

Abstract
Aggregates or oligomeric forms of many intrinsically disordered proteins (IDPs), including α-synuclein, are hallmarks of neurodegenerative diseases, like Parkinson’s and Alzheimer’s disease, and key contributors to their pathogenesis. Due to their disordered nature and therefore lack of defined drug-binding pockets, IDPs are difficult targets for traditional small molecule drug design and are often referred to as “undruggable”. The 20S proteasome is the main protease that targets IDPs for degradation and therefore small molecule 20S proteasome enhancement presents a novel therapeutic strategy by which these undruggable IDPs could be targeted. The concept of 20S activation is still relatively new, with few potent activators having been identified thus far. Herein, we synthesized and evaluated a library of dihydroquinazoline analogues and discovered several promising new 20S proteasome activators. Further testing of top hits revealed that they can enhance 20S mediated degradation of α-synuclein, the IDP associated with Parkinson’s disease.
Keywords: Proteasome, Activation, Neurodegenerative diseases, Parkinson’s disease, Undruggable
Graphical Abstract
Neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are predicted to become the second-most prevalent cause of death in the next 20 years, making the discovery of disease-modifying therapies an urgent need.1,2 Currently there are no such treatments for any neurodegenerative diseases, further exacerbating the problem.3–5 Although the pathogenesis of these diseases is unclear, the accumulation and aggregation of intrinsically disordered proteins (IDPs) in affected cells is a common feature among them. IDPs are a class of soluble proteins that generally function as regulatory and signalling proteins and are unique in their ability to interact with numerous binding partners due to their disordered nature.6,7 Unfortunately, when these IDPs accumulate within neurons these same features contribute to their aggregation, resulting in harmful signalling events and neurotoxicity.8–12 Additionally, IDPs are difficult to target using traditional small molecule drug design because of a lack of defined binding pockets.
The proteasome is a large enzyme complex that is responsible for the proteolytic degradation of misfolded, redundant and damaged proteins within the cell.13–17 The proteasome exists in equilibrium between the 26S and 20S forms and both serve important roles in maintaining cellular homeostasis. This equilibrium is dictated by the reversible docking of 19S regulatory units (caps) on the 20S core particle to form the fully assembled 26S proteasome. These caps give the 26S proteasome the ability to recognize and unfold ubiquitinylated proteins followed by their proteolytic degradation. The 20S proteasome lacks these 19S caps and constitutes the catalytic core particle of the proteasome. This core particle consists of four stacked concentric rings, two α-rings and two β-rings, each made up of 7 subunits.18,19 The top and bottom α-rings act as a gating mechanism to restrict access to the inner catalytic core of the proteasome.20,21 This inner core is made up of the two β-rings and contains 6 total catalytic sites with two chymotryptic-like, two tryptic-like and two caspase-like threonine protease activities. The 20S proteasome is, unlike the 26S, unable to recognize ubiquitinylated proteins, nor can it unfold and degrade structured proteins.18–21 Consequently, the 20S is restricted to the degradation of IDPs and other unfolded proteins. As a result, the 20S plays a critical role in the regulation of free cytosolic levels of IDPs.13,17,22
IDP levels can become dysregulated as we age due to oxidative stress, reduced proteasome activity and changes in IDP production.23–29 The result is accumulation and aggregation of IDPs like α-synuclein (α-syn) or amyloid beta (Aβ) as seen in PD and AD, respectively.10,12,30–38 Due to this role in the regulation of cellular IDP levels, enhancing 20S proteasome mediated proteolysis has recently emerged as a potential therapeutic strategy for the treatment of neurodegenerative diseases.39–44
Our lab recently reported the discovery of a novel 20S proteasome activator, TCH-165, which induces a conformational change in the α-ring of the 20S proteasome.45,46 This leads to an “open-gate” conformation that allows easier access of IDP substrates to the catalytic core of the 20S, thereby enhancing the rate of their degradation. This method for 20S proteasome enhancement shows potential as a new therapeutic strategy by which accumulation and subsequent aggregation of IDPs can be prevented.40,45,47 Despite findings like these, there are still relatively few reported small molecule activators of the 20S proteasome48 and many still suffer from limitations, such as low potency, off-target effects and poor drug-like properties.40,45,49–54 The continued exploration of 20S proteasome activation as a therapeutic method for neurodegenerative diseases will require additional molecular scaffolds to be explored to identify new lead molecules for testing in model systems. As part of an effort to expand upon the imidazoline-mediated allosteric proteasome modulators,55–57 we screened a range of structural motifs for new 20S proteasome enhancers. Herein, we report on the structure activity relationship (SAR) and efficacy of a novel class of dihydroquinazoline-based 20S proteasome activators.
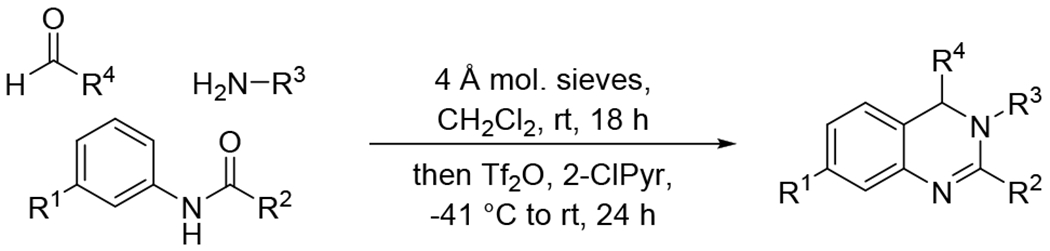
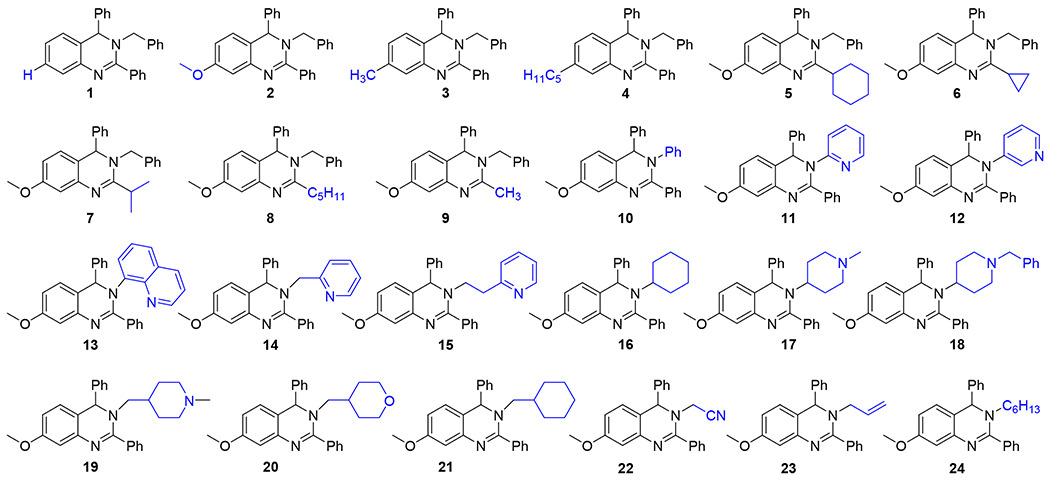
The synthesis of dihydroquinazolines is accomplished via our recently reported one-pot multicomponent reaction of amides, amines, and aldehydes (Scheme 1).58 This method involves in situ imine formation from an amine and an aldehyde in the presence of molecular sieves, followed by tandem assembly of the heterocyclic ring through successive Tf2O-mediated amide dehydration, imine insertion, and Pictet-Spengler-like cyclization. The multicomponent nature of the method permits the construction of highly diverse dihydroquinazolines due to the compatibility of a wide range of simple starting materials. A small library of dihydroquinazolines was generated using the multicomponent method to probe the ability of members of this class of compounds to activate the 20S proteasome. Compounds chosen to populate the library differed in their structural features at the 7-, 2-, and 3-positions (R1, R2, and R3, respectively) of the heterocyclic scaffold (Figure 1). Variation at the 7- and 2-positions was accomplished using select amides (e.g. 1 – 9), while the substituents at the 3-position were introduced using chosen amines (e.g. 10 – 24). R1 and R2 groups introduced from the starting amides provided preliminary structure activity relationship information which was utilized for the construction of the remaining members of the compound library in which the R3 group was varied. Simple alkyl and alkoxy substituents were explored at R1, along with the absence of any additional group at this location (e.g. 1 – 4), and the investigated R2 substituents included alkyl and cycloalkyl groups to compare them to the aromatic counterpart (e.g. 5 – 9 vs 2). A range of R3 substituents were installed to include aryl and heteroaryl groups (e.g. 10 – 13), tethered heteroaryl groups (e.g. 14 – 15), and alkyl groups with varying ring and heteroatom placement (e.g. 16 – 24).
Scheme 1.
Multicomponent synthesis of dihydroquinazolines.
Figure 1.
Structures of dihydroquinazoline analogues.
Screening of this small library of dihydroquinazolines was performed in two stages. In the first stage, each compound was screened at 3 concentrations (3, 10 and 30 μM) to select lead agents. Secondly, lead agents were further analysed using full concentration responses (6-point titration ranging from 1.25 μM-40 μM) for each of the three proteolytic activities of the 20S proteasome and a combination thereof. The proteolytic activity of the 20S proteasome can be monitored in vitro by measuring the cleavage of fluorogenic peptide substrates for the different catalytic sites as an increase in 7-amino-4-methylconmarin (AMC) fluorescence over time.40,43–45,49,59 A combination of chymotrypsin-like (CT-L, Suc-LLVY-AMC), trypsin-like (Tryp-L, Boc-LRR-AMC) and caspase-like (Casp-L, Z-LLE-AMC) peptide substrates were used in equal amounts to screen compounds 1–24 for overall 20S activity. Pure human 20S proteasome was pre-treated with 3, 10 or 30 μM of one of the analogues or DMSO (vehicle control) for 15 minutes at 37°C. To each sample was then added a mixture of the three substrates (13.3 μM each). The release of AMC was monitored as fluorescence overtime for 1 hour and the resulting 20S activity changes were determined by comparing to the untreated 20S and calculating the fold-increase in activity for each analogue at a given concentration (Table 1, 30 μM and Table S1, 3 and 10 μM).
Table 1.
Compounds 1–24 ranked by fold enhancement of 20S activity at 30 μM
| Compound | Fold increase over vehicle (30 μM) |
|---|---|
| 10 | 9.5 |
| 21 | 8.2 |
| 3 | 8.1 |
| 2 | 7.9 |
| 18 | 7 |
| 11 | 6.9 |
| 16 | 6.7 |
| 5 | 6.5 |
| 12 | 5.9 |
| 13 | 5.3 |
| 1 | 4 |
| 8 | 2.8 |
| 15 | 2.8 |
| 4 | 2.8 |
| 6 | 2.7 |
| 22 | 2.7 |
| 7 | 2.3 |
| 23 | 2.3 |
| 14 | 2 |
| 9 | 1.8 |
| 24 | 1.6 |
| 20 | 1.6 |
| 19 | 1.2 |
| 17 | 1 |
The data collected from this screen (Table 1 and see Table S1 for 3 and 10 μM results) shows a few insightful trends in the SAR of the dihydroquinazolines. Small changes in substitution at the 7-position appear to have a significant effect on activity of the dihydroquinazolines. Compound 1, which displays a 4-fold (i.e. 400%) increase over background 20S activity, lacks a substituent at the 7-position but is otherwise identical to compounds 2 (7.9-fold increase) and 3 (8.1-fold increase). This small change results in a reduction in 20S activity from 8-fold enhancement down to a 4-fold enhancement at 30 μM. Similarly, the addition of a longer alkyl chain on compound 4 resulted in a steep drop in 20S activity to 2.8-fold at 30 μM. Changes at the 2-position show similar effects to that of the 7-position, where most substitutions other than a phenyl group (compounds 5–9) caused marked decreases in 20S activity. All have less than 3-fold activation at 30 μM, apart from compound 5 (6.5-fold increase).
Substitutions at the 3-position showed more flexibility to changes than either the 7- or 2-positions, while still having a significant effect on the relative 20S activities of the analogues. Many of the most potent analogues, like compounds 10, 3, 2 and 5 (9.5, 8.1, 7.9 and 6.5-fold increase of 20S activity, respectively), contain a phenyl or benzyl functionality at the 3-position. Other similarly sized and shaped substituents like cyclohexane (compound 16 (6.7-fold)) and pyridine compounds 11 (6.9-fold) and 12 (5.9-fold)) also provided some of the most potent analogues. Interestingly, larger substituents at the 3-position as seen in compounds 18 (7-fold) and 13 (5.3-fold) also yielded potent analogues, suggesting that additional functionalities may be incorporated here for further optimization if necessary. The substitution of the phenyl or benzyl groups for some other heterocycles such as N-methyl piperidine (compounds 17 (1-fold) and 19 (1.2-fold)), tetrahydropyran (compound 20 (1.6-fold)) or even a pyridine linked by a methyl group in compound 14 (2-fold) lead to significant decreases in 20S activity. This suggests that placement of heteroatoms at the 3-position may disrupt hydrophobic interactions in that region. The difference in activity shown between 17 and 18 could be caused by a disruption of hydrophobic interactions with the addition of the piperidine nitrogen, which could then be replaced by new interactions made by the phenyl group in 18. The addition of non-cyclic substituents at the 3-position (compounds 22, 23 and 24) resulted in very little 20S activity (2.7, 2.3 and 1.6-fold increase in 20S activity, respectively) in all cases suggesting that larger hydrophobic groups at the 3-position are likely required for 20S activity.
After analysing the results in Table 1, three of the most promising analogues were selected for further studies into their 20S activity. Compounds 10, 2 and 18 were selected to be carried forward based on their fold increase (Table S1, fold 20S activity increase > 200% at 3.0 μM) and the highest Max-Fold activities (Table S1, at high dose of 30μM). Compound 17 was also carried forward to use as a negative control since it had no discernible activity towards the 20S. These compounds were then tested to obtain a full concentration-response (Figure 2) of their activities towards the 20S proteasome using each of the three substrates for the three catalytic sites individually and the combination of the three substrates. This was done to ensure that each of the selected compounds activate the 20S proteasome at all three catalytic sites, which is critical for effective IDP degradation, as these proteins are likely to contain multiple cleavage sites for each. Previously identified 20S proteasome activators that were only able to activate a single catalytic site showed poor enhancement of IDP degradation in vitro when compared to those that activated all three catalytic sites.40
Figure 2.

Concentration response (0–40 μM) from fluorogenic peptide digestions with compounds 2, 10, 17 and 18. Error bars denote standard deviation. These data were collected in triplicate.
Using the data in Figure 2, the concentration at which 20S activity was doubled (AC200) was calculated for each compound using each substrate and the combination of the three substrates (Table 2). Because of variations in the maximum fold enhancement between 20S enhancers, AC200 values allow for easy comparisons to be made between activators. It was found that each of the active compounds (10, 2 and 18) achieved both high maximum fold increases (>500%) in 20S activity and doubled 20S activity in the combination at low μM concentrations.
Table 2.
Detailed analysis of 20S activation by select dihydroquinazoline analogues
| Combo | CT-L | Casp-L | T-L | |||||
|---|---|---|---|---|---|---|---|---|
| Compound | AC200 (μM) | Max Fold | AC200 (μM) | Max Fold | AC200 (μM) | Max Fold | AC200 (μM) | Max Fold |
| 10 | 2.3 | 11.1 | 8.1 | 6.7 | 5.8 | 15.3 | 2.5 | 15.0 |
| 2 | 2.0 | 10.1 | 12.9 | 5.6 | 5.6 | 13.4 | 5.0 | 11.7 |
| 18 | 1.3 | 5.5 | 10.5 | 3.8 | 2.8 | 10.6 | 1.7 | 8.7 |
| 17 | N/A | 1.4 | N/A | 1.1 | N/A | 0.8 | N/A | 1.0 |
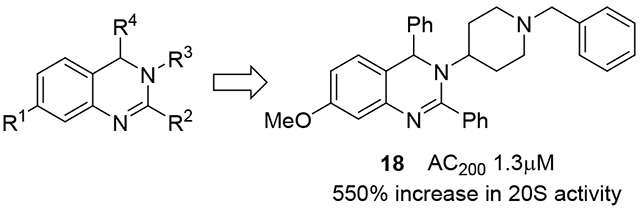
Although the three compounds showed near equipotent activities, compound 18 was selected to move forward with due to its lowest overall AC200 values (Table 2: combo, AC200 1.3μM) and the individual activities of the catalytic sites of the 20S. The efficacy of compound 18 was tested by observing its ability to enhance 20S mediated degradation of α-synuclein, the IDP associated with the development of Parkinson’s Disease. Briefly, the 20S proteasome was incubated with compound 18, followed by addition of pure human α-synuclein. This mixture was then incubated for 4 hours. The digestions were analysed using silver stain and quantified (Figure 3 and Figure S1) to determine the ability of compound 18 to enhance IDP degradation by the 20S. It was found that compound 18 effectively enhanced the rate of degradation of α-synuclein by the 20S in vitro in a concentration dependant manner. As a control, we used the proteasome inhibitor, bortezomib, which prevented α-synuclein degradation, confirming that the clearance of α-synuclein in Figure 3 is a proteasome mediated event. This shows that the prior peptide substrate-based results are likely to translate to the degradation of full IDPs and that dihydroquinazolines represent a promising new lead from which potent 20S activators that enhance IDP degradation can be developed.
Figure 3:
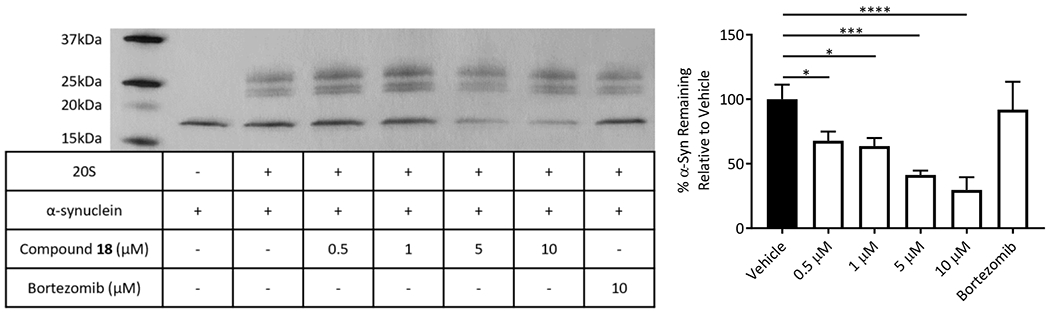
In vitro digestion of purified α-synuclein with human 20S proteasome and enhancement by compound 18. Representative silver stain of these α-synuclein digestions and quantification of three trials (see Figure S1 for other replicates). 20S proteasome subunits were used as a loading control in the quantifications (right). These data were collected in triplicate (n=3). Error bars denote standard deviation. Ordinary one-way ANOVA statistical analysis was used to determine statistical significance (ns=not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
In conclusion, this study demonstrates that dihydroquinazolines represent a promising scaffold from which potent 20S activators can be developed. Additionally, recently developed synthetic methods allow for access to a broad scope of dihydroquinazoline analogues, allowing for exploration of a variety of different substituents and substitution patterns. Among the analogues tested, we found several active compounds and a few of the most potent 20S activators identified to date. Further optimization and testing of dihydroquinazoline analogues may yield even more potent and drug-like leads, which can assist in the exploration of 20S activation as a novel therapeutic method.
Supplementary Material
Acknowledgments
The authors gratefully appreciate assistance from Thomas Dexheimer at the Assay Development and Drug Repurposing Core Facility at Michigan State University. The authors gratefully acknowledge financial support for this work from the National Institutes of Aging 1R01 AG066223-01A1 (JJT), National Institute of General Medical Sciences, T32GM092715 (TF) of the National Institutes of Health, and the National Science Foundation (MRI award 1626523).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare no competing interest.
Supplemental data
Supplemental information for this paper includes full compound characterization, detailed experimental procedures, and reproducibility of experiments.
References
- 1.Gammon K, Neurodegenerative disease: brain windfall, Nature, 2014, 515, 299–300. [DOI] [PubMed] [Google Scholar]
- 2.Gitler AD, Dhillon P and Shorter J, DMM Dis. Model. Mech, 2017, 10, 499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang CV, Reddy EP, Shokat KM and Soucek L, Drugging the ‘undruggable’ cancer targets, Nat. Rev. Cancer, 2017, 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazo JS and Sharlow ER, Drugging Undruggable Molecular Cancer Targets, Annu. Rev. Pharmacol. Toxicol, 2016, 56, 23–40. [DOI] [PubMed] [Google Scholar]
- 5.Hu G, Wu Z, Wang K, Uversky VN and Kurgan L, Untapped Potential of Disordered Proteins in Current Druggable Human Proteome, Curr. Drug Targets, 2016, 17, 1198–1205. [DOI] [PubMed] [Google Scholar]
- 6.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC and Obradovic Z, Intrinsically disordered protein, J. Mol. Graph. Model, 2001, 19, 26–59. [DOI] [PubMed] [Google Scholar]
- 7.DeForte S, Uversky V, DeForte S and Uversky VN, Order, Disorder, and Everything in Between, Molecules, 2016, 21, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babu MM, van der Lee R, de Groot NS and Gsponer J, Curr. Opin. Struct. Biol, 2011, 21, 432–440. [DOI] [PubMed] [Google Scholar]
- 9.Berrocal R, Vasquez V, KRS SR, Gadad BS and KS R, Mol Neurobiol, 2015, 51, 1417–1431. [DOI] [PubMed] [Google Scholar]
- 10.Korsak M and Kozyreva T, Springer, Cham, 2015, pp. 401–421. [DOI] [PubMed] [Google Scholar]
- 11.Uversky VN, Na I, Landa KS and Schenck RO, Highly disordered proteins in prostate cancer, Curr. Protein Pept. Sci, 2017, 18, 453–481. [DOI] [PubMed] [Google Scholar]
- 12.Uversky VN, Oldfield CJ and Dunker AK, Intrinsically Disordered Proteins in Human Diseases: Introducing the D 2 Concept, Annu. Rev. Biophys, 2008, 37, 215–246. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Nissan G and Sharon M, Regulating the 20S proteasome ubiquitin-independent degradation pathway., Biomolecules, 2014, 4, 862–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung T, Höhn A and Grune T, The proteasome and the degradation of oxidized proteins: Part III—Redox regulation of the proteasomal system, Redox Biol, 2014, 2, 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Höhn TJA and Grune T, Redox Biol, 2014, 2, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka K, Mizushima T and Saeki Y, in Biological Chemistry, 2012, vol. 393, pp. 217–234. [DOI] [PubMed] [Google Scholar]
- 17.Kumar Deshmukh F, Yaffe D, Olshina M, Ben-Nissan G and Sharon M, The Contribution of the 20S Proteasome to Proteostasis, Biomolecules, 2019, 9, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budenholzer L, Cheng CL, Li Y and Hochstrasser M, J. Mol. Biol, 2017, 429, 3500–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD and Huber R, Structure of 20S proteasome from yeast at 2.4 Å resolution, Nature, 1997, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- 20.Finley D, Chen X and Walters KJ, Gates, Channels, and Switches: Elements of the Proteasome Machine., Trends Biochem. Sci, 2016, 41, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH and Finley D, A gated channel into the proteasome core particle, Nat. Struct. Biol, 2000, 7, 1062–1067. [DOI] [PubMed] [Google Scholar]
- 22.Erales J and Coffino P, Ubiquitin-independent proteasomal degradation, Biochim. Biophys. Acta - Mol. Cell Res, 2014, 1843, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding Q, Cecarini V and Keller JN, Interplay between protein synthesis and degradation in the CNS: physiological and pathological implications, Trends Neurosci, 2007, 30, 31–36. [DOI] [PubMed] [Google Scholar]
- 24.Ding Q, Dimayuga E and Keller JN, Proteasome Regulation of Oxidative Stress in Aging and Age-Related Diseases of the CNS, Antioxid. Redox Signal, 2006, 8, 163–172. [DOI] [PubMed] [Google Scholar]
- 25.Chondrogianni N, Sakellari M, Lefaki M, Papaevgeniou N and Gonos ES, Proteasome activation delays aging in vitro and in vivo, Free Radic. Biol. Med, 2014, 71, 303–320. [DOI] [PubMed] [Google Scholar]
- 26.Chondrogianni N, Georgila K, Kourtis N, Tavernarakis N and Gonos Efstathios S, Enhanced proteasome degradation extends Caenorhabditis elegans lifespan and alleviates aggregation-related pathologies, Free Radic. Biol. Med, 2014, 75, S18. [DOI] [PubMed] [Google Scholar]
- 27.Bulteau A-L, Szweda LI and Friguet B, Age-Dependent Declines in Proteasome Activity in the Heart, Arch. Biochem. Biophys, 2002, 397, 298–304. [DOI] [PubMed] [Google Scholar]
- 28.Gonos E, in Advances in experimental medicine and biology, 2015, vol. 821, pp. 7–7. [DOI] [PubMed] [Google Scholar]
- 29.Keller JN, Gee J and Ding Q, The proteasome in brain aging., Ageing Res. Rev, 2002, 1, 279–93. [DOI] [PubMed] [Google Scholar]
- 30.Longhena F, Spano P and Bellucci A, Springer, Cham, 2017, pp. 85–110. [Google Scholar]
- 31.Uversky VN, Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators, Front. Mol. Biosci, 2014, 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky VN, Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept., Expert Rev. Proteomics, 2010, 7, 543–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uversky VN, Synuclein Misfolding and Neurodegenerative Diseases, 2008, vol. 9. [DOI] [PubMed] [Google Scholar]
- 34.Levine ZA, Larini L, LaPointe NE, Feinstein SC and Shea J-E, Regulation and aggregation of intrinsically disordered peptides., Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 2758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raychaudhuri S, Majumder P, Sarkar S, Giri K, Mukhopadhyay D and Bhattacharyya NP, Huntingtin interacting protein HYPK is intrinsically unstructured, Proteins Struct. Funct. Bioinforma, 2007, 71, 1686–1698. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z and Dunker AK, Unfoldomics of human diseases: linking protein intrinsic disorder with diseases., BMC Genomics, 2009, 10 Suppl 1, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uversky VN, in Protein Folding and Misfolding: Neurodegenerative Diseases, Springer; Netherlands, Dordrecht, 2009, pp. 21–75. [Google Scholar]
- 38.Midic U, Oldfield CJ, Dunker AK, Obradovic Z and Uversky VN, Protein disorder in the human diseasome: unfoldomics of human genetic diseases, BMC Genomics, 2009, 10, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Njomen E and Tepe JJ, Proteasome Activation as a New Therapeutic Approach to Target Proteotoxic Disorders, J. Med. Chem, 2019, 62, 6469–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones CL, Njomen E, Sjögren B, Dexheimer TS and Tepe JJ, Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins, ACS Chem. Biol, 2017, 12, 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Opoku-Nsiah KA and Gestwicki JE, Transl. Res, 2018, 198, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones CL and Tepe JJ, Molecules, 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coleman RA and Trader DJ, Methods to Discover and Evaluate Proteasome Small Molecule Stimulators, Molecules, 2019, 24, 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zerfas BL, Coleman RA, Salazar-Chaparro AF, Macatangay NJ and Trader DJ, Fluorescent Probes with Unnatural Amino Acids to Monitor Proteasome Activity in Real-Time, ACS Chem. Biol, 2020, 15, 2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Njomen E, Osmulski PA, Jones CL, Gaczynska M and Tepe JJ, Small Molecule Modulation of Proteasome Assembly, Biochemistry, 2018, 57, 4214–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Njomen E and Tepe JJ, Regulation of Autophagic Flux by the 20S Proteasome, Cell Chem. Biol, 2019, 26, 1283–1294.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Njomen E, Lansdell TA, Vanecek A, Benham V, Bernard MP, Yang Y-T, Schall PZ, Isaac D, Alkharabsheh O, Al-Janadi A, Giletto MB, Ellsworth E, Taylor C, Tang T, Lau S, Bailie M, Bernard JJ, Yuzbasiyan-Gurkan V and Tepe JJ, Enhancing c-MYC degradation via 20S proteasome activation induces in vivo anti-tumor efficacy, bioRxiv, 2020, 2020.08.24.265470. [Google Scholar]
- 48.Coleman RA, Muli CS, Zhao Y, Bhardwaj A, Newhouse TR and Trader DJ, Analysis of chain length, substitution patterns, and unsaturation of AM-404 derivatives as 20S proteasome stimulators, Bioorg. Med. Chem. Lett, 2019, 29, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trader DJ, Simanski S, Dickson P and Kodadek T, Establishment of a suite of assays that support the discovery of proteasome stimulators, Biochim. Biophys. Acta - Gen. Subj, 2017, 1861, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee B-H, Lee MJ, Park S, Oh D-C, Elsasser S, Chen P-C, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW and Finley D, Enhancement of proteasome activity by a small-molecule inhibitor of USP14, Nature, 2010, 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B, Zaal EA, van de Kooij B, Laufer S, Heck AJR, Borst J, Scheper W, Berkers CR and Ovaa H, Proteasome Activation by Small Molecules, Cell Chem. Biol, 2017, 24, 725–736.e7. [DOI] [PubMed] [Google Scholar]
- 52.Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL and Duff KE, Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling, Nat. Med, 2016, 22, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krahn JH, Kaschani F and Kaiser M, Cell Chem. Biol, 2017, 24, 653–655. [DOI] [PubMed] [Google Scholar]
- 54.Trippier PC, Zhao KT, Fox SG, Schiefer IT, Benmohamed R, Moran J, Kirsch DR, Morimoto RI and Silverman RB, Proteasome activation is a mechanism for pyrazolone small molecules displaying therapeutic potential in amyotrophic lateral sclerosis, ACS Chem. Neurosci, 2014, 5, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lansdell TA, Hurchla MA, Xiang J, Hovde S, Weilbaecher KN, Henry RW and Tepe JJ, Noncompetitive modulation of the proteasome by imidazoline scaffolds overcomes bortezomib resistance and delays MM tumor growth in vivo, ACS Chem. Biol, 2013, 8, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahlon DK, Lansdell TA, Fisk JS and Tepe JJ, Structural-activity relationship study of highly-functionalized imidazolines as potent inhibitors of nuclear transcription factor-κB mediated IL-6 production, Bioorganic Med. Chem, 2009, 17, 3093–3103. [DOI] [PubMed] [Google Scholar]
- 57.Azevedo LM, Lansdell TA, Ludwig JR, Mosey RA, Woloch DK, Cogan DP, Patten GP, Kuszpit MR, Fisk JS and Tepe JJ, Inhibition of the Human Proteasome by Imidazoline Scaffolds, J. Med. Chem, 2013, 56, 5974–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magyar CL, Wall TJ, Davies SB, Campbell MV, Barna HA, Smith SR, Savich CJ and Mosey RA, Triflic anhydride mediated synthesis of 3,4-dihydroquinazolines: A three-component one-pot tandem procedure, Org. Biomol. Chem, 2019, 17, 7995–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaczynska M and Osmulski PA, Methods Enzymol, 2005, 398, 425–438. [DOI] [PubMed] [Google Scholar]
- 60.Xie S, Zhang Y, Ramström O and Yan M, Base-catalyzed synthesis of aryl amides from aryl azides and aldehydes, Chem. Sci, 2016, 7, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braddock DC, Lickiss PD, Rowley BC, Pugh D, Purnomo T, Santhakumar G and Fussell SJ, Tetramethyl Orthosilicate (TMOS) as a Reagent for Direct Amidation of Carboxylic Acids, Org. Lett, 2018, 20, 950–953. [DOI] [PubMed] [Google Scholar]
- 62.Mei C and Lu W, Palladium(II)-Catalyzed Oxidative Homo- and Cross-Coupling of Aryl ortho -sp2 C-H Bonds of Anilides at Room Temperature, J. Org. Chem, 2018, 83, 4812–4823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.