

Abstract
Serum response factor (SRF), a member of the Mcm1, Agamous, Deficiens, and SRF (MADS) box transcription factor, is widely expressed in all cell types and plays a crucial role in the physiological function and development of diseases. SRF regulates its downstream genes by binding to their CArG DNA box by interacting with various cofactors. However, the underlying mechanisms are not fully understood, therefore attracting increasing research attention due to the importance of this topic. This review's objective is to discuss the new progress in the studies of the molecular mechanisms involved in the activation of SRF and its impacts in physiological and pathological conditions. Notably, we summarized the recent studies on the interaction of SRF with its two main types of cofactors belonging to the myocardin families of transcription factors and the members of the ternary complex factors. The knowledge of these mechanisms will create new opportunities for understanding the dynamics of many traits and disease pathogenesis especially, cardiovascular diseases and cancer that could serve as targets for pharmacological control and treatment of these diseases.
Keywords: binding, cofactors, mechanisms, myocardin, serum response factor, transcription
Serum response factor is a key transcription factor that contributes to multiple cellular functions. It controls the expression of downstream genes through binding to two main cofactors, myocardin‐related transcription factors and ternary complex factors, in a cell‐specific manner. The interaction between SRF and the cofactor is regulated by various signaling upon extracellular stimuli and participates in cell development and functions. This process also has an overall effect on health outcomes, notably cardiovascular diseases and cancers.
Abbreviations
- ChIP
chromatin immunoprecipitation
- cIAP2
cellular inhibitor of apoptosis protein 2
- CVD
cardiovascular diseases
- DDR2
discoidin domain receptor 2
- DNMTs
DNA methyltransferases
- ECM
extracellular matrix
- Elk‐1
ETS like‐1 protein
- ERK1/2
extracellular signal‐regulated protein kinase 1/2
- FOXO3
forkhead box O3
- GI
gastrointestinal tract
- GSK‐3
glucose synthase kinase‐3
- IEG
immediate early gene
- KLF4
Kruppel‐like transcription factor 4
- MAPK
mitogen‐activated protein kinase
- MDM 4
mouse double minute 4 protein
- MKL
megakaryoblastic leukemia
- MRTFs
myocardin families of transcription factors
- PCNA
proliferating cell nuclear antigen
- PDGF
platelet‐derived growth factor
- ROCK
RhoA kinase
- SIRT 1 and 2
sirtuins 1 and 2
- Skp2
S‐phase kinase‐associated protein 2
- SMC
smooth muscle cell
- SRE
serum response element
- SRF
serum response factor
- STARs
striated muscle activator of Rho signaling
- TCFs
ternary complex factors
- TGF‐β
transforming growth factor‐β
- VSMCs
vascular smooth muscle cells
Introduction
Serum response factor (SRF) is a member of the Mcm1, Agamous, Deficiens, and SRF (MADS) box transcription factor widely expressed in all cell types. SRF participates in multiple biological functions in many cells, such as muscle cells (cardiac, skeletal, and smooth), endothelial cells, fibroblasts, hepatocytes, immune cells, and neurons [1, 2, 3], and plays a crucial role in the tissue development of gastrointestinal tracts (GI), and cardiovascular and immune systems [4, 5, 6, 7, 8]. SRF is also involved in various diseases' pathogenesis, including multiple types of cardiovascular diseases (CVD) and cancers [1, 9].
Although the importance of SRF in these conditions is widely recognized, the mechanisms involved remain largely unknown and need to be investigated. Several studies have suggested that the CArG box [CC(A/T)6GG] DNA sequences within the promoters of some genes are critically responsible for the transcriptional effects of SRF [1, 2, 10]. CArG element is a component of the serum response element (SRE) that is present in the promoter of c‐fos, one of the immediate early genes (IEG) [11]. CArG box is vital for the process of serum induction of the promoter when stimulated by growth factors. It is commonly known as the consensus binding site for the SRF [11]. For example, evidence indicates that the binding between SRF and CArG box is vital for the expression of smooth muscle cell (SMC) genes that mediate the cellular differentiation and proliferation under physiological conditions and also play critical roles in the development of vascular diseases [11]. Importantly, studies have demonstrated that the transcription effect of SRF on these downstream genes relies on its interaction with the diverse cofactors to constitute a functional SRF/cofactor complex controlling the downstream gene expression [1, 11].
This SRF/cofactor interaction varies depending upon different stimuli and mediates a distinct effect in a cell‐specific manner, resulting in a high diversity of SRF functions. Due to the importance of these regulations of SRF in both physiological and pathological conditions, increasing attention has been focused on this research area. Numerous studies are being conducted to reveal the underlying mechanisms responsible for the SRF/cofactors interaction and their potential roles in the pathogenesis of the diseases [1].
In this review, we summarized the new progress in the studies related to the effects of SRF, focusing on the molecular basis and regulatory mechanisms of the interaction between SRF and its main cofactors. With a comprehensive search of the PubMed database, we collected the published articles on SRF/cofactor interactions and the health outcomes, especially in CVD and cancers. Specifically, we mainly included the recent studies and highlighted the new information in the following aspects; first, we discussed the molecular basis of SRF and its main cofactors, as well as the regulations of their interaction; Secondly, we highlighted the effects of SRF/cofactors at multiple levels, including the molecular level on the expressions of the downstream genes, cellular functions, tissue development, and physiological function. Thirdly, we presented the implications of SRF/cofactors interactions on various diseases focusing on CVD and cancers. Also, we presented the perspectives on future research direction on the related areas.
The main cofactors of SRF
Serum response factor regulates numerous gene expressions through its association with various accessory cofactors, among which the most well‐reported ones are myocardin‐related transcription factors (MRTFs) and the members of the ternary complex factors (TCFs) [2, 3, 11] (Fig. 1). Although other potential cofactors, such as GATA and NK2 homeobox 5 family of transcription factors, are reported, they are involved to a lesser extent [12].
Fig. 1.
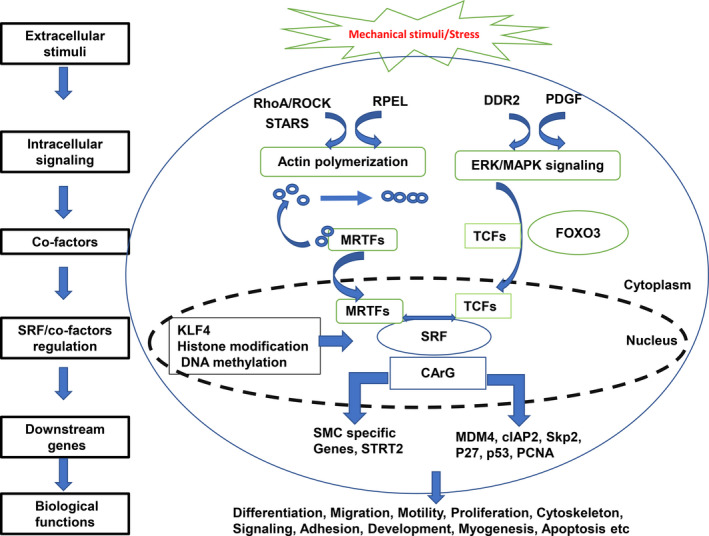
Overview of the molecular mechanisms governing SRF binding to cofactors and the subsequent transcription of target genes in cells. SRF, serum response factor; ROCK, RhoA kinase; STARs, striated muscle activator of Rho signaling; MRTFs, myocardin families of transcription factors; DDR2, discoidin domain receptor 2; PDGF, platelet‐derived growth factor; ERK, extracellular signal‐regulated protein kinase; MAPK, mitogen‐activated protein kinase; TCFs, ternary complex factors; FOXO3, forkhead box O3; KLF4, Kruppel‐like transcription factor 4; MDM 4, Mouse Double Minute 4 protein; cIAP2, cellular inhibitor of apoptosis protein 2; Skp2, S‐phase kinase‐associated protein 2; PCNA, proliferating cell nuclear antigen.
Myocardin‐related transcription factors, including myocardin, MRTF‐A/MKL1/MAL, and MRTF‐B/MKL2, comprise a family of related transcriptional co‐activators with multiple biological functions which appear in other reviews [2, 13]. MRTFs physically associate with SRF and synergistically activate transcription, which regulates cellular differentiation by activating the downstream genes through their interactions with the CArG box [11, 14, 15, 16]. The expression of myocardin is specific to the cardiac and vascular SMCs, while MKL1 and MKL2 are more broadly expressed [3, 11]. By interacting with SRF, myocardin induces the expression of SMC marker genes. At the same time, the MKL1 and MKL2, on the other hand, are involved in actin dynamics resulting in the control of SMC‐specific contractile genes during actin polymerization [3, 17]. However, they do not bind directly to DNA sequences but associate directly with SRF to control SMC gene transcription [3, 18]. SRF connects with the Rho‐actin cytoskeleton to initiate transcription response through its binding interactions with the MRTFs [19, 20]. This can also alternatively be made possible in response to cell proliferative growth factor stimulation with the consequent displacement of myocardin in favor of ETS like‐1 protein (Elk‐1) interaction and the increased expression of IEGs, for instance, c‐fos [21, 22].
The TCFs, including the ETS‐like proteins (Elk1, Elk3, and Elk4), associate with SRF through the mitogen‐activated protein kinase (MAPK or MAP kinase) in response to serum or growth factors and regulate IEGs [3, 13, 19, 23]. Interactions between the TCFs and SRF result in a simultaneous binding to ETS‐binding site adjacent to the CArG box [3]. Also, the TCFs have been reported to have the capacity to be independent of SRF [24, 25, 26].
The MKLs and TCFs interact with SRF in a mutually exclusive manner and compete for SRF DNA‐binding domain. MKLs are recently known to be involved in the regulation of some IEG expressions under serum induction [3, 13, 17, 27]. Some IEGs have been reported to be coupled to one pathway or another in fibroblasts, while in SMCs, platelet‐derived growth factor (PDGF) induces cofactor exchange [22, 24]. However, it is uncertain whether the cofactor competition is commonly associated with SRF regulation in vivo.
Numerous studies have implicated the TCFs in cell proliferation and cancer; however, the extent to which the transcription of IEGs is TCF‐dependent and the target genes are involved are still unknown [13, 17, 24]. MRTFs, on the other hand, have been shown to mediate the morphogenetic, adhesive, and motile processes [13, 24]. It was recently demonstrated that the transcription of much of the serum‐induced IEGs is MRTF/SRF‐dependent but the role of TCF‐SRF signaling could not be determined due to the lack of specific TCF inhibitors as well as the poor quality of TCF chromatin immunoprecipitation (ChIP) results [24, 28]. It is generally considered that the TCFs are antagonistic of MRTF‐dependent SRF target genes and compete directly for SRF‐binding sites. Consequently, this competition is mainly responsible for the balance between the proliferative and contractile gene expressions [24].
The activation of SRF by interacting with its cofactors
The SRF binding to CArG box enhances its ability to act as an anchoring protein by binding to other cofactors to effect regulation of target gene transcription [29]. However, several mechanisms govern these interactions between SRF and the cofactors, and these remain primarily unexplained [30]. It is a potent transcriptional regulator of target genes, with numerous experiments suggesting over 200 of such genes regulated by SRF [31]. The ability of SRF to regulate these different sets of downstream target genes is a function of the promoter context and its interactions with cofactors [31]. SRF controls the transcription of several IEGs and associates mainly with two families of signal‐regulated cofactors, the ERK‐regulated TCFs and the Rho‐actin controlled MRTFs [30, 31]. Some of the mechanisms associated with these SRF interactions with cofactors are briefly discussed here.
The interaction between SRF and MRTFs
As stated above, SRF transcription is activated mainly by MRTFs when they translocate into the nucleus where they interact with SRF. Therefore, the ability of the MRTFs to regulate this SRF transcription is dependent upon their nuclear translocation [32]. It has been shown that this process is regulated by the RhoA signaling (Rho family of small GTPases) and the subsequent actin polymerization [33, 34]. The RhoA signaling increases the F‐actin/G‐actin ratio in different types of cells through multiple pathways, for instance, by promoting F‐actin assembly in fibroblasts and activation of RhoA kinase (ROCK) in vascular smooth muscle cells (VSMCs) [34, 35, 36]. This results in the release of myocardin from G‐actin and transfers to the nucleus, enabling it to form complexes with SRF to activate the transcription of the downstream genes [37]. This binding is also influenced by RPEL actin‐binding domains that enable the MRTFs to bind monomeric G‐actin, leading to their retention in the cytoplasm [13, 32]. Following stimulation under mechanical stress and actin polymerization into filamentous F‐actin, the MRTFs will relocalize to the nucleus with a subsequent increase in SRF transcriptional activity [17, 32]. Changes in actin dynamics related to SM physiology also associate with the expression of myocardin [34].
The role of striated muscle activator of Rho signaling (STARS) in promoting nuclear localization of MRTF‐A and MRTF‐B has also been described, indicating the likelihood of competing with their RPEL motif for actin binding [34, 38]. Additionally, the sequestration of MRTF‐A results in actin polymerization due to the RhoA signaling pathway and the subsequent activation of SRF caused by the translocation of MRTF‐A/MAL from the cytoplasm to the nucleus [17, 34, 39]. The role of SRF in actin dynamics is responsible for the regulatory loop in which actin synthesis is promoted by changes in cell shape that may influence the cytoskeletal structure [34].
The possibility of a common mechanism regulating SIRT2 and SRF during serum stimulation has also been reported [40]. The SIRT2 gene is upregulated during conditions of serum deprivation in similar ways that the SRF gene also responds to serum deprivation and/or serum restoration following deprivation [40, 41]. SRF binding to SIRT2 is associated with a CArG element in the SIRT2 promoter gene. Here, serum deprivation was reported to induce SIRT2 expression while SRF and SRF‐binding protein, p49/STRAP on the other hand, repressed SIRT2 expression [40]. The Rho/SRF inhibitor, CCG‐1423, also suppressed the expression of the SIRT2 gene, suggesting that the SIRT2 gene is a downstream target of the Rho/SRF signaling mechanism [40].
The interaction between SRF and TCFs
SRF transcription is also controlled by the transcription cofactors TCFs that are activated through MAPK signaling pathways. Recently, a study reported novel results that mitogen‐activated cardiac fibroblast utilizes the mechanism related to collagen receptor, discoidin domain receptor 2 (DDR2)‐dependent activation of extracellular signal‐regulated protein kinase 1/2 (ERK1/2) MAPK, and SRF for coordinated regulation of resistance to apoptosis and cell cycle progression [42]. This is achieved through enhanced expression of apoptotic cellular inhibitor of apoptosis protein 2 (cIAP2) in cardiac fibroblasts with the consequent protection against oxidative injury [42]. Additionally, the transcription process upregulates S‐phase kinase‐associated protein 2 (Skp2), leading to post‐translational degradation of the cyclin‐dependent kinase inhibitor, p27, responsible for cell cycle arrest, and promoting G1‐S transition, Rb phosphorylation, increased proliferating cell nuclear antigen (PCNA), and flow cytometry [42]. Finally, DDR2‐dependent activation of ERK1/2 MAPK also led to the suppression of forkhead box O3, FOXO3a‐mediated transcriptional induction of p27 [42].
Interactions between SRF and ETS domain transcription factors have also been reported to be one of the mechanisms for the regulation of the transcription of the mouse double minute 4 protein (MDM 4) oncogene in hepatocellular carcinoma (HCC) [43]. The MDM 4 protein is known to be a p53‐negative transcription regulator that inhibits the transcriptional activities of p53. Its protein and mRNA are upregulated in human HCC due to copy number alterations and post‐transcriptional mechanisms associated with the AKT/mTOR signaling [43]. Using in silico analysis, SRF, ELK1, and ELK4 were reported to be putative transcription factors binding to the MDM 4 promoter region. Also, there was a strong positive correlation between SRF and MDM 4 expression and high mRNA levels of MDM4, SRF, and ELK4 associated with reduced survival of HCC patients following liver resection. On the other hand, inhibition of the transcription factors caused a reduction in the mRNA levels of MDM 4, suggesting the critical roles of SRF and its cofactors in promoting the oncogenic function of MDM 4 in HCC [43]. Therefore, targeting the transcription of MDM 4 may offer a promising therapeutic approach for the treatment of liver cancer patients [43].
In addition, a new mechanism has been reported involving the repression of the expression of multiple SMC genes by Kruppel‐like transcription factor 4 (KLF4) and platelet‐derived PDGF‐BB [44, 45]. First, KLF4 repressed the myocardin‐induced activation of SMCs and the expression of myocardin itself [44]. Then, the upregulation of KLF4 under PDGF‐BB stimulation reduced SRF binding to CArG‐containing regions of intact chromatin [44]. The association suggests that KLF4 represses the expression of SMCs by downregulating expression of myocardin and preventing the SRF/myocardin cofactor interactions in their association with the promoter region of SMCs [44].
The competition between MRTFs and TCFs and their cell/tissue specificity
The TCFs become phosphorylated when MAPK signaling pathway is activated, and Elk‐1 interacts with SRF by binding to the short peptide motif called B‐box [34]. On the other hand, myocardin and MRTFs’ SRF binding are similar to the predicted secondary structure of the B‐box. However, it differs from that of Elk‐1 by the absence of direct amino acid homology [34]. Therefore, the deletion of this myocardin region hinders the ability of myocardin to interact with SRF to activate SRF‐dependent target genes. However, these functions are often reversed when Elk‐1 B‐box replaces this binding region [22, 34]. As such, myocardin and Elk‐1 compete for this SRF‐binding site in a mutually exclusive manner to create a switch that facilitates the regulation of SMCs by growth factors [17, 22, 34].
Stimulation of SMCs by PDGF results in Elk‐1 phosphorylation by MAPK signaling pathway to cause it to interact with SRF and displace myocardin [22, 34]. This change in Elk‐1 binding to SRF due to repression of myocardin results in an overall reduction in the expression of SMCs because Elk‐1 is relatively weaker than myocardin [34]. Conversely, reduction in the levels of endogenous Elk‐1 in SMCs will increase the expression of SMC target genes due to derepression of the SRF–myocardin binding [34, 46]. Phosphorylation of SRF can also lead to modification and alteration of its affinity for DNA binding [47].
Chromatin immunoprecipitation assay together with human promoter microarrays has been used to identify over 200 SRF‐binding sites downstream, including many other new sites in three different human cell lines (Jurkat cells, T/G HA‐VSMC, and Be(2)‐C cell line) [4]. A genome‐wide view of SRF occupancy at its different binding sites with differing cell types was also used along with PCR validations at over half of the binding sites to make deductions of the results [4]. Binding of ELK4 cofactor and epigenetic modifications were reported to be the fundamental mechanisms responsible for tissue‐specific SRF binding [4]. ELK4 interacts with SRF to activate the transcription of downstream genes [4]. The interactions of SRF with its cofactors can also be specific to different tissues within the human body [4, 48]. It is known that epigenetic mechanisms are critically involved in the regulation of chromatin structure and remodeling, suggesting that they are crucial mediators in cell‐type‐specific gene expression during growth and disease conditions [49]. Histone modification and DNA methylation are the most extensively studied epigenetic changes. While histone modifications alter the packaging of chromatin, DNA methylation occurs at the 5′ position of the cytosine ring due to DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) [49].
The epigenomic regulation of transcriptional control of SRF on their downstream genes
Identifying the genes that SRF regulates is critical to understanding the functional roles it plays in health and diseases [4]. SRF regulation and its target genes demonstrate a typical example of how diverse genes are controlled by a single DNA‐binding protein and the significance of cofactors in this molecular regulation of gene expression [34]. Many target genes of SRF regulation are involved in cell proliferation and muscle differentiation, with muscle genes being repressed by growth factor, and are, therefore, not activated until myoblasts are absent from the cell cycle [34].
In a study, several genes were reported to be directly regulated by SRF, with half of them being experimentally validated, and are mainly involved in cell growth, migration, cytoskeletal organization, and myogenesis [34, 50]. A common example of SRF target gene that is involved in cell growth is the IEG, c‐fos which is controlled by SRE, acting together with the surrounding cis elements in the promoter [34]. There is specificity in the expression of CArG box‐dependent SRF target muscle genes, with some of the genes being expressed only in one type of muscle cell, for instance, smooth, skeletal, or cardiac muscle cells while others are expressed in multiple muscle cells [34]. Though the molecular mechanisms responsible for this have not been fully elucidated, it has, however, been suggested to possibly involve both positive and negative controls of proteins as well as gene‐specific action of SRF [34]. As SRF and MRTFs regulate the transcription of SMC‐specific genes through the interactions with the conserved CArG elements within the promoters of the SMCs [20, 51], however, the fact that these transcription factors are also present in other non‐SMCs demonstrates the possibility of other mechanisms being associated with the expression of these genes [20].
Previous studies have suggested that SRF binding to CArG box DNA sequences within the context of intact chromatin induced the expression of these SMC genes [1, 11, 19, 20]. Chromatin structures determine the permissiveness of DNA sequences to transcription factor binding, and it could offer a glimpse into the regulation of SMCS by SRF [1]. For instance, histone modifications that promote gene expression such as H3 and H4 acetylation, H3K4 methylation, and H3K9 demethylation/acetylation were previously reported at the SMC‐specific promoters in SMC [1, 10, 20]. MRTFs have also been shown to enhance the modification of chromatin by using histone‐modifying enzymes [1, 20, 52]. DNA methylation has been reported to be another mechanism equally responsible for the transcription of SMC‐specific genes [20].
The ability of SRF to regulate SMCS also involves other mechanisms that may likely control chromatin structure and access to SMC‐specific target gene promoters [1, 51]. SRF binding to these target gene promoters has been reported to correlate with positive chromatin marks [51]. Chromatin structure and function are greatly influenced by histone proteins post‐translational modifications, and they regulate the permissiveness of chromatin to DNA transcription factor binding by either acetylation or methylation [1]. MRTFs have been shown in several studies to interact with chromatin modifiers [1, 51, 53, 54]. High expression of SRF induced by several agonists, especially transforming growth factor‐β (TGF‐β), promotes increased SRF binding to the CArG elements present within the promoters of specific genes [51]. In addition to these, phosphorylation of Ser103 by kinases has also been reported to cause increased affinity of SRF to CArG elements [47, 51, 55].
The association between SRF and CArG is also responsible for the transcriptional repression of these genes during disease conditions owing to changes in environmental conditions both in vitro and in vivo [1, 3]. It is equally essential for cell differentiation and repression under both physiological and pathological conditions, respectively. However, the mechanisms responsible for this association are still not clearly understood [1].
It has been demonstrated in macrophages using genome‐wide location analysis that SRF binding is not only enriched at target gene promoters but also occur at distal inter‐ and intragenic locations [48]. This is contrary to previous studies, suggesting that SRF binding is mainly at the proximal sites because almost all functional CArG boxes were shown to be located within 4 kb of the transcription start site [48, 50, 56]. Functional studies also established that PU.1, an E26 transformation‐specific family of transcription factor, is required to activate these target genes, thereby providing better understanding into the molecular mechanisms regulating cell‐specific programs of SRF‐dependent gene expression [48].
Participation of SRF in cellular functions and tissue development
Serum response factor is a highly versatile transcription factor encoded by a single gene that is ubiquitously expressed in different cell types [13]. It regulates the transcription of various target genes that perform diverse essential molecular and biological functions of multiple cells including muscle cells, endothelial cells, fibroblasts, hepatocytes, and neurons. It is involved in the development of gastrulation, heart, vascular system, and liver as well as the immune system and neurons by regulating cell proliferation, differentiation, cell growth, and regeneration [13]. SRF also contributes to the regulation in cell survival [13, 57].
By using the strategies of either the downregulation or overexpression of SRF, several cell culture and animal experiments have revealed the significant roles of SRF in serum‐dependent cell growth and skeletal muscle differentiation [34, 58, 59, 60]. In addition, SRF‐deficient phenotypes exhibit defective development and maintenance of the heart and GI. However, the survival of the animal varies and may depend on the time of knockout and the promoter that drives the expression of Cre recombinase [60]. For example, although SRF knockout has been demonstrated to be lethal in congenital knockout systems, exhibiting cardiac or GI SM defects [5, 60, 61], Myh11 knockout of SRF resulted in a more extended survival compared to other promoters such as SM22α. Inducible knockout experiments of the genes in adult SMCs also caused severe GI dilation and thinning of the SM layers but survived longer than the congenital knockout model [60]. The phenotypic similarities between congenital and inducible knockout animals suggest the importance of SRF in cardiac and SM development in embryos and maintenance in adults [60].
Furthermore, the importance of SRF in cell growth and skeletal muscle differentiation was also demonstrated in cell culture experiments in which SRF was downregulated [34, 58]. This resulted in the blockage of coronary SMC differentiation in chick embryos and disruption of skeletal and cardiac muscle differentiation in transgenic mice [34, 59]. Moreover, in a similar way, a homozygous SRF‐null mutation in mice had lethal effect at gastrulation, indicating the essential role of SRF in regulating genes involved in cell migration and adhesion needed for gastrulation [8, 34].
It is well known that embryonic stem cells that are deficient in SRF exhibit this abnormality due to a loss of actin stress fibers and a consequent loss of the genes associated with components of actin stress fibers such as vinculin, talin, and an actin isoform [34, 57]. Conditional SRF deletion from cardiac muscle led to significant disruption in sarcomeric structure and abnormal muscle gene regulation [5, 62, 63]. In SMCs, SRF deletion led to reduction in the number of differentiated SMCs near the dorsal aorta, while the few that survived had visible cytoskeletal defects [5, 34]. In the skeletal muscle, SRF deletion caused perinatal lethality resulting from hypoplasia [34, 64]. SRF may also play a critical role in muscle development; however, the early lethality of SRF‐null mice makes the study difficult [34].
Serum response factor is also found to be important for the regulation of the development of axons in the mammalian brain [65]. Conditional knockout mice experiments have demonstrated that SRF plays an important cell‐autonomous role in axonal growth [65, 66]. Although the mechanisms responsible for these SRF regulatory activities in the neurons are not properly understood, some studies linked it to the phosphorylation of SRF by glucose synthase kinase‐3 (GSK‐3) which increased SRF binding with MKL1 and MKL2 [65]. More importantly, it was discovered that vinculin (an actin‐binding protein and SRF target gene) is involved in promoting axon growth in SRF‐deficient and GSK‐3‐inhibited neurons, suggesting that SRF is important for GSK‐3‐mediated axonal growth [65]. However, other conflicting reports also showed that blocking GSK‐3 activity increases the expression of SRF target genes, suggesting that SRF alone can promote axonal growth in the absence of GSK‐3 signaling [65].
In addition, the association of SRF with MRTFs has been reported to be critical for megakaryocyte (Mk) maturation [33]. SRF conditional knockout mice with Mk lineage have been observed to display abnormal Mk maturation and thrombocytopenia, while those with MRTF‐A knockout showed blocked Mk maturation [33, 67, 68]. These conditions become more severe when both MRTF‐A and MRTF‐B are knocked out in the mice [33, 69].
The role of SRF in the regulation of apoptosis has also recently come to light, especially in SMCs where massive apoptosis was observed in a knockout mouse model accompanied by an abnormal increase in apoptotic proteins and a deficiency of anti‐apoptotic miRNA [60, 70]. SRF depletion/deficiency and inhibition have also been associated with apoptosis in the embryonic heart [62], lung [71], SH‐J1 cells [72], and the GI [60]. These studies and others indicate that SRF plays an anti‐apoptotic role and is essential for promoting cell survival [60].
Implications of SRF on various diseases
Since SRF is widely expressed in various cells and regulates numerous genes, it has also been linked with the development of many human diseases [4] (Table 1). In this review, we focused on two of the highest risk diseases: cancer and CVD [4, 73, 74, 75]. By interacting with its cofactors, SRF controls the expression of most of the genes associated with contractile apparatus and actin cytoskeleton [76, 77]. These SRF target genes are involved in numerous processes in the body including contractility, cell movement, and cell growth signaling that are required for the normal development and functioning of the heart and vessels [50, 78]. As such, deficiency in the transcription of these SRF‐dependent genes can cause various diseases of the cardiovascular system including congenital heart and vascular defects and other cardiomyopathy such as hypertrophy, heart failure, atherosclerosis, and restenosis [13]. Excessive overexpression of SRF may also be pathogenic to the cardiovascular system, suggesting the need for cardiac homeostasis in SRF signaling pathway [13].
Table 1.
Summary of the studies associated with SRF/cofactors interaction in different diseases.
| S/N | Model | Function | Mechanism | Outcome | Reference |
|---|---|---|---|---|---|
| 1 | Cardiac fibroblasts isolated from young adult male Sprague Dawley rats. | Anti‐apoptosis and resistance to oxidative injury | ERK1/2 MAPK‐activated SRF | Activation of DDR2‐mediated ERK1/2 MAPK regulates cell survival and cell cycle progression in cardiac fibroblasts via SRF | [42] |
| 2 | Human hepatocellular carcinoma (HCCs) | Liver cancer | Transcription of the MDM 4 oncogene | SRF, ELK1, and ELK4 were reported to be putative transcription factors binding to the MDM 4 promoter region and were associated with reduced survival of HCC patients following liver resection. | [43] |
| 3 | SRF (‐/‐) embryonic stem cells | Cell migration | Actin cytoskeletal structure | Downregulation of FA proteins in ES cells lacking SRF led to inefficient activation of the FA signaling kinase FAK and reduced overall actin expression levels in Srf (‐/‐) ES cells. These changes were accompanied by an offset treadmilling equilibrium, resulting in lowered F‐actin levels. | [57] |
| 4 | SRF knockout mice (cardiomyocytes and SMCs) | Cardiovascular development (growth and muscle differentiation) | Actin contractile and cytoskeletal structure | SRF mutant mice displayed structural defects in the heart and vasculature which coincided with decreases in SRF‐dependent gene expression and death. | [5] |
| 5 | SRF mutant mice | Skeletal muscle development | Actin cytoskeletal muscle growth and maturation | SRF deletion resulted in formation of muscle fibers without hypertrophic growth after birth leading to death during the perinatal period from severe skeletal muscle hypoplasia. | [64] |
| 6 | SRF‐f/f mice | Axon growth in mammalian brain | GSK‐3‐activated SRF phosphorylation | Phosphorylation and activation of SRF by GSK‐3 that is critical for SRF‐dependent axon growth in mammalian central neurons. | [65] |
| 7 | SRFf/f mice | Axon and neuron development | Actin cytoskeleton | SRF mutant mice exhibited deficits in cortical axonal projections with a variable loss of the corpus callosum. The number of proliferative cells in the ventricular zone increased during development. These changes were also observed in the developing excitatory neurons of neocortex and hippocampus. | [66] |
| 8 | SMC‐restricted Srf‐inducible knockout mice | Anti‐apoptosis | SRF‐dependent miRNAs | Mice exhibited severe degeneration of SMCs with reduced expression of apoptosis‐associated miRNAs, high level of SMC death, and myopathy in the intestinal muscle layers. These suggest that SMC degeneration via anti‐apoptotic miRNA deficiency resulting from SRF deficiency may be responsible. | [61] |
| 9 | Cross‐sectional study of CTD patients | Heart development | Impaired SRF transcription | Two novel mutations of SRF were identified in the DNA from the peripheral leukocyte cells. There were no differences between the mutants and wild‐type SRF in their protein expression and mRNA transcription. However, both SRF mutants had impaired SRF transcriptional activity at the SRF promoter and atrial natriuretic factor (ANF) promoter as well as reduced synergism with GATA4. | [29] |
| 10 | SHR and WKY rats | Aortic VSMC stiffening | Extracellular dysregulation (integrin β1 and BMP1/LOX via SRF/myocardin signaling) | Reconstituted vessel segments from SHR VSMCs were stiffer, had different morphologies, and less adaptable to stretch than WKY VSMCs. Also, SHR VSMCs had increased synthesis of collagen and induced collagen in reconstituted vessels in addition to higher levels of active integrin β1 and bone morphogenetic protein 1 (BMP1)‐mediated proteolytic cleavage of lysyl oxidase (LOX). These changes were attenuated by an SRF/myocardin. | [79] |
| 11 | Alzheimer’s disease patients | Cognitive decline and dementia in Alzheimer’s Disease | SRF/myocardin overexpression | There was overexpression of several SRF/myocardin‐regulated contractile proteins with hypercontractile phenotype in AD VSMC. Also, overexpression of myocardin in control human cerebral VSMC caused an AD‐like hypercontractile phenotype and reduced endothelial‐dependent and endothelial‐independent relaxation in the mouse aorta ex vivo. However, silencing SRF normalized and reversed these changes. | [82] |
| 12 | Intestinal cells and human colon cell line | Tumorigenesis | Alternatively spliced variants and isoforms of SRF | Full‐length SRF was discovered to be the predominant form of SRF in all 3 cells used (rat IEC‐6 cells, normal human colonic mucosa, and HT‐29 cells). However, the colon cancer cell lines from poorly differentiated tumors had SRFΔ5 as the predominant isoform expressed. IEC‐6 cells transfected with SRFΔ5 also had higher survival than the parental cells | [84] |
Serum response factor inactivation in cells is associated with defective local homeostasis and eventual death in most cases [76]. For instance, its genetic inactivation in developing vascular SMC leads to reduced expression in contractile genes as well as the recruitment of newly developing SMC to the dorsal aorta, eventually causing midgestation arrest of the mouse [5, 76]. In SRF knockout mice in the heart‐forming region, appearance of rhythmic beating myocytes which is considered to be one of the earliest cardiac defects was blocked, suggesting the role of SRF during early cardiac myocyte commitment and differentiation [5, 76].
Mutations in SRF have also been associated with conotruncal heart disease, a group of congenital heart malformations which causes abnormal cardiac outflow tracts [29]. SRF is traditionally known to be a critical factor in heart development, being strongly expressed in the myocardium of the developing mouse and chicken hearts [29]. Loss of SRF arising from inactivation especially during heart development can have lethal consequences and defects in the myocardium of developing mice [29]. The SRF mutants were shown to display impaired SRF transcriptional activity at both the SRF and atrial natriuretic factor promoter, suggesting that they may have potential pathogenic effects [29].
Recent studies have also established a link between upregulation of SRF/myocardin pathways and the pathogenesis of aortic stiffness in age‐related hypertension [37, 79, 80, 81]. Aortic stiffness is known as an independent risk factor for hypertension and cardiovascular morbidity in the elderly, and it is associated with intrinsic mechanical properties of VSMCs [79]. The underlying molecular mechanisms contributing to this condition is not known. Recent studies discovered that the RhoA/ROCK/SRF/myocardin plays a major role in the onset and progression of aortic stiffness and the development of hypertension by mediating a series of alterations including the VSMC intrinsic mechanical property, extracellular matrix (ECM) remodeling, and interaction between VSMC and ECM [37, 79, 80, 81]. Importantly, these regulations by SRF are specific in VSMCs in large conduct vessel but not in small arteries. Pharmacological inhibition of this signaling pathway selectively attenuates pathological aortic stiffening but did not affect the aortic function in normal condition, suggesting that this could be a novel therapeutic strategy for the treatment of age‐related hypertension by targeting these cellular contributors to this condition in the elderly [81].
In Alzheimer's disease, overexpression of SRF and myocardin in small cerebral arteries was shown to contribute to the pathogenesis of the condition as they increase arterial contractility and reduced blood flow due to the activation of SRF‐dependent SM contractile genes [13, 82]. It has also been implicated in pathological SMC proliferation in response to injuries leading to atherosclerosis and restenosis [9, 13]. Suppression of SRF‐dependent gene transcription by the upregulation of other transcription factors such as FOXO4 and KLF4 dedifferentiation of VSMCs also contributes to this phenotypic switch [13, 44]. This is because suppression of myocardin and MRTF activities causes SMC proliferation, especially in atherosclerosis and restenosis, suggesting the importance of SRF/myocardin as a sensor under mechanical stress and growth factor signaling to regulate such phenotypic switches in SMCs [13].
Since SRF was found to be involved in the expression of the genes controlling cell proliferation such as Fos, Junb, Fosb, and Egr1 [4], various studies have associated SRF with tumor formation and cancer metastasis but this role can be either positive, which causes tumor proliferation, or negative, which suppresses tumor cells depending on the specific pathways involved [13, 60]. This suggests a dual role of SRF in the pathogenesis of tumor formation [60]. For example, in gastric carcinoma, the promoter and exon 1 of SRF gene become hypermethylated leading to the downregulation of the mRNA expression [60, 83]. In colon cancer, abnormal overexpression of a truncated SRF isoform is linked with increased cell survival, suggesting that it may contribute to the pathogenesis of colon cancer though it remains uncertain whether the truncation alone is responsible for induction of cell growth or it simply regulated the effect of SRF [60, 84]. The oncogene four‐and‐a‐half LIM domain 2, a potent epithelial–mesenchymal transition inducer, has also been implicated in the pathogenesis of cancer cells, especially in prostate and colon cancer. It is a cell cycle and growth modulator that is required for cancer cell invasion, migration, and adhesion to ECM, and its expression is induced by SRF [13, 60]. Although there is a relationship between actin/MRTF/SRF circuit with human cancer development, suggesting the involvement of MRTF/SRF neoplastic process, there is no definitive evidence to establish the causative association to clinically reported carcinogenesis [13].
Conclusion and future directions
As summarized in Fig. 1, the information presented in this review indicates that SRF is a critical transcriptional factor with diverse biological functions in cells and plays an essential role in the development and maintenance of the normal physiological function in multiple important tissues. It is also involved in the pathogenesis of some diseases that cause high mortality. Mechanistically, SRF confers its transcriptional effects by selectively interacting with its distinct cofactors in a cell‐specific manner which is regulated by the different upstream signaling of these cofactors. Although there is a lot that still needs to be known regarding the effect of SRF, the evidence from the current study highlights the importance of this factor and brings new insights into the understanding of cellular dynamics of so many functional traits and disease pathogenesis, especially CVD and cancer. These molecular mechanisms of SRF binding and gene transcription regulation can be used as molecular targets for the pharmacological control, intervention, and treatment of these diseases and many other conditions, thus opening new ways and opportunities for future studies.
Based on the known information regarding SRF functions, there are a few critical research areas that need to be addressed. First, since the interaction between SRF and its cofactors is the key determinant of its activity, future research should be focused on the regulatory mechanism that controls this interaction. Although some of the molecular mechanisms regulating the interactions with cofactors TCF and MRTF families have been reported, the controls of these SRF/cofactor interactions are far from fully understood. In addition, other unrevealed cofactors and their functions as well as their biological roles need to be investigated. The efforts on these researches will increase the understanding of the molecular mechanisms underlying the diverse functions of SRF and lead to new strategies to treat the SRF‐associated diseases, especially through inhibiting or activating the SRF/cofactor interactions. Secondly, it is notable that SRF plays its role in a cell‐specific manner. It is important to discover the mechanisms of the cell‐selective effect and their specific regulatory signaling and target genes. These studies will lead to the discovery of the distinct therapeutic targets for different diseases and avoid the side effects due to the broad impacts of SRF and its wide distribution. Thirdly, the binding sites of SRF to its downstream target genes are also not fully identified, and its potential regulatory mechanism remains largely unknown. Particularly, it will be important to investigate the epigenomic network that regulates the binding of SRF/cofactor complex by using advanced techniques to discover the new mechanisms involved in cancer and CVD.
Finally, considering the importance of SRF to the control of numerous biological functions in multiple cells, the development of a novel approach for the prevention of pathological conditions associated with its expression [60] could be of tremendous potential clinical application in the treatment of disease conditions associated with SRF deficiency and overexpression. In addition, some in vitro and in vivo animal studies have shown that some drug compounds have been found to be effective in the treatment of pathological conditions related to the upregulation of SRF. For example, a group of small‐molecule inhibitors of RhoA transcriptional signaling (CCG‐100602, CCG‐203971, CCG‐1423, CCG222740, and CCG222740) has been found to be able to inhibit MRTF/SRF‐mediated upregulation of the gene transcription caused by several environmental (mechanical stress) and cytokine (TGF‐β) stimuli and repressed fibrosis and ECM stiffness as well as the VSMC stiffness [37, 79, 81, 85, 86, 87, 88, 89]. Although the results remain contradictory and the mechanisms involved are still not fully identified, they provide a promising strategy for the development of a therapeutic drug for clinical application. Efforts should be made to explore further the targets of these compounds and the mechanisms involved and the strategies to reduce potential side effects.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
JOO wrote the initial draft of the manuscript, and HQ revised the prepared manuscript for publication.
Acknowledgement
This work is partially supported by grants from the National Institute of Health (NIH) (HQ: RO1HL115195, HL142291, and HL137962).
[Correction added on 04 November 2020, after first online publication: The title of the article has been updated.]
References
- 1. McDonald OG, Wamhoff BR, Hoofnagle MH & Owens GK (2006) Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo . J Clin Investig 116, 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miano JM (2003) Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 35, 577–593. [DOI] [PubMed] [Google Scholar]
- 3. Xie L (2014) MKL1/2 and ELK4 co‐regulate distinct serum response factor (SRF) transcription programs in macrophages. BMC Genom 15, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper SJ, Trinklein ND, Nguyen L & Myers RM (2007) Serum response factor binding sites differ in three human cell types. Genome Res 17, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miano JM, Ramanan N, Georger MA, de Mesy Bentley KL, Emerson RL, Balza RO Jr, Xiao Q, Weiler H, Ginty DD & Misra RP (2004) Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci USA 101, 17132–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary‐Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D et al. (2005) Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 112, 2930–2939. [DOI] [PubMed] [Google Scholar]
- 7. Knoll B, Kretz O, Fiedler C, Alberti S, Schutz G, Frotscher M & Nordheim A (2006) Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci 9, 195–204. [DOI] [PubMed] [Google Scholar]
- 8. Arsenian S, Weinhold B, Oelgeschlager M, Ruther U & Nordheim A (1998) Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J 17, 6289–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Owens GK, Kumar MS & Wamhoff BR (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84, 767–801. [DOI] [PubMed] [Google Scholar]
- 10. Manabe I & Owens GK (2001) CArG elements control smooth muscle subtype‐specific expression of smooth muscle myosin in vivo . J Clin Investig 107, 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hendrix JA, Wamhoff BR, McDonald OG, Sinha S, Yoshida T & Owens GK (2005) 5' CArG degeneracy in smooth muscle alpha‐actin is required for injury‐induced gene suppression in vivo . J Clin Investig 115, 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gau D & Roy P (2018) SRF'ing and SAP'ing ‐ the role of MRTF proteins in cell migration. J Cell Sci 131, jcs218222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olson EN & Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J, Kitchen CM, Streb JW & Miano JM (2002) Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34, 1345–1356. [DOI] [PubMed] [Google Scholar]
- 15. Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK & Parmacek MS (2003) Myocardin is a critical serum response factor co‐factor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol 23, 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA & Olson EN (2001) Activation of cardiac gene expression by myocardin, a transcriptional co‐factor for serum response factor. Cell 105, 851–862. [DOI] [PubMed] [Google Scholar]
- 17. Miralles F, Posern G, Zaromytidou AI & Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342. [DOI] [PubMed] [Google Scholar]
- 18. Zaromytidou AI, Miralles F & Treisman R (2006) MAL and ternary complex factor use different mechanisms to contact a common surface on the serum response factor DNA‐binding domain. Mol Cell Biol 26, 4134–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hipp L, Beer J, Kuchler O, Reisser M, Sinske D, Michaelis J, Gebhardt JCM & Knoll B (2019) Single‐molecule imaging of the transcription factor SRF reveals prolonged chromatin‐binding kinetics upon cell stimulation. Proc Natl Acad Sci USA 116, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rozenberg JM, Tesfu DB, Musunuri S, Taylor JM & Mack CP (2014) DNA methylation of a GC repressor element in the smooth muscle myosin heavy chain promoter facilitates binding of the Notch‐associated transcription factor, RBPJ/CSL1. Arterioscler Thromb Vasc Biol 34, 2624–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pagiatakis C, Gordon JW, Ehyai S & McDermott JC (2011) A novel RhoA/ROCK‐CPI‐17‐MEF2C signaling pathway regulates vascular smooth muscle cell gene expression. J Biol Chem 287, 8361–8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A & Olson EN (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428, 185–189. [DOI] [PubMed] [Google Scholar]
- 23. Gineitis D & Treisman R (2001) Differential usage of signal transduction pathways defines two types of serum response factor target gene. J Biol Chem 276, 24531–24539. [DOI] [PubMed] [Google Scholar]
- 24. Gualdrini F, Esnault C, Horswell S, Stewart A, Matthews N & Treisman R (2016) SRF Co‐factors control the balance between cell proliferation and contractility. Mol Cell 64, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boros J, Donaldson IJ, O'Donnell A, Odrowaz ZA, Zeef L, Lupien M, Meyer CA, Liu XS, Brown M & Sharrocks AD (2009) Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res 19, 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Costello P, Nicolas R, Willoughby J, Wasylyk B, Nordheim A & Treisman R (2010) Ternary complex factors SAP‐1 and Elk‐1, but not net, are functionally equivalent in thymocyte development. J Immunol 185, 1082–1092. [DOI] [PubMed] [Google Scholar]
- 27. Lee SM, Vasishtha M & Prywes R (2010) Activation and repression of cellular immediate early genes by serum response factor co‐factors. J Biol Chem 285, 22036–22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N & Treisman R (2014) Rho‐actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28, 943–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mengmeng X, Yuejuan X, Sun C, Yanan L, Fen L & Kun S (2020) Novel mutations of the SRF gene in Chinese sporadic conotruncal heart defect patients. BMC Med Genet 21, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esnault C, Gualdrini F, Horswell S, Kelly G, Stewart A, East P, Matthews N & Treisman R (2017) ERK‐induced activation of TCF family of SRF cofactors initiates a chromatin modification cascade associated with transcription. Mol Cell 65, 1081–1095. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coletti D, Daou N, Hassani M, Li Z & Parlakian A (2016) Serum response factor in muscle tissues: from development to ageing. Eur J Transl Myol 26, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solagna F, Nogara L, Dyar KA, Greulich F, Mir AA, Turk C, Bock T, Geremia A, Baraldo M, Sartori R et al. (2020) Exercise‐dependent increases in protein synthesis are accompanied by chromatin modifications and increased MRTF‐SRF signaling. Acta Physiol 230, e13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rahman NT, Schulz VP, Wang L, Gallagher PG, Denisenko O, Gualdrini F, Esnault C & Krause DS (2018) MRTFA augments megakaryocyte maturation by enhancing the SRF regulatory axis. Blood Adv 2, 2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pipes GC, Creemers EE & Olson EN (2006) The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev 20, 1545–1556. [DOI] [PubMed] [Google Scholar]
- 35. Geneste O, Copeland JW & Treisman R (2002) LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol 157, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Copeland JW & Treisman R (2002) The diaphanous‐related formin mDia1 controls serum response factor activity through its effects on actin polymerization. Mol Biol Cell 13, 4088–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou N, Lee JJ, Stoll S, Ma B, Costa KD & Qiu H (2017) Rho kinase regulates aortic vascular smooth muscle cell stiffness via Actin/SRF/Myocardin in hypertension. Cell Physiol Biochem 44, 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuwahara K, Barrientos T, Pipes GC, Li S & Olson EN (2005) Muscle‐specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol 25, 3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Posern G, Miralles F, Guettler S & Treisman R (2004) Mutant actins that stabilise F‐actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J 23, 3973–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang X, Azhar G & Wei JY (2017) SIRT2 gene has a classic SRE element, is a downstream target of serum response factor and is likely activated during serum stimulation. PLoS One 12, e0190011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang W, Gao F, Zhang P, Pang S, Cui Y, Liu L, Wei G & Yan B (2017) Functional genetic variants within the SIRT2 gene promoter in acute myocardial infarction. PLoS One 12, e0176245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Titus AS, Harikrishnan V & Kailasam S (2020) Co‐ordinated regulation of cell survival and cell cycle pathways by DDR2‐dependent SRF transcription factor in cardiac fibroblasts. Am J Physiol Heart Circ Physiol 318, H1538–H1558. [DOI] [PubMed] [Google Scholar]
- 43. Pellegrino R, Thavamani A, Calvisi DF, Neumann A, Geffers R, Pinna F, Schirmacher P, Nordheim A & Longerich T (2019) The interaction of Serum Response Factor (SRF) and ETS domain transcription factors regulate the transcription of the MDM 4 oncogene in HCC. Z Gastroenterol 57, 29. [Google Scholar]
- 44. Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH & Owens GK (2005) Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem 280, 9719–9727. [DOI] [PubMed] [Google Scholar]
- 45. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC et al. (2015) KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou J, Hu G & Herring BP (2005) Smooth muscle‐specific genes are differentially sensitive to inhibition by Elk‐1. Mol Cell Biol 25, 9874–9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iyer D, Chang D, Marx J, Wei L, Olson EN, Parmacek MS, Balasubramanyam A & Schwartz RJ (2006) Serum response factor MADS box serine‐162 phosphorylation switches proliferation and myogenic gene programs. Proc Natl Acad Sci USA 103, 4516–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sullivan AL, Benner C, Heinz S, Huang W, Xie L, Miano JM & Glass CK (2011) Serum response factor utilizes distinct promoter‐ and enhancer‐based mechanisms to regulate cytoskeletal gene expression in macrophages. Mol Cell Biol 31, 861–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu R, Leslie KL & Martin KA (2015) Epigenetic regulation of smooth muscle cell plasticity. Biochim Biophys Acta 1849, 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ Jr & Miano JM (2006) Defining the mammalian CArGome. Genome Res 16, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mack CP (2011) Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol 31, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manabe I & Owens GK (2001) Recruitment of serum response factor and hyperacetylation of histones at smooth muscle‐specific regulatory regions during differentiation of a novel P19‐derived in vitro smooth muscle differentiation system. Circ Res 88, 1127–1134. [DOI] [PubMed] [Google Scholar]
- 53. Zhou J, Zhang M, Fang H, El‐Mounayri O, Rodenberg JM, Imbalzano AN & Herring BP (2009) The SWI/SNF chromatin remodeling complex regulates myocardin‐induced smooth muscle‐specific gene expression. Arterioscler Thromb Vasc Biol 29, 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lockman K, Taylor JM & Mack CP (2007) The histone demethylase, Jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ Res 101, e115–e123. [DOI] [PubMed] [Google Scholar]
- 55. Blaker AL, Taylor JM & Mack CP (2009) PKA‐dependent phosphorylation of serum response factor inhibits smooth muscle‐specific gene expression. Arterioscler Thromb Vasc Biol 29, 2153–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shen X, Walsh B, Li JJ, Pang HX, Wang WJ & Tao SH (2009) The correlations of the function and positional distribution of the cis‐elements CArG around the TSS in the genes of Mus musculus. Genome 52, 217–221. [DOI] [PubMed] [Google Scholar]
- 57. Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O & Nordheim A (2002) Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol 156, 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaplan‐Albuquerque N, Van Putten V, Weiser‐Evans MC & Nemenoff RA (2005) Depletion of serum response factor by RNA interference mimics the mitogenic effects of platelet derived growth factor‐BB in vascular smooth muscle cells. Circ Res. 97, 427–433. [DOI] [PubMed] [Google Scholar]
- 59. Zhang X, Chai J, Azhar G, Sheridan P, Borras AM, Furr MC, Khrapko K, Lawitts J, Misra RP & Wei JY (2001) Early postnatal cardiac changes and premature death in transgenic mice overexpressing a mutant form of serum response factor. J Biol Chem 276, 40033–40040. [DOI] [PubMed] [Google Scholar]
- 60. Ro S (2016) Multi‐phenotypic role of serum response factor in the gastrointestinal system. J Neurogastroenterol Motil 22, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Park C, Lee MY, Park PJ, Ha SE, Berent RM, Fuchs R, Miano JM, Becker LS, Sanders KM & Ro S (2015) Serum response factor is essential for prenatal gastrointestinal smooth muscle development and maintenance of differentiated phenotype. J Neurogastroenterol Motil 21, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Parlakian A, Tuil D, Hamard G, Tavernier G, Hentzen D, Concordet JP, Paulin D, Li Z & Daegelen D (2004) Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol Cell Biol 24, 5281–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niu Z, Yu W, Zhang SX, Barron M, Belaguli NS, Schneider MD, Parmacek M, Nordheim A & Schwartz RJ (2005) Conditional mutagenesis of the murine serum response factor gene blocks cardiogenesis and the transcription of downstream gene targets. J Biol Chem 280, 32531–32538. [DOI] [PubMed] [Google Scholar]
- 64. Li S, Czubryt MP, McAnally J, Bassel‐Duby R, Richardson JA, Wiebel FF, Nordheim A & Olson EN (2005) Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue‐specific gene deletion in mice. Proc Natl Acad Sci USA 102, 1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li CL, Sathyamurthy A, Oldenborg A, Tank D & Ramanan N (2014) SRF phosphorylation by glycogen synthase kinase‐3 promotes axon growth in hippocampal neurons. J Neurosci 34, 4027–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lu PP & Ramanan N (2011) Serum response factor is required for cortical axon growth but is dispensable for neurogenesis and neocortical lamination. J Neurosci 31, 16651–16664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Halene S, Gao Y, Hahn K, Massaro S, Italiano JE Jr, Schulz V, Lin S, Kupfer GM & Krause DS (2010) Serum response factor is an essential transcription factor in megakaryocytic maturation. Blood 116, 1942–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cheng EC, Luo Q, Bruscia EM, Renda MJ, Troy JA, Massaro SA, Tuck D, Schulz V, Mane SM, Berliner N et al. (2009) Role for MKL1 in megakaryocytic maturation. Blood 113, 2826–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith EC, Thon JN, Devine MT, Lin S, Schulz VP, Guo Y, Massaro SA, Halene S, Gallagher P, Italiano JE Jr et al. (2012) MKL1 and MKL2 play redundant and crucial roles in megakaryocyte maturation and platelet formation. Blood 120, 2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Park C, Lee MY, Slivano OJ, Park PJ, Ha S, Berent RM, Fuchs R, Collins NC, Yu TJ, Syn H et al. (2015) Loss of serum response factor induces microRNA‐mediated apoptosis in intestinal smooth muscle cells. Cell Death Dis 6, e2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, Kim KK, Keshamouni VG, White ES, Zhou Y et al. (2015) Inhibition of myocardin‐related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol 185, 969–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bae JS, Noh SJ, Kim KM, Jang KY, Chung MJ, Kim DG & Moon WS (2014) Serum response factor induces epithelial to mesenchymal transition with resistance to sorafenib in hepatocellular carcinoma. Int J Oncol 44, 129–136. [DOI] [PubMed] [Google Scholar]
- 73. Chang J, Wei L, Otani T, Youker KA, Entman ML & Schwartz RJ (2003) Inhibitory cardiac transcription factor, SRF‐N, is generated by caspase 3 cleavage in human heart failure and attenuated by ventricular unloading. Circulation 108, 407–413. [DOI] [PubMed] [Google Scholar]
- 74. Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, Trent JM, Staudt LM, Hudson J Jr, Boguski MS et al. (1999) The transcriptional program in the response of human fibroblasts to serum. Science 283, 83–87. [DOI] [PubMed] [Google Scholar]
- 75. Nelson TJ, Balza R Jr, Xiao Q & Misra RP (2005) SRF‐dependent gene expression in isolated cardiomyocytes: regulation of genes involved in cardiac hypertrophy. J Mol Cell Cardiol 39, 479–489. [DOI] [PubMed] [Google Scholar]
- 76. Long X, Slivano OJ, Cowan SL, Georger MA, Lee TH & Miano JM (2011) Smooth muscle calponin: an unconventional CArG‐dependent gene that antagonizes neointimal formation. Arterioscler Thromb Vasc Biol 31, 2172–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miano JM, Long X & Fujiwara K (2007) Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292, C70–C81. [DOI] [PubMed] [Google Scholar]
- 78. Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, Nordheim A & Schwartz RJ (2008) Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci USA 105, 17824–17829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hays TT, Ma B, Zhou N, Stoll S, Pearce WJ & Qiu H (2018) Vascular smooth muscle cells direct extracellular dysregulation in aortic stiffening of hypertensive rats. Aging Cell 17, e12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lacolley P, Li Z, Challande P & Regnault V (2017) SRF/myocardin: a novel molecular axis regulating vascular smooth muscle cell stiffening in hypertension. Cardiovasc Res 113, 120–122. [DOI] [PubMed] [Google Scholar]
- 81. Zhou N, Lee JJ, Stoll S, Ma B, Wiener R, Wang C, Costa KD & Qiu H (2017) Inhibition of SRF/myocardin reduces aortic stiffness by targeting vascular smooth muscle cell stiffening in hypertension. Cardiovasc Res 113, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM & Zlokovic BV (2007) Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci USA 104, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Z, Zhang J, Gao Y, Pei L, Zhou J, Gu L, Zhang L, Zhu B, Hattori N, Ji J et al. (2014) Large‐scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin Cancer Res 20, 4598–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Patten LC, Belaguli NS, Baek MJ, Fagan SP, Awad SS & Berger DH (2004) Serum response factor is alternatively spliced in human colon cancer. J Surg Res 121, 92–100. [DOI] [PubMed] [Google Scholar]
- 85. Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR & Higgins PD (2014) Novel Rho/MRTF/SRF inhibitors block matrix‐stiffness and TGF‐beta‐induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis 20, 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bond JE, Kokosis G, Ren L, Selim MA, Bergeron A & Levinson H (2011) Wound contraction is attenuated by fasudil inhibition of Rho‐associated kinase. Plastic Reconstr Surg 128, 438e–450e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Okumura N, Koizumi N, Ueno M, Sakamoto Y, Takahashi H, Hirata K, Torii R, Hamuro J & Kinoshita S (2011) Enhancement of corneal endothelium wound healing by Rho‐associated kinase (ROCK) inhibitor eye drops. Br J Ophthalmol 95, 1006–1009. [DOI] [PubMed] [Google Scholar]
- 88. Sandbo N, Kregel S, Taurin S, Bhorade S & Dulin NO (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF‐beta. Am J Respir Cell Mol Biol 41, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yu‐Wai‐Man C, Spencer‐Dene B, Lee RMH, Hutchings K, Lisabeth EM, Treisman R, Bailly M, Larsen SD, Neubig RR & Khaw PT (2017) Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci Rep 7, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]