

Abstract
Sandhoff disease (SD) is an autosomal recessive lysosomal storage disease caused by defects in the β-subunit of β-N-acetylhexosaminidase (Hex), the enzyme that catabolizes GM2 ganglioside (GM2). Hex deficiency causes neuronal storage of GM2 and related glycoconjugates, resulting in progressive neurodegeneration and death, typically in infancy. No effective treatment exists for human patients. Adeno-associated virus (AAV) gene therapy led to improved clinical outcome and survival of SD cats treated before the onset of disease symptoms. Most human patients are diagnosed after clinical disease onset, so it is imperative to test AAV gene therapy in symptomatic SD cats to provide a realistic indication of therapeutic benefits that can be expected in humans. In this study, AAVrh8 vectors injected into the thalamus and deep cerebellar nuclei of symptomatic SD cats resulted in widespread central nervous system enzyme distribution, although a substantial burden of storage material remained. Cats treated in the early symptomatic phase showed delayed disease progression and a significant survival increase versus untreated cats. Treatment was less effective when administered later in the disease course, although therapeutic benefit was still possible. Results are encouraging for the treatment of human patients and provide support for the development AAV gene therapy for human SD.
Introduction
The GM2 gangliosidoses are recessively inherited lysosomal storage diseases (LSDs) characterized by neuronal accumulation of GM2 ganglioside (GM2), progressive neurodegeneration, and early death. Lysosomal β-N-acetylhexosaminidase (Hex. EC 3.2.1.52) cleaves the terminal sugar residue from GM2 ganglioside and related glycoconjugates. Catabolism requires the concerted action of three gene products: the Hex α-subunit, Hex β-subunit, and the GM2 activator protein (GM2AP). Deficiencies in the gene products result in Tay-Sachs disease (TSD), Sandhoff disease (SD), and GM2AP deficiency, respectively. The two major Hex isozymes, HexA (αβ) and HexB (ββ), have overlapping substrate specificities, but only HexA can cleave charged substrates, such as GM2 ganglioside. A third isoform, HexS (αα), is unstable and has no appreciable catalytic activity in humans1.
GM2 gangliosidosis occurs as three clinical disease forms distinguished by the age of symptom onset and progression of clinical symptoms, which is largely determined by the residual level of Hex activity2. The most common infantile-onset form is characterized by relentless neurodegeneration and premature death, often by age 3-5 years old. Later-onset juvenile and adult disease forms have slower progression and a more heterogeneous clinical course. Typical symptoms of adult-onset disease include extrapyramidal signs, spinocerebellar disease, lower motor neuron disease, and psychosis, but intellect is usually preserved. TSD and SD are clinically similar, but SD patients may have subtle peripheral manifestations resulting from additional accumulation of neutral substrates due to combined HexA and HexB deficiency1.
Several treatment approaches have been tested in humans, including bone marrow transplantation3, substrate reduction therapy4-7, and chemical chaperone therapy8, 9, but there remains no effective treatment. As Hex deficiency globally affects the CNS, treatment strategies must target widespread areas of the brain and spinal cord. Adeno-associated virus (AAV) gene therapy has the potential to generate a permanent source of Hex in the CNS. Following transduction, a proportion of lysosomal enzyme can be secreted and “cross-correct” neighboring cells9, 10. In addition to local diffusion11, enzyme is more widely distributed by axonal transport12-14 and cerebrospinal fluid (CSF) flow15, 16.
First described in 1977, the feline SD model (β-subunit deficient) is highly analogous to the human infantile-onset disease form and provides an ideal large animal model to test therapies for translation to humans. A naturally occurring mutation results in <0.01 fold normal CNS Hex activity, neuronal GM2 storage, and progressive neurodegeneration with death at 4.4 months old17, 18, 19. AAV gene therapy in pre-symptomatic SD murine and feline models demonstrated widespread CNS Hex distribution resulting in significant gains in clinical function and lifespan15, 20, 21-23. Although the outcome is encouraging, the average delay from symptom onset to diagnosis for GM2 gangliosidosis humans is currently 6.7 - 9.6 months24, 25. Therefore, most patients are in the symptomatic disease stage at the time of diagnosis. Studies in SD mice demonstrated that enzyme restoration and clearance of storage material were independent of treatment age. However, clinical outcome declined with increasing treatment age, likely due to irreversible pathological changes, such as demyelination and neuronal apoptosis26. The current study tested post-symptomatic AAV gene therapy in the feline SD model, with a size, complexity, longevity, and enzyme biochemistry that more closely resemble humans. Results will be important to inform inclusion criteria for patients in future clinical trials.
Materials and Methods
AAV vector design and preparation
The feline hexosaminidase (Hex) α- and β- subunit cDNA sequences have GenBank accession numbers JF899596 and JF899597, respectively. AAV vectors were produced as previously described by triple transfection of 293T cells with vector plasmid (derived from the plasmid pAAV-CBA-MBG-W by replacing MBG with cDNA for feline HEXA or feline HEXB27), a mini adenovirus helper plasmid pFΔ6, and AAVrh8 helper plasmid pAR828. The ITRs in the vectors are derived from AAV2. Transgene expression is controlled by CBA, and vectors carry the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE).
Surgeries and Treatment Groups
The feline SD breeding colony is maintained at Auburn University whose institutional animal care and use committee approved the research described herein. Affected SD cats were treated during the early symptomatic disease stage (1.6 – 1.8 months old; early postsymptomatic (EPS) group) defined by subtle tremors but no gait defects (score of 10 with mild tremors according to the scale in Figure 1a), or the late symptomatic disease stage (2.1 – 2.7 months old; late postsymptomatic (LPS) group) defined by overt whole body tremors and ataxia, with or without instability (score of 6 or 7 according to the scale in Figure 1a). Animals were assigned to treatment cohorts randomly, with segregation to a cohort determined by birth order and sex. General anesthesia was induced with ketamine (10 mg/kg) and dexmedetomidine (0.04 mg/kg) through an IV catheter and maintained using isofluorane (0.5-1.5 %) in oxygen delivered through an endotracheal tube. For intracranial injections, cats were positioned sternally using a Horsley-Clark stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). Cats received injections of AAV vectors bilaterally in the thalamus and deep cerebellar nuclei (DCN) as previously described21. Animals were given AAVrh8-fHEXA+AAVrh8-fHEXB in a 1:1 ratio in a total injected dose of 4.4 × 1012 vector genomes (vg; equivalent to 2.0 × 1014 vg/kg brain weight or 6.3 × 1012 vg/kg body weight). AAV-treated SD cats (n = 7) were euthanized at humane endpoint (defined by a score of 3 on the clinical rating scale in Figure 1a). Controls included untreated SD cats (n = 5 for lysosomal enzyme analysis, n = 4 for storage analysis, and n = 14 for clinical analysis), and age matched normal cats for the EPS and LPS cohorts (for each cohort, n = 5 for enzyme analysis, n = 4 for storage analysis, and n = 7 for clinical analysis). For AAV-treated cats, sample size was chosen based on the availability of animals and on previous studies in pre-symptomatic cats using the same AAV vectors, in which 3-4 animals per treatment group were sufficient to generate statistically meaningful results. For controls, we used as many age-matched animals as were available.
Figure 1.

Survival and clinical progression of post-symptomatic AAV-treated SD cats and untreated SD cats. (a) Age of symptom onset is shown for untreated SD cats (mean ± s.d, n = 14). The scale is based on gait defects, which ultimately define humane endpoint, with initial deficits at 2.1 months old. However, disease onset begins at 1.3 months old with subtle tremors of the head/tail that progress to overt whole-body tremors at 2.4 months old. Neurologic humane endpoint is reached at a score of 3. (b) Kaplan-Meier survival curves for untreated SD cats (solid black line, n = 14) and SD cats treated post-symptomatically. Survival of the EPS cohort is significantly increased compared to untreated (P = 0.0010, dashed black line, n = 4) and compared to the LPS cohort (P = 0.048). Survival of the LPS cohort is not significantly increased compared to untreated (P < 0.28, solid grey line, n = 3). Normal controls (not shown) remained alive and healthy throughout the study. (c, d) Composite clinical scores are shown for untreated SD cats (n = 14) while EPS (c) and LPS (d) treated cats are depicted separately. Open symbols denote the presence of subtle tremors, and checkered symbols denote the presence of overt whole body tremors, which were absent in all EPS treated cats, but present in all LPS treated cats. Scores were assigned using two separate, independent readouts, as described in materials and methods. Normal controls (not shown) scored a 10 on the clinical rating scale and had no tremors for the duration of the study.
Tissue Preparation
For biochemical analysis, brains were divided into coronal blocks of 0.6 cm from the frontal pole through the cerebellum and seven spinal cord blocks of 0.6 cm were taken from the cervical (3 blocks), thoracic (1 block), and lumbar (3 blocks) spinal cord. Coronal blocks from the right brain hemisphere and each spinal cord block were frozen in optimum cutting temperature (OCT) medium or directly in liquid nitrogen and used for the following analyses: lysosomal enzyme distribution by histochemical staining, lysosomal enzyme specific activity by 4-methylumbelliferone (4MU) enzyme assays (performed in duplicate for each block), AAV vector distribution by SYBR Green quantitative PCR (performed in triplicate for each block), and sialic acid storage by Periodic Acid Schiff staining. Coronal blocks from the left hemisphere were halved to 0.3 cm and stored at −80 °C, along with each spinal cord block, for analysis of lipid storage by resorcinol-based assay (performed in duplicate). Tissue was also taken from sciatic nerve, liver, skeletal muscle (quadriceps) and heart and stored at −80°C.
Determination of lysosomal enzyme activity and distribution
For brain and spinal cord tissue extraction, several frozen sections (50 μm) were cut from each coronal block. For sciatic nerve and other non-nervous peripheral tissue extraction, a sample was taken from tissue stored at −80 °C. Tissue was homogenized manually in 50 mM citrate phosphate buffer, pH 4.4 (50 mM citric acid, 50 mM Na2HPO4, 10 mM NaCl) containing 0.1% TritonX and 0.05% bovine serum albumin (BSA), followed by 2 freeze-thaw cycles and centrifugation at 15,700 g for 5 minutes at 4 °C. Cerebrospinal fluid (CSF) samples were analyzed directly from −80 °C. The activity of Hex A, total Hex and α-mannosidase were measured as previously described using synthetic 4MU fluorogenic substrates29. Specific activity was expressed as nmol 4MU/mg/hr after normalization to protein concentration by the Lowry method. Samples were analyzed in duplicate and the average value reported.
To qualitatively assess the distribution of Hex activity, frozen sections at 40 μM were thawed and then fixed, washed and stained with AS-BI-N-acetyl-B-D-glucosaminide (naphthol) as previously described30. Naphthol staining was performed once for each cat.
Periodic Acid Schiff (PAS) staining for ganglioside storage material
Storage material was assessed qualitatively with PAS staining, as previously described31, which detects the oligosaccharide side chain of ganglioside. PAS staining was performed once for each cat.
Lipid extraction and sialic acid quantification
Punch biopsies of 8 mm diameter were taken from representative areas of the brain and spinal cord and lyophilized overnight. Next day, 10-25 mg of dry weight sample was rehydrated in 0.5 ml water and lipid extraction and sialic acid quantification were performed as previously described31. Samples were analyzed in duplicate and the average value reported.
Quantitative PCR for vector genomes
Vector copy number was quantified as previously described21, 31 by quantitative PCR using SYBR Green-based reactions (Applied Biosystems, Warrington, UK) with primers specific for WPRE in the vector (forward 5′-AGTTG TGGCCCGTTGTCA-3′; reverse 5′-GAGGGGGAAAGCGAAAGT-3′). Concentration of genomic DNA samples was 20 ng for spinal cord, 50 ng for brain and 25 ng for peripheral tissues. Background amplification was determined on DNA from untreated normal and untreated SD cats. The assay limit of detection above background was 20 AAV copies per reaction. Samples were analyzed in triplicate and the average value reported.
Magnetic resonance imaging (MRI)
Data were acquired as previously described31.
Clinical rating scores and neurological exam performance
Cats were assigned a clinical rating score from the scale in Figure 1a using two separate, independent readouts: (1) neurological exams performed by a veterinarian at 2–4 week intervals and (2) video footage, which was retrospectively analyzed by a second, independent investigator. With the personnel available, it was not possible for evaluators to be blinded to treatment status, though their observations were performed independently. During neurological exams, deficits in performing common neurological tests were recorded by the veterinarian.
Statistical Analysis
Data is expressed as mean (± standard deviation) throughout the text and figures. Statistics were performed using SAS 9.2 software, and a P value ≤ 0.05 was considered significant for all tests. The Wilcoxon signed rank test was used for pairwise comparisons of lysosomal enzyme activity, AAV vector distribution, and sialic acid levels since a normal distribution could not be assumed with the animal numbers in each group. One-sided testing is reported throughout the text and figures to determine directional significance (i.e., significantly higher or lower values), which would often not be realized using two-sided testing due to low animal numbers when using a feline model. The logrank test was used for survival comparison between groups. Survival is our primary outcome measure, and standard deviation in survival for EPS and LPS cohorts (3.7 months versus 4.2 months) is similar. All data meet the assumptions of the tests used. No data was excluded from analysis and, as outliers are hard to determine with small sample sizes, no data points in the study were defined as outliers.
Code Availability
The SAS 9.2 software codes used to generate survival curves, logrank test survival statistics and Wilcoxon signed rank statistics can be provided upon request by contacting the corresponding author.
Results
Treatment groups and clinical and survival outcome
Seven SD cats were treated after the average age of clinical disease onset with bilateral thalamus and DCN injections of monocistronic AAVrh8 vectors encoding feline Hex α- and Hex β-subunits (1:1 ratio). The total dose of 4.4 x 1012 vg equates to 2.0 × 1014 vg/kg brain weight or 6.3 × 1012 vg/kg body weight. Vectors were the same as those that proved successful in previous studies in pre-symptomatic SD cats21, 23. Treatment groups are detailed in Table 1. Stereotypical disease progression in untreated SD cats (n = 14) begins at 1.3 ± 0.2 months old with fine tremors of the head and tail. Symptoms progress according to the clinical rating scale in Figure 1a until cats cannot stand, which defines the humane endpoint at 4.4 (± 0.6) months old. Four SD cats were treated at the stage of subtle tremors but no gait defects (early post-symptomatic (EPS) group), and 3 SD cats were treated at the stage of overt whole body tremors and ataxia, with or without occasional falling (late post-symptomatic (LPS) group).
Table 1.
Treatment groups for post-symptomatic AAV-treated SD cats
| Treatment group |
Cat | Gender | Tx age (mo.) |
CRS score at txA |
Age at endpoint (mo.) |
Survival (mo.) (mean, s.d.) |
|---|---|---|---|---|---|---|
| Late post-symptomatic | 11-994 | F | 2.2 | 7.5** | 11.5 | 6.6 (4.2) |
| 7-866 | M | 2.1 | 7.5** | 4.4 | ||
| 7-801 | F | 2.7 | 6.5** | 4.0 | ||
| Early post-symptomatic | 7-877 | F | 1.6 | 10* | 10.6 | 15.4 (3.7) |
| 11-869 | F | 1.6 | 10* | 14.3 | ||
| 11-868 | M | 1.6 | 10* | 17.7 | ||
| 11-831 | M | 1.8 | 10* | 18.9 |
CRS score relates to Figure 1a.
Mild tremors.
Overt whole body tremors.
Abbreviations: CRS = clinical rating score; tx = treatment; mo = months
Disease progression was delayed for all cats in the EPS cohort, which demonstrated a 2.4 - 4.3-fold survival increase over untreated SD cats (mean survival, 15.4 ± 3.7 months, P = 0.0010, Figure 1b). Treated cats eventually lost the ability to stand due to hind limb weakness but debilitating whole body tremors characteristic of untreated SD cats did not develop (Figure 1c). In accordance with improved clinical performance, the EPS cohort also showed delayed acquisition of deficits on common neurological tests compared to untreated SD cats (Figure 2).
Figure 2.
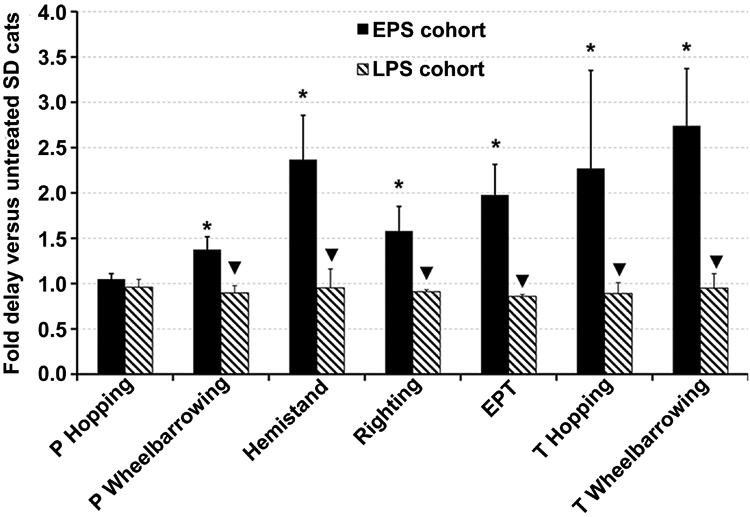
Neurological exam performance of post-symptomatic AAV-treated SD cats versus untreated SD cats. Cats were given a neurological examination twice monthly and deficits were recorded. * Onset of deficit was significantly delayed compared to untreated SD cats (P ≤ 0.011); ▼, onset of deficit was significantly earlier compared to the EPS cohort (P ≤ 0.026). In the LPS cohort, pelvic hopping deficits and pelvic wheelbarrowing deficits were already apparent at the time of treatment for cat 7-866 and all deficits except thoracic wheelbarrowing were apparent at the time of treatment for cat 7-801. The first deficit to appear in untreated SD cats is pelvic hopping at 2.2 ± 0.7 months, and the last deficit to appear is thoracic wheelbarrowing at 3.0 ± 0.7 months. Abbreviations: P = pelvic limb; T = thoracic limb; EPT = extensor postural thrust; LPS = late post-symptomatic; EPS = early post-symptomatic. Data is expressed as mean ± s.d. No deficits were recorded in normal control cats.
Survival of the LPS cohort was not significantly different versus untreated SD cats (6.6 ± 4.2 months, P = 0.28, Figure 1b), and it was significantly lower than survival of the EPS cohort (P = 0.048). Two LPS cats (7-801 and 7-866) continued to develop clinical symptoms at the stereotypical age, including progressively worsening whole body tremors, and reached humane endpoint at 4.0 and 4.4 months old, respectively. Disease progression in cat 11-994 stabilized approximately 1 month after surgery despite the continued presence of whole body tremors, and cat 11-994 lived to 11.5 months old: 2.6 times longer than untreated SD cats (Figure 1d). Therefore, mean survival was not significantly improved in LPS treated cats, but treatment can be beneficial in some individuals. Acquisition of neurological deficits on common tests was not delayed in the LPS cohort versus untreated SD cats although several deficits were already apparent at the time of treatment for cats 7-866 and 7-801 (Figure 2).
On T2 weighted MRI, white matter is hypointense to gray matter in the cortex and cerebellum of normal cats (Figures 3a and b) but becomes hyperintense with disease progression in untreated SD cats (Figures 3c and d). AAV treated cat 11-994 at 11.5 months old showed some preservation of white matter intensity in the cortex (Figure 3e), but the white matter area of the DCN was hyperintense to the surrounding cerebellar gray matter (Figure 3f). Brain atrophy in cat 11-994 was comparable to the 4-month old untreated SD cat in terms of widening of cerebral sulci, ventricles and cerebellar fissures (Figures 3e and f). Cat 11-994 was treated in the LPS disease stage, and it is likely, but unverified, that demyelination and atrophy had begun at the time of treatment, which is supported clinically by the fact that cat 11-994 had overt whole body tremors at the time of treatment, indicative of cerebellar disease. Also, we know from later studies using a 7T magnet that there is already pathology by two months in untreated SD cats (unpublished data). Cats in a concurrent study treated in the pre-symptomatic stage showed good preservation of brain architecture up to 24 months old23. MRI analysis was not performed on the other six treated cats.
Figure 3.
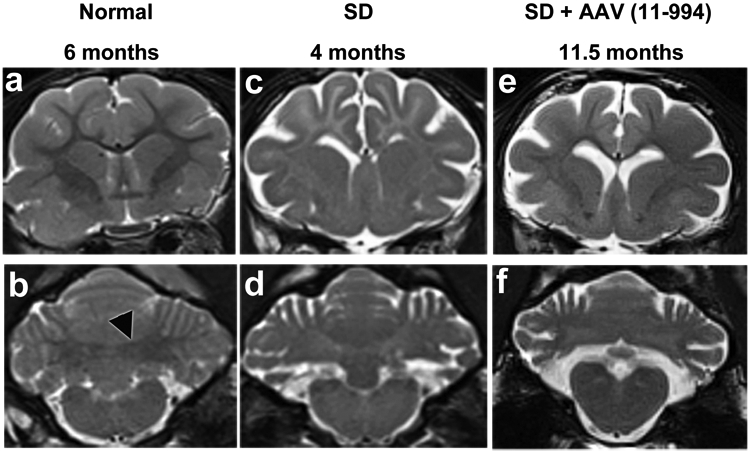
MRI evaluation of late post-symptomatic AAV-treated SD cats and untreated controls. T2-weighted MR images (3 Tesla) were taken at the level of the caudate nucleus (a, c and e) and DCN (b, d and f). Cortical white matter is hypointense to (darker than) gray matter in normal cats but hyperintense to (lighter than) gray matter in untreated SD cats. Also, the DCN area is hypointense to surrounding gray matter in normal cats (outlined black arrowhead in panel b) but becomes hyperintense with disease progression in untreated SD cats. In LPS cat 11-994 at humane endpoint, hypointensity of cortical white to gray matter is improved compared to untreated but is not normal. However, the DCN area has turned hyperintense to gray matter, similar to untreated SD cats. At the time of imaging cat 11-994 was on a clinical rating score of 3 (Figure 1a). There were no appreciable differences between a normal cat brain at 6 months old and at older ages. Brain atrophy, indicated by the amount of CSF (bright white area), in cat 11-994 was comparable to the untreated SD cat.
Although disease progression in SD cats is defined by neurological deterioration, Hex deficiency also affects non-nervous peripheral tissues. In SD mice15 and pre-symptomatic SD cats23 treated with AAV gene therapy, improved neurologic outcome allowed the emergence of peripheral tissue disease that was subclinical in untreated SD animals. EPS and LPS AAV-treated SD cats in the present study also developed peripheral disease manifestations (Supplementary Table S1). However, peripheral disease did not limit survival of any subjects in this study as all cats reached neurologic humane endpoint (score of 3 in Figure 1a).
Therapeutic enzyme distribution
Postmortem histochemical staining showed Hex activity in all brain blocks of both cohorts, but distribution varied between animals, and activity was not uniformly distributed within blocks (Figures 4b and d). In both cohorts, the extent of Hex distribution correlated with survival. Staining was most intense at the thalamus and DCN injection sites in the EPS cohort (blocks D and G in Figure 4b). In the LPS cohort, staining was also intense at the thalamus injection site (block D in Figure 4d), but a focal area of high activity was not as apparent at the cerebellar injection site (block G in Figure 4d) in two of the three cats. In both cohorts, Hex activity in the cervical spinal cord (blocks I – K in Figures 4c and e) was restricted to the dorsal funiculus with minimal activity detected in gray matter. In the thoracolumbar spinal cord (blocks M – O in Figures 4c and e), staining was detected in the dorsal gray matter and also extended into the ventral gray matter.
Figure 4.

Therapeutic enzyme distribution in the CNS of post-symptomatic AAV-treated SD cats. Tissues from post-symptomatic AAV-treated SD cats were collected at humane endpoint. (a) Shown are injection sites (white circles) and 0.6 cm coronal blocks of the brain (A-H) and spinal cord (I-O) as depicted in a previous study21. Brain blocks were halved, and the right half was assayed for enzyme activity. Lysosomal Hex activity (red) was visualized throughout the brain (b) and spinal cord (c) of early post-symptomatic AAV-treated SD cat 11-831 (SD + AAV) at 18.9 months old. Similarly, Hex activity was visualized throughout the brain (d) and spinal cord (e) of late post-symptomatic AAV-treated SD cat 11-994 (SD + AAV) at 11.5 months old. Representative control sections are shown from untreated normal cats along with untreated SD cats, which express ≤0.02 fold normal HexA activity in the brain and spinal cord. Corresponding HexA activity against MUGS substrate is shown below each block as fold normal level (fold N). Each block from treated and untreated SD cats is normalized to the corresponding block from age-matched normal controls (the mean value from 5 normal cats). HexA specific activity in normal control cats ranged from 28.1 ± 7.5 to 57.4 ± 5.7 nmol 4MU/mg protein/hr in the brain and from 6.7 ± 0.8 to 17.5 ± 3.2 nmol 4MU/mg/hr in the spinal cord.
Hex activity was measured quantitatively with the Hex α-subunit preferred substrate, MUGS. As we have shown previously21, 27, the AAVrh8 vectors used herein produce primarily HexB (ββ) and HexA (αβ) isozymes, with little or no HexS (αα). Thus, the MUGS substrate is an accurate measure of HexA in our system. Residual HexA activity against MUGS in untreated SD cats was ≤0.01 fold normal in the brain and ≤0.02 fold normal in the spinal cord. In both AAV-treated cohorts, mean HexA activity was significantly higher than untreated SD cats in all brain and spinal cord blocks (Table 2). Mean brain HexA activity ranged from 1.1 - 22-fold normal in the EPS cohort, with 4 brain regions significantly higher than normal (Table 2) and 1.4 - 9.9-fold normal in the LPS cohort, with 1 brain region significantly higher than normal (Table 2). Although it did not reach significance, HexA activity was lower in all cerebellar blocks (F – H) of the LPS cohort versus the EPS cohort. HexA activity in the spinal cord ranged from 0.83 - 4.7-fold normal for the EPS group and 0.34 - 2.8-fold normal for the LPS group. HexA activity was significantly lower in the LPS cohort versus the EPS cohort in spinal cord blocks M and O, although mean activity for these blocks was above normal for both cohorts. AAV vector was detected by quantitative PCR in all CNS blocks from AAV-treated SD cats indicating widespread dissemination from injection sites (Supplementary Table S2). In accordance with the lower HexA activity, vector copy number was significantly lower in blocks M - O of the LPS group versus the EPS group.
Table 2.
Hex activity in the nervous system, CSF, liver, skeletal muscle, and heart of AAV-treated and untreated SD cats
| Region | Block | Fold normal HexA (mean, s.d.) |
||
|---|---|---|---|---|
| Treatment group | ||||
| EPSB | LPSC | SD no txD | ||
| CNS and PNS | ||||
| Cerebrum | A | 2.8 (2.5) | 1.7 (2.0) | 0.00 (0.0) |
| B | 1.1 (0.48) | 1.6 (2.1) | 0.00 (0.0) | |
| C | 2.0 (2.2) | 1.5 (2.1) | 0.00 (0.0) | |
| D | 6.5 (4.0)* | 2.9 (3.7) | 0.00 (0.0) | |
| E | 2.8 (2.4)* | 9.9 (5.4)* | 0.00 (0.0) | |
| Cerebellum | F | 9.0 (8.4)* | 1.4 (1.1) | 0.00 (0.0) |
| G | 22 (15) | 1.4 (1.4) | 0.00 (0.01) | |
| H | 12 (9.4)* | 3.6 (3.8) | 0.00 (0.0) | |
| Spinal cord | I | 0.83 (0.38) | 0.34 (0.12)* | 0.00 (0.0) |
| J | 0.92 (0.28) | 0.54 (0.10)* | 0.00 (0.0) | |
| K | 1.2 (0.33) | 0.68 (0.27) | 0.00 (0.01) | |
| L | 1.8 (0.55) | 0.89 (0.25) | 0.02 (0.03) | |
| M | 2.6 (0.73)* | 1.4 (0.31)▼ | 0.02 (0.04) | |
| N | 4.7 (2.5)* | 1.8 (1.0)* | 0.01 (0.02) | |
| O | 3.9 (0.36)* | 2.8 (0.99)*▼ | 0.01 (0.01) | |
| CSF | - | 2.3 (1.3) | 2.7 (2.0) | 0.03 (0.04) |
| Sciatic nerve | - | 2.2 (1.3) | 0.76 (0.40) | 0.02 (0.01) |
| Periphery | ||||
| Liver | - | 0.45 (0.56) | 0.13 (0.02) | 0.02 (0.00) |
| Skeletal muscle | - | 0.09 (0.12) | 0.02 (0.02) | 0.02 (0.00) |
| Heart | - | 0.05 (0.04) | 0.04 (0.00) | 0.00 (0.00) |
HexA values are shown with the exception of CSF for which only total Hex was measured due to limited sample volume. HexA specific activity in normal control cats ranged from 28.1 ± 7.5 to 57.4 ± 5.7 nmol 4MU/mg protein/hr in the brain and from 6.7 ± 0.8 to 17.5 ± 3.2 nmol 4MU/mg/hr in the spinal cord.
n = 4, HexA activity was significantly higher than untreated SD cats (n = 5) in A-H (P ≤ 0.017 for each block), I-O (P ≤ 0.018 for each block), CSF (P = 0.01), sciatic nerve (P = 0.0097), liver (P = 0.033), and heart (P = 0.0090).
n = 3, HexA activity was significantly higher than untreated SD cats (n = 5) in A-H (P ≤ 0.026 for each block), I-O (P ≤ 0.033 for each block), CSF (P = 0.041, n = 2, no sample available for 11-994), sciatic nerve (P = 0.018), and heart (P = 0.016).
HexA activity was significantly lower in untreated SD cats (n = 5) versus normal cats (n = 5) in A-O (P ≤ 0.0097 for each block), sciatic nerve (P = 0.0060), liver (P = 0.0061), skeletal muscle (P = 0.0060), and heart (P = 0.0056).
HexA specific activity was significantly higher/lower than normal cats (n = 5) (P ≤ 0.037)
HexA specific activity was significantly lower versus the EPS cohort (P = 0.026).
Abbreviations: no tx = no treatment; CSF = cerebrospinal fluid; CNS = central nervous system; PNS = peripheral nervous system; EPS = early posy-symptomatic; LPS = late post-symptomatic. Samples were analyzed in duplicate and the average value is reported.
During intraparenchymal injection, vector can leak into and be distributed by CSF and the vasculature15, 32, suggesting a mechanism for distribution to peripheral organs. In addition, Hex activity in CSF of the EPS and LPS cohorts was 2.3 and 2.7-fold normal, respectively, indicating that CSF transport may have contributed to enzyme dissemination (Table 2). HexA activity was also detected at near or above normal levels in sciatic nerve of both cohorts (Table 2). In other peripheral tissues, mean HexA activity was significantly higher than untreated in the heart of both cohorts and in the liver of the EPS, but not the LPS cohort. HexA activity was not significantly different from untreated in skeletal muscle. Vector copies were also detected in liver and heart tissue of both groups indicating that a portion of HexA activity was due to vector transduction (Table S2).
In some cases, vector copy number did not appear to correlate with HexA activity in peripheral tissues, partly due to the varying level of activity from one tissue type to another. Since HexA activity is expressed relative to normal levels (fold normal), the same amount of specific activity may appear higher or lower based on the endogenous activity of the control tissue. (Also, CBA promoter activity may vary across tissues, as may the capacity to process and glycosylate the Hex subunits.) Finally, some tissues (such as sciatic nerve) had low or undetectable levels of vector but relatively high HexA activity, which may be due to enzyme transport from distant transfected sites and/or cross-correction from adjacent tissues (Table 2, Table S2).
Clearance of storage material
Total ganglioside sialic acid accumulates to several fold normal in the brain of SD cats17, 33. As glycosphingolipids with complex oligosaccharide side chains containing one or more sialic acid residues34, gangliosides can be detected colorimetrically by Periodic Acid Schiff (PAS) staining. To evaluate the clearance of stored ganglioside after treatment, PAS staining was performed throughout the CNS. Storage levels were normalized in areas corresponding to Hex restoration, but ganglioside accumulation was apparent in areas that lacked Hex activity (Figure 5a).
Figure 5.

Storage in the CNS of post-symptomatic AAV-treated SD cats. (a) Storage in untreated SD cats is visualized by dark PAS staining in gray matter. In treated brains, ganglioside storage was present in Hex deficient areas (see corresponding naphthol staining for the same cat, 11-831, in Figures 4b and c), such as the temporal lobe (blocks d and e) and cervical spinal cord (block k). (b) Sample sites for sialic acid quantitation (circles) in brain (A-H) and spinal cord (K and O; half of each block was used). (c) Sialic acid levels were measured in untreated SD cats (n = 4) and in the EPS (n = 4) and LPS (n = 3) cohorts for comparison to normal cats (n = 4 for each cohort). **, all samples from untreated cats were significantly higher than normal (P = 0.015 for each block) in the cerebrum (2.6 – 4.9 fold normal), brainstem and cerebellum (2.0 - 3.8 fold normal) and spinal cord (3.2 - 3.8 fold normal); *, samples from treated cats that were not significantly higher than normal. Samples without this symbol were significantly higher than normal (P ≤ 0.030 for each block); ▼, samples from treated cats that were significantly lower than untreated (P ≤ 0.030); ▲, sample from treated cats that was significantly higher than untreated (P = 0.015); ●, sample that was significantly lower in the LPS cohort versus the EPS cohort (P = 0.037). Abbreviations: EPS = early post-symptomatic; LPS = late post-symptomatic; tx = treatment. Data is expressed as mean ± s.d. Samples were analyzed in duplicate and the average value is reported.
When quantitative sialic acid assays were performed on 15 samples throughout the CNS (Figure 5b), cats from the LPS cohort had significant storage reductions versus untreated cats in 5/9 cerebral samples, the middle and caudal cerebellum, and the lumbar spinal cord (Figure 5c). In the EPS cohort, significant storage reductions were found in all cerebellar samples and the lumbar spinal cord. Some samples from the EPS cerebrum had higher storage levels than the LPS cohort, even exceeding that of untreated SD cats in some cases. For example, storage in the temporal cortex of the EPS cohort was significantly higher than that of untreated SD cats (sample D3 in Figure 5c), probably due to the much longer life span of AAV-treated cats. Also, the temporal lobe is consistently the region treated least effectively by thalamic injections of AAV21, 31.
Activity of a secondary lysosomal biomarker
In LSDs, activity of non-mutant lysosomal hydrolases is often elevated36. In untreated SD cats, mannosidase activity was significantly elevated up to 8.4-fold normal in the brain, 11-fold normal in the spinal cord, and 4-fold normal in liver. In AAV-treated EPS and LPS SD cats, mannosidase activity was significantly reduced in most CNS blocks compared to untreated SD cats (Figure 6). However, mannosidase activity remained significantly higher than normal in most brain and spinal cord blocks, as well as in liver, indicating only partial normalization of this secondary lysosomal biomarker.
Figure 6.
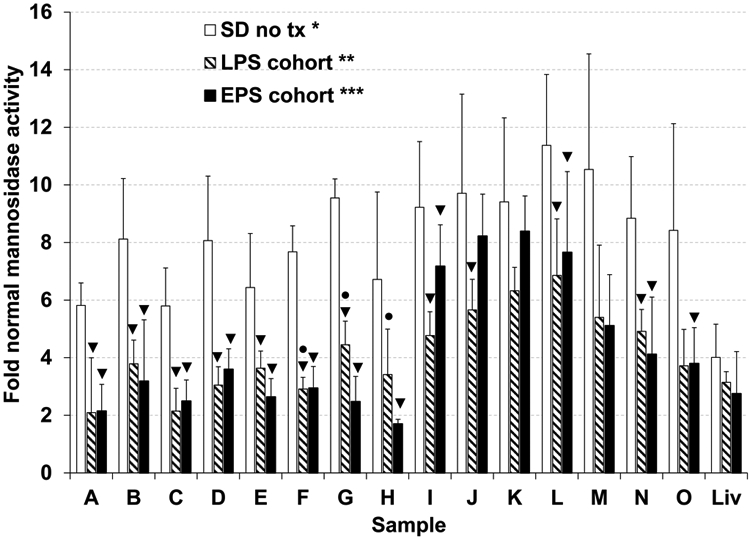
Normalization of lysosomal mannosidase activity in the CNS and liver of post-symptomatic AAV-treated SD cats. Cats from the EPS (n = 4, black bars) and LPS (n = 3, striped bars) cohorts were euthanized at humane endpoint and lysosomal mannosidase activity was compared to untreated SD cats (n = 5, gray bars) and normal cats (n = 5) in the brain (A-H), spinal cord (I-O), and liver (Liv). CNS sample lettering corresponds to Figure 4a. *, all samples from untreated SD cats were significantly higher than normal (A-H, I-O, and liver, P = 0.0061 for each block); **, samples B-F, H, I-O, and liver from the LPS cohort remained significantly higher than normal (P ≤ 0.037 for each block); ***, all CNS samples from the EPS cohort remained significantly higher than normal (P ≤ 0.019 for each block), but liver was not significantly higher than normal; ▼, samples from AAV-treated SD cats that were significantly lower than untreated (P ≤ 0.037); ●, samples that were significantly higher in the LPS cohort versus the EPS cohort (P ≤ 0.040). Data is expressed as mean ± s.d. Samples were analyzed in duplicate and the average value is reported.
Discussion
The GM2 gangliosidoses, TSD and SD, are progressive, fatal, neurodegenerative lysosomal storage diseases with no effective treatment. To date AAV-mediated gene therapy has proven the most successful experimental treatment approach in SD murine15, 20, 26 and feline models21-23, 27 as well as in the closely related GM1 gangliosidosis feline model31. Delivery of therapeutic AAV-vectors to the cerebrum and DCN of pre-symptomatic gangliosidosis mice and cats resulted in widespread enzyme distribution in the CNS as well as dissemination to some peripheral tissues. Lifespan was near normalized in treated SD mice15 and increased by an average of 4.3 fold in treated SD cats versus untreated SD cats23.
These unprecedented survival gains occurred in animals that were treated before clinical disease onset, which does not accurately reflect the clinical status at diagnosis for most human patients who will be the eventual recipients of therapy. Prenatal screening for GM2 gangliosidosis is only routinely performed in high risk populations, such as those with Jewish ancestry37, so most human patients are not diagnosed until after the appearance of clinical symptoms24, 38. Currently, average definitive diagnosis of infantile TSD and SD occurs at 13.1 and 14.7 months old, respectively, which is in the early symptomatic disease stage following average onset of symptoms at 5.2 and 4.4 months old, respectively. At the time of diagnosis, most patients have elevated noise sensitivity and may display other neurological signs, such as loss of sitting, hypotonia, loss of head movement, spasticity, and diminished eyesight24. Therefore, it is important to determine clinical benefit from AAV gene therapy in post-symptomatic animals.
Studies in SD mice, as well as other LSD animal models, determined that clinical benefit decreases with increasing treatment age26, 39-41, and that a therapeutic window exists beyond which vector administration provides no clinical benefit26. In this study we applied the thalamus and DCN treatment approach from pre-symptomatic23 to post-symptomatic SD cats. In a SD mouse study, AAV-vector was administered to the striatum and cerebellum at progressively older ages, but SD mice were not clinically symptomatic at any treatment time point. At the oldest time point tested (1 week before average symptom onset), no clinical benefit was achieved26. In contrast, in the present study, all SD cats were treated after the age of clinical disease onset, and clinical benefit was demonstrated in 5/7 cats. This highlights the importance of using large animal models that more closely resemble human disease to inform preclinical studies.
All SD cats treated in the EPS stage demonstrated significant survival gains. Although average survival was lower than cats treated pre-symptomatically23 (mean survival, EPS group 15.4 months; pre-symptomatic group 19.1 months), 2 of 4 EPS treated cats survived longer than 5 of 9 pre-symptomatic treated cats, so there was survival overlap between groups. Unlike the post-symptomatic SD mouse study26, this study using the feline model was unable to identify a point beyond which treatment is ineffective as one cat in the LPS cohort (11-994) demonstrated survival benefit that overlapped with the EPS cohort. However, treatment did not result in survival benefit for the other 2 of 3 LPS cats even though cat 7-866 was only treated 0.3 months later than the longest living EPS cat (11-831) and 0.1 months before the longest living LPS cat. This demonstrates that there may be a narrow window of time during which pathological disease progresses beyond a critical point that is not amenable to therapy. Although feline SD progression is stereotypical, some variation is associated with the disease course as evidenced by the attainment of humane endpoint between 3.8 - 5.2 months old in untreated cats (Figure 1b). At the time of treatment, the disease progression of cat 11-994 (LPS cohort) was likely still within the critical threshold amenable to therapy. Although cats 7-866 and 11-994 in the LPS cohort were both treated whilst on a score of 7.5 according to the clinical rating scale in Figure 1a, the scale does not incorporate subtle deficits such as those in Figure 2. Cat 7-866 had subtle neurological exam deficits in addition to those of 11-994 at the time of treatment, indicating that disease progression may have been slightly more advanced. It will be important in future to investigate whether certain deficits mark a critical point in disease progression beyond which therapy may not be beneficial.
While disease is still amenable to therapy, clinical outcome likely depends on the ability of vector to produce and distribute sufficient therapeutic enzyme. Previous studies showed that enzyme and vector biodistribution were not affected by pathologic disease status at the time of treatment (although clinical outcome was), which suggests that preexisting storage material does not affect enzyme dissemination from affected neurons26, 40. In this study, enzyme activity was at least normal in all brain regions and in the lumbar spinal cord of both cohorts, which is an achievement in itself. While restoration of ~ normal levels of Hex activity may be interpreted to produce only moderate therapeutic benefit, it is important to consider how broadly Hex is distributed throughout each CNS block. Enzyme activity values represent a homogenate of the whole block depicted in Figure 4a. With naphthol staining, it is evident that activity is not evenly distributed within a block, with some regions having high activity and other regions having little to no activity. The uneven distribution of enzyme is likely the result of two factors: (1) dissemination of vector in a relatively limited zone around the injection site, and (2) axonal transport of the enzyme, which depends on the axonal connections between neurons at the injection site and other brain regions12 - 14. Enzyme distribution was variable between subjects in both the EPS and LPS cohorts. Variation in enzyme distribution within each cohort likely occurred due to slight differences in anatomy between cats as well as variation in the precise site of injection within the thalamus and DCN. For example, the thalamic injection site was in block D or E in Figure 4a depending on the animal, and the DCN injection site was in block F, G or H in Figure 4a.
In most blocks of the CNS, mean Hex activity was higher in the EPS cohort than in the LPS cohort, which may be related to survival differences. However, enzyme distribution and vector copy number were only significantly lower in the LPS versus EPS cohort in the thoracolumbar spinal cord. Although they did not reach significance, enzyme activity and vector copy number were also lower in all cerebellar samples from the LPS cohort versus the EPS cohort. A possible reason for the lower Hex activity in the LPS cohort is that cells may already have been dying in those regions or perhaps were unhealthy and more susceptible to the possible toxic effects of Hex overexpression42.
As the longest surviving cats in each cohort in the present study demonstrated the most widespread enzyme distribution with naphthol staining, it is important to investigate ways to further enhance enzyme distribution, such as adding additional intraparenchymal injection sites or testing novel AAV capsids. We have already tested adding an AAV injection into the CSF compartment in addition to bilaterial thalamic and DCN injections, but treated cats did not show improved enzyme distribution or survival versus cats treated with bilaterial thalamic and DCN injection alone (unpublished data). However, it is also true that AAV efficacy after thalamus and lateral ventricular injection is equivalent to that of thalamus and DCN injection in presymptomatic SD cats22. Injection of AAV into the lateral ventricle treats the cerebellum efficiently enough and avoids the high risk of hemorrhage associated with direct injection of the DCN.
Clearance of pathologic storage material was shown to be reversible in mice and not dependent on treatment age26, 40, 43. In this study, areas positive for Hex activity with naphthol staining also showed clearance of ganglioside storage upon PAS staining. However, Hex deficient areas showed dark, dense accumulations of storage material. Therefore, remaining storage was probably related to insufficient Hex activity in those areas. Quantitative measurement of sialic acid revealed substantial storage material remaining in most samples from both cohorts except for the middle cerebellum and lumbar spinal cord for which both groups showed significantly normalized storage. The storage burden correlated with the incomplete reduction of lysosomal mannosidase levels.
Cat 11-831 in the EPS group and cat 11-994 in the LPS group experienced seizure activity, well controlled by medication, with onset at 7.8 and 8.4 months old, respectively. Seizures are a feature of late stage feline SD35, but no untreated SD cats in the present study had seizures as they occur beyond the humane endpoint presently in use. As previously reported in AAV-treated cats with the related disorder, GM1 gangliodidosis31, seizure activity may be related to lack of disease correction in the temporal lobe, which was deficient in enzyme activity in cats 11-831 and 11-994 (Figures 4b and d) and contained significant storage material.
In pre-symptomatic AAV-treated SD cats in a concurrent study, partial correction of CNS disease allowed longer lifespan with emergence of previously subclinical peripheral disease23. In the present study, cats in the EPS cohort also suffered peripheral disease manifestations due to their longer survival, despite significant Hex correction in liver and low but significant Hex correction in heart. Though it remains largely subclinical in untreated SD cats, peripheral disease is well-established and involves the liver44, heart and valves, bone, cornea and other organs45. It is unlikely that peripheral disease is caused by the vector itself. For example, the same vectors were used to treat 4 SD heterozygote cats27 and 6 normal cats (unpublished data), which showed no evidence of peripheral organ toxicity. Seven of the 10 treated cats were followed for ≥18 months.
Therefore, whilst some enzyme/vector is transported to the periphery, future studies must more effectively address treatment of peripheral organs. AAV delivered through IV injections is one potential way to more effectively correct peripheral tissues in large animal models46, 47 and is currently being tested in clinical trials for MPS IIIa and IIIb (ClinicalTrials.gov identifier NCT02716246 and NCT03315182, respectively). Following peripheral gene therapy in Pompe disease mice, muscle was refractory to vector transduction in old mice compared to young mice, but liver and heart were not48. In the current study, mean peripheral tissue HexA activity was lower in treated LPS cats versus EPS cats, but values did not reach significance.
In LSD mouse models, clinical disease is stabilized, but not improved, following AAV-treatment in post-symptomatic animals. Lack of improvement is due to progressive development of pathologic changes, such as myelin abnormalities, persistence of ubiquitinated inclusions, neuronal loss and neuroinflammation26, 39, 40. In previous studies, we demonstrated pronounced microgliosis in untreated SD cats that was abrogated almost entirely for at least 4 months after AAVrh8 treatment. Similar reductions in microgliosis were reported in SD mice treated by intracranial injection of AAV1 vectors expressing Hex, whether mice were injected at early or late disease stages26. Ectopic dendrite formation also occurs in feline gangliosidosis models51, and was refractory to therapy in an induced α-mannosidosis feline model52. Therefore, improving enzyme distribution and clearance of storage material in the CNS and periphery is important to achieve further survival gains in EPS SD cats, but development of irreversible pathologic changes may still limit the degree of functional rescue.
In cats with naturally occurring α-mannosidosis (not the induced model described above), AAV1 gene therapy in post-symptomatic cats resulted in reversal of clinical disease and improvements in myelination53. Like results in SD mice, clinical disease stabilized in post-symptomatic AAV-treated SD cats in the present study, but did not improve, so the capacity for clinical improvement may be disease specific rather than species specific. However, other factors like AAV serotype and injection route may also play a role in the different capacities for improvement reported in different studies. At humane endpoint, myelination in cat 11-994 was better in the cortex compared to untreated cats. However, with no control MRI images available at the 2-month time point post-treatment, it cannot be determined if myelin abnormalities were reversed or if demyelination/dysmyelination was stabilized. As cat 11-994 did not show reversal of clinical symptoms following treatment, it is unlikely that myelination abnormalities were reversed.
This study in post-symptomatic SD cats demonstrates that AAV-gene therapy is therapeutic when administered during the EPS disease stage. Therapy may also provide disease stabilization when administered in the LPS stage, though it is less likely. These results are highly encouraging for the prospects of treating human patients, who are usually diagnosed during the early symptomatic disease stage24. If gene therapy, or any other treatment approach, is efficacious in patients, then addition of Sandhoff disease to the Lysosomal Storage Disorders Newborn Screening Panel or rapid diagnosis following symptom onset would be needed if patients are to be treated during the most beneficial early symptomatic stage. Also encouraging is the prospect of treatment for juvenile and adult onset disease forms. Though diagnosis of patients with attenuated disease forms often takes longer than infantile patients, disease progression is slower, so there is a larger window of opportunity to treat late onset patients during the early symptomatic stage.
Supplementary Material
Acknowledgements
All contributors have been acknowledged by authorship.
Funding
This work was funded by a grant from the US National Institutes of Health (U01NS064096 to MSE and DRM) and contributions from the Scott-Ritchey Research Center, National Tay-Sachs and Allied Diseases Association, the Jewish Community Endowment Fund, and the Cure Tay-Sachs Foundation.
Footnotes
Conflict of Interest
The authors are beneficiaries of a licensing agreement with Axovant Gene Therapies (New York City) based partly on this technology. Drs. Sena-Esteves and Martin are shareholders in Lysogene (Neuilly-sur-Seine, France).
References
- 1.Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K, Suzuki K. The GM2 Gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease. 8th edn. (McGraw-Hill, New York, 2001) vol. 3, chapter 153. [Google Scholar]
- 2.Conzelmann E, Kytzia H, Navon R, Sandhoff K. Ganglioside GM2 N-acetyl-beta-D-galactosaminidase activity in cultured fibroblasts of late-infantile and adult GM2 gangliosidosis patients and of healthy probands with low hexosaminidase level. Am J Hum Genet. 1983; 35: 900–914. [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobs J, Willemsen M, Groot-Loonen J, Wevers R, Hoogerbrugge P. Allogeneic BMT followed by substrate reduction therapy in a child with subacute Tay-Sachs disease. Bone Marrow Transplant. 2005; 36: 925–926. [DOI] [PubMed] [Google Scholar]
- 4.Bembi B, Marchetti F, Guerci V, Ciana G, Addobbati R, Grasso D, et al. Substrate reduction therapy in the infantile form of Tay-Sachs disease. Neurology. 2006; 66: 278–280. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny EH. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med. 2009; 11: 425–433. [DOI] [PubMed] [Google Scholar]
- 6.Maegawa GH, Banwell BL, Blaser S, Sorge G, Toplak M, Ackerley C, et al. Substrate reduction therapy in juvenile GM2 gangliosidosis. Mol Genet Metab. 2009; 98: 215–224. [DOI] [PubMed] [Google Scholar]
- 7.Tallaksen C, Berg J. Miglustat therapy in juvenile Sandhoff disease. J Inherit Metab Dis. 2009; 32: 289–293. [DOI] [PubMed] [Google Scholar]
- 8.Clarke JTR, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay–Sachs or Sandhoff variants). Mol Genet Metab. 2011; 102: 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osher E, Fattal-Valevski A, Sagie L, Urshanski N, Amir-Levi Y, Katzburg S, et al. Pyrimethamine increases β-hexosaminidase A activity in patients with Late Onset Tay Sachs. Mol Genet Metab. 2011; 102: 356–363. [DOI] [PubMed] [Google Scholar]
- 10.Neufeld EF, Fratantoni JC. Inborn errors of mucopolysaccharide metabolism. Science. 1970; 169: 141–146. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RM, Wolfe JH. Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector-corrected fibroblasts secreting high levels of beta-glucuronidase. Nat Med. 1997; 3: 771–774. [DOI] [PubMed] [Google Scholar]
- 12.Passini MA, Lee EB, Heuer GG, Wolfe JH. Distribution of a lysosomal enzyme in the adult brain by axonal transport and by cells of the rostral migratory stream. J Neurosci. 2002; 22: 6437–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cearley CN, Wolfe JH. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. J Neurosci. 2007; 27: 9928–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broekman M, Tierney L, Benn C, Chawla P, Cha J, Sena-Esteves M. Mechanisms of distribution of mouse β-galactosidase in the adult GM1-gangliosidosis brain. Gene Ther. 2008; 16: 303–308. [DOI] [PubMed] [Google Scholar]
- 15.Cachón-González MB, Wang SZ, McNair R, Bradley J, Lunn D, Ziegler R, et al. Gene Transfer Corrects Acute GM2 Gangliosidosis—Potential Therapeutic Contribution of Perivascular Enzyme Flow. Mol Ther. 2012; 20: 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haurigot V, Marco S, Ribera A, Garcia M, Ruzo A, Villacampa P, et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J of Clin Invest. 2013; 123: 3254–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cork L, Munnell JF, Lorenz MD, Murphy JV, Baker HJ, Rattazzi MC. GM2 ganglioside lysosomal storage disease in cats with beta-hexosaminidase deficiency. Science. 1977; 196: 1014–1017. [DOI] [PubMed] [Google Scholar]
- 18.Cork LC, Munnell JF, Lorenz MD. The pathology of feline GM2 gangliosidosis. Am J Pathol. 1978; 90: 723–734. [PMC free article] [PubMed] [Google Scholar]
- 19.Martin DR, Krum BK, Varadarajan G, Hathcock TL, Smith BF, Baker HJ. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant 0. Exp Neurol. 2004; 187: 30–37. [DOI] [PubMed] [Google Scholar]
- 20.Cachón-González MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. PNAS. 2006; 103: 10373–10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCurdy VJ, Rockwell HE, Arthur JR, Bradbury AM, Johnson AK, Randle AN, et al. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 2015; 22: 181–189. [DOI] [PubMed] [Google Scholar]
- 22.Rockwell HE, McCurdy VJ, Eaton SC, Wilson DU, Johnson AK, Randle AN, et al. AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro. 2015; 7(2): 1759091415569908. doi: 10.1177/1759091415569908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradbury AM, McCurdy VJ, Johnson AK, Gray-Edwards H, Brunson BL, Randle AN, et al. Intracranial AAV Gene Therapy Extends the Life Span of GM2 Gangliosidosis Cats >Four-Fold with No Clinical Evidence of Vector Toxicity. Mol Ther, 2013; 21; S226 (abstract 590). [Google Scholar]
- 24.Bley AE, Giannikopoulos OA, Hayden D, Kubilus K, Tifft CJ, Eichler FS. Natural history of infantile GM2 gangliosidosis. Pediatrics. 2011: 128: e1233–e1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith NJ, Winstone A, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012; 54: 176–182. [DOI] [PubMed] [Google Scholar]
- 26.Cachon-Gonzalez MB, Wang SZ, Ziegler R, Cheng SH, Cox TM. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum Mol Genet. 2014; 23: 730–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradbury AM, Cochran JN, McCurdy VJ, Johnson AK, Brunson BL, Gray-Edwards H, et al. Therapeutic Response in Feline Sandhoff Disease Despite Immunity to Intracranial Gene Therapy. Mol Ther. 2013; 21: 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broekman M, Comer L, Hyman B, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or-2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006; 138: 501–510. [DOI] [PubMed] [Google Scholar]
- 29.Bradbury AM, Morrison NE, Hwang M, Cox NR, Baker HJ, Martin DR. Neurodegenerative lysosomal storage disease in European Burmese cats with hexosaminidase beta-subunit deficiency. Mol Genet Metab. 2009; 97: 53–57. [DOI] [PubMed] [Google Scholar]
- 30.Lacorazza H, Jendoubi M. In situ assessment of beta-hexosaminidase activity. Biotechniques. 1995; 19: 434–440. [PubMed] [Google Scholar]
- 31.McCurdy VJ, Johnson AK, Gray-Edwards HL, Randle AN, Brunson BL, Morrison NE. Sustained normalization of neurologic disease after intracranial gene therapy in a feline model. Science Translational Medicine. 2014; 6(231): 231ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, et al. Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh. 10 rhesus macaque-derived adeno-associated virus vector. Mol Ther. 2006; 15: 481–491. [DOI] [PubMed] [Google Scholar]
- 33.Baek RC, Martin DR, Cox NR, Seyfried TN. Comparative analysis of brain lipids in mice, cats, and humans with Sandhoff disease. Lipids. 2009; 44: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolter T, Proia RL, Sandhoff K. Combinatorial ganglioside biosynthesis. J Biol Chem. 2002; 277: 25859–25862. [DOI] [PubMed] [Google Scholar]
- 35.Baker HJ, Reynolds GD, Walkley SU, Cox NR, Baker GH. The Gangliosidoses: Comparitive Features and Research Applications. Vet Pathol. 1979; 16: 635–649. [DOI] [PubMed] [Google Scholar]
- 36.Cressant A, Desmaris N, Verot L, Bréjot T, Froissart R, Vanier MT, et al. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. Journal Neurosci. 2004; 24: 10229–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lew RM, Proos AL, Burnett L, Delatycki M, Bankier A, Fietz MJ. Tay Sachs disease in Australia: reduced disease incidence despite stable carrier frequency in Australian Jews. Medical J Aust. 2012; 197: 652–654. [DOI] [PubMed] [Google Scholar]
- 38.Smith NJ, Winstone A, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012; 54: 176–182. [DOI] [PubMed] [Google Scholar]
- 39.Ahmed SS, Li H, Cao C, Sikoglu EM, Denninger AR, Su Q, et al. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS Gene therapy in canavan mice. Mol Ther. 2013; 21: 2136–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, et al. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther. 2007; 15: 1782–1788. [DOI] [PubMed] [Google Scholar]
- 41.Ellinwood NM, Ausseil J, Desmaris N, Bigou S, Liu S, Jens JK, et al. Safe, efficient, and reproducible gene therapy of the brain in the dog models of Sanfilippo and Hurler syndromes. Mol Ther. 2011; 19: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golebiowski D, van der Bom IMJ, Kwon C, Miller AD, Petrosky K, Bradbury AM, et al. Direct Intracranial Injection of AAVrh8 Encoding Monkey β-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum Gene Ther. 2017; 28: 510 – 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elliger S, Elliger C, Aguilar C, Raju N, Watson G. Elimination of lysosomal storage in brains of MPS VII mice treated by intrathecal administration of an adeno-associated virus vector. Gene Ther. 1999; 6: 1175–1178. [DOI] [PubMed] [Google Scholar]
- 44.Bradbury AM, Gray-Edwards H, Shirley JL, McCurdy VJ, Colaco AN, Randle AN, et al. Biomarkers for Disease Progression and AAV Therapeutic Efficacy in Feline Sandhoff Disease. Exp Neurol. 2015; 263: 102 – 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gray-Edwards HL, Brunson BL, Holland M, Hespel AM, Bradbury AM, McCurdy VJ. Mucopolysaccharidosis-like Phenotype in Feline Sandhoff Disease and Partial Correction after AAV Gene Therapy. Mol Genet Metab. 2015; 116: 80 – 87. [DOI] [PubMed] [Google Scholar]
- 46.Bradbury AM, Bagel JH, Brisson BK, Marshall MS, Pesayco Salvador J, et al. AAVrh10 Gene Therapy Ameliorates Central and Peripheral Nervous System Disease in Canine Globoid Cell Leukodystrophy (Krabbe Disease). Hum Gene Ther. 2018; 29:785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurda BL, De Guilhem De Lataillade A, Bell P, Zhu Y, Yu H, Wang P, et al. (2016) Evaluation of AAV-mediated Gene Therapy for Central Nervous System Disease in Canine Mucopolysaccharidosis VII. Mol Ther 24(2):206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun B, Zhang H, Bird A, Li S, Young SP, Koeberl DD. Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression. Journal Gene Med. 2009; 11: 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d'Azzo A. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003; 126: 974 – 87. [DOI] [PubMed] [Google Scholar]
- 50.Bradbury AM, Peterson TA, Gross AL, Wells SZ, McCurdy VJ, Wolfe KG. AAV Mediated Gene Delivery Attenuates Neuroinflammation in Feline Sandhoff Disease. Neuroscience. 2016; 340: 117 – 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walkley SU. Cellular pathology of lysosomal storage disorders. Brain Pathol. 1998; 8: 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walkley S, Wurzelmann S, Siegel D. Ectopic axon hillock-associated neurite growth is maintained in metabolically reversed swainsonine-induced neuronal storage disease. Brain Res. 1987; 410: 89–96. [DOI] [PubMed] [Google Scholar]
- 53.Vite CH, McGowan JC, Niogi SN, Passini MA, Drobatz KJ, Haskins ME, et al. Effective gene therapy for an inherited CNS disease in a large animal model. Ann Neurol. 2005; 57: 355–364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.