

Abstract
Ischemia-reperfusion (IR) injury to the renal epithelia is associated with endoplasmic reticulum stress (ERS) and mitochondria dysfunction, which lead to oxidative stress-induced acute kidney injury (AKI). X-box binding protein 1 (XBP1), an ERS response protein, could play a prominent role in IR-induced AKI. In this study, we revealed that XBP1 and its downstream target HRD1 participated in the crosstalk between ERS and mitochondrial dysfunction via regulation of NRF2/HO-1-mediated reactive oxidative stress (ROS) signaling. Mice with reduced expression of XBP1 (heterozygous Xbp1±) were resistant to IR-induced AKI due to the enhanced expression of NRF2/HO-1 and diminished ROS in the kidney. Downregulation of XBP1 in renal epithelial cells resulted in reduced HRD1 expression and increased NRF2/HO-1 function, accompanied with enhanced antioxidant response. Furthermore, HRD1 served as an E3-ligase to facilitate the downregulation of NRF2 through ubiquitination-degradation pathway, and the QSLVPDI motif on NRF2 constituted an active site for its interaction with HRD1. Thus, our findings unveil an important physiological role for XBP1/HRD1 in modulating the antioxidant function of NRF2/HO-1 in the kidney under stress conditions. Molecular therapeutic approaches that target XBP1-HRD1-NRF2 pathway may represent potential effective means to treat renal IR injury.
Subject terms: Ubiquitylation, Endoplasmic reticulum, Acute kidney injury, Apoptosis
Introduction
Renal ischemia-reperfusion injury (IRI) is one of the leading causes of acute kidney injury (AKI), which contributes to high morbidity and mortality rates worldwide1,2. Several concomitant factors, such as reactive oxygen species (ROS) production, abnormal protein synthesis and increased cell death, can exacerbate the progression of renal IRI3–5. However, substantial improvement of efficacy of renal IRI treatment has yet to be achieved2,6, due to limited knowledge of the pathogenesis of IRI. Indeed, a hypothesis has been proposed based on the recent researches on endoplasmic reticulum stress (ERS)7–9. Previous studies from our and other groups have suggested that ERS acts as a major contributor to the exacerbation of IRI10–13. Thus, molecular remodeling of ERS pathway may serve as a new therapeutic strategy for renal IRI10,14,15.
Cellular adaptation to ERS is achieved by the activation of unfolded protein response (UPR), which involves three principal ERS sensors, such as inositol-requiring enzyme 1α (IRE1α), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6)16–18. Notably, the most highly conserved branch of UPR is IRE1α-X-box-binding protein 1 (XBP1) signaling19–21. IRE1α autophosphorylation activates the endoribonuclease activity to splice Xbp1 mRNA (unspliced Xbp1 or Xbp1u, a nonactive form of ERS), thereby changing the reading frame of Xbp1 to encode a potent transcription factor (termed as XBP1s, the active form of ERS)22. XBP1s plays a crucial role in cell survival and death by upregulating UPR-related genes involved in the entrance of proteins into endoplasmic reticulum23. Apart from its multiple roles in neurodegenerative and metabolic diseases20, IRE1α-XBP1 pathway has been reported to cause AKI in patients receiving cardiac surgery24. Furthermore, XBP1s also plays a role in sepsis-induced AKI and inflammation25, and inhibition of ERS (including XBP1) protects kidney against rhabdomyolysis-induced AKI26. However, sepsis-induced AKI (LPS-related) has been driven by markedly different molecular pathologic characteristics from IR-induced AKI25, and the association between XBP1 and significant IR-induced AKI still remains to be elucidated25,27.
Hypoxia and reoxygenation can result in a massive ROS release28, which promotes mitochondrial damage and participates in the crosstalk between ERS and mitochondrial dysfunction29, leading to tubular cell damage and renal injury30. Apart from regulating the main effector molecules as reported in our previous work31, ROS attenuation remains an effective option to protect kidney against IRI via nuclear factor erythroid 2-related factor 2 (NRF2) and heme oxygenase-1 (HO-1)32. The transcription factor NRF2 is responsible for the regulation of various antioxidant and antiapoptotic genes, primarily in response to oxidative stress, and thus increases the levels of endogenous antioxidants and ameliorates cell apoptosis33. HO-1 is modulated by NRF2 activation, which acts and functions as a rate-limiting enzyme of heme catabolism for attenuating ROS production34,35. Hitherto, the current knowledge on NRF2 modulation remains grossly inadequate36. It is believed that NRF2 can be dual-regulated by Kelch-like ECH-associated protein 1 (Keap1) and glycogen synthase kinase 3 beta (GSK-3β), in response to oxidative stress37–39. As a key downstream component of XBP1, HRD1 acts as the core structural component of a large ER membrane-embedded protein complex40, and is initially characterized as an E3 ubiquitin ligase that coordinates the destruction of folding-defective proteins in the early secretory pathway to prevent ERS-associated degradation41. A recent study on liver cirrhosis has found that HRD1 can suppress NRF2-mediated cellular protection40. However, no information on the specific motifs of NRF2 that interact with HRD1 has been reported. Therefore, whether and how XBP1-HRD1 and NRF2-HO-1 exhibit intrinsic relevance and manipulate the crosstalk between ERS and mitochondrial dysfunction in kidney await further clarification.
In this study, we investigated the roles of XBP1 on IR-induced AKI and revealed the mechanisms of NRF2-HO-1 mediated regulation of XBP1-HRD1. The findings provide novel insights into the therapeutic implications of the XBP1-HRD1-NRF2 pathway for treating AKI.
Results
XBP1 is involved in the crosstalk between mitochondrial damage and ERS in renal IRI
After 24 h of IR exposure, the ultrastructural changes in renal tissues were analyzed by transmission electron microscopy (TEM). Both mitochondrial damage (mitochondrial swelling, mitochondrial cristae and membrane disappearance) and ERS (obvious swelling and vacuolization, and ribosome detachment from rough ER) were observed (Fig. 1A). The level of ROS, which reflects mitochondrial damage, was significantly elevated in IRI group compared to sham group (205.75 ± 33.72 vs. 46.40 ± 8.53, respectively; P < 0.01; Fig. 1B). The expression of ATM and P53 were significantly enhanced in IRI group than in sham group (3.00 ± 0.63 vs. 0.83 ± 0.40, 5.83 ± 0.40 vs. 1.00 ± 0.63, respectively; P < 0.01; Fig. 1D). The number of apoptotic cells was significantly higher in IRI group than in sham group (36.17 ± 4.40 vs. 4.50 ± 1.05, respectively; P < 0.01; Fig. 1D). Latterly, the expression levels of ERS-related molecules (BiP, XBP1u, XBP1s, and downstream HRD1) were determined by western blotting. Their relative fold changes in IRI kidneys were significantly increased compared to those in normal kidneys (2.45 ± 0.32, 2.75 ± 0.56, 1.48 ± 0.24, and 2.07 ± 0.30, respectively; P < 0.01; Fig. 1E). Among which, the upregulation of XBP1s was prominent. Collectively, these findings indicate the presence of crosstalk between mitochondrial damage and severe ERS in renal IRI, and the prominent high expression levels of XBP1s suggest that XBP1 may play a unique role in the crosstalk between mitochondrial damage and ERS during renal IRI.
Fig. 1. XBP1 is involved in the crosstalk between mitochondrial damage and ERS in renal IRI.

A Mitochondrial damage and severe ERS occurred simultaneously. TEM analysis revealed mitochondrial swelling, mitochondrial cristae and membrane disappearance, as well as the obvious swelling and vacuolization of ER and detachment of ribosomes from rough ER in IRI group. Normal ER is denoted by red arrows, swelling ER is represented by orange arrows, and swelling mitochondria is indicated by white arrows. Scale bar = 1 μm; ×7000 magnification. B IRI significantly elevated the levels of ROS in IRI group compared to sham group. DHE staining; ×400 magnification; scale bar = 50 μm. **P < 0.01 vs. sham group (n = 6). C The expression of ATM and P53 were raised in IRI group than in sham group. Immunohistochemistry; ×400 magnification; scale bar = 50 μm. **P < 0.01 vs. sham group (n = 6). D The number of apoptotic cells was higher in IRI group than in sham group. TUNEL staining; ×400 magnification; scale bar = 50 μm. **P < 0.01 vs. sham group (n = 6). E The fold changes in the expression of ERS-related molecules in the renal tissues after IRI. The expression levels of BiP, XBP1u, XBP1s, and HRD1 were measured by western blotting. Compared to sham group, the expression levels of ERS-related molecules were all increased in IRI group, especially XBP1s. **P < 0.01 vs. sham group (n = 6).
Mice with Xbp1 downregulation are resistant to IR-induced AKI due to the enhanced expression of NRF2/HO-1 and diminished ROS in kidneys
We next tested the potential role of XBP1 in the mouse model of renal IRI. Twenty-four hours after IR, XBP1 downregulation significantly prolonged the survival of heterozygous Xbp1+/− mice, with a higher rate of 66.66% compared to wild-type (WT) mice (P < 0.05; Fig. 2A). Pathological examination revealed the severe diffuse acute tubular necrosis, sloughing of epithelial cells and tubular basement membrane damage in WT renal tissue exposed to IR. The pathological score (106.83 ± 24.39) of Xbp1+/− mice was significantly lower than that (238.83 ± 20.38) of WT mice (P < 0.01; Fig. 2B), indicating that XBP1 downregulation can alleviate pathological damage in kidneys. Moreover, the function of kidney was determined by assessing the serum levels of Cr and NGAL 24 h after IR. The serum levels of Cr were 10.15 ± 6.10, 216.92 ± 23.13, and 114.33 ± 10.75 μmoI/L in sham, WT and Xbp1+/− groups, respectively (P < 0.01; Fig. 2C), while those of NGAL were 19.42 ± 3.66, 613.01 ± 46.51, and 68.56 ± 7.44 ng/mL in the three groups, respectively (P < 0.01; Fig. 2C), suggesting Xbp± kidneys were more resistant to IR-induced dysfunction than those of WT mice.
Fig. 2. Mice with reduced Xbp1 are resistant to IRI-induced AKI due to the enhanced expression of NRF2/HO-1 and diminished ROS in kidneys.

A XBP1 downregulation significantly prolonged the survival of Xbp1+/− mice. *P < 0.05 vs. IRI group (n = 6). B XBP1 downregulation significantly alleviated acute renal damage and decreased pathological damage scores. H&E staining; scale bar = 50 μm, ×400 magnification. **P < 0.01 vs. IRI group (n = 6). Tubular necrosis is denoted by light blue arrows, sloughing of epithelial cells are represented by white arrows, and tubular basement membrane damage are indicated by black arrows. H&E staining; ×400 magnification; scale bar = 50 μm. **P < 0.01 vs. the sham group (n = 6). C XBP1 downregulation significantly reduced the sera levels of Cr and NGAL. **P < 0.01 vs. IRI group (n = 6). D XBP1 downregulation significantly decreased the expression levels of XBP1 and HRD1, while increased the expression levels of NRF2 and HO-1. Western blot; **P < 0.01 vs. IRI group (n = 6). E IR-induced ROS production was significantly attenuated in the kidneys of Xbp1+/− mice, as shown by both MFI of DHE. Scale bar = 50 μm; ×400 magnification. **P < 0.01 vs. IRI group (n = 6). F XBP1 downregulation significantly reduced the number of apoptotic cells. TUNEL assay; scale bar = 50 μm, ×400 magnification. **P < 0.01 vs. IRI group (n = 6).
The kidneys of WT and Xbp1+/− mice were harvested for Western blot analysis, in order to determine the expression levels of XBP1/HRD1 and NRF2/HO-1 proteins. Compared to WT controls, Xbp1+/− renal tissues exhibited significant decreases in the expression levels of XBP1 and HRD1, while significant increases in those of NRF2 and HO-1 (Fig. 2D). In addition, the ROS level in the kidneys of Xbp1+/− mice was markedly reduced compared to WT controls (99.62 ± 8.02 vs. 205.75 ± 33.72, respectively; P < 0.01; Fig. 2E). These data show that XBP1 downregulation effectively modulates the expression of XBP1, HRD1, NRF2, and HO-1, and consequently lead to ROS suppression. Thereafter, we verified whether the downregulated expression of XBP1 can attenuate cell apoptosis. As shown in Fig. 2F, the number of apoptotic cells was remarkably lower (P < 0.01) in Xbp1+/− group (17.67 ± 2.50) than in WT group (36.17 ± 4.40). Taken together, we found that downregulation of XBP1 protected against renal IRI via inhibiting ROS formation and promoting cell survival. The molecular mechanisms underlying downregulation of XBP1 might include downregulation of a known target of XBP1, HRD1 and upregulation of NRF2 and HO-1.
XBP1 downregulation protects tubular cells against hypoxia/reoxygenation (H/R) damage via control of HRD1, NRF2, and HO-1
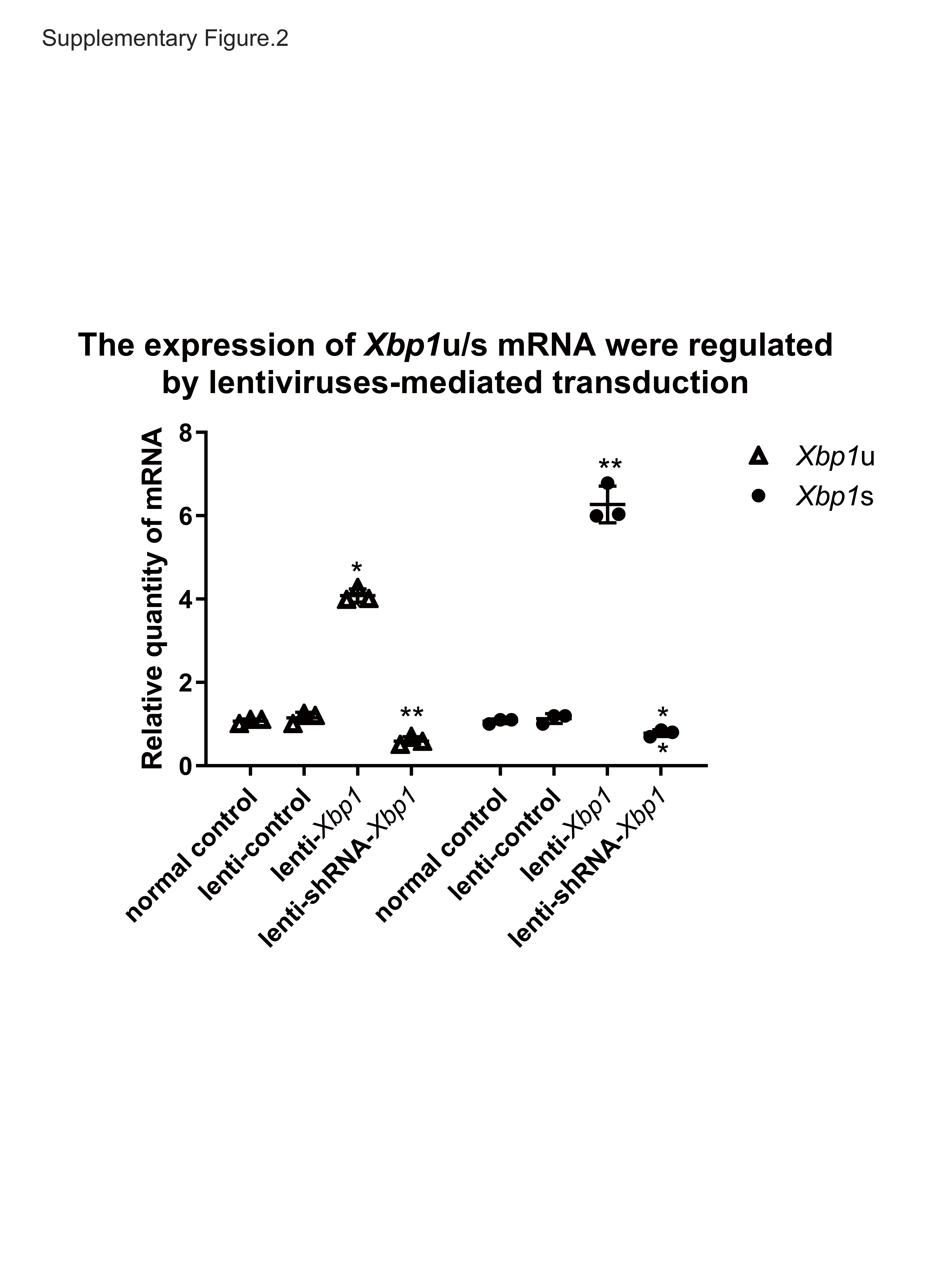
Next, we sought to confirm the molecular actions of XBP1 in IRI. First, we manipulated expression of XBP1 with both lentivirus mediated overexpression and siRNA knockdown in mouse kidney epithelial cell line (TCMK-1) (Fig. S2, S3). Then, different cell lines were subjected to H/R injury. As shown in Fig. 3A, the levels (MFI of DCF) of ROS were reduced in XBP1 knockdown cells and increased in XBP1 overexpression cells as compared to control cells (11.32 ± 1.38 in lenti-control H/R, 16.03 ± 1.32 lenti-Xbp1 and 6.59 ± 0.71 in lenti-shRNA-Xbp1). Meanwhile, the expression of P53, ATM, and cleavage caspase 3 were descended in XBP1 knockdown cells and enhanced in XBP1 overexpression cells as compared to control cells (Fig. 3B). Similarly, flow cytometric analysis revealed that the apoptotic cells were small reduced in XBP1 knockdown cells as compared to that in control cells (11.12 ± 0.86 % in in lenti-control H/R and 3.02 ± 1.30% in lenti-shRNA-Xbp1, Fig. 3C). Additionally, the proliferative capacity of H/R-exposed TCMK-1 cells was significantly enhanced in lenti-shRNA-Xbp1 group (Fig. 3D). These findings demonstrated that XBP1 downregulation may ameliorate H/R injury in vitro.
Fig. 3. XBP1 downregulation protects TCMK-1 cells against H/R via regulation of HRD1, NRF2, and HO-1.

A The level of ROS was attenuated by lenti-shRNA-Xbp1 transduction in H/R-exposed TCMK-1 cells. DCFH-DA assay; **P < 0.01 vs. lenti-control H/R group at each time point (n = 6). B P53, ATM, and cleavage caspase 3 expression were ascended by lenti-Xbp1 transduction and descended by lenti-shRNA-Xbp1 transduction in H/R-exposed TCMK-1 cells. Western blot; **P < 0.01 vs. lenti-control H/R group (n = 6). C The apoptotic proportion of H/R-exposed TCMK-1 cells was decreased by lenti-shRNA-Xbp1 transduction, as revealed by flow cytometry. **P < 0.01 vs. lenti-control H/R group at each time point (n = 6). D The BrdU incorporation (proliferative capability) of H/R-exposed TCMK-1 was enhanced by lenti-shRNA-Xbp1 transduction. *P < 0.05 vs. lenti-control H/R group at each time point (n = 6). E HO-1 expression was negatively modulated by XBP1 expression following lenti-Xbp1 and lenti-shRNA-Xbp1 transduction. Western blot; **P < 0.01 vs. lenti-control H/R group (n = 6). F XBP1 positively and negatively regulated HRD1 and NRF2, respectively. *P < 0.05, **P < 0.01 vs. lenti-control H/R group (n = 6).
To further explore the molecular action of XBP1 in response to H/R injury, the expression levels of HO-1 were examined, and the results showed that HO-1 was significantly downregulated in lenti-Xbp1 and upregulated in lenti-shRNA-Xbp1 cells as compared to that in control cells (P < 0.01; Fig. 3E), suggesting that XBP1 negatively regulates HO-1 expression. Furthermore, western blot analysis revealed that XBP1 downregulation suppressed HRD1 expression and induced NRF2 expression, while XBP1 upregulation increased HRD1 expression and decreased NRF2 expression (Fig. 3F). These findings show that XBP1 positively and negatively regulates HRD1 and NRF2/HO-1, respectively.
HRD1 modulates NRF2 through ubiquitination degradation
As it was shown that HRD1 served as an E3 ubiquitin ligase for NRF2 in liver cirrhosis40, we performed the following experiments to test whether HRD1 could regulate NRF2 in the setting of renal IR injury as well. First, confocal images showed colocalization between HRD1 and NRF2 was increased in H/R-exposed TCMK-1 cells compared to normal cells, with the Pearson’s coefficient values of (1708.00 ± 130.70) × 10−4 and (5.55 ± 0.53) × 10−4, respectively, (P < 0.01; Fig. 4A), indicating an enhanced endogenous interaction between HRD1 and NRF2 after H/R exposure. Next, co-immunoprecipitation analysis demonstrated that HRD1 and NRF2 apparently coexisted in the immune-precipitated complexes in H/R-exposed TCMK-1 cells (Fig. 4B). Moreover, neither upregulation nor downregulation of HRD1 could alter XBP1 expression, while HRD1 negatively regulated NRF2 expression (Fig. 4C). In contrast, alteration of NRF2 had no effects on the expression of XBP1 nor HRD1 (Fig. S4). Furthermore, ubiquitylation assay was used to determine the ubiquitylation of NRF2 by HRD1 in HEK-293T cells. Overexpression of HRD1 promoted NRF2 ubiquitylation, thus reducing the level of NRF2 via degradation (Fig. 4D). Taken together, HRD1 is an E3 ubiquitin ligase of NRF2 in IR conditions.
Fig. 4. HRD1 modulates NRF2 through ubiquitination degradation.

A Colocalization of HRD1 and NRF2 in H/R-exposed TCMK-1 cells. Confocal microscopy analysis; Pearson’s correlation coefficient; scale bar = 50 μm; ×600 magnification. **P < 0.01 vs. normal control group (n = 6). B HRD1 and NRF2 coexisted in the precipitated complexes of H/R-exposed TCMK-1 cells. IP immunoprecipitation, IB immunoblot,. C Silencing of HRD1 led to an increase in NRF2 level, while overexpression of HRD1 caused a reduction in NRF2 level. D HRD1 overexpression promoted NRF2 ubiquitylation and degradation, and subsequently reduced NRF2 level in HEK-293T cells. E The QSLVPDI motif on NRF2 constituted an active site for its interaction with HRD1. F The deletion of QSLVPDI amino acids sequence (Δ125–131) remarkably decrease ubiquitylation of NRF2 mediated by HRD1. G Graphical illustration of the working mechanism: XBP1/HRD1 is involved in IR-induced AKI through the regulation of NRF2/HO-1-mediated ROS signaling.
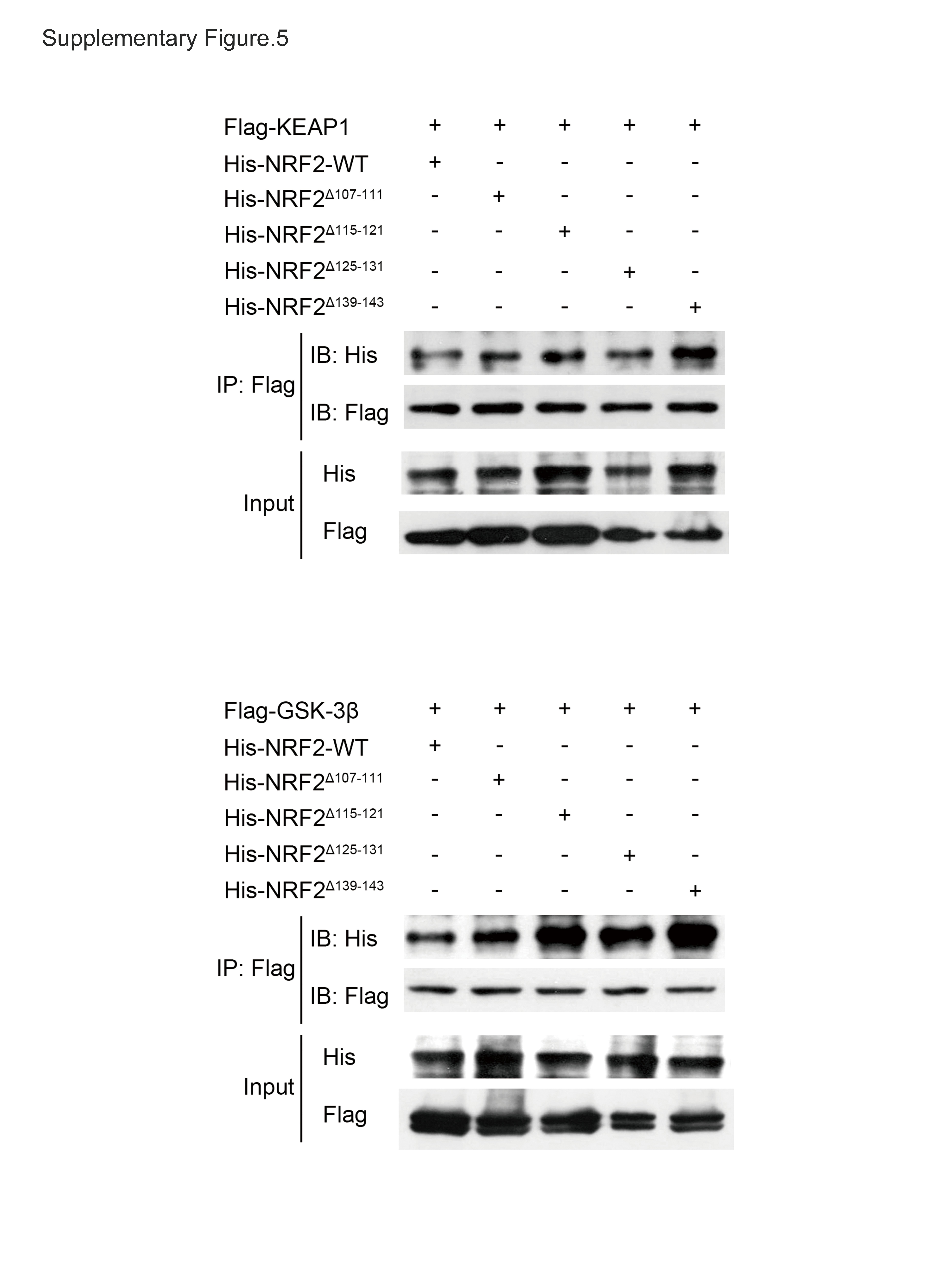
Recent data have suggested a direct interaction between NRF2 Neh4-5 domains and HRD140. Through a bioinformatic approach, we identified four conserved sequences (Δ107–111, Δ115–121, Δ125–131, and Δ139–143) in Neh4-5 domains of NRF2 that potentially bind with HRD1 (Fig. 4E). To further verify whether these sequences can act as the motifs of NRF2 that interact with HRD1, an in vitro binding assay was performed by using HA-tagged ubiquitin proteins, Flag-tagged HRD1 proteins and His-tagged NRF2 proteins with deletion mutants in Δ107–111, Δ115–121, Δ125–131, or Δ139–143 domain and immunoprecipitated from HEK-293T cells. The results showed that the deletion of QSLVPDI amino acids sequence (Δ125–131) remarkably diminished the interaction between NRF2 and HRD1 and ubiquitylation of NRF2, meaning HRD1 binded to the motif-QSLVPDI (Δ125–131) on NRF2 to ubiquitylate NRF2 (Fig. 4E, F). In addition, motif-QSLVPDI (Δ125–131) was proven to have no effect on Keap1 and GSK-3β (Fig. S5). In summary, our study suggested that renal IRI could induce expression of XBP1 and its target HRD1, the latter serving as an E3-ligase of NRF2 to inhibit NRF2/HO-1 signaling in mitochondria (as illustrated in Fig. 4G).
Discussion
In the kidneys undergoing IR-induced AKI, at ultrastructural level, TEM observed mitochondrial damage and severe ERS, supporting the co-presence of mitochondrial dysfunction and ERS after renal IRI. IRI involves a complex pathological process where the tissue injury is initiated by H/R, leading to damaged electron transport chain and high ROS production in mitochondria42. With recovery of oxidation, excessive ROS can be generated from the oxidative damage43. We demonstrated a large amount of ROS (denoting the severity of mitochondrial damage) and elevated apoptosis following IRI Based on the current knowledge that ERS can crosstalk with ROS-induced mitochondrial damage in an infection model29, XBP1s pathway is associated with mitochondrial function44,45, and N-acetylcysteine abrogates mitochondrial ROS through ERS and thus alleviates myocardial apoptosis46, our findings indicate a potential role of XBP1 in crosstalk of ERS and mitochondrial damage in renal IRI. Indeed, modulation of XBP1 led to complex alteration of both the ERS signaling factor (HRD1) and mitochondrial damage factors (NRF2/HO-1).
The protective effects of XBP1 downregulation on kidney against IRI were subsequently tested. Due to mouse kidney’s high tolerability on IR, mild IR injury by the reported 20-min ischemia and 24-h reperfusion failed to affect XBP1 expression in mice kidney25, and exerted no significant impact on renal function and survival. Therefore, we used a severe IR injury model (45-min ischemia and 24-h reperfusion) to archive clinical significance. Though the previous study has shown that XBP1s hyperactivity is detrimental to kidney25, XBP1 is necessary for survival. Complete knockout of XBP1 resulted in embryonic lethality47,48, and deletion of an XBP1 allele in mice led to enhanced ERS resulting in systemic insulin resistance and insulin receptor substrate-1 degradation47,49,50. Therefore, in this work, heterozygous Xbp1+/− mice were used as an animal model for XBP1 downregulation. Following IRI, the survival rate of heterozygous Xbp1+/− mice was significantly increased compared to WT mice. The IR-exacerbated pathological damages and kidney function (sera Cr and NGAL) had shown significant improvement in heterozygous Xbp1+/− mice. The underlying molecular events were further clarified. XBP1 expression was shown to be downregulated in the renal tissues of Xbp1+/− mice after IRI. Consequently, XBP1 downregulation decreased HRD1 expression and increased the expression of NRF2 and HO-1. XBP1 has been recently reported positive regulation on NRF2 expression in retinal pigment epithelium51. As results, both ROS levels and apoptosis in the renal tissues were attenuated. In addition, researchers also observed the effects of XBP1 downregulation on the innate and adaptive immune systems during renal IRI process1,52,53, which await further investigation into the underlying mechanism. Collectively, mice with Xbp1 downregulation are resistant to IR-induced AKI due to the enhanced expression of NRF2/HO-1 and diminished ROS in kidneys.
To precisely describe the mechanism of XBP1 on renal IRI, TCMK-1 cells were used. XBP1 is recognized as a stress-inducible transcriptional activator of ERS54–56. Our study demonstrated that lenti-Xbp1 transduction effectively upregulated the expression of XBP1s (the active form), while lenti-shRNA-Xbp1 transduction remarkably downregulated its expression. Moreover, we found that XBP1 upregulation elevated ROS levels, increased apoptosis, and reduced proliferation in H/R-exposed tubular cells, while XBP1 downregulation exhibited the opposite effects. Therefore, modulation of XBP1 can regulate ROS production and apoptosis of tubular cells undergoing H/R.
It is well accepted that HO-1 is modulated by NRF2, and HRD1 represents the key downstream molecules of XBP157. Our studies suggested XBP1 could regulate NRF2/HO-1 signaling via regulating HRD1 expression. In turn, mitochondrial ROS formation was altered by XBP1.
Next, we sought to determine the mechanisms underlying HRD1-mediated regulation of NRF2. First, a strong colocalization between HRD1 and NRF2 was detected in the cytoplasm of H/R-exposed tubular cells. Second, co-immunoprecipitation analysis revealed a molecular conjunction between HRD1 and NRF2. As an E3 ubiquitin ligase58, HRD1 may degrade NRF2 by identifying protein substrates and facilitating or directly catalyzing the binding of ubiquitin after H/R exposure. The results of ubiquitylation assay showed that HRD1 overexpression promoted NRF2 ubiquitylation and reduced the expression level of NRF2 protein. Furthermore, HRD1 was demonstrated to bind with the motif-QSLVPDI (Δ125–131), but not other motifs (Fig. S6), on both NRF2 and ubiquitylated NRF2.
In summary, our study unveils that ERS signaling axis, XBP1/HRD1, participates in the crosstalk between ERS and mitochondrial dysfunction through control of NRF2/HO-1-mediated ROS signaling. IR increases XBP1-HRD1 expression and promotes interaction between HRD1 and NRF2, which in turn induce ubiquitylation and degradation of NRF2. The interaction domain within NRF2 is the motif-QSLVPDI (Δ125–131). Downregulation of NRF2 induces ROS production, impairs kidney function and shortens animal survival. Thus, downregulation of XBP1 can effectively protect kidney against IRI, which may be of great importance in the development of novel XBP1-based therapeutic strategies for kidney protection against IRI.
Materials and methods
Experimental mice
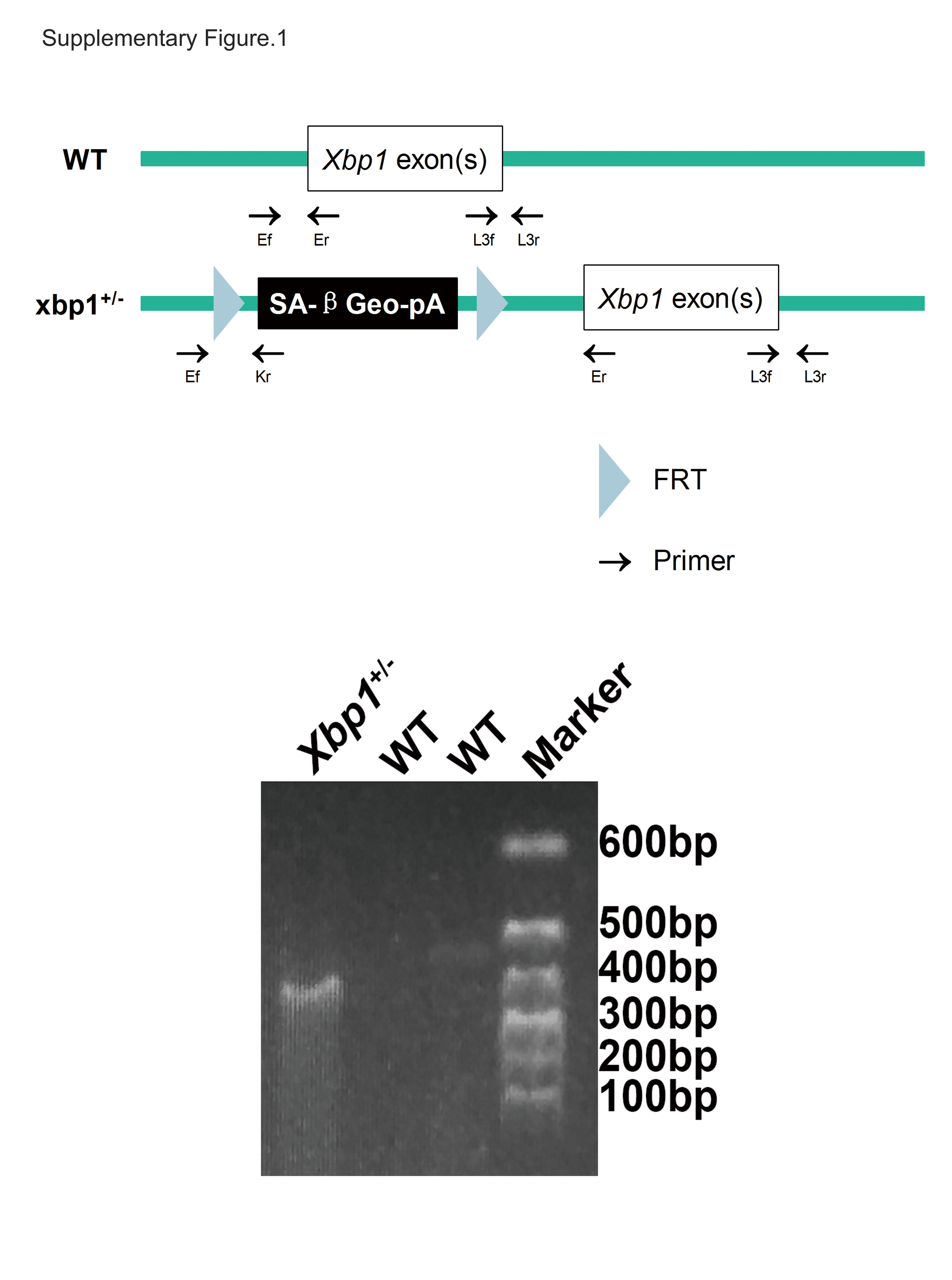
Wild-type (WT) C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (China). Heterozygous Xbp1+/− mice (C57BL/6NTac-Xbp1 < tm1a(EUCOMM)Wtsi > /IcsOrl mice) were obtained from the European Mouse Mutant Cell Repository (Phil Avner, Paris, France) via Beijing Vital River Laboratory Animal Technology Co., Ltd. (China). The Xbp1 allele contained a trapping cassette SA-βgeo-pA (splice acceptor-beta-geo-polyA) flanked by flippase (Flp) recombination target (FRT) site upstream of exon. This resulted in the truncation of endogenous transcript, and thus forming a constitutive null mutation to knockdown XBP1 expression. The FRT-flanked region of heterozygous Xbp1+/− mice was detected by genotyping using PCR assay (Fig. S1).
All animals were housed in specific pathogen-free facilities and maintained under controlled conditions (22 °C, 55% humidity and 12 h day/night cycle) at Huazhong University of Science and Technology animal center. All animal experiments were approved by the Institutional Animal Care and Use Committee at Tongji Hospital, and carried out in accordance with the Experimental Animal Management Ordinance (National Science and Technology Committee of China).
Renal IRI model
Renal IRI was induced on female mice aged 8–10 weeks as described previously59. The left renal pedicle was occluded for 45 min, followed by reperfusion. After that, right kidney nephrectomy was performed. The mice were randomized into the following groups: (1) sham group: mice received sham surgery; (2) WT group: mice with left kidney IRI; and (3) Xbp1+/− group: Xbp1+/− mice with left kidney IRI. Six animals in each group were studied for their survival rates, while three animals were selected for mechanistic study for six times. The animals were sacrificed 24 h after reperfusion, in order to collect serum and left kidney samples.
TEM
The left kidneys in sham and IRI groups were prepared for semi-thin sections (0.5–1 mm in thickness). The sections were stained with methylene blue and examined under transmission electron microscope (Hitachi, Tokyo, Japan).
Measurement of ROS levels in renal tissues
Left kidneys were dissected 24 h after IRI, rapidly frozen in liquid nitrogen, cut at 2 μm in a cryostat (CryoStar NX50, Thermo Fisher Scientific, Inc., Waltham, MA, USA), and stained with dihydroethidium (DHE; Sigma–Aldrich; Merck KGaA, Darmstadt, Germany). The stained sections were observed under a fluorescence microscope (ECLIPSE C1, NIKON, Tokyo, Japan). ROS production in renal tissues was evaluated by determining the mean fluorescence intensities (MFI) of DHE.
Immunohistochemistry
Immunohistochemistry of ATM and P53 was performed on formalin-fixed and paraffin-embedded sections according to the previously described procedure60. Images were captured under the Olympus BX-51 light microscope. The staining intensity and proportion of positive cells were calculated as previously described61. The intensity was scored as follows, 0 = negative, 1 = weak, 2 = moderate, 3 = strong. Proportion scores are points assigned based on the percentage of positive cells as follows: <10% for 0 point, 10–25% for 1 point. 26–50% for 2 point, 51–75% for 3 point, and >75% for 4 point. A total score was obtained by multiplying intensity score × proportion score. The total score ranged from 0 to 12.
Terminal dUTP nick end labeling (TUNEL)
Cell apoptosis in paraffin sections was evaluated by TUNEL assay using the In Situ Cell Death Detection Kit, POD (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Sections of formalin-fixed and paraffin-embedded kidney tissue were dewaxed by washing in xylene and rehydrated through a gradual series of ethanol and distilled water. Proteinase K-permeabilized sections were subjected to enzymatic in situ labeling of DNA strand breaks using TUNEL technique as it is indicated in manufacturer instruction. Apoptosis was assessed using the Olympus BX-51 light microscope, six images were captured in randomly selected fields from each section and the number of TUNEL-positive cells were counted.
Histology staining
Left kidneys were dissected from mice 24 h after IRI and fixed in 10% formalin. The formalin-fixed kidneys were embedded in paraffin and sectioned at 4 μm thickness. The sections were then subjected to hematoxylin and eosin (H&E) staining. Semi-quantitative analysis was performed as previously described62.
Random cortical fields were observed using a ×20 objective. A graticule grid (25 squares) was used to determine the number of line intersects involving tubular profiles. One hundred intersections were examined for each kidney, and a score from 0 to 3 was given for each tubular profile involving an intersection: 0 = normal histology; 1 = tubular cell swelling, brush border loss, nuclear con-densation, with up to one third of the tubular profile showing nuclear loss; 2 = same as for score 1, but greater than one third and less than two thirds of the tubular profile show nuclear loss; and 3 = greater than two thirds of the tubular profile showing nuclear loss. The total score for each kidney was calculated by the addition of all 100 scores with a maximum score of 300.
Images were recorded under a light microscope (Olympus BX-51, Tokyo, Japan), and then analyzed using ImageJ software.
Sera measurement
Serum creatinine (Cr) concentrations were measured at the clinical laboratory of Tongji Hospital (Wuhan, China). The concentrations of neutrophil gelatinase associated lipocalin (NGAL) were determined using LEGEND MAX™ Mouse NGAL ELISA kit (Biolegend Inc., San Diego, CA, USA).
Cells
The mouse kidney epithelial cell line (TCMK-1) and human embryonic kidney cells (HEK-293T) were purchased from ATCC (Manassas, VA, USA). TCMK-1 cells were cultured in RPMI 1640 (HyClone; GE Healthcare, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA). HEK-293T cells were cultured in high glucose Dulbecco’s modified eagle medium (DMEM; HyClone; GE Healthcare, Logan, UT, USA) supplemented with 10% FBS. The cultured cells were maintained at 37 °C in a humidified incubator containing 5% CO2.
Plasmids, small interfering RNA (siRNA), and lentiviruses
Plasmids expressing mouse Hrd1, human Flag-HRD1, human Flag-KEAP1, human Flag-GSK-3β, human His-NRF2, and HA-ubiquitin (human) as well as control plasmid were purchased from Vigene Bioscience Co., Ltd. (Rockville, MD, USA).
The conserved sequences between the Neh4-5 domain of Nrf2 and the residues 31–81 of NRF1 were predicated using PRALINE (http://www.ibi.vu.nl/programs/pralinewww/). The deletions of Δ107–111, Δ115–121, Δ125–131, and Δ139–143 were constructed within pcDNA3.1/ His-NRF2 by site-directed mutagenesis (SDM) using the primers listed in Table S1.
Mouse Hrd1 siRNA and control siRNA were purchased from Guangzhou Ribobio Co., Ltd. (Guangzhou, China). Mouse Xbp1 lentivirus, mouse Xbp1 shRNA lentivirus and control lentivirus were purchased from Hanbio Co., Ltd. (Shanghai, China). The sequences of siRNA and shRNA are presented in Table S2.
Transfection of plasmid and siRNA was performed using Lipofectamine 2000 Transfection Reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. Transduction of lentiviruses into TCMK-1 cells was carried out as described previously63.
Inhibitor and activator of NRF2
To downregulate NRF2 expression, ML385 (Sigma–Aldrich; Merck KGaA, Darmstadt, Germany), a specific NRF2 inhibitor, was added into the culture medium at a concentration of 5 μM, followed by incubation for 48 h. Meanwhile, CDDO-Me (Sigma–Aldrich; Merck KGaA, Darmstadt, Germany), a specific NRF2 activator, was used to upregulate NRF2 expression by incubating for 48 h at a concentration of 0.5 μM.
Cell hypoxia/reoxygenation (H/R) injury
For hypoxic induction, TCMK-1 and HEK-293T cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2, 1% O2 and 94% N2 for 24 h. Then, the cells were reoxygenated under normal conditions for 2 h. The details of TCMK-1 cell grouping are summarized in Table S3.
Flow cytometry
Cell apoptosis was evaluated after labeling with propidium iodide (10 μL; Biolegend Inc., San Diego, CA, USA) and Annexin V (5 μL; Biolegend Inc., San Diego, CA, USA). The production of ROS was measured using the Total ROS Assay Kit (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cell proliferation was evaluated by BrdU Cell Proliferation Assay Kit (MultiSciences, Hangzhou, China).
Immunofluorescence
TCMK-1 cell culture slides and renal tissue sections were incubated with primary antibodies at 4 °C overnight. Secondary antibodies were then added and incubated at 37 °C for 50 min. Cell nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI). The details of antibodies are provided in Table S4. Images were obtained using a fluorescence microscope (ECLIPSE C1, NIKON, Tokyo, Japan). A confocal laser scanning microscope (FV1000, Olympus, Tokyo, Japan) was used to examine the colocalization between HRD1 and NRF2 molecules. Images processing and analysis were performed using with ImageJ software.
Quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was isolated from TCMK-1 cells and kidney samples using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Total RNA was reverse-transcribed into cDNA using the PrimeScript™ RT reagent kit (Perfect Real Time; Takara Bio Inc., Otsu, Shiga, Japan) following the manufacturer’s protocol. qRT-PCR was conducted using TB Green Premix Ex Taq (Tli RNaseH Plus) and ROX plus (Takara Bio Inc., Otsu, Shiga, Japan). The fold changes in gene expression were calculated upon data normalization. The sequences of the primers are listed in Table S5.
Western blot
TCMK-1 cells and renal tissues were lysed in radio-immunoprecipitation assay buffer containing phosphatase and protease inhibitors. Total protein concentration was determined using bicinchoninic acid protein assay (Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. Equal amounts of total proteins were loaded into 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. The membranes were blocked with nonfat milk for 2 h at 37 °C and then incubated with primary antibodies at 4 °C overnight. Subsequently, the membranes were washed in Tris-buffered saline-0.1% Tween-20 and incubated with secondary antibodies at 37 °C for 1.5 h. After incubation, the immunoblot bands were visualized using enhanced chemiluminescence reagent (Beyotime Institute of Biotechnology). Band intensities were quantified by densitometric analysis using ImageJ 1.6.0_20 (National Institutes of Health, Bethesda, MD, USA) after normalized with β-actin (loading control). The details of antibodies are shown in Table S4.
Co-immunoprecipitation
Briefly, H/R-exposed HEK-293T cells were lysed in sample lysis buffer. Total protein lysates (1 mg) were incubated with 1 mg of antibodies against specific proteins or the indicated rabbit polyclonal IgG control, followed by rotation for 4 h at 4 °C. Next, 20 mL of resuspended volume of Protein A/G Plus-Agarose (Santa Cruz Biotechnology, Dallas, TX, USA) was added to the samples and rotation was continued for 2 h. The resulting complexes were eluted according to the manufacturer’s instructions, and subjected to western blot analysis. The details of antibodies can be found in Table S4.
Ubiquitylation assay
The ubiquitylation assay was carried out according to a previously described protocol24. Briefly, HEK-293T cells were co-transfected with plasmids for HA-Ub and the indicated proteins for 48 h, and treated with 10 μM MG132 (Sigma–Aldrich; Merck KGaA, Darmstadt, Germany) for 4 h to inhibit protein degradation. Subsequently, the protein lysates of HEK-293T cells were precipitated and determined by western blot analysis using LC3 and HA antibodies. The details of antibodies are listed in Table S4.
Statistical analysis
Statistical analyses were performed using PRISM 6 (GraphPad Software, La Jolla, CA). All data were presented as mean ± standard error of the mean (SEM). Survival rates were analyzed by a log-rank test, while other data accorded with normal distribution and homogeneity of variance were compared using Student’s t-test or one-way ANOVA. P-values of less than 0.05 were considered statistically significant.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Funding
The present study was supported by the grants from the National Natural Science Foundation of China (grant no. 81873623, 81570678, and 81500573), the Major State Basic Research Development Program of China (grant no. 2013CB530803), the Clinical Research Physician Program of Tongji Medical College, HUST and the Non-Profit Central Research Institute Fund of Chinese Academy of Medical Sciences (grant no. 2018PT32018).
Conflict of interest
The authors declare no cometing interests.
Footnotes
Edited by Maria Victoria Niklison Chirou
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ji Zhang, Jiasi Zhang
Supplementary information
The online version contains supplementary material available at 10.1038/s41420-021-00425-z.
References
- 1.Zuk A, Bonventre JV. Acute kidney injury. Annu. Rev. Med. 2016;67:293–307. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng BQ, et al. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3β signaling. Proc. Natl Acad. Sci. USA. 2017;114:12608–12613. doi: 10.1073/pnas.1705615114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gang GT, et al. Protection of NAD(P)H:quinone oxidoreductase 1 against renal ischemia/reperfusion injury in mice. Free Radic. Biol. Med. 2014;67:139–149. doi: 10.1016/j.freeradbiomed.2013.10.817. [DOI] [PubMed] [Google Scholar]
- 4.Thiele JR, et al. A conformational change in C-reactive protein enhances leukocyte recruitment and reactive oxygen species generation in ischemia/reperfusion injury. Front. Immunol. 2018;9:675. doi: 10.3389/fimmu.2018.00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailly-Maitre B, et al. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA. 2006;103:2809–2814. doi: 10.1073/pnas.0506854103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemmner S, Bachmann Q. Normothermic machine perfusion may prevent regulated cell death following renal ischemia-reperfusion injury. Am. J. Transpl. 2019;19:1245. doi: 10.1111/ajt.15189. [DOI] [PubMed] [Google Scholar]
- 7.Ling Q, et al. Roles of the exogenous H2S-mediated SR-A signaling pathway in renal ischemia/reperfusion injury in regulating endoplasmic reticulum stress-induced autophagy in a rat model. Cell Physiol. Biochem. 2017;41:2461–2474. doi: 10.1159/000475915. [DOI] [PubMed] [Google Scholar]
- 8.Hosszu A, et al. σ1-receptor agonism protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2017;28:152–165. doi: 10.1681/ASN.2015070772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017;13:681–696. doi: 10.1038/nrneph.2017.129. [DOI] [PubMed] [Google Scholar]
- 10.Fan Y, et al. Inhibition of reticulon-1A-mediated endoplasmic reticulum stress in early AKI attenuates renal fibrosis development. J. Am. Soc. Nephrol. 2017;28:2007–2021. doi: 10.1681/ASN.2016091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, et al. Mesencephalic astrocyte-derived neurotrophic factor as a urine biomarker for endoplasmic reticulum stress-related kidney diseases. J. Am. Soc. Nephrol. 2016;27:2974–2982. doi: 10.1681/ASN.2014100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, et al. A novel organ preservation solution with efficient clearance of red blood cells improves kidney transplantation in a canine model. Cell Biosci. 2018;8:28. doi: 10.1186/s13578-018-0226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y, et al. Bone marrow-derived mesenchymal stem cells protect islet grafts against endoplasmic reticulum stress-induced apoptosis during the early stage after transplantation. Stem Cells. 2018;36:1045–1061. doi: 10.1002/stem.2823. [DOI] [PubMed] [Google Scholar]
- 14.Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum-new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014;10:369–378. doi: 10.1038/nrneph.2014.67. [DOI] [PubMed] [Google Scholar]
- 15.Tajiri S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 16.Zhao N, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Invest. 2018;128:1283–1299. doi: 10.1172/JCI95873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madhusudhan T, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat. Commun. 2015;6:6496. doi: 10.1038/ncomms7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han CY, Lim SW, Koo JH, Kim W, Kim SG. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut. 2016;65:1377–1388. doi: 10.1136/gutjnl-2014-308506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tohmonda T, et al. IRE1α/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. J. Clin. Invest. 2015;125:3269–3279. doi: 10.1172/JCI76765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang D, Niwa M, Koong AC. Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015;33:48–56. doi: 10.1016/j.semcancer.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sha H, et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao G, et al. Unspliced XBP1 confers VSMC homeostasis and prevents aortic aneurysm formation via FoxO4 interaction. Circ. Res. 2017;121:1331–1345. doi: 10.1161/CIRCRESAHA.117.311450. [DOI] [PubMed] [Google Scholar]
- 23.Choi, S. I. et al. Melatonin reduces endoplasmic reticulum stress and corneal dystrophy-associated TGFBIp through activation of endoplasmic reticulum-associated protein degradation. J. Pineal Res.10.1111/jpi.12426 (2017). [DOI] [PubMed]
- 24.Fohlen B, et al. Real-time and non-invasive monitoring of the activation of the IRE1α-XBP1 pathway in individuals with hemodynamic impairment. EBioMedicine. 2018;27:284–292. doi: 10.1016/j.ebiom.2017.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrè S, et al. Renal tubular cell spliced X-box binding protein 1 (Xbp1s) has a unique role in sepsis-induced acute kidney injury and inflammation. Kidney Int. 2019;96:1359–1373. doi: 10.1016/j.kint.2019.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Y, et al. Selective histone deacetylase 6 inhibitor 23BB alleviated rhabdomyolysis-induced acute kidney injury by regulating endoplasmic reticulum stress and apoptosis. Front. Pharmacol. 2018;9:274. doi: 10.3389/fphar.2018.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding C, et al. miR-124/IRE-1α affects renal ischemia/reperfusion injury by regulating endoplasmic reticulum stress in renal tubular epithelial cells. Acta Biochim. Biophys. 2020;52:160–167. doi: 10.1093/abbs/gmz150. [DOI] [PubMed] [Google Scholar]
- 28.Nezu M, et al. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017;91:387–401. doi: 10.1016/j.kint.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 29.Liao Y, et al. Endoplasmic reticulum stress induces macrophages to produce IL-1β during Mycobacterium bovis infection via a positive feedback loop between mitochondrial damage and inflammasome activation. Front. Immunol. 2019;10:268. doi: 10.3389/fimmu.2019.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee PK, et al. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney Int. 2000;58:658–673. doi: 10.1046/j.1523-1755.2000.00212.x. [DOI] [PubMed] [Google Scholar]
- 31.He, W. et al. Hypothermic oxygenated perfusion (HOPE) attenuates ischemia/reperfusion injury in the liver through inhibition of the TXNIP/NLRP3 inflammasome pathway in a rat model of donation after cardiac death. FASEB J. 10.1096/fj.201800028RR (2018). [DOI] [PubMed]
- 32.Zhang X, et al. Stress protein activation by the cyclopentenone prostaglandin 15-deoxy-delta12,14-prostaglandin J2 in human mesangial cells. Kidney Int. 2004;65:798–810. doi: 10.1111/j.1523-1755.2004.00454.x. [DOI] [PubMed] [Google Scholar]
- 33.Noel S, et al. T lymphocyte-specific activation of Nrf2 protects from AKI. J. Am. Soc. Nephrol. 2015;26:2989–3000. doi: 10.1681/ASN.2014100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83:1029–1041. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muhammad, T., Ikram, M., Ullah, R., Rehman, S. U. & Kim, M. O. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients10.3390/nu11030648 (2019). [DOI] [PMC free article] [PubMed] [Retracted]
- 36.Fratta Pasini AM, et al. Nrf2 expression is increased in peripheral blood mononuclear cells derived from mild-moderate ex-smoker COPD patients with persistent oxidative stress. Int. J. Chron. Obstruct Pulmon Dis. 2016;11:1733–1743. doi: 10.2147/COPD.S102218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuadrado A, Kügler S, Lastres-Becker I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018;14:522–534. doi: 10.1016/j.redox.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chowdhry S, et al. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben Mkaddem S, et al. Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ. 2010;17:1474–1485. doi: 10.1038/cdd.2010.26. [DOI] [PubMed] [Google Scholar]
- 43.Bos EM, et al. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. 2013;24:759–770. doi: 10.1681/ASN.2012030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015;40:141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duplan E, et al. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 2013;126:2124–2133. doi: 10.1242/jcs.127340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He Y, Zhou L, Fan Z, Liu S, Fang W. Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by N‑acetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell Death Dis. 2018;9:568. doi: 10.1038/s41419-018-0593-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 48.Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003;194:29–38. doi: 10.1034/j.1600-065X.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 49.Drori A, Misaghi S, Haimovich J, Messerle M, Tirosh B. Prolonged endoplasmic reticulum stress promotes mislocalization of immunoglobulins to the cytoplasm. Mol. Immunol. 2010;47:1719–1727. doi: 10.1016/j.molimm.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 50.Sozen E, Ozer NK. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: an updated mini-review. Redox Biol. 2017;12:456–461. doi: 10.1016/j.redox.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen C, et al. Regulation of Nrf2 by X box-binding protein 1 in retinal pigment epithelium. Front. Genet. 2018;9:658. doi: 10.3389/fgene.2018.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huen SC, Cantley LG. Macrophages in renal injury and repair. Annu. Rev. Physiol. 2017;79:449–469. doi: 10.1146/annurev-physiol-022516-034219. [DOI] [PubMed] [Google Scholar]
- 53.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015;11:88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 54.Cubillos-Ruiz JR, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527–1538. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu P, et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamamoto K, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 58.Amano T, et al. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003;17:2436–2449. doi: 10.1101/gad.1096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jankowski J, et al. Epithelial and endothelial pannexin1 channels mediate AKI. J. Am. Soc. Nephrol. 2018;29:1887–1899. doi: 10.1681/ASN.2017121306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y-Q, et al. Genetically engineered distal airway stem cell transplantation protects mice from pulmonary infection. EMBO Mol. Med. 2020;12:e10233–e10233. doi: 10.15252/emmm.201810233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu S, et al. RBCK1 promotes p53 degradation via ubiquitination in renal cell carcinoma. Cell Death Dis. 2019;10:254–254. doi: 10.1038/s41419-019-1488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chatterjee PK, et al. Calpain inhibitor-1 reduces renal ischemia/reperfusion injury in the rat. Kidney Int. 2001;59:2073–2083. doi: 10.1046/j.1523-1755.2001.00722.x. [DOI] [PubMed] [Google Scholar]
- 63.Xiao J, et al. Downregulation of Blimp1 inhibits the maturation of bone marrow-derived dendritic cells. Int. J. Mol. Med. 2019;43:1094–1104. doi: 10.3892/ijmm.2018.4000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.