

Abstract
Innate immune system plays an essential role in combating infectious diseases by recognizing invading pathogens and activating host defense response. Inflammasomes complexes are a central component of the cytosolic innate immune surveillance and are vital in host defense against bacterial pathogens. Bacterial products or pathogen-induced modifications in the intracellular environment are sensed by the inflammasome receptors that form complexes that serve as a platform for caspase-1- or caspase-11-dependent induction of pyroptosis and secretion of cytokines, IL-1β and IL-18. However, several pathogenic bacteria have developed strategies to evade inflammasome activation. This review highlights the recent advances in the mechanism of inflammasome activation by bacterial pathogens and some of the bacterial evasion strategies of inflammasome activation.
Introduction
The innate immune system plays a crucial role in detecting pathogens and mounting host defense response. Germ-line encoded pattern recognition receptors (PRRs) sense pathogen associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs) [1]. PRRs can either be membrane bound or cytosolic. The cytosolic multiprotein complexes, inflammasomes, have emerged as essential elements of innate immune defense [2]. The assembly of inflammasome complexes is initiated by a class of cytosolic receptors belonging to the nucleotide-binding domain and leucine rich repeat containing protein (NLR) family, AIM2 (absent in melanoma 2)-like receptor (ALR) family, or by a protein, pyrin. Upon sensing of pathogens or danger signals, these proteins recruit an adaptor protein ASC (apoptosis-associated speck like protein containing CARD), which links the binding of NLR/ALR/Pyrin to the proform of a protease, caspase-1 [3]. Alternatively, certain receptors could directly bind to caspase-1 [2]. Subsequently, the inflammasome complex undergoes oligomerization resulting in the autoproteolysis of caspase-1 into enzymatically active caspase-1, which cleaves proforms of the inflammatory cytokines IL-1β and IL-18 into their mature forms [3]. In addition, inflammasome associated caspases (caspase-1, caspase-11, human caspase-4/5) induce cleavage of gasdermin D (GSDMD), releasing its N-terminal fragment, which oligomerizes on the plasma membrane, resulting in pore formation and inflammatory cell death called pyroptosis (Figure 1) [4–7]. Inflammasome activation also requires an upstream ‘first signal’ that is initiated by recognition of ligands by toll-like receptors (TLRs) and the subsequent NF-κB- or IRF3-dependent transcription of certain inflammasome components and pro-IL-1β, and the sensor caspase-11, respectively [2].
Figure 1. Recent advances in the mechanisms of inflammasome activation by bacterial pathogens.
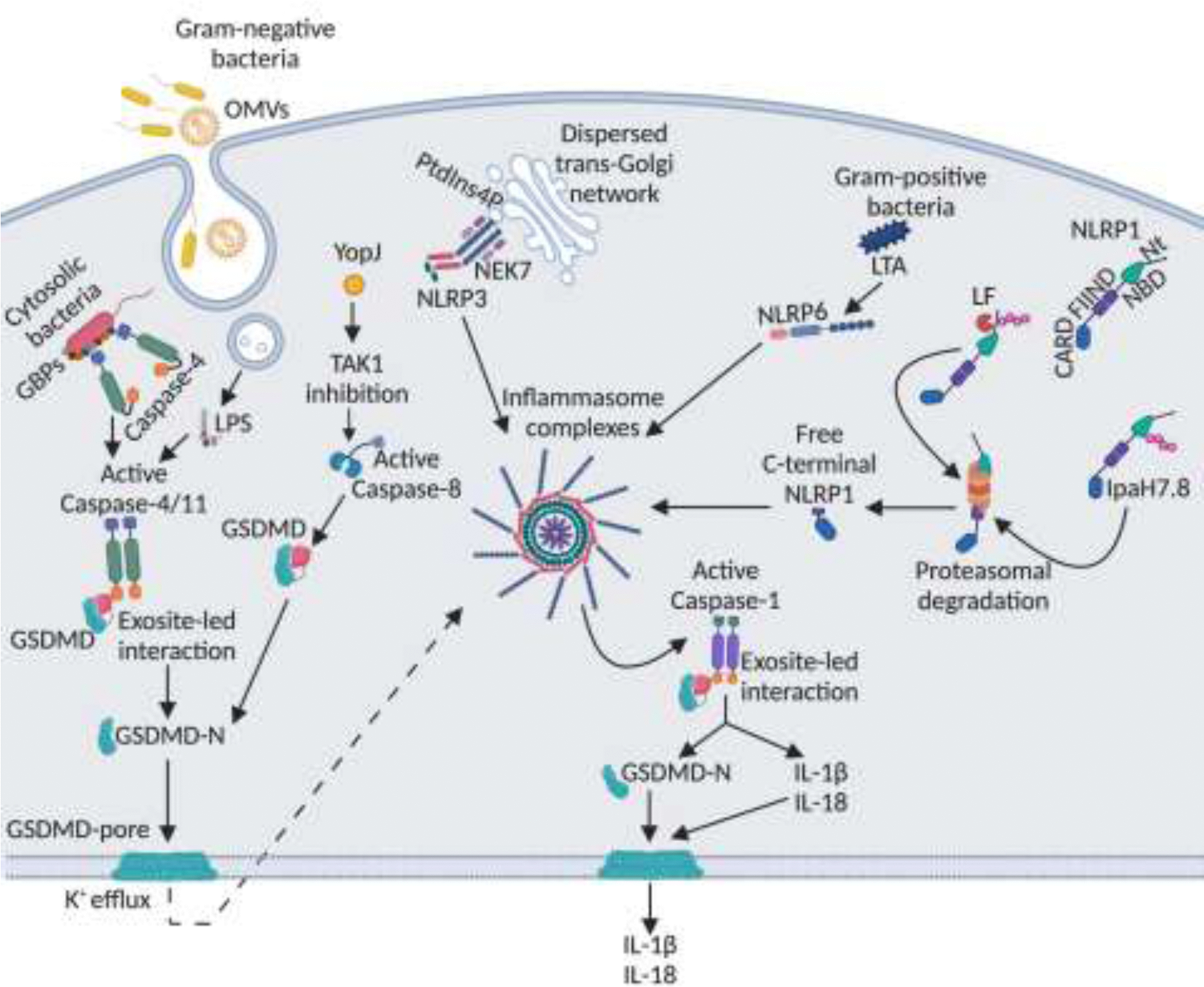
LPS from Gram-negative bacteria enters the cytosol from OMV-containing endosomes or directly from cytosolic bacteria. GBPs coat cytosolic bacteria and serve as a platform for caspase-4 recruitment and activation. Active caspase-4/11 interacts with the C-terminus of GSDMD in an exosite-dependent manner leading to GSDMD cleavage into N-terminal fragment, which forms pores on the plasma membrane resulting in pyroptosis. K+ efflux via GSDMD pores leads to NLRP3 activation. YopJ protein of Yersinia inhibits TAK1 leading to activation of caspase-8, which cleaves GSDMD leading to pyroptosis and NLRP3 activation. Certain NLRP3 activators trigger disassembly of trans-Golgi network exposing PtdIns4P, which recruits NLRP3 leading to its aggregation and activation. NEK7 also aids in the NLRP3 oligomerization. Lipoteichoic acid (LTA) of Gram-positive bacteria binds to NLRP6 leading to inflammasome activation. The FIIND domain of NLRP1 undergoes constitutive autoproteolysis during unstimulated conditions, however, the resulting C- and N-terminal polypeptides remain bound together. Anthrax lethal factor (LF) cleaves the N-terminal peptide leading to its ubiquitination. Shigella IpaH7.8 directly ubiquitinates the N-terminal peptide. Ubiquitinated N-terminal peptide undergoes proteasomal degradation releasing a free NLRP1 C-terminal fragment, which assembles into an inflammasome complex. Once the inflammasome complex is assembled, procaspase-1 is cleaved into active caspase-1, which cleaves pro-forms of IL-1 cytokines into their active forms. Active caspase-1 also interacts with the C-terminus of GSDMD in an exosite-dependent manner leading to GSDMD cleavage, pyroptosis, and release of IL-1 cytokines via GSDMD pores.
The major inflammasome pathways involved in sensing bacterial infections include, NLRP1b, NLRP3, caspase-11/4, NLRC4, AIM2, Pyrin, and NLRP6 [8]. A wide range of bacterial products induce the formation of these inflammasome complexes and the downstream signaling cascades. However, it has also been well-established that numerous bacterial pathogens have developed strategies to evade inflammasome activation. This review highlights the inflammasome pathways that are activated by bacterial pathogens, the underlying mechanisms of activation, and the inflammasome evasion strategies employed by pathogenic bacteria.
NLRP1b inflammasome
NLRP1 is the first NLR protein reported to form an inflammasome complex [9]. Unlike other NLRs, NLRP1 is characterized by its distinct domain structure with a C-terminal function-to-find (FIIND) domain followed by a CARD domain (Figure 1) [10]. Humans only have one NLRP1 protein whereas mice have three paralogs of NLRP1 (NLRP1a-c) [9,11]. Among these, mouse NLRP1b is the most well characterized and is known to be activated by the lethal factor of anthrax toxin and by IpaH7.8, an E3 ubiquitin ligase effector secreted by Shigella flexneri [10]. Although the molecular mechanism of NLRP1b inflammasome activation remained unclear, multiple studies have now provided crucial insights into this [10,12–17]. During unstimulated conditions, the NLRP1b FIIND domain undergoes a constitutive autoproteolysis resulting in the generation of two polypeptides. Interestingly, these N-terminal and C-terminal peptides remain bound by a noncovalent interaction [10,17]. When cells are exposed to anthrax lethal toxin, the protective antigen enables translocation of lethal factor into the host cell cytoplasm. The enzymatically active lethal factor cleaves the N-terminal fragment of NLRP1b leading to its ubiquitination and proteasomal degradation resulting in the release of the C-terminal fragment. The CARD domain of C-terminal fragment then recruits caspase-1 and assembles into an inflammasome complex (Figure 1). Similarly, Shigella ligase, IpaH7.8, directly ubiquitinates NLRP1b, leading to the proteasomal degradation of N-terminal fragment and subsequent C-terminal-mediated inflammasome complex formation (Figure 1) [10,17]. These observations demonstrated that cleavage and functional degradation of N-terminal fragment is an essential initiating step in NLRP1b inflammasome activation. Notably, mice carrying a cleavage-resistant variant of NLRP1b fail to sense B. anthracis infection and are highly susceptible to the pathogen [18,19]. Listeria monocytogenes have also been reported to activate NLRP1 through metabolic stress but the exact mechanism is unknown [20].
Canonical NLRP3 inflammasome
NLRP3 is the best-studied inflammasome and is activated by a broad range of triggers [9]. NLRP3 is not known to interact with these ligands directly, instead these stimuli induce certain downstream cellular perturbations such as the efflux of potassium ions (K+), generation of mitochondrial reactive oxygen species (ROS), release of mitochondrial DNA into the cytosol, and cathepsin release due to lysosomal disruption that trigger the NLRP3 activation [8,21]. Additionally, certain NLRP3 activators trigger disassembly of the trans-Golgi network [22]. This dispersed trans-Golgi network interacts with the polybasic region of NLRP3 via its phosphatidylinositol-4-phosphate (PtdIns4P) and facilitates NLRP3 activation by serving as a platform for NLRP3 aggregation (Figure 1) [22]. Notably, interaction with a kinase, NIMA-related kinase 7 (NEK7), is also required for NLRP3 activation [23,24]. NLRP3 bound NEK7 interacts with a neighboring NLRP3 to mediate NLRP3 oligomerization (Figure 1) [25]. Several Gram-positive bacterial pathogens such as Streptococcus pyogenes, Staphylococcus aureus, and Listeria monocytogenes activate the NLRP3 inflammasome via pore-forming toxins such as streptolysin O, hemolysins, listeriolysin O, and pneumolysin [26]. ADP-ribosylating and vacuolating toxin of Mycoplasma pneumoniae mediates NLRP3 inflammasome activation by direct ADP-ribosylation of NLRP3 [27]. Interestingly, Yersinia type III secretion system effector protein, YopJ, activates NLRP3 via a novel mechanism; YopJ inhibits TAK1, which leads to a caspase-8-dependent cleavage of GSDMD and subsequent K+ efflux-mediated NLRP3 activation (Figure 1) [28]. Additionally, purified bacterial RNA and bacterial RNA:DNA hybrids have been shown to trigger NLRP3 activation [29,30]. The NLRP3 inflammasome is crucial for protection against multiple bacterial infections. For example, mice lacking NLRP3 exhibit higher bacterial loads and increased mortality upon infection with Streptococcus pneumonia [31]. Similarly, Nlrp3−/− mice infected with M. pneumoniae fail to produce IL-1β and exhibit delayed bacterial clearance [32]. In contrast, NLRP3 inflammasome has a detrimental role during infection with B. cereus in vivo [26]. Nlrp3−/− mice and mice administered with pharmacological inhibitor of NLRP3 are protected from B. cereus induced lethality [26].
Noncanonical NLRP3 inflammasome or the cytosolic LPS sensing pathway
The non-canonical inflammasome, also known as the cytosolic LPS sensing pathway, is activated primarily by Gram-negative bacteria such as Escherichia coli, Shigella flexneri, Citrobacter rodentium, Vibrio cholerae, and Burkholderia thailandensis [33]. Unlike the canonical inflammasome pathways, the activation of the cytosolic LPS sensing pathway is mediated by a set of inflammatory caspases; caspase-11 in mice and caspase-4 or 5 in humans [34–37]. Caspase-11/4/5 directly binds to the lipid A moiety of cytosolic LPS via its CARD domain resulting in the self-oligomerization of these caspases [34]. This leads to a site-specific cleavage of caspase-11/4/5 generating an enzymatically active p10 form of the corresponding caspase [38]. The p10 fragment binds to the C-terminal domain of GSDMD with high affinity via an exosite mediated interaction, which results in a tetrapeptide-independent GSDMD cleavage, generation of N-terminal GSDMD fragment, and pyroptosis (Figure 1) [38]. Additionally, the formation of plasma membrane pores by N-terminal GSDMD leads to K+ efflux-mediated activation of NLRP3 inflammasome and subsequent secretion of IL-1 cytokines [39].
Several mechanisms have been described for cytosolic access of LPS, which is an important pre-requisite for caspase-11 activation. In the case of cytosolic bacteria and bacteria residing in the phagosomal vacuoles, members of the guanylate-binding protein (GBP) and immunity related GTPase (IRG) family such as IRGB10 enable the release of LPS [40–42]. Recent studies have shown that GBP1 functions as a cytosolic PRR that binds to LPS on the surface of cytosol-invading pathogens such as Salmonella and Shigella. Upon sensing of LPS, GBP1 initiates the assembly of a GBP2–4 platform on the bacterial surface, which recruits caspase-4 leading to its activation (Figure 1) [43–45]. On the other hand, during infections with extracellular bacteria, LPS delivery into the cytosol is mediated by outer membrane vesicles (OMVs) secreted by the bacteria. OMVs enter the cells via clathrin-mediated endocytosis and LPS is released into the cytosol from early endosome (Figure 1) [46]. Other proteins such as HMGB1 and SCGB3A2 have also been implicated in LPS delivery into the cytosol. Upon sensing of extracellular LPS by TLR4, hepatocytes release HMGB1, which directly binds to LPS and facilitates LPS entry into the cells via RAGE-receptor mediated endocytosis [47]. HMGB1 then permeabilizes the endosomal membrane leading to the release of LPS into cytoplasm [47].
The cytosolic LPS sensing pathway has been demonstrated to have a protective or detrimental role depending on the type of bacterial infection. Mice deficient in caspase-11 are highly susceptible to Burkholderia thailandensis or B. pseudomallei and display high bacterial burdens [48]. Caspase-11 is also required for effective bacterial clearance in lungs during Acinetobacter baumannii or Klebsiella pneumoniae infection, in intestine during Salmonella Typhimurium infection, in kidney during uropathogenic Escherichia coli (UPEC) infection, and in spleen during Brucella abortus infection [49–52]. In contrast to these, cytosolic LPS sensing by caspase-11 plays a detrimental role during sepsis. Here, the inflammasome responses go unchecked resulting in tissue damage and lethality in host [53,54]. Caspase-11 mediated cleavage of GSDMD and the resulting pyropotosis appears to be the primary mediator of sepsis pathogenesis. Consistent with this, mice deficient in caspase-11 or GSDMD are protected from sepsis induced by high dose LPS as well as cecal ligation and puncture [5,37].
NLRC4 inflammasome
The NLRC4 inflammasome detects bacterial pathogens like S. flexneri, Salmonella Typhimurium, Pseudomonas aeruginosa, L. pnuemophila and is triggered by flagellin and type III (T3SS) and IV (T4SS) secretion system components that enter the cytosol [55]. Interestingly, NLRC4 does not bind to the ligands directly, instead members of another NLR family of proteins namely neuronal apoptosis inhibitory proteins (NAIPs) act as sensors of the ligands and directly bind to them. NAIP-ligand binding is followed by NAIP-NLRC4 interaction which further leads to inflammasome assembly [55]. Phosphorylation of NLRC4 by protein kinase Cδ and/or leucine rich repeat-containing kinase-2 (LRRK2) at S533 residue is critical for its activation [56]. There are seven NAIP genes in mice whereas humans have a single NAIP gene encoding two functional isoforms [57]. Murine NAIP1 recognizes T3SS needle proteins whereas NAIP2 interacts with T3SS basal rod proteins. NAIP5 and 6 recognize flagellin [57] . Some bacteria like L. monocytogenes escape the pathogen-containing vacuole, in order to invade the cytoplasm and replicate, thereby directly introducing flagellin into the cytoplasm. On the other hand, in the case of bacteria that remain in a pathogen-containing vacuole such as S. Typhimurium, flagellin is injected into the cytoplasm via T3SS or T4SS apparatus [57]. One isoform of human NAIP binds to the T3SS needle subunit of Chromobacterium violaceum whereas the other isoform of recognizes flagellin and T3SS inner rod and needle proteins of S. Typhimurium, S. flexneri, and Burkholderia spp., resulting in NLRC4 inflammasome activation in macrophages [57]. NLRC4 inflammasome plays a crucial role in host defense against many bacterial pathogens. NLRC4-deficient mice exhibit increased susceptibility to oral infection with Salmonella Typhimurium [58]. Similarly, NLRC4 deficient mice also succumb to oral infection with Citrobacter rodentium and intratracheal infection with K. pneumoniae [55,59,60].
AIM2 inflammasome
AIM2 inflammasome detects double-stranded DNA (dsDNA) from intracellular pathogens like Francisella tularensis, F.novicida, and L. monocytogenes [9]. AIM2 directly binds to DNA via its HIN-200 DNA binding domain and induces caspase-1 activation in an ASC-dependent manner [9]. The mechanism by which the bacterial DNA is released into the cytoplasm of the host cells has been well studied in F. novicida infection. Shortly after phagocytosis by the host cell, F. novicida escape the pathogen containing vacuole utilizing its T6SS and invade the cytoplasm. Subsequently, the GBPs, GBP2, GBP5 and the interferon-inducible protein, IRGB10, bind to the cytosolic bacteria and promote bacterial lysis and DNA release into the cytoplasm [42]. The expression of GBPs and IRGB10 are controlled by type I interferon signaling cascade. Cytosolic dsDNA is also recognized by cGAS (cyclic GMP-AMP synthase), which in turn triggers STING-dependent activation of type I IFNs. Notably, AIM2-mediated cleavage of GSDMD and the resulting K+ efflux suppresses this cGAS-mediated type I IFN activation during Francisella infection [61]. AIM2-deficient mice are highly susceptible to Francisella and Mycobacterium tuberculosis infection [42,61,62]. GBP and IRGB10 deficient mice are also highly susceptible to Francisella infection suggesting that they are critical for the protective role of AIM2 [42].
Pyrin inflammasome
Pyrin is a non-NLR inflammasome protein encoded by MEFV gene which has been associated with familial Mediterranean fever [63]. Pyrin does not directly bind to PAMPs but is activated in response to modifications of host proteins, Rho GTPases, by bacterial toxins and effector proteins [63]. During homeostatic conditions, GTPase, RhoA, activates the serine-threonine protein kinases, PKN1 and PKN2, that bind and phosphorylate pyrin at Ser208 and Ser242. This phosphorylated pyrin interacts with chaperone proteins 14–3-3ε and 14–3-3τ and remains in an inactive state [64]. Several bacterial proteins like TcdA/B of Clostridium difficile, C3 toxin of C. botulinum, TecA of Burkholderia cenocepacia, VopS of Vibrio parahaemolyticus, YopE and YopT of Yersinia, and pertussis toxin from Bordetella pertussis modify RhoA and prevent pyrin phosphorylation, thereby promoting pyrin inflammasome activation [63,65,66]. Pyrin has a host protective role in certain bacterial infections; it has been shown that TecA-mediated pyrin activation decreases bacterial loads and virulence of B. cenocepacia during systemic infection in mice [65].
NLRP6 inflammasome
NLRP6 is a relatively new member of the NLR family involved in inflammasome activation. NLRP6 is highly expressed in the intestine and plays a crucial role in maintaining intestinal homeostasis by regulating the composition of microbiome [8]. A recent study identified lipoteichoic acid (LTA) from Gram-positive bacteria, L. monocytogenes, as a ligand for NLRP6. Binding of LTA to NLRP6 induced caspase-11 activation leading to processing of caspase-1 and IL-18 secretion (Figure 1) [67]. Interestingly, Nlrp6−/− mice upon Listeria infection, show reduced bacterial burdens and better survival, thus suggesting a detrimental role for NLRP6 in bacterial infection. Similarly, NLRP6 also acts as a negative regulator of host defense in mice infected with Staphylococcus aureus, or S. Typhimurium [68,69]. On the other hand, NLRP6 plays a protective role in murine model of Citrobacter-induced enteritis indicating that the role of NLRP6 vary depending on the type of bacterial infection [70].
Inflammasome evasion by bacterial pathogens
The mechanisms by which the pathogens evade inflammasome defense mechanisms can be broadly categorized into two strategies: a) escaping inflammasome sensing by modifying or suppressing the expression of ligands and b) active suppression of inflammasomes using inhibitory proteins. Pathogens can alter the structure of an inflammasome-activating ligand. Heliobacter pylori, Yersinia pestis, and F. novicida modify their LPS into a tetra-acylated form to avoid detection by both TLR4 and caspase-11 [71]. Another strategy adapted by pathogens is to minimize the expression of inflammasome ligand. For example, during gastrointestinal infection, Salmonella expresses high levels of flagellin and rod protein PrgJ, which are sensed by NLRC4, whereas during intracellular replication during a systemic infection, the expression of flagellin is suppressed and rod protein SsaI is expressed, which is not detected by NLRC4 [72,73]. When Salmonella are manipulated to maintain flagellin expression during systemic infection, it resulted in an effective NLRC4-dependent bacterial clearance. This explains why Nlrc4−/− mice exhibit increased susceptibility to infection with Salmonella introduced orally but not intraperitoneally [72].
Numerous Gram-negative bacterial pathogens are equipped with special secretion systems such as the T3SS that resemble a syringe that deliver bacterial effector proteins across the cell membrane into the cytosol. Many of these effectors efficiently interfere with inflammasome activation (Figure 2). A well-studied example is Yersinia spp. (Y. pestis, Y. enterocolitica, Y. pseudotuberculosis) T3SS effector proteins known as Yops (Yersinia outer proteins). YopM protein of Yersinia directly binds to caspase-1 and prevents its interaction with ASC [74]. YopM also inhibits activation of pyrin inflammasome by binding to host kinases, PRK1 and PRK2 [75,76] (Figure 2). Another effector YopK interacts with the T3SS translocon and blocks the leakage of flagellin or PAMPs into the host cell cytoplasm, thus evading NLRP3 and NLRC4 inflammasome activation [77]. P. aeruginosa secretes T3SS effector exoenzymes, ExoU and ExoS, that interfere with NLRC4 inflammasome activation [78]. The intracellular pathogenic bacterium Edwardsiella tarda inhibits NLRP3 inflammasome via a type VI secretion system effector EvpP. EvpP blocks elevated intracellular Ca2+-mediated Jnk activation, which is essential for ASC phosphorylation and subsequent oligomerization [79]. An effector protein, OspC3, from Shigella binds to p19 subunit of caspase-4 and interferes with caspase-4 p19/p10 heterodimerization and activation to delay epithelial cell death and promote infection [80]. Legionella pneumophila T4SS effector SdhA stabilizes the Legionella-containing vacuole membrane, prevents the release of bacterial DNA into the cytosol, and thereby blocks AIM2 inflammasome activation [81]. Similarly, Salmonella T3SS effector SifA maintains the stability of pathogen-containing vacuole to prevent the activation of caspase-11 [48]. T4SS effector IcaA of Coxiella burnetii hampers the interaction between LPS and caspase-11 and inhibits the activation of the cytosolic LPS sensing pathway [82]. Overall, it is becoming increasingly clear that bacterial pathogens have developed a diverse range of strategies to interfere with inflammasome activation.
Figure 2. Inflammasome inhibition by effector proteins of bacterial secretion systems.
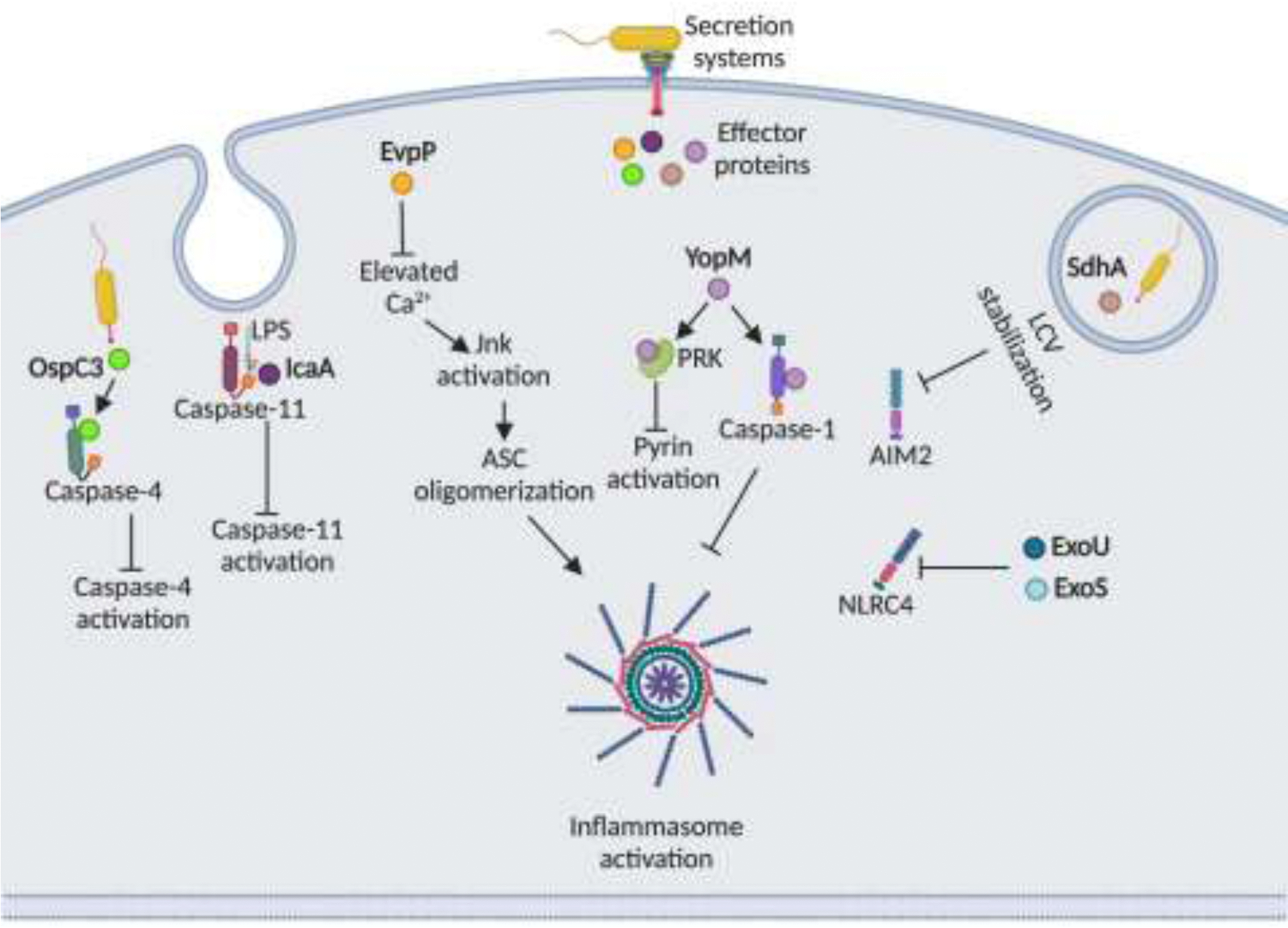
Effectors of T3SS/T4SS suppress inflammasome activation by varying mechanisms. OspC3 of Shigella binds to the p19 subunit of caspase-4 and inhibits its interaction with p10 subunit thereby suppressing caspase-4 activation. IcaA of C. burnetti interferes with LPS-caspase-11 interaction to inhibit caspase-11 activation. EvpP of E. tarda blocks elevation of intracellular Ca2+ levels, which is required for JnK activation and ASC oligomerization. Yersinia YopM protein directly binds to caspase-1 and prevents caspase-1-ASC interaction. YopM also inhibits pyrin inflammasome by binding to host PRK kinases, which are required for pyrin activation. SdhA of L. pneumophila maintains the integrity of Legionella-containing vacuole (LCV) to block the escape of dsDNA into the cytosol and subsequent AIM2 activation. ExoU and ExoS exoenzymes of P. aeruginosa inhibits NLRC4 inflammasome activation.
Conclusions
Over the recent years, much progress has been made towards understanding the mechanisms of inflammasome activation by bacterial pathogens and their components and how this activation shapes up the host defense against these pathogens. Yet, detailed elucidation of molecular mechanism of inflammasome activation in some cases needs to be identified. We have highlighted a few of examples of how the bacterial pathogens counteract and escape the inflammasome activation. Further studies on how various bacterial pathogens subvert inflammasome activation is required. Similarly, studies that focus on the host regulatory mechanisms that keep inflammasome responses under control are limited. Uncovering the mechanisms of inflammasome regulation may lead to development of therapeutics for the treatment of diseases causes by unchecked inflammasome responses such as sepsis.
Highlights.
Functional degradation is an essential step in NLRP1b inflammasome activation.
Site-specific cleavage of caspase-11/4/5 generates an enzymatically active fragment that cleaves gasdermin D.
Lipoteichoic acid of Listeria monocytogenes activates the NLRP6 inflammasome.
Bacterial pathogens employ secretion system effectors to evade inflammasome activation.
Acknowledgements
The authors acknowledge many investigators in the field whose primary data could not be cited in this review because of space limitations. Figures were created with BioRender.com. This work was supported by the National Institutes of Health grant AI132850 to SKV.
Footnotes
Declaration of interests
The authors have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Fitzgerald KA, Kagan JC: Toll-like Receptors and the Control of Immunity. Cell 2020, 180:1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathinam VAK, Fitzgerald KA: Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165:792 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell 2002, 10:417 426. [DOI] [PubMed] [Google Scholar]
- 4.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F: Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526:660–665. [DOI] [PubMed] [Google Scholar]
- 5.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. : Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526:666–671. [DOI] [PubMed] [Google Scholar]
- 6.He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z-H, Zhong C-Q, Han J: Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 2015, 25:1285 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, Shao F: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535:111–116. [DOI] [PubMed] [Google Scholar]
- 8.Vanaja SK, Rathinam VAK, Fitzgerald KA: Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 2015, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathinam VAK, Vanaja SK, Fitzgerald KA: Regulation of inflammasome signaling. Nat Immunol 2012, 13:333 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE: Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364:eaau1330.**Along with Chui et al., this study provided crucial insights into the molecular mechanism of NLRP1b activation. They determined that anthrax lethal factor cleaves the N-terminal fragment of NLRP1b leading to its ubiquitination and proteasomal degradation resulting in the release of the C-terminal fragment. C-terminal fragment then assembles into an inflammasome complex thus showing that functional degradation is an essential step in NLRP1b activation.
- 11.von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE: Recognition of Bacteria by Inflammasomes. Annu Rev Immunol 2013, 31:73–106. [DOI] [PubMed] [Google Scholar]
- 12.Frew BC, Joag VR, Mogridge J: Proteolytic Processing of Nlrp1b Is Required for Inflammasome Activity. Plos Pathog 2012, 8:e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M: Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. Plos Pathog 2012, 8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavarría-Smith J, Vance RE: The NLRP1 inflammasomes. Immunol Rev 2015, 265:22–34. [DOI] [PubMed] [Google Scholar]
- 15.Chavarría-Smith J, Vance RE: Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. Plos Pathog 2013, 9:e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung AL, Stoiber C, Herkt CE, Schulz C, Bertrams W, Schmeck B: Legionella pneumophila - Derived Outer Membrane Vesicles Promote Bacterial Replication in Macrophages. Plos Pathog 2016, 12:e1005592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, et al. : N-terminal degradation activates the NLRP1B inflammasome. Sci New York N Y 2019, 364:82–85.**Along with Sandstrom et al., this study provided crucial insights into the molecular mechanism of NLRP1b activation. Using a genome-wide knock out screen, they demonstrated the requirement of proteasomal degradation of N-terminal fragment in the activation of NLRP1b inflammasome thus showing that functional degradation is an essential step in NLRP1b activation.
- 18.Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL, LeVine SM, Bradley KA: Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol Baltim Md 1950 2009, 184:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, Liu S, Sastalla I, Leppla SH: Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. Plos Pathog 2010, 6:e1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neiman-Zenevich J, Stuart S, Abdel-Nour M, Girardin SE, Mogridge J: Listeria monocytogenes and Shigella flexneri Activate the NLRP1B Inflammasome. Infect Immun 2017, 85:e00338–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Platnich JM, Muruve DA: NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys 2019, 670:4–14. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Chen ZJ: PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Zeng MY, Yang D, Motro B, nunez G: NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530:354 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. : NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 2015, 17:250 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Núñez G, Mao Y, et al. : Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathur A, Feng S, Hayward JA, Ngo C, Fox D, Atmosukarto II, Price JD, Schauer K, Märtlbauer E, Robertson AAB, et al. : A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat Microbiol 2018, 4:362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bose S, Segovia JA, Somarajan SR, Chang T-H, Kannan TR, Baseman JB: ADP-ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. Mbio 2014, 5:e02186–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. : Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362:1064–1069.**This study demonstrated that the Yersinia T3SS effector protein, YopJ, suppresses TAK1–IκB kinase signaling and thereby activates caspase-8, which cleaves GSDMD. This study unraveled a new mechanism of cleavage of GSDMD during bacterial infection.
- 29.Kanneganti T-D, Ozören N, Body-Malapel M, Amer A, Park J-H, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. : Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440:233 236. [DOI] [PubMed] [Google Scholar]
- 30.Vanaja SK, Rathinam VAK, Atianand MK, Kalantari P, Skehan B, Fitzgerald KA, Leong JM: Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc National Acad Sci 2014, 111:7765–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, et al. : The NLRP3 Inflammasome Is Differentially Activated by Pneumolysin Variants and Contributes to Host Defense in Pneumococcal Pneumonia. J Immunol 2011, 187:434–440. [DOI] [PubMed] [Google Scholar]
- 32.Segovia JA, Chang T-H, Winter VT, Coalson JJ, Cagle MP, Pandranki L, Bose S, Baseman JB, Kannan TR: NLRP3 Is a Critical Regulator of Inflammation and Innate Immune Cell Response during Mycoplasma pneumoniae Infection. Infect Immun 2017, 86:e00548–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rathinam VAK, Zhao Y, Shao F: Innate immunity to intracellular LPS. Nat Immunol 2019, 20:527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F: Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514:187. [DOI] [PubMed] [Google Scholar]
- 35.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, et al. : Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341:1246–1249. [DOI] [PubMed] [Google Scholar]
- 36.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA: Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341:1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. : Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479:117. [DOI] [PubMed] [Google Scholar]
- 38.Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, Li Z, Wang Y, Zhao Q, Shao F, et al. : Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180:941–955.e20.**This study provided crucial insights into the molecular mechanism of caspase-11/4/5 dependent cytosolic LPS sensing pathway. Using crystal structure analysis and other methods, they showed that LPS binding leads to a site-specific cleavage of caspase-11/4/5 generating an enzymatically active p10 form of the corresponding caspase, which binds to the C-terminus of GSDMD leading to its cleavage and generation of the N-terminal pore-forming fragment.
- 39.Rühl S, Broz P: Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 2015, 45:2927 2936. [DOI] [PubMed] [Google Scholar]
- 40.Meunier E, Dick MS, Dreier RF, Schürmann N, Broz DK, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, et al. : Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014, 509:366 370. [DOI] [PubMed] [Google Scholar]
- 41.Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J: Guanylate binding proteins promote caspase-11–dependent pyroptosis in response to cytoplasmic LPS. Proc National Acad Sci 2014, 111:6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, et al. : IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 2016, 167:382 396.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wandel MP, Kim B-H, Park E-S, Boyle KB, Nayak K, Lagrange B, Herod A, Henry T, Zilbauer M, Rohde J, et al. : Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat Immunol 2020, 21:880–891.**Along with Santos et al., and Kutsch et al., this study showed that GBPs bind to the surface of cytosolic bacteria where they serve as a platform for caspase-4 recruitment and activation.
- 44.Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, Heilig R, Chen KW, Lim RYH, Broz P: Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun 2020, 11:3276.**Along with Wandel et al., and Kutsch et al., this study showed that GBPs bind to the surface of cytosolic bacteria where they serve as a platform for caspase-4 recruitment and activation.
- 45.Kutsch M, Sistemich L, Lesser CF, Goldberg MB, Herrmann C, Coers J: Direct binding of polymeric GBP1 to LPS disrupts bacterial cell envelope functions. Embo J 2020, 39:e104926.**Along with Wandel et al., and Santos et al., this study showed that GBPs bind to the surface of cytosolic bacteria where they serve as a platform for caspase-4 recruitment and activation.
- 46.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VAK: Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165:1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. : The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49:740 753.e7.*This study demonstrated a mechanism of LPS delivery into the cytosol during sepsis. Using inhibition/neutralization of HMGB1 or RAGE-deficiency, they showed that upon sensing LPS, hepatocytes release HMGB1, which directly binds to LPS and facilitates LPS entry into the cells via RAGE-receptor mediated endocytosis. Further, HMGB1 permeabilizes the lysosomal membrane leading to release of LPS into the cytosol.
- 48.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, et al. : Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science 2013, 339:975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Shao Y, Li S, Xin N, Ma T, Zhao C, Song M: Caspase-11 Plays a Protective Role in Pulmonary Acinetobacter baumannii Infection. Infect Immun 2017, 85:e00350–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, Zhao C: Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol 2017, 307:490 496. [DOI] [PubMed] [Google Scholar]
- 51.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA: Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 2014, 16:249 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimarães ES, Morais SB, Broz P, Zamboni DS, Oliveira SC: Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. Plos Pathog 2018, 14:e1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russo AJ, Behl B, Banerjee I, Rathinam VAK: Emerging Insights into Noncanonical Inflammasome Recognition of Microbes. J Mol Biol 2018, 430:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. : Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 2017, 127:4124 4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao Y, Shao F: The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol Rev 2015, 265:85–102. [DOI] [PubMed] [Google Scholar]
- 56.Liu W, Liu X, Li Y, Zhao J, Liu Z, Hu Z, Wang Y, Yao Y, Miller AW, Su B, et al. : LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med 2017, 214:3051–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz VMR, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, Yan BM, Boyer MA, Pensinger DA, Sauer J-D, Shin S: Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc National Acad Sci 2017, 114:13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, Shaw MH, Kim Y-G, Núñez G: NLRC4-driven production of IL-1[beta] discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol 2012, 13:449 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S: NLRC4 inflammasome-mediated production of IL-1β modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol Baltim Md 1950 2012, 188:5623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duncan JA, Canna SW: The NLRC4 Inflammasome. Immunol Rev 2018, 281:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Ménoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, et al. : Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49:413 426.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, Takeda K: Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 2012, 24:637–644. [DOI] [PubMed] [Google Scholar]
- 63.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong N, Peng X, Xi JJ, Chen S, et al. : Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513:237. [DOI] [PubMed] [Google Scholar]
- 64.Park YH, Wood G, Kastner DL, Chae JJ: Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 2016, 17:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aubert DF, Xu H, Yang J, Shi X, Gao W, Li L, Bisaro F, Chen S, Valvano MA, Shao F: A Burkholderia Type VI Effector Deamidates Rho GTPases to Activate the Pyrin Inflammasome and Trigger Inflammation. Cell Host Microbe 2016, 19:664–674. [DOI] [PubMed] [Google Scholar]
- 66.Loeven NA, Medici NP, Bliska JB: The pyrin inflammasome in host-microbe interactions. Curr Opin Microbiol 2020, 54:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, Núñez G: The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175:1651–1664.e14.**This study showed for the first time that during Listeria monocytogenes infection lipoteichoic acid activates the NLRP6 inflammasome. Using knock out mice they also showed that NLRP6 has a detrimental role in Listeria infection.
- 68.Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti T-D: NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S: NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. Plos Pathog 2018, 14:e1007308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang J-P, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. : NLRP6 inflammasome orchestrates the colonic hostmicrobial interface by regulating goblet cell mucus secretion. Cell 2014, 156:1045 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, Gudino CV, Hamilton JA, Darveau RP, Genco CA: Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. Plos Pathog 2014, 10:e1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A: Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 2010, 11:1136 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A: Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc National Acad Sci 2010, 107:3076–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LaRock CN, Cookson BT: The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 2012, 12:799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ratner D, Orning MPA, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, et al. : The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. Plos Pathog 2016, 12:e1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, Chae JJ, Bliska JB: The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe 2016, 20:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R: A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 2010, 7:376–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA: Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Medicine 2007, 204:3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen H, Yang D, Han F, Tan J, Zhang L, Xiao J, Zhang Y, Liu Q: The Bacterial T6SS Effector EvpP Prevents NLRP3 Inflammasome Activation by Inhibiting the Ca(2+)-Dependent MAPK-Jnk Pathway. Cell Host Microbe 2017, 21:47–58. [DOI] [PubMed] [Google Scholar]
- 80.Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart J-M, et al. : The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 2013, 13:570–83. [DOI] [PubMed] [Google Scholar]
- 81.Ge J, Gong Y-N, Xu Y, Shao F: Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc National Acad Sci 2012, 109:6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS: Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun 2015, 6:10205. [DOI] [PMC free article] [PubMed] [Google Scholar]