and non-dependent mice, respectively (Fig 1. Described in detail in the methods). We first assessed the full heterogeneity of brain immune cells using multiparameter flow cytometry and unsupervised hierarchical clustering (Rphenograph) based on the expression of 24 preselected, informative markers. We identified a total of 19 different clusters in non-dependent mice (Fig. 2A). Notably, alcohol dependence differentially altered specific clusters including significantly increasing (e.g. 1, 5, 6) and decreasing (e.g. 11, 18, 19) clusters while having no significant effect on other clusters. Of the other clusters significantly increased by alcohol dependence, cluster 1 was especially interesting due to its high expression of cytokines including IL-10 and IFNγ as well as major histocompatibility complex class two (MCH II) and EGF module-containing mucin-like receptor (F4/80), possibly representing an activated microglial population (Bo et al. 1994; Ulvestad et al. 1994; Wyss-Coray and Mucke 2002). Cluster 1 also had a high expression of programmed cell death protein 1 (PD-1), making it distinct from its nearest related cluster 4, which was unaltered by alcohol dependence. Cluster 5 was increased by alcohol dependence and had the greatest expression of forkhead box protein P3 (FoxP3), a specific marker for T-regulatory cells, compared to all the other clusters. Cluster 6 was also increased following alcohol dependence, which highly expressed glial fibrillary acidic protein (GFAP) and MHC II, suggesting a possible activated astrocytic population. Interestingly, all the significantly decreased clusters had high expression of GFAP, an astrocyte marker. Overall, these data highlight the heterogeneity of immune populations in the brain and the specificity of the effects of alcohol dependence on distinct immune populations, suggesting heightened microglial activation and neuroimmune signaling in dependent mice.
Figure 2. Whole brain flow cytometry analysis of IL-10 and IL-10 receptor expressing cells.
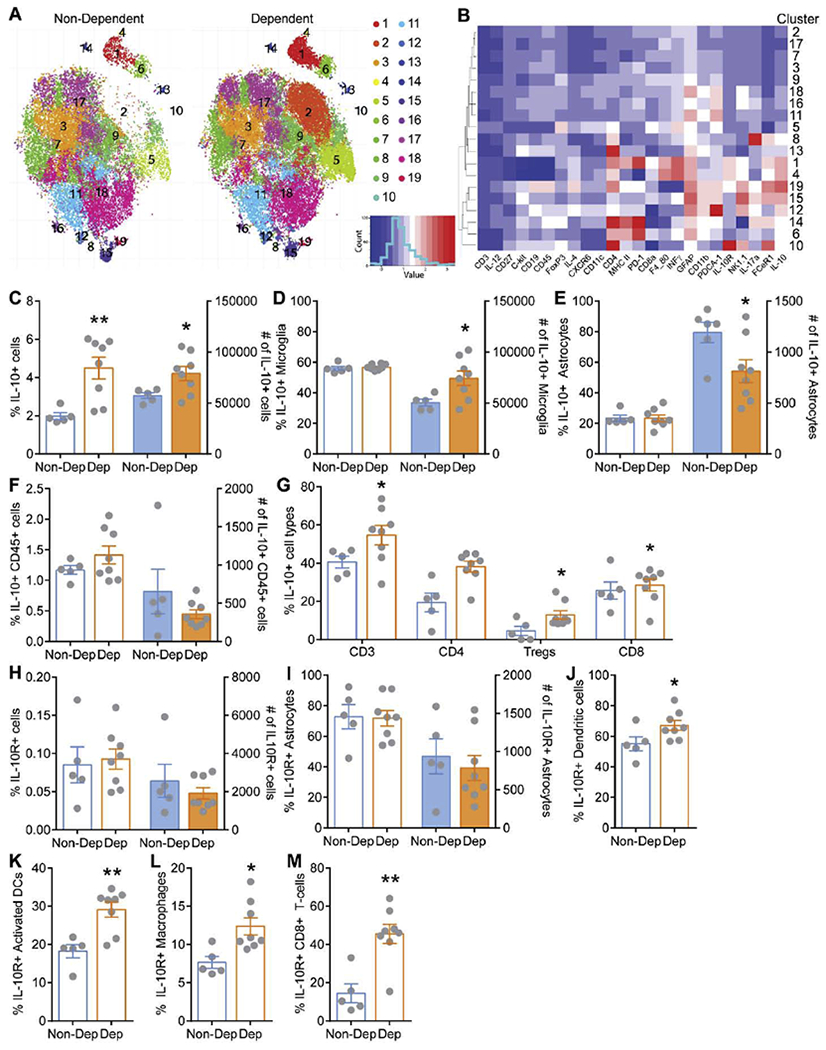
A. Rphenograph clustering plots from multiparameter flow cytometry analysis for non-dependent and alcohol dependent mice. B. Heat plot depicting marker expression across clusters. C. Percent (open bars) and total number (filled bars) of brain IL-10+ cells from all live, single cells isolated from non-dependent (Non-Dep, blue) and dependent (Dep, orange) mice. Open bars correspond to left y-axis and filled bars correspond to right y-axis. D. Percent and total number of brain IL-10+ microglia (CD45dim CD11b+). E. Percent and total number of brain IL-10+ astrocytes (CD45− GFAP+). F. Percent and total number of brain IL-10+ hematopoietic cells (CD45high). G. Percent of brain IL-10+ subsets of hematopoietic cells including T cells (CD45high CD3), helper T-cells (CD45high CD4), regulatory T cells (CD45high Foxp3+), and cytotoxic T cells (CD45high CD8). H. Total percent and number of brain IL-10 receptor expressing (IL-10R+) from all live, single cells isolated from Non-dep and Dep mice. I. Percent and total number of brain IL-10R+ astrocytes (CD45− GFAP+). J-M. Percent brain IL-10R+ dendritic cells (CD45+ C3− CD19-NK1.1− CD11c+), activated dendritic cells (CD45+ C3− CD19− NK1.1− CD11c+ MHC-II+), macrophages (CD45+ CD3− CD19− NK1.1− F4/80+), and cytotoxic T-cells (CD45+ CD3+ CD4− CD8a+) in Non-Dep and Dep mice. *, p<0.05; **, p<0.01 by t-test; N=5-8.
To date, most studies have predominantly focused on the role of pro-inflammatory mediators in alcohol dependence. Importantly, pro-inflammatory mediators can stimulate the production of IL-10 (Williams et al. 1996; Ishii et al. 2013; Veroni et al. 2010), which critically limits inflammation. Consistent with this, clustering analysis also revealed dysregulation of brain immune populations expressing IL-10 (e.g. cluster 1) by alcohol dependence, which may lead to compromised anti-inflammatory signaling. We therefore specifically investigated the brain immune cells expressing IL-10 (i.e. GFP expressing cells) and its cognate IL-10 receptor in alcohol dependent and non-dependent mice. We found a significantly higher percentage and total number of IL-10-producing cells in the brain of alcohol dependent compared to non-dependent control mice (Fig. 2C). The large majority of IL-10 producing cells were microglia (CD45dim CD11b+), and while their percentages were similar in the two groups, a significantly greater number of microglia produced IL-10 in alcohol dependent compared to non-dependent brains (Fig. 2D). In contrast, while there was no difference in the overall percentage of IL-10 producing astrocytes, a significantly fewer number of astrocytes produced IL-10 in alcohol dependent compared to non-dependent control brains (Fig. 2E). We further examined IL-10-producing lymphocytes, conventional macrophages, and dendritic cells. While the overall percentage and total numbers of IL-10-producing hematopoietic cells (CD45+) was similar (Fig. 2F), further examination of individual immune cell types revealed that the percentage of IL-10 producing T cells, specifically T helper cells and amongst them T-regulatory cells increased in alcohol dependent compared to non-dependent control brains (Fig. 2G). Nevertheless, astrocytes and hematopoietic immune cells were much less frequent in numbers compared to microglia. B cells and natural killer cells were also analyzed but did not produce significant amounts of IL-10.
Next, we identified the IL-10-responsive cell types by evaluating IL-10R expressing cells. We found that the overall percentage and total number of IL-10R expressing cells were much less prevalent than IL-10 producing cells and did not change between non-dependent and alcohol dependent mice (Fig. 2H). Notably, most astrocytes expressed IL-10R, but did so equally in both groups (Fig. 2I). Interestingly, the percentage of IL-10R expressing dendritic cells (DCs; CD45+ CD3− CD19− CD11c+) was significantly elevated in alcohol dependent mice, and this difference was even more pronounced when activated DC (MHCII+) were examined (Fig. 2J–K). Macrophages (CD45+ CD3− CD19− F4/80+), who are also antigen presenting cells, and cytotoxic T lymphocytes (CD3+ CD4− CD8+) likewise presented with a higher percentage of IL-10R expression in alcohol dependent mice compared to non-dependent mice (Fig. 2L–M). Of note, 10% of all IL-10R expressing cells were B cells, but the percentage of IL-10R expressing B cells did not change with chronic alcohol exposure. NK cells did not express significant amounts of IL-10R in either group. In summary, microglia are the most prominent immune cell type in the brain and the major producers of IL-10, which are increased in alcohol dependent compared to non-dependent mice along with IL-10 producing T-regulatory cells. In contrast, overall IL-10R expressing immune cells are unaltered by alcohol dependence, but a higher frequency of DCs, activated DCs, macrophages, and cytotoxic T lymphocytes expressed IL-10R.
CeA IL-10 abundance is decreased following chronic alcohol exposure.
Since alcohol dependence induces global alterations in IL-10 and IL-10R producing cells in the brain, we next asked whether this leads to changes in local CeA IL-10 and IL-10R levels. As mentioned, the CeA is the major output nucleus of the amygdala complex and has been critically implicated in both anxiety-like behavior and alcohol dependence (Roberto, Gilpin, and Siggins 2012; Fadok et al. 2018). We used a multiplex assay to measure several cytokine levels in the CeA from naïve, non-dependent and dependent mice (Suppl. Table 2). We found that overall IL-10 abundance was significantly decreased in alcohol dependent compared to naive mice (Fig. 3A), suggesting that CeA IL-10 signaling is compromised following chronic alcohol exposure. Previously we have reported increased CeA IL-1β levels, a prominent pro-inflammatory cytokine that initiates the neuroimmune response, in non-dependent and alcohol dependent mice compared to naive (Patel et al. 2019), and we observed similar trends towards an increase in IL-1β levels in this study (Suppl. Table 2), supporting the hypothesis that alcohol exposure induces an ongoing neuroimmune response in critical addiction-related brain regions.
Figure 3. IL-10 and IL-10 receptor levels in the central amygdala.
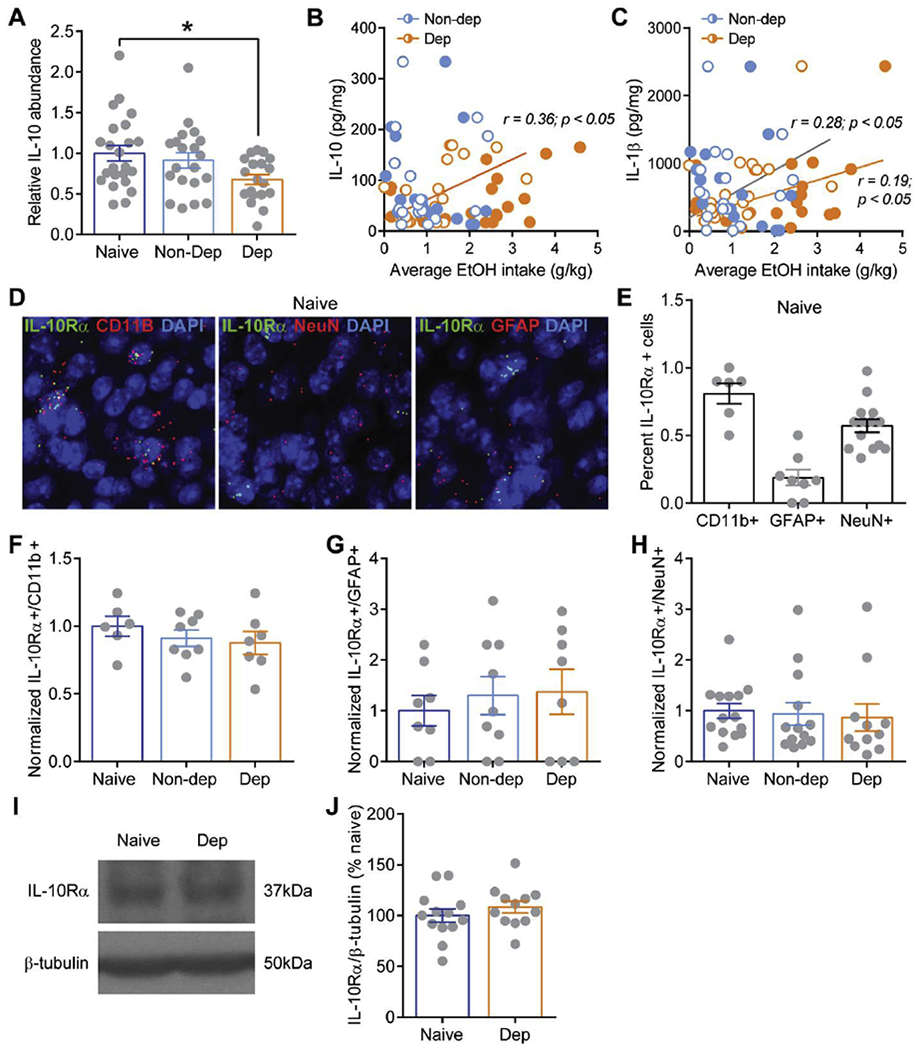
A. Normalized IL-10 protein abundance in the central amygdala of naïve (dark blue), non-dependent (Non-Dep; blue), and dependent (Dep; orange) mice. B, C. Correlation of IL-10 and IL-1β protein abundance, respectively, and average alcohol intake during baseline (open circles) and following CIE (filled circles) for Non-Dep (blue) and Dep (orange) mice. D. Representative medial subdivision of the CeA images from in situ hybridization analysis of Il-10ra (i.e. IL-10Rα) co-localization with Itgam (i.e. CD11b, left), Rbfox3 (i.e. NeuN, middle), and Gfap (i.e. GFAP, right) labeling putative microglia, neurons, and astrocytes, respectively. Scale bar=10um E. Percent of Il-10ra+ expressing nuclei co-labeled with Itgam, Gfap, and Rbfox3. F-H. Percent of Il-10ra+ nuclei co-labeled with Itgam, Gfap, and Rbfox3 in naïve, Non-dep and Dep mice relative to control naïve. I. Representative western blot images of IL-10R (top) and β-tubulin from a naïve and Dep mouse. J. Summary graph of western blot analysis showing normalized ratios of IL-10R/β-tubulin from naïve and Dep mice. *, p<0.01 by one-way ANOVA.
We also examined the correlation between CeA cytokine levels and the average alcohol intake during the four weeks of baseline two bottle choice testing, average alcohol intake during the four weeks of two bottle choice testing following chronic intermittent ethanol (CIE) vapor or air exposure, and the average escalation in alcohol intake between baseline and following CIE/air calculated as the average baseline intake divided by the average CIE/air intake (Fig. 1 and Suppl. Table 1). Of the significantly correlated cytokines (e.g. IL-1α, IL-2, IL-6, IL-12, KC, and LIF) identified in non-dependent mice, the majority are generally classified as pro-inflammatory and were all negatively correlated with alcohol intake (Suppl. Table 3). In contrast, all significantly correlated pro-inflammatory cytokines (e.g. IL-1β, IL-7, IL-12, TNFα) identified in dependent mice were positively correlated to alcohol intake levels. Notably, IL-10 levels directly correlate with baseline alcohol intake, but not CIE intake or escalation, and only in dependent mice (Fig. 3B). Thus, CeA IL-10 levels may be a predictive marker for later development of alcohol dependence and provides further support that CIE may compromise IL-10 signaling as it is no longer correlated to alcohol intake following CIE. Of note, IL-1β showed direct correlations with baseline as well as CIE alcohol intake only in dependent mice (Fig. 3C), potentially driving the continual neuroimmune response thought to contribute to the transition and maintenance of alcohol dependence. These data show that alcohol dependence induces a specific cytokine response in the CeA, wherein anti-inflammatory signaling is compromised.
Using in situ hybridization, we also examined Il-10ra (i.e. IL-10Rα) expression co-localization with Itgam (i.e.CD11b), Gfap (i.e. GFAP), and Rbfox3 (i.e. NeuN) to identify putative microglia, astrocytes, and neurons, respectively in the medial subdivision of the CeA. We found that Il-10ra is expressed in all three cellular populations, but predominantly in microglia (Fig. 3D–E). Interestingly, we found no change in Il-10ra expression in non-dependent and dependent mice compared to naïve in any of the three cellular populations (Fig. 3F–H), consistent with our flow cytometry analysis. Using western blot analysis to measure CeA IL-10Rα protein, we confirmed that there was no difference in CeA IL-10Rα levels in alcohol dependent mice compared to naïve (Fig. 3I–J). Overall, based on our flow cytometry data and CeA measurements of IL-10 and IL-10Rα, alcohol dependence appears to have a greater impact specifically on IL-10 rather than the IL-10 receptor.
Amygdala IL-10 overexpression decreases anxiety-like behavior.
Chronic alcohol exposure alters CeA IL-10, however it is still unknown if local IL-10 production in discrete brain regions impacts behavior. To address this, we used viral-mediated overexpression of IL-10 or control GFP in the amygdala of naïve mice and measured anxiety-like behaviors, which has been shown to be mediated in part by the CeA (Fadok et al. 2018), on well-established tests including novelty-suppressed feeding test (NSFT), digging, and elevated plus maze test (EPM) (Fig. 4). IL-10 levels were significantly increased in the CeA of IL-10 mice (194.7 ± 38.3% of control; n = 7; p < 0.05 by unpaired t-test) compared to control mice (100.0 ± 10.7% of control; n = 7). In NSFT, we found a significant interaction effect F(1,36)=4.78; p < 0.05 by two-way ANOVA with a significant decrease in latency to feed in the novel, open arena in amygdala IL-10 overexpressing mice compared to control (Fig. 4C), suggesting a greater drive for approach compared to avoidance behaviors. Note, there was no difference in latency to feed in the home cage, suggesting that the difference in latency to feed in the open arena was not due to changes in motivational drive to eat (i.e. hunger levels). Amygdala IL-10 overexpression leads to a trend and significantly decreases the number of digging bouts and total time digging, respectively (Fig. 4D–F). Lastly, in EPM, amygdala IL-10 overexpression significantly increased the percent time spent in open arms, number of entries past the midpoint on the open arms (i.e. went further towards the end of arms), and the percent of open arm entries compared to control with no difference in total arm entries (Fig. 4G–K), suggesting reduced avoidance behaviors and greater exploratory drive. Note, no difference in total arm entries was observed between the groups, suggesting no change in locomotor activity. Overall, amygdala IL-10 overexpression consistently decreases anxiety-like behaviors (increases approach and exploratory behaviors and reduces avoidance behaviors) in all three ethological behavioral tests, and therefore is sufficient to modulate anxiety-like behaviors.
Figure 4. Amygdala IL-10 overexpression effects on anxiety-like behavior in naïve mice.
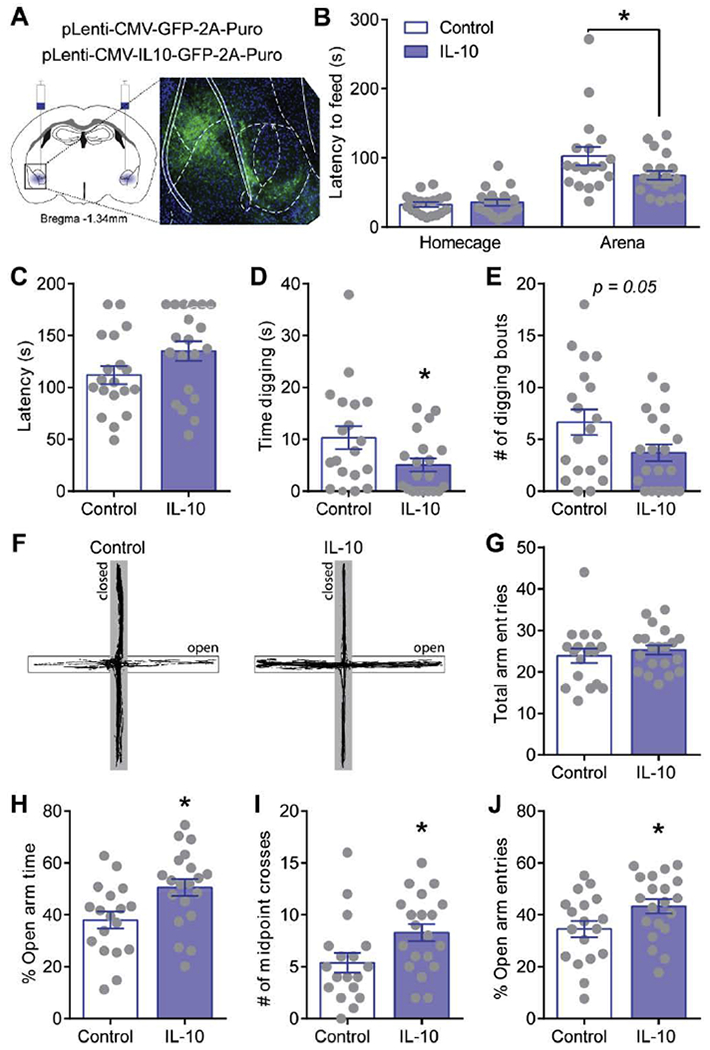
A. Schematic of amygdala viral injection sites and representative image of virus expression of copGFP in the amygdala. B. Latency to feed in homecage and open arena during novelty-suppressed feeding test. C-E. Latency to first digging bout, total time digging, and total number of digging bouts in control (open bars) and amygdala IL-10 overexpressing (filled bars) naïve mice. F. Representative behavioral track tracing on elevated plus maze from a control (left) and amygdala IL-10 overexpressing (right) naïve mouse. Closed arms are presented in greyed area. G-J. Total arm entries, percent open arm time, open arm entries, and number of open arm midpoint crosses during elevated plus maze test. *, p<0.05 by t-test; N=18-20.
IL-10 decreases CeA vesicular GABA release via IL-10 receptors, PI3K and p38 MAPK.
To gain mechanistic insight into how amygdala IL-10 regulates anxiety-like behavior, we asked if IL-10 can directly modulate neuronal activity, specifically CeA GABA transmission. To test this, we recorded miniature inhibitory postsynaptic currents (mIPSCs), to specifically isolate activity at GABA synapses from spontaneous action potential driven network activity, in the medial subdivision of the CeA (Fig. 5). In naïve male and female mice, IL-10 consistently decreased mIPSC frequency (Fig. 5B; Suppl. Fig. 2), suggesting decreased GABA release from presynaptic terminals. This effect was prevented by pre-treatment with an IL-10R neutralizing antibody and either phosphoinositide-3-kinase (PI3K) inhibitors or mitogen-activated protein kinase (MAPK) inhibitors, canonical IL-10R signaling pathways (Fig. 5C). Thus, IL-10 serves as a neuromodulator by mechanistically acting at presynaptic GABAergic terminals to decrease GABA release through its cognate receptor, PI3K, and p38 MAPK pathways.
Figure 5. IL-10 effects on CeA miniature inhibitory postsynaptic currents.
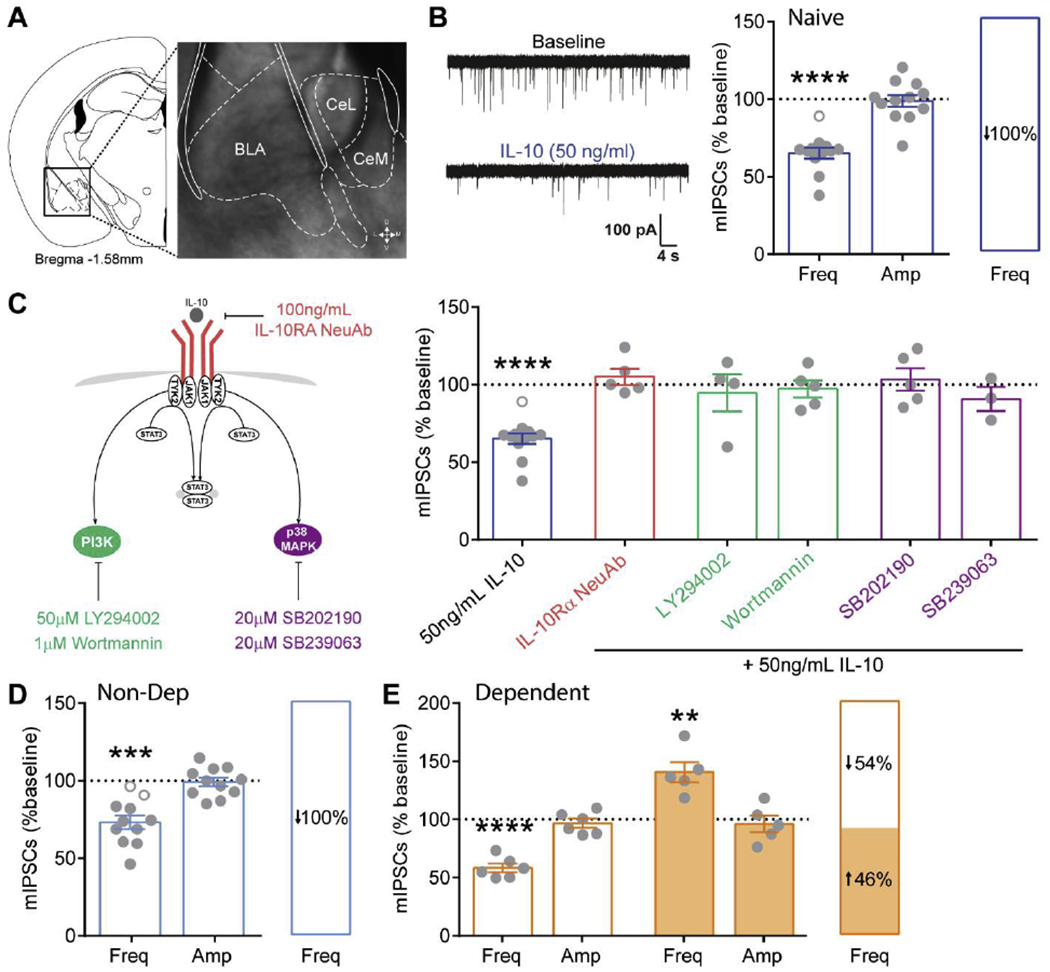
A. Coronal mouse brain atlas (Franklin 2008) image of the CeA and corresponding 4X magnification, brightfield image of boxed region depicting the medial subdivision of the CeA (CeM). B. Representative miniature inhibitory postsynaptic current (mIPSC) traces before (baseline) and during IL-10 application from a naïve mouse (left). Average normalized effect of IL-10 on mIPSC frequency and amplitude (middle). Distribution of IL-10’s effect on mIPSC frequency, showing that all cells responded to IL-10 with a decrease in mIPSC frequency (right). Open grey circles represent cells that responded with <15% change to IL-10. C. Schematic of canonical IL-10/IL-10 receptor signaling pathways (left). Effect of IL-10 on mIPSCs frequency following pretreatment with PI3K and p38 MAPK inhibitors in naïve mice. Note, IL-10 per se effects on mIPSCs coincide with the same group of neurons from panel B for comparison. D, E. Average normalized effect of IL-10 on mIPSC frequency and amplitude in non-dependent (blue) and dependent (orange) mice, respectively, and the distribution of IL-10’s effect on mIPSC frequency. **, p<0.01; ***, p<0.001, ****, p<0.0001 by one-sample t-test; n represented by grey data points in figure.
Similar to naïve mice, IL-10 decreased mIPSC frequency in non-dependent mice (Fig. 5D). Interestingly, IL-10 had dual effects on mIPSC frequency in alcohol dependent mice with 54% of cells displaying a decrease and 46% an increase in mIPSC frequency (Fig. 5E). Taken together with the fact that amygdala IL-10R expression was unchanged by chronic alcohol exposure, this suggests that alcohol dependence induced neuroadaptations in downstream IL-10R signaling altering its action at presynaptic GABAergic terminals.
IL-10 normalizes chronic alcohol induced-aberrant CeA GABAergic transmission.
To determine the overall impact of IL-10 on CeA activity, we then examined the effects of IL-10 on spontaneous inhibitory postsynaptic currents (sIPSCs), where network activity remains intact (Fig. 6). We found that IL-10 has dual effects on sIPSC frequency with 46% of cells displaying a decrease and 54% an increase compared to baseline in naïve mice (Fig. 6A). Similarly, dual effects were observed in non-dependent mice with 43% of cells displaying an increase in sIPSC frequency and 57% a decrease (Fig. 6B). In contrast, IL-10 significantly decreases sIPSC frequency in all cells recorded from alcohol dependent mice (Fig. 6C), suggesting that IL-10 dampens alcohol dependence-induced aberrant CeA GABAergic activity by decreasing action-potential dependent GABA release. Indeed, alcohol dependent mice had a significantly higher baseline sIPSC frequency compared to naïve and non-dependent mice (Fig. 6D), similar to that previously observed in rat and non-human primate models of alcohol dependence (Jimenez et al. 2019; Roberto et al. 2004).
Figure 6. IL-10 effects on CeA spontaneous inhibitory postsynaptic currents and firing.
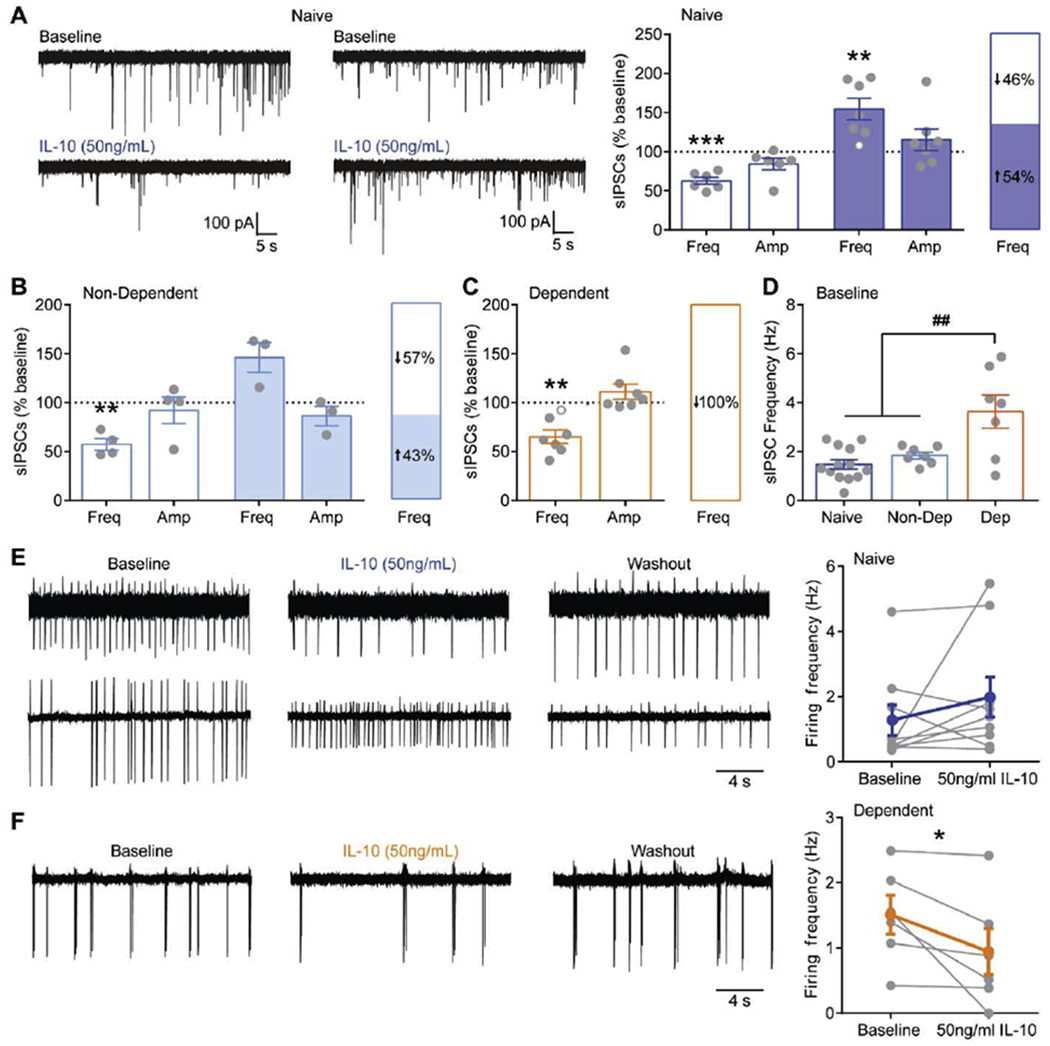
A. Left, Representative traces of spontaneous inhibitory postsynaptic currents (sIPSCs) before (baseline) and after IL-10 application in cells that respond with decreased (top, left) and increased (top, right) sIPSC frequency in naïve mice. Right, Summary of the dual effects of IL-10 on sIPSC frequency and distribution of responses in naïve mice. Open grey circles represent cells that responded with <15% change to IL-10. B. Summary of the dual effects of IL-10 on sIPSC frequency and distribution of responses in non-dependent mice. C.
Summary of IL-10’s effect on sIPSC frequency and amplitude and the distribution of responses in dependent mice. D. Baseline sIPSC frequency in naïve, non-dependent and dependent mice. E. Representative traces of cell-attached spontaneous firing recordings from cells that responded to IL-10 with a decrease (top row) and increase (bottom row) in spontaneous firing in naïve mice. Right, Summary of IL-10’s effects on spontaneous firing in naïve mice where individual cells are displayed in grey and the overall average effect is presented in blue. F. Representative traces of cell-attached spontaneous firing recordings from a dependent mouse. Right, Summary of IL-10’s effects on spontaneous firing in dependent mice. *, p<0.01 by paired t-test; **, p<0.01; ***, p<0.001 by one sample t-test; ##, p<0.01 by one-way ANOVA; n represented by grey data points in figure.
Differential effects of IL-10 on sIPSC (increase and decrease) and mIPSC (decrease) frequency in naïve, suggests that IL-10 also modulates network activity. To examine changes in network activity, we used cell-attached recordings to measure spontaneous firing, which provides the advantages of allowing cytosolic contents to remain intact and prevents changes in firing induced by dialyzing the cell cytosol with an artificial internal solution. We found that IL-10 increased, decreased, or had no effect on firing in naïve mice (Fig. 6E), consistent with the downstream dual effects observed on sIPSC frequency (i.e. GABA release). Note, amplitude of events does not necessarily reflect true changes in membrane voltage and are uninterpretable under cell-attached measurements. Interestingly, IL-10 significantly decreased firing in all cells from alcohol dependent mice (Fig. 6F), consistent with the observed decrease in sIPSC frequency in dependent mice. This suggests that IL-10’s effect on network activity override the effect of IL-10 at local presynaptic GABAergic terminals, where dual effects on mIPSC frequency were observed in dependent mice. Thus, IL-10 modulates CeA network activity contributing to changes in GABA release. In addition, alcohol dependence induces neuroadaptations in IL-10 signaling leading to altered effects of IL-10 at both the presynaptic terminals and on the network level – both of which are consistent with an overall decrease in CeA GABAergic transmission.
CeA IL-10 overexpression blocks chronic alcohol-induced escalation in alcohol intake.
Acute and chronic alcohol potentiate CeA GABAergic transmission (Roberto et al. 2003; Roberto et al. 2004) and modulation of CeA GABAergic transmission alters alcohol drinking and other dependence-induced behaviors (Gilpin and Roberto 2012; Roberto et al. 2010; Hyytia and Koob 1995). Given IL-10’s neuromodulatory effects on GABAergic transmission in the CeA, we hypothesized that IL-10 may regulate alcohol drinking behavior. To test this hypothesis, we virally overexpressed IL-10 or control GFP in the amygdala of mice prior to undergoing the 2BC-CIE paradigm described above (Fig. 1). We observed no differences in water intake (Fig. 7A–B) and blood ethanol levels achieved by CIE exposure (Fig. 7C) between IL-10 overexpressing and control GFP mice, suggesting that IL-10 does not alter water drinking behavior during 2BC or systemic alcohol metabolism. Moreover, no difference in baseline alcohol intake was observed between IL-10 overexpressing and control mice (Fig. 7D). CIE exposure escalated alcohol intake compared to air controls (Fig. 7D), a hallmark of alcohol dependence mimicked in this animal model. Notably, IL-10 overexpression significantly decreases alcohol intake compared to control GFP dependent mice, abolishing CIE-induced escalation in alcohol intake (Fig. 7D). In summary, amygdala IL-10 specifically regulates dependence-induced escalation of alcohol intake.
Figure 7. Amygdala IL-10 overexpression effects on dependence-induced escalated alcohol intake.
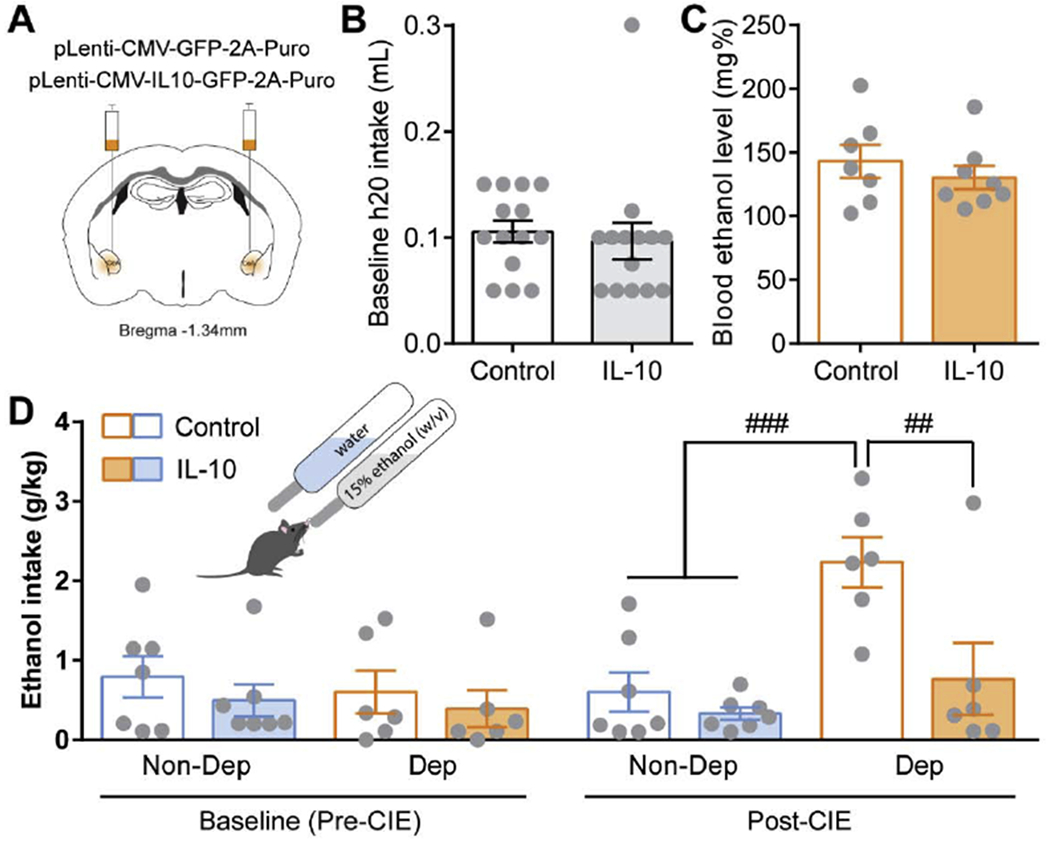
A. Schematic of amygdala viral injection sites. B. Average baseline water intake during 2-bottle choice (2BC) sessions. C. Average blood alcohol level achieved during CIE weeks in control (open bar) and IL-10 overexpressing (filled bar) dependent mice. D. Average alcohol intake pre-CIE (baseline) and post-CIE (following CIE) in control (open bars) and amygdala IL-10 overexpressing (filled bars) non-dependent (blue) and dependent (orange) mice. ##, p<0.01 and ###, p<0.001 by repeated measures, two-way ANOVA and multiple comparisons. N=6-8.
Discussion
In summary, we identified for the first time important global changes in the heterogeneous brain immune cell populations and local neuroimmune adaptations in the central amygdala that critically contribute to the regulation of anxiety-like and alcohol dependence-induced behaviors. We found that alcohol dependence-induced alterations in the landscape of brain immune cells including upregulating IL-10 expressing microglia, the most prominent immune cell type in the brain, and T-regulatory cells. Despite the global increase in IL-10 producing cells, local CeA IL-10 levels were significantly decreased following alcohol dependence. Local changes in IL-10 levels directly impact behavior, given our findings that amygdala IL-10 overexpression is sufficient to decrease anxiety-like behaviors in naïve mice. Mechanistically, IL-10 serves as a neuromodulator of GABAergic transmission by 1) directly decreasing presynaptic GABA release and 2) indirectly by altering spontaneous firing. Importantly, alcohol dependence induced neuroadaptations in CeA IL-10 signaling leads to IL-10-induced decreased GABA release and spontaneous firing, which dampened aberrant GABAergic transmission in this region observed following chronic alcohol exposure. Most notably, amygdala overexpression of IL-10 abolished the escalation of alcohol intake in dependent mice without altering baseline alcohol or water intake. Taken together, these findings highlight the importance of IL-10 signaling in the alcohol-induced neuroimmune response potentially underlying anxiety-like behavior and escalated alcohol intake.
The data-driven approach taken to examine the full heterogeneity of brain immune populations and their response to chronic alcohol exposure identified increases in three clusters that may represent activated microglia, T-regulatory cells and astrocytic populations. Microglial activation is associated with MHC II induction (Almolda, Gonzalez, and Castellano 2011), which was highly expressed in the cluster, and typically occurs during pathological conditions (Wyss-Coray and Mucke 2002). For example, microglial MHC II is upregulated in neurodegenerative diseases such as Alzheimer’s disease and is inversely correlated with cognition (Parachikova et al. 2007). Moreover, it has been reported that MHC II+ IL-10+ microglia are potent inducers of Foxp3+ T-regulatory cells (Ebner et al. 2013). In the periphery, Foxp3+ T-regulatory cells secrete anti-inflammatory cytokines including IL-10 and alter activity of antigen presenting cells (Bettini and Vignali 2009). The ability of microglia to induce T-regulatory cells is dependent on the amount of IFNγ present (Ebner et al. 2013). Consistent with this, a significant increase in the percentage of IFNγ expressing immune cells in alcohol dependent compared to non-dependent mice was observed (data not shown), which has also been observed in the medial prefrontal cortex of humans with an AUD (Johnson et al. 2015). T-regulatory cells are major contributors to the anti-inflammatory response during infection, autoimmune disease, and cancer (Zhao et al. 2011; Lowther and Hafler 2012; Ito et al. 2019). Thus, microglia and T-regulatory immune populations along with IFNγ and IL-10 may represent a common neuroimmune axis induced during chronic CNS disorders including alcohol dependence.
While the overall number of IL-10 producing cells are increased following alcohol dependence, it remains unclear whether there is greater production and release of IL-10 from these cells. IL-10 has important regulatory roles within the CNS including: 1) reducing the antigen-presenting astrocytes and microglia consequently lowering T cell responses and 2) inhibition of pro-inflammatory cytokine production – which are both increased following chronic alcohol exposure. In line with this, we observed a significant increase in IFNγ producing cells in the brain of dependent mice, and IFNγ can stimulate activation of microglia and astrocytes to express MHC II and pro-inflammatory mediators (John, Lee, and Brosnan 2003). This suggests that despite the presence of more IL-10 producing cells, anti-inflammatory IL-10 signaling is compromised and cannot temper alcohol dependence-induced neuroimmune signaling. Overall, the ongoing brain neuroimmune response induced by alcohol dependence engages both pro- and anti-inflammatory mechanisms, and how this balance is fine-tuned in distinct brain regions regulating specific behaviors needs further exploration.
Regional changes may differ from global alterations in immune cell populations. Indeed, we found diminished IL-10 protein levels specifically in the CeA. It has previously been reported that loss of IL-10 promotes differentiation of microglia into an M1 (i.e. pro-inflammatory) phenotype (Laffer et al. 2019). It is possible that these regional differences may shift the local neuroimmune response to more pro-inflammatory signaling driving continual aberrant activity in specific brain regions. Interestingly, of the significant alcohol drinking correlated CeA cytokines identified in non-dependent mice, the majority are generally classified as pro-inflammatory and were all negatively correlated with alcohol intake. In contrast, significant cytokines identified in dependent mice were all positively correlated with drinking. This may be due to differential effects of acute versus chronic exposure to alcohol. In vitro studies in human monocytes show decreased inflammatory signaling following short-term ethanol exposure and increased inflammatory signaling following long-term ethanol exposure (Mandrekar et al. 2009). Furthermore, the innate immune gene polymorphisms associated with increased risk of an AUD in humans generally lead to increased pro-inflammatory responses either by increasing expression of pro-inflammatory mediators or by decreasing anti-inflammatory signaling (Mayfield, Ferguson, and Harris 2013). This suggests that increased pro-inflammatory mediators contribute to the neurobiology of alcohol dependence.
IL-10 is a prominent anti-inflammatory mediator, and its immunological roles are much better understood compared to its neuromodulatory roles in the CNS. IL-10 has been shown to be important for limiting inflammatory signaling and promoting neuronal survival. We found here that IL-10 can serve as a neuromodulator of GABA transmission and spontaneous firing in the CeA of naïve mice. IL-10 has previously been shown to decrease GABA release in the hippocampus (Suryanarayanan et al. 2016) and have dual effects on gonadotropin releasing hormone neuron excitability both depolarizing some neurons and hyperpolarizing others (Barabas et al. 2018), consistent with the effects of IL-10 in the CeA observed in this study. Notably, these effects demonstrate region-specificity, as we did not observe an effect of IL-10 on GABAergic transmission in the medial prefrontal cortex of naive or dependent mice despite similar expression of IL-10 receptors (Suppl. Fig. 3).
Given the heterogenous neuronal populations in the CeA and distributed expression of IL-10 receptors across cell types (Wolfe et al. 2019), the observed dual effects of IL-10 on spontaneous GABA release and firing are unsurprising. Previous findings from our lab and others have shown that distinct amygdala cellular populations (i.e. corticotropin-releasing factor receptor 1 expressing and non-expressing neurons) have differing membrane properties and divergent chronic intermittent ethanol-induced adaptions in GABA transmission (Herman et al. 2013; Herman, Contet, and Roberto 2016; Agoglia et al. 2020). However, we did not observe any correlations between the effect of IL-10 on GABA transmission or firing and the membrane properties of the recorded cell, suggesting a more complex circuit and cell type interaction of IL-10’s effects. One possibility is that differential expression, of downstream conical signaling cascades (i.e. PI3K and p38 MAPK pathways), receptor localization, and receptor coupling may account for the dual effects of IL-10. The CeA also has a dense expression of many neuromodulator systems, which can regulate GABA transmission and neuronal activity (Roberto, Gilpin, and Siggins 2012). It is also possible that IL-10 induced decrease GABA release (as seen in mIPSCs) leads to increased activity of CeA neurons when the network is intact. This could cause indirect effects of neuromodulator release and consequent changes in CeA activity, although this remains to be directly tested. Moreover, we found that alcohol dependence induced adaptations in the neuromodulatory effects of IL-10 on GABAergic transmission and spontaneous firing. Interestingly, IL-10R expression was not significantly altered based on our flow cytometry, in situ hybridization, and western blot studies, suggesting dysregulation of downstream IL-10R signaling. Alcohol exposure directly affects PI3K, a canonical IL-10R signaling pathway, in the brain (Cozzoli et al. 2009; Neasta et al. 2011), which may contribute to the observed effects. Thus, a complex interaction between signaling and circuit level effects of IL-10 simultaneously shape activity in the CeA to regulate behavior, and further dissection of chronic ethanol-induced signaling and circuit disruptions in IL-10 modulation may provide further insight into the behavioral adaptations.
It is becoming increasingly evident that neuroimmune mediators have important roles outside of pathological conditions in regulating physiological functions (Nimmerjahn, Kirchhoff, and Helmchen 2005). There is significant basal expression of IL-10 and IL-10R in naïve mice, and we found that amygdala IL-10 modulates innate anxiety-like behaviors in naïve mice in the absence of a neuroimmune response or disease state. These findings support a previously unknown role for IL-10 in regulating physiological neuronal activity and behavior. It is still unclear which cellular populations mediate the observed behavioral effects of IL-10. Interestingly, acute ethanol is also anxiolytic and increases GABA release in the CeA (Cunningham, Fidler, and Hill 2000; Roberto et al. 2003). While we found that IL-10 has dual effects on spontaneous GABA release, it is possible that IL-10 increases GABA release, like acute ethanol, on the subset of CeA neurons regulating anxiety-like behaviors. In addition, we identified a critical role for IL-10 in specifically regulating alcohol dependence-induced behaviors. The progressive increase in the amount and frequency of drug use is a defining feature of AUD (Diagnostic and Statistical Manual of Mental Disorders, 5th Edition). This is mimicked in the 2-bottle choice-chronic intermittent ethanol vapor exposure paradigm used to induce alcohol dependence in mice, highlighting the face and construct validity of this animal model. We found that amygdala IL-10 overexpression was sufficient to selectively abolish escalated ethanol intake in dependent mice without altering baseline ethanol or water intake, suggesting that IL-10 specifically impacts the motivation for alcohol following chronic alcohol exposure. While it was not directly tested in this study, it is also possible that decreased amygdala IL-10 observed in alcohol dependent mice contributes to heightened anxiety-like behaviors and promotes alcohol drinking behaviors. IL-10 could potentially reduce negative affective states thought to drive escalation in alcohol intake during the development of dependence (Koob 2015). These findings highlight the potential of IL-10 as a novel therapeutic approach for AUD.
It is important to note that this study primarily used male mice. Though, we did find similar effects of IL-10 mediated decreases in CeA GABA release in naïve male and female mice, suggesting that IL-10 may play a similar role in anxiety-like behavior and dependence-induced drinking in females. However, given the known sexual dimorphism in anxiety-like behaviors, alcohol-induced behavioral, neural, and immune responses (Bangasser and Wiersielis 2018; Donner and Lowry 2013; Becker and Koob 2016; Bekhbat and Neigh 2018), a parallel study to examine the role of amygdala IL-10 in anxiety-like behaviors and dependence-induced drinking in females is warranted to identify potential sex differences.
Enhancing anti-inflammatory processes with drugs including minocycline, topiramate, and pioglitazone has been shown to decrease alcohol drinking in preclinical models, suggesting a promising avenue for therapeutic discovery (Mayfield, Ferguson, and Harris 2013). While targeting IL-10 signaling has not yet been attempted for AUD, recombinant IL-10, agonists enhancing IL-10 production, viral delivery of IL-10, and promotion of IL-10 producing B- and T-cells have been used to treat other CNS disorders including EAE, ischemia, Parkinson’s and Alzheimer’s disease (Lobo-Silva et al. 2016). However, these attempts have had mixed results, beneficial in some cases and detrimental to disease progression in others, partly due to the lack of a complete understanding of the spatial and temporal actions of IL-10 during the disease course. The findings from this study suggests that enhancing IL-10 may be an efficacious treatment strategy for AUD. Specifically, enhancing IL-10 producing microglia and T-regulatory cells in limbic brain regions may decrease negative affective states and motivation for alcohol in individuals with an AUD. These findings offer a new avenue of exploration for treating AUD.
Supplementary Material
Acknowledgements
We would like to thank Dr. Nhan Nguyen for assistance with the IL-10 Luminex assay. This work was supported by the National Institute on Alcohol Abuse and Alcoholism [AA021491, AA013498, AA006420, AA017447, AA015566, AA027700, AA026765, AA007456]; and the Pearson Center for Alcoholism and Addiction Research. This is Scripps Manuscript number 29951.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
None.
References
- Agoglia AE, Zhu M, Ying R, Sidhu H, Natividad LA, Wolfe SA, Buczynski MW, Contet C, Parsons LH, Roberto M, and Herman MA. 2020. ‘Corticotropin-Releasing Factor Receptor-1 Neurons in the Lateral Amygdala Display Selective Sensitivity to Acute and Chronic Ethanol Exposure’, eNeuro, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almolda B, Gonzalez B, and Castellano B. 2011. ‘Antigen presentation in EAE: role of microglia, macrophages and dendritic cells’, Front Biosci (Landmark Ed), 16: 1157–71. [DOI] [PubMed] [Google Scholar]
- Anchang B, Hart TD, Bendall SC, Qiu P, Bjornson Z, Linderman M, Nolan GP, and Plevritis SK. 2016. ‘Visualization and cellular hierarchy inference of single-cell data using SPADE’, Nat Protoc, 11: 1264–79. [DOI] [PubMed] [Google Scholar]
- Augier E, Barbier E, Dulman RS, Licheri V, Augier G, Domi E, Barchiesi R, Farris S, Natt D, Mayfield RD, Adermark L, and Heilig M. 2018. ‘A molecular mechanism for choosing alcohol over an alternative reward’, Science, 360: 1321–26. [DOI] [PubMed] [Google Scholar]
- Bajo M, Montgomery SE, Cates LN, Nadav T, Delucchi AM, Cheng K, Yin H, Crawford EF, Roberts AJ, and Roberto M. 2016. ‘Evaluation of TLR4 Inhibitor, T5342126, in Modulation of Ethanol-Drinking Behavior in Alcohol-Dependent Mice’, Alcohol Alcohol, 51: 541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Patel RR, Hedges DM, Varodayan FP, Vlkolinsky R, Davis TD, Burkart MD, Blednov YA, and Roberto M. 2019. ‘Role of MyD88 in IL-1beta and Ethanol Modulation of GABAergic Transmission in the Central Amygdala’, Brain Sci, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser DA, and Wiersielis KR. 2018. ‘Sex differences in stress responses: a critical role for corticotropin-releasing factor’, Hormones (Athens), 17: 5–13. [DOI] [PubMed] [Google Scholar]
- Barabas K, Barad Z, Denes A, Bhattarai JP, Han SK, Kiss E, Sarmay G, and Abraham IM. 2018. ‘The Role of Interleukin-10 in Mediating the Effect of Immune Challenge on Mouse Gonadotropin-Releasing Hormone Neurons In Vivo’, eNeuro, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC, and Lopez MF. 2004. ‘Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice’, Alcohol Clin Exp Res, 28: 1829–38. [DOI] [PubMed] [Google Scholar]
- Becker JB, and Koob GF. 2016. ‘Sex Differences in Animal Models: Focus on Addiction’, Pharmacol Rev, 68: 242–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhbat M, and Neigh GN. 2018. ‘Sex differences in the neuro-immune consequences of stress: Focus on depression and anxiety’, Brain Behav Immun, 67: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini M, and Vignali DA. 2009. ‘Regulatory T cells and inhibitory cytokines in autoimmunity’, Curr Opin Immunol, 21: 612–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo L, Mork S, Kong PA, Nyland H, Pardo CA, and Trapp BD. 1994. ‘Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions’, J Neuroimmunol, 51: 135–46. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Muller C, and Luthi A. 2010. ‘Encoding of conditioned fear in central amygdala inhibitory circuits’, Nature, 468: 277–82. [DOI] [PubMed] [Google Scholar]
- Cozzoli DK, Goulding SP, Zhang PW, Xiao B, Hu JH, Ary AW, Obara I, Rahn A, Abou-Ziab H, Tyrrel B, Marini C, Yoneyama N, Metten P, Snelling C, Dehoff MH, Crabbe JC, Finn DA, Klugmann M, Worley PF, and Szumlinski KK. 2009. ‘Binge drinking upregulates accumbens mGluR5-Homer2-PI3K signaling: functional implications for alcoholism’, J Neurosci, 29: 8655–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Fidler TL, and Hill KG. 2000. ‘Animal models of alcohol’s motivational effects’, Alcohol Res Health, 24: 85–92. [PMC free article] [PubMed] [Google Scholar]
- Donner NC, and Lowry CA. 2013. ‘Sex differences in anxiety and emotional behavior’, Pflugers Arch, 465: 601–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, and Pare D. 2014. ‘Amygdala microcircuits controlling learned fear’, Neuron, 82: 966–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V, Schueler J, Stubbe T, Ellinghaus A, Meisel C, Sawitzki B, and Nitsch R. 2013. ‘Microglial activation milieu controls regulatory T cell responses’, J Immunol, 191: 5594–602. [DOI] [PubMed] [Google Scholar]
- Fadok JP, Markovic M, Tovote P, and Luthi A. 2018. ‘New perspectives on central amygdala function’, Curr Opin Neurobiol, 49: 141–47. [DOI] [PubMed] [Google Scholar]
- Fickenscher H, Hor S, Kupers H, Knappe A, Wittmann S, and Sticht H. 2002. ‘The interleukin-10 family of cytokines’, Trends Immunol, 23: 89–96. [DOI] [PubMed] [Google Scholar]
- Franklin K and Paxinos G . 2008. ‘The Mouse Brain in Stereotaxic Coordinates’.
- Gilpin NW, and Roberto M. 2012. ‘Neuropeptide modulation of central amygdala neuroplasticity is a key mediator of alcohol dependence’, Neurosci BiobehavRev, 36: 873–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Luthi A, and Anderson DJ. 2010. ‘Genetic dissection of an amygdala microcircuit that gates conditioned fear’, Nature, 468: 270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Contet C, Justice NJ, Vale W, and Roberto M. 2013. ‘Novel subunit-specific tonic GABA currents and differential effects of ethanol in the central amygdala of CRF receptor-1 reporter mice’, J Neurosci, 33: 3284–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Contet C, and Roberto M. 2016. ‘A Functional Switch in Tonic GABA Currents Alters the Output of Central Amygdala Corticotropin Releasing Factor Receptor-1 Neurons Following Chronic Ethanol Exposure’, J Neurosci, 36: 10729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitron-Resendiz S, Nadav T, Krause S, Cates-Gatto C, Polis I, and Roberts AJ. 2018. ‘Effects of Withdrawal from Chronic Intermittent Ethanol Exposure on Sleep Characteristics of Female and Male Mice’, Alcohol Clin Exp Res, 42: 540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytia P, and Koob GF. 1995. ‘GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats’, Eur J Pharmacol, 283: 151–9. [DOI] [PubMed] [Google Scholar]
- Ishii H, Tanabe S, Ueno M, Kubo T, Kayama H, Serada S, Fujimoto M, Takeda K, Naka T, and Yamashita T. 2013. ‘ifn-gamma-dependent secretion of IL-10 from Th1 cells and microglia/macrophages contributes to functional recovery after spinal cord injury’, Cell Death Dis, 4: e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, Sakai R, Matsuo K, Nakayama T, Yoshie O, Nakatsukasa H, Chikuma S, Shichita T, and Yoshimura A. 2019. ‘Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery’, Nature, 565: 246–50. [DOI] [PubMed] [Google Scholar]
- Janak PH, and Tye KM. 2015. ‘From circuits to behaviour in the amygdala’, Nature, 517: 284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez VA, Herman MA, Cuzon Carlson VC, Walter NA, Grant KA, and Roberto M. 2019. ‘Synaptic adaptations in the central amygdala and hypothalamic paraventricular nucleus associated with protracted ethanol abstinence in male rhesus monkeys’, Neuropsychopharmacology, 44: 982–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John GR, Lee SC, and Brosnan CF. 2003. ‘Cytokines: powerful regulators of glial cell activation’, Neuroscientist, 9: 10–22. [DOI] [PubMed] [Google Scholar]
- Johnson S, Duncan J, Hussain SA, Chen G, Luo J, McLaurin C, May W, Rajkowska G, Ou XM, Stockmeier CA, and Wang JM. 2015. ‘The IFNgamma-PKR pathway in the prefrontal cortex reactions to chronic excessive alcohol use’, Alcohol Clin Exp Res, 39: 476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwilasz AJ, Grace PM, Serbedzija P, Maier SF, and Watkins LR. 2015. ‘The therapeutic potential of interleukin-10 in neuroimmune diseases’, Neuropharmacology, 96: 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF 2015. ‘The dark side of emotion: the addiction perspective’, Eur J Pharmacol, 753: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffer B, Bauer D, Wasmuth S, Busch M, Jalilvand TV, Thanos S, Meyer Zu Horste G, Loser K, Langmann T, Heiligenhaus A, and Kasper M. 2019. ‘Loss of IL-10 Promotes Differentiation of Microglia to a M1 Phenotype’, Front Cell Neurosci, 13: 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, and Li B. 2013. ‘Experience-dependent modification of a central amygdala fear circuit’, Nat Neurosci, 16: 332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Frelinger J, Jiang W, Finak G, Seshadri C, Bart PA, Pantaleo G, McElrath J, DeRosa S, and Gottardo R. 2015. ‘Identification and visualization of multidimensional antigen-specific T-cell populations in polychromatic cytometry data’, Cytometry A, 87: 675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Silva D, Carriche GM, Castro AG, Roque S, and Saraiva M. 2016. ‘Balancing the immune response in the brain: IL-10 and its regulation’, J Neuroinflammation, 13: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther DE, and Hafler DA. 2012. ‘Regulatory T cells in the central nervous system’, Immunol Rev, 248: 156–69. [DOI] [PubMed] [Google Scholar]
- Mandrekar P, Bala S, Catalano D, Kodys K, and Szabo G. 2009. ‘The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes’, J Immunol, 183: 1320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos M, Pastor I, Gonzalez-Sarmiento R, and Laso FJ. 2008. ‘Interleukin-10 gene polymorphism is associated with alcoholism but not with alcoholic liver disease’, Alcohol Alcohol, 43: 523–8. [DOI] [PubMed] [Google Scholar]
- Marshall SA, McKnight KH, Blose AK, Lysle DT, and Thiele TE. 2017. ‘Modulation of Binge-like Ethanol Consumption by IL-10 Signaling in the Basolateral Amygdala’, J Neuroimmune Pharmacol, 12: 249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J, Ferguson L, and Harris RA. 2013. ‘Neuroimmune signaling: a key component of alcohol abuse’, Curr Opin Neurobiol, 23: 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows JR, Parker C, Gilbert KM, Blossom SJ, and DeWitt JC. 2017. ‘A single dose of trichloroethylene given during development does not substantially alter markers of neuroinflammation in brains of adult mice’, J Immunotoxicol, 14: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neasta J, Ben Hamida S, Yowell QV, Carnicella S, and Ron D. 2011. ‘AKT signaling pathway in the nucleus accumbens mediates excessive alcohol drinking behaviors’, Biol Psychiatry, 70: 575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenov MN, Konakov MV, Teplov IY, and Levin SG. 2019. ‘Interleukin-10 Facilitates Glutamatergic Synaptic Transmission and Homeostatic Plasticity in Cultured Hippocampal Neurons’, Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F. 2005. ‘Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo’, Science, 308: 1314–8. [DOI] [PubMed] [Google Scholar]
- Pahng AR, Paulsen RI, McGinn MA, Edwards KN, and Edwards S. 2017. ‘Neurobiological Correlates of Pain Avoidance-Like Behavior in Morphine-Dependent and Non-Dependent Rats’, Neuroscience, 366: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, and Cotman CW. 2007. ‘Inflammatory changes parallel the early stages of Alzheimer disease’, NeurobiolAging, 28: 1821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Khom S, Steinman MQ, Varodayan FP, Kiosses WB, Hedges DM, Vlkolinsky R, Nadav T, Polis I, Bajo M, Roberts AJ, and Roberto M. 2019. ‘IL-1beta expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala’, Brain Behav Immun, 75: 208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, and Mayfield RD. 2012. ‘Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence’, J Neurosci, 32: 1884–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, and Parsons LH. 2010. ‘Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence’, Biol Psychiatry, 67: 831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, and Siggins GR. 2012. ‘The central amygdala and alcohol: role of gamma-aminobutyric acid, glutamate, and neuropeptides’, Cold Spring Harb Perspect Med, 2: a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, and Siggins GR. 2003. ‘Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons’, Proc Natl Acad Sci U S A, 100: 2053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, and Siggins GR. 2004. ‘Increased GABA release in the central amygdala of ethanol-dependent rats’, J Neurosci, 24: 10159–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Patel RR, and Bajo M. 2017. ‘Ethanol and Cytokines in the Central Nervous System’, Handb Exp Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A. 2012. ‘Fiji: an open-source platform for biological-image analysis’, Nat Methods, 9: 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryanarayanan A, Carter JM, Landin JD, Morrow AL, Werner DF, and Spigelman I. 2016. ‘Role of interleukin-10 (IL-10) in regulation of GABAergic transmission and acute response to ethanol’, Neuropharmacology, 107: 181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R Core. 2018. ‘R: A language and environment for statistical computing’, Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, and Deisseroth K. 2011. ‘Amygdala circuitry mediating reversible and bidirectional control of anxiety’, Nature, 471: 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulvestad E, Williams K, Bo L, Trapp B, Antel J, and Mork S. 1994. ‘HLA class II molecules (HLA-DR, -DP, -DQ) on cells in the human CNS studied in situ and in vitro’, Immunology, 82: 535–41. [PMC free article] [PubMed] [Google Scholar]
- van Unen V, Hollt T, Pezzotti N, Li N, Reinders MJT, Eisemann E, Koning F, Vilanova A, and Lelieveldt BPF. 2017. ‘Visual analysis of mass cytometry data by hierarchical stochastic neighbour embedding reveals rare cell types’, Nat Commun, 8: 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, Columba-Cabezas S, Arico E, Aloisi F, and Agresti C. 2010. ‘Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways’, Mol Cell Neurosci, 45: 234–44. [DOI] [PubMed] [Google Scholar]
- Williams K, Dooley N, Ulvestad E, Becher B, and Antel JP. 1996. ‘IL-10 production by adult human derived microglial cells’, Neurochem Int, 29: 55–64. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Sidhu H, Patel RR, Kreifeldt M, D’Ambrosio SR, Contet C, and Roberto M. 2019. ‘Molecular, Morphological, and Functional Characterization of Corticotropin-Releasing Factor Receptor 1-Expressing Neurons in the Central Nucleus of the Amygdala’, eNeuro, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, and Wu Z. 2010. ‘Exploration, visualization, and preprocessing of high-dimensional data’, Methods Mol Biol, 620: 267–84. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, and Mucke L. 2002. ‘Inflammation in neurodegenerative disease--a double-edged sword’, Neuron, 35: 419–32. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhao J, Fett C, Trandem K, Fleming E, and Perlman S. 2011. ‘IFN-gamma- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis’, J Exp Med, 208: 1571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.