

Abstract
Increasing evidence in substantiating the roles of endoplasmic reticulum stress, oxidative stress, and inflammatory responses and their interplay is evident in various diseases. However, an in-depth mechanistic understanding of the crosstalk between the intracellular stress signaling pathways and inflammatory responses and their participation in disease progression has not yet been explored. Progress has been made in our understanding of the cross talk and integrated stress signaling network between endoplasmic reticulum stress and oxidative stress towards the pathogenesis of diabetic nephropathy. In this present study, we studied the crosstalk between the endoplasmic reticulum stress and oxidative stress by understanding the role of protein disulfide isomerase and endoplasmic reticulum oxidase 1α, a key player in redox protein folding in the endoplasmic reticulum. We had recruited a total of 90 subjects and divided into three groups (control (n = 30), type 2 diabetes mellitus (n = 30), and diabetic nephropathy (n = 30)). We found that endoplasmic reticulum stress markers, activating transcription factor 6, inositol-requiring enzyme 1α, protein kinase RNA-like endoplasmic reticulum kinase, C/EBP homologous protein, and glucose-regulated protein-78; oxidative stress markers, thioredoxin-interacting protein and cytochrome b-245 light chain; and the crosstalk markers, protein disulfide isomerase and endoplasmic reticulum oxidase-1α, were progressively elevated in type 2 diabetes mellitus and diabetic nephropathy subjects. The association between the crosstalk markers showed a positive correlation with endoplasmic reticulum stress and oxidative stress markers. Further, the interplay between endoplasmic reticulum stress and oxidative stress was investigated in vitro using a human leukemic monocytic cell line under a hyperglycemic environment and examined the expression of protein disulfide isomerase and endoplasmic reticulum oxidase-1α. DCFH-DA assay and flow cytometry were performed to detect the production of free radicals. Further, phosphorylation of eIF2α in high glucose–exposed cells was studied using western blot. In conclusion, our results shed light on the crosstalk between endoplasmic reticulum stress and oxidative stress and significantly contribute to the onset and progression of diabetic nephropathy and therefore represent the major therapeutic targets for alleviating micro- and macrovascular complications associated with this metabolic disturbance.

Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s12192-020-01176-z.
Keywords: Endoplasmic reticulum stress, Oxidative stress, Diabetic nephropathy, Crosstalk, Protein disulfide isomerase, ER oxidase 1α
Introduction
Diabetic nephropathy (DN), the most prevalent cause of end-stage renal disease, constitutes to 20–30% of renal transplantation worldwide (Hakim and Pflueger 2010). DN is characterized by abnormal glomerular filtration rate and albuminuria and the progression is described with five different stages: glomerular hyperfiltration, early nephropathy, microalbuminuria, evident proteinuria, and end-stage renal disease (Mogensen et al. 1983). Chronic hyperglycemia is the major cause of metabolic and vascular abnormalities in DN and recent research has unraveled the combination of genetic and epigenetic factors that, in turn, regulate diverse cellular signaling networks and thereby manifest the pathology associated with DN. The elevated level of reactive oxygen species was evident in diabetes and found to be a major causative factor towards the pathogenesis of diabetic complications. Oxidative stress (OS) causes podocyte injury, endothelial cell dysfunction, mesangial cell injury, the elevated level of transforming growth factor-β, microalbuminuria, and glomerular apoptosis and accelerates the progression of DN which leads to end-stage renal disease (Singh et al. 2011). Apart from oxidative stress–mediated diabetic complications, endoplasmic reticulum stress (ERS) is also evident in diverse renal diseases, including primary glomerulonephritis, diabetic nephropathy, acute kidney injury, chronic kidney disease, and renal fibrosis (Cybulsky 2017).The endoplasmic reticulum (ER) is complex and well-orchestrated, and the principal organelle involved in folding of the nascent polypeptide chain and subsequent protein trafficking (Liu and Li 2019). ER stress inducers such as hyperglycemia, free fatty acids, and lipoproteins disturb the proteostasis which leads to the accumulation of unfolded/misfolded proteins in the ER lumen, alters calcium homeostasis, and initiates the unfolded protein response (UPR) (Iwawaki and Oikawa 2013). Under acute stress, UPR is triggered by three ER transmembrane proteins or ER-resident sensors, activating transcription factor 6 (ATF6), inositol-requiring enzyme 1α (IRE1α), and protein kinase RNA-like endoplasmic reticulum kinase (PERK), which in turn trigger activation of their downstream target genes resulting in endoplasmic reticulum-associated degradation (Needham et al. 2019). In contrast, chronic ER stress leads to the activation of pro-apoptotic genes and its downstream mediators and drives the cell towards apoptosis with the help of ER-resident stress sensors. Mediators of apoptosis include C/EBP homologous protein (CHOP) induced via PERK and ATF4 (Wang et al. 2019). Alternatively, IRE1α can induce apoptosis via activation of caspase 4 or caspase 12 (Cybulsky 2017). Since ER plays a pivotal role in protein folding and maintaining a native stable conformational state is paramount, two major enzymes, namely, protein disulfide isomerase (PDI) and ER oxidase 1α (ERO1A) govern this important function (Tu and Weissman 2004).
Recently, researchers shed light on the intracellular stress signaling mechanism and suggest that crosstalk between ER and oxidative stress is evidenced in many pathophysiological conditions. The enzymes PDI and ERO1A are involved in forming disulfide bridges between the cysteine residues of the protein and ERO1 mediates the transfer of electrons from PDI to molecular oxygen, leading to the production of reactive oxygen species in the ER lumen (Cuozzo and Kaiser 1999). Under hyperglycemic condition, there is an increase in the formation of non-native disulfide bridges, which results in glutathione consumption as a defensive mechanism. Thus, lowering cellular glutathione under constant hyperglycemia fuels excessive reactive oxygen species generation and enhances oxidative stress (van der Vlies et al. 2003). In the face of mounting oxidant damage, elevated CHOP, in turn, regulates the expression of ERO1A, triggering inositol-1,4,5-trisphosphate receptor-mediated Ca2+ release from the ER. The mitochondria are a prime target of elevated cytosolic Ca2+ that is already grappling with hyperglycemia-induced elevated reactive oxygen species (Gorlach et al. 2006). In this present study, we examined the status of ERS and OS by studying the levels of its markers in the progression of DN subjects and compared with type 2 diabetes mellitus and healthy controls. Further, the association of crosstalk markers with ERS and OS has also been investigated. In addition, the role of crosstalk markers (PDIA2 and ERO1A) in the activation of ERS and OS in human monocytic cells has been studied under the diabetic niche and put forth a promising approach towards the molecular switch between ERS and OS.
Materials and methods
Study population
Ninety study subjects of 40–65 years were divided into three groups: healthy control subjects (n = 30; M/F, 15/15), subjects with type 2 diabetes mellitus (T2DM) (n = 30; M/F, 15/15), and subjects with diabetic nephropathy (DN) (n = 30; M/F, 17/13). In brief, healthy control refers to subjects with no history of diabetes, cardiovascular diseases, and renal diseases and were randomly selected those having fasting plasma glucose less than 5.6 mmol/l and 2 h postprandial plasma glucose value less than 7.8 mmol/l (140 mg/dl) during an oral glucose tolerance test, whereas T2DM subjects had fasting plasma glucose level of more than 7.0 mmol/l (≥ 126 mg/dl) and postprandial plasma glucose level of more than 11.1 mmol/l (≥ 200 mg/dl). All the study samples were collected from M.V. Hospital for Diabetes, Royapuram, Chennai, India. This study was approved by the institutional ethics committee (IEC/N-003/02/2020) and all methods were performed in accordance with the relevant guidelines and regulations of the institution. This study was also explained in their native languages with the aid of both written instructions in their mother tongue and also orally for easy understanding, after which they were given adequate time to consider their participation in this study. The informed consent was collected from all study participants. The inclusion criteria for the recruitment of DN subjects were of age between 30 and 60 years, glycated hemoglobin level should be greater than 7 or 7.5, duration of diabetes should be more than 10 years, and the estimated glomerular filtration rate should be in the range of ≥ 90 to ≤ 15 (ml/min/1.73 m2). The exclusion criteria included subjects with a history of inflammatory or infectious diseases, autoimmune and rheumatic diseases, cancer, hematological diseases, and type 1 DM. Subjects who are under the administration of anti-inflammatory drugs and pregnant or nursing mothers were also excluded from the present study.
Anthropometric measurements and biochemical parameters
Anthropometric parameters, including weight, height, and body mass index, were verified using standard techniques. The fasting blood samples were obtained from the study subjects. The body mass index was calculated as the weight in kilograms divided by the square of height in meters. Fasting plasma glucose, serum cholesterol, serum triglycerides, high-density lipoprotein cholesterol, and creatinine were measured using commercially available standard kits by Hitachi-912 AutoAnalyzer (Hitachi, Mannheim, Germany). Glycated hemoglobin A1c was estimated by high-pressure liquid chromatography (Bio-Rad, Hercules, CA).
Isolation of PBMC from peripheral blood by the Ficoll-Paque density gradient centrifugation
Peripheral blood mononuclear cells (PBMCs) were isolated from fresh peripheral blood of study subjects. About 2 ml of blood was drawn from the selected subjects and carefully layered on 3 ml of Ficoll to a 15ml falcon tube. The contents were centrifuged at 4200 rpm for 30 min at 25 °C. Four layers were separated, which consisted of plasma, PBMCs, Ficoll, and erythrocytes, respectively. Plasma was separated and stored in a separate 2ml centrifuge tube. The PBMC (buffy coat) layer was taken carefully and transferred to another centrifuge tube. The layer was washed with 1x PBS and centrifuged at 2500 rpm for 10 min at 25 °C. A thick pellet was observed at the bottom of the tube, which had PBMCs. The pellet was then stored at − 80 °C and further used for mRNA isolation.
Total RNA isolation and cDNA conversion
Total RNA was isolated using the manual extraction method. About 300 μl of chloroform and TRIzol (RNAiso Plus, Takara) was added to the PBMC pellet. The sample was then incubated at 4 °C for about 10 min, followed by centrifugation for 20 min at 12,000 rpm at 4 °C. The upper aqueous layer containing RNA was transferred to a fresh centrifuge tube and equal volume of isopropanol was added and incubated overnight at − 20 °C. The mixture was centrifuged at 12,000 rpm for 20 min at 4 °C to pellet down the RNA. The pellet was then washed with 70% ethanol and air dried, and 20 μl nuclease-free water was added and stored at − 80 °C. About 1 μg of RNA was reverse transcribed to cDNA using a commercial kit as per the manufacturer’s instructions (Qiagen, CA, USA). The resulting cDNA was used for gene expression studies using qRT-PCR (Bio-Rad CFX connect systems, Bio-Rad, PA, USA). The primer sequences for the target genes are given in Table S1.
Human monocytic cell line (THP-1) culture and high glucose treatment
In order to understand the ER-OS-crosstalk mechanism, we put forth a mechanistic approach to understand the intracellular stress signaling cascade in human leukemic monocytic cell line (THP-1) cells. THP-1 cells were used because monocytes are key players involved in inflammatory response and inflammation is evident in diabetes and its associated complications. THP-1 cells were purchased from the National Centre of Cell Sciences, Pune, India, and cultured in the Roswell Park Memorial Institute Medium-1640 (Invitrogen Life Technologies, Carlsbad, CA, USA) medium supplemented with fetal bovine serum (10%) (FBS; Invitrogen), 2 mM glutamine (Sigma, USA), 1 mM sodium pyruvate, 2 g/l sodium bicarbonate in 25-cm2 culture flask and incubated at 37 °C in an atmosphere of 5% CO2. Once the cells reached confluence, they were serum-starved overnight, followed by high glucose treatment (33.3 mM) at varied time intervals (30 min to 24 h). After each time points, the cells were harvested and washed with 1x PBS and subjected to RNA isolation for subsequent analysis of gene expression of ER, OS and crosstalk makers.
Western blotting
THP-1 cells were treated with high glucose (33.3 mM) for different time intervals, the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Abcam, USA), the whole-cell lysates were collected, and protein concentration was determined using the Bradford assay (Bio-Rad, PA, USA). 30 μg of whole protein lysate was resolved using SDS-PAGE and blotted to nitrocellulose membrane (Bio-Rad, PA, USA). Primary antibodies against eIF2α (C.No. 9722; Cell Signaling Technology, USA), p-eIF2α (S51) (C.No. 9721; Cell Signaling Technology, USA), and β-actin (sc-47,778; Santa Cruz, USA) were probed, followed by exposure to secondary antibodies. Expression was detected by the enhanced chemiluminescence (ECL) kit (Bio-Rad, PA, USA) and documented using the ChemiDoc system (GBOX, Syngene, UK). The data was normalized to the housekeeping gene, β-actin, and represented as relative fold change compared to the control group.
Measurement of intracellular oxidative stress using DCFDA (2′,7′-dichlorofluoresceindiacetate) dye
Reactive oxygen species was measured using the H2DCFDA assay and detected using flow cytometry. H2DCFDA is de-acetylated by intracellular esterases to 2′,7′-dichlorodihydrofluorescein (H2DCF), which directly detects free radicals, which then oxidizes non-fluorescent H2DCF to fluorescent DCF (Karlsson et al. 2010). The DCF assay estimates the total amount of intracellular reactive oxygen species generated in the cells. THP-1 cells were treated with high glucose (33.3 mM) at different time intervals. At the end of the experimental period, the cells were washed with ice-cold PBS twice and incubated with 10 μM DCFDA at 37 °C for 30 min in the dark. The fluorescent intensity was measured at 488 nm excitation/530 nm emission through flow cytometry (BD FACSCalibur, USA).
Statistical analysis
Data analysis was performed using GraphPad Prism version 6.00 (GraphPad Software, La Jolla California USA). Data are represented as mean ± SD. For a non-normal distribution, we used the Kruskal-Wallis H test, followed by Dunn’s post hoc test, among two or more groups. Spearman’s correlation analysis was performed to find out the association of crosstalk markers with ERS and OS markers in the DN subjects (IBM SPSS Statistics, version 20.0).
Results
Clinical and biochemical characteristics of the study subjects
The clinical and biochemical characteristics of the study subjects are presented in Table 1. Compared to control, T2DM and DN subjects showed a significant increase in vital parameters such as fasting plasma glucose, glycated hemoglobin, total serum cholesterol, and estimated glomerular filtration rate. Also, DN subjects showed a significant increase in postprandial plasma glucose, HDL cholesterol, urea, and creatinine compared to the control group. However, there was no significant change observed in body mass index, postprandial plasma glucose, HDL cholesterol, urea, and creatinine in T2DM subjects when compared to control groups.
Table 1.
Clinical and biochemical characteristics of the study subjects
| Clinical parameters | Control (n = 30) | T2DM (n = 30) | DN (n = 30) |
|---|---|---|---|
| Gender (M/F) | 15/15 | 15/15 | 17/13 |
| Age (years) | 45.3 ± 6.9 | 52.7 ± 8.4 | 55.1 ± 11.4 |
| Body mass index (kg/m2) | 22.7 ± 1.4 | 27.1 ± 3.3 | 28.2 ± 4.1‡ |
| Fasting plasma glucose (mg/dl) | 95.6 ± 7.1 | 168.4 ± 68.7† | 175.5 ± 53.6‡‡‡ |
| Postprandial plasma glucose (mg/dl) | 108.2 ± 15.9 | 212.6 ± 46.4 | 301.3 ± 98.9‡ |
| Glycated hemoglobin (%) | 5.5 ± 0.2 | 8.9 ± 2.5† | 10.9 ± 2.2‡‡ |
| Total serum cholesterol (mg/dl) | 158.5 ± 31.1 | 180.4 ± 34.8† | 187.5 ± 45.4‡‡‡ |
| HDL cholesterol (mg/dl) | 43.6 ± 8.4 | 44.2 ± 6.9 | 38.4 ± 12.4‡‡‡ |
| Urea (mg/dl) | 23.2 ± 4.0 | 24.5 ± 6.8 | 42.3 ± 15.3‡ |
| Creatinine (mg/dl) | 0.8 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.5‡‡ |
| Estimated glomerular filtration rate (ml/min per 1.73 m2) | 129.9 ± 14.7 | 98.1 ± 12.5† | 79.6 ± 20.1‡‡‡ |
p values were calculated using the Kruskal-Wallis H test on the GraphPad Prism software
*p < 0.05, **p < 0.01, ***p < 0.001. † indicates comparison was made between control and T2DM. ‡ indicates comparison was made between control and DN
Gene expression of endoplasmic reticulum stress pathway markers as assessed by qRT-PCR
A progressive increase in the expression ER stress markers such as PERK, ATF6, and IRE1α was observed in PBMCs of DN subjects when compared to T2DM and control group (Fig. 1). There was a 9.0- (p < 0.001, Dunn’s post hoc test), 5.8- (p < 0.01, Dunn’s post hoc test), and 4.0- (p < 0.01, Dunn’s post hoc test) fold increase and statistically significant in gene expression of PERK, IRE1α, and ATF6 respectively in DN patients when compared to control group. ER chaperone protein GRP78 and pro-apoptotic protein CHOP were also significantly increased up to 5.4- (p < 0.001, Dunn’s post hoc test) and 6.9- (p < 0.01, Dunn’s post hoc test) fold respectively in DN subjects compared to control subjects. We also observed a significant increase in PERK expression, IRE1α, and CHOP in DN subjects compared to T2DM subjects.
Fig. 1.

The expression levels of ER stress markers such as PERK, IRE1, ATF6, ER chaperone protein GRP78, and pro-apoptotic protein CHOP in PBMCs of study subjects were measured using qRT-PCR. A total of twenty-seven samples were analyzed for gene expression studies. Data are expressed as fold change over control and presented as mean ± SD of three independent experiments. Statistical analysis was performed by the Kruskal-Wallis test. *p < 0.05, **p < 0.01, and ***p < 0.001
Gene expression of oxidative stress markers as assessed by qRT-PCR
As shown in Fig. 2, the expression levels of oxidative stress markers in PBMCs isolated from DN subjects revealed a progressive increase in the oxidative stress markers thioredoxin-interacting protein (TXNIP) and p22pHox by 4.0- (p < 0.05, Dunn’s post hoc test) and 5.04- (p < 0.01, Dunn’s post hoc test) fold respectively in DN subjects when compared to control samples. We also observed a significant increase in the expression of p22pHox in DN subjects (p < 0.05, Dunn’s post hoc test) compared to T2DM subjects.
Fig. 2.
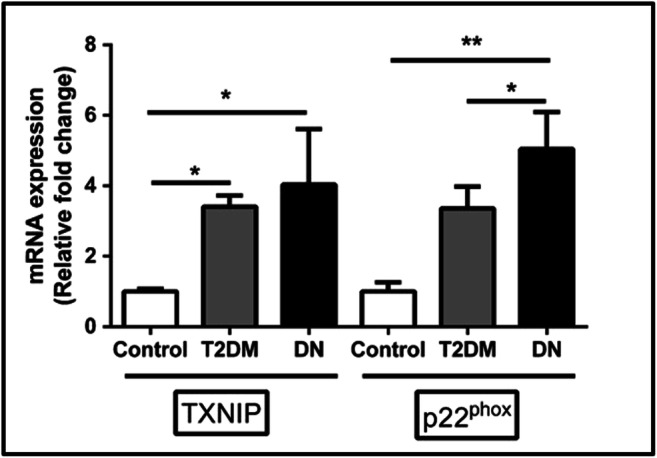
The expression levels of selected oxidative stress markers TXNIP and p22pHox in PBMCs of study subjects were measured using the qRT-PCR analysis. A total of twenty-seven samples were analyzed for gene expression studies. Data are expressed as fold change over control and presented as mean ± SD of three independent experiments. Statistical analysis was performed by the Kruskal-Wallis test. *p < 0.05, **p < 0.01, and ***p < 0.001
Gene expression of crosstalk markers as assessed by qRT-PCR
We performed the gene expression of crosstalk markers PDIA2 and ERO1A in the PBMCs of study subjects to assess their potential mediation in their contribution to overall oxidative stress (Fig. 3). Interestingly, the gene expression of PDIA2 and ERO1A were found to significantly increase; especially, the fold change between the DN and control subjects of PDIA2 was found to be 9.31-fold (p < 0.001, Dunn’s post hoc test) and 6.53-fold (p < 0.05, Dunn’s post hoc test) compared to control and T2DM subjects. On the other hand, the PDI mediator, ERO1A, which governs the redox folding by releasing reactive oxygen species, was also significantly increased among DN subjects 3.5- (p < 0.01, Dunn’s post hoc test) and 6.45- (p < 0.001, Dunn’s post hoc test) fold compared to T2DM subjects and healthy control group respectively. However, we had also observed a significant increase in the expression of PDIA2 (p < 0.01, Dunn’s post hoc test) in T2DM compared to control groups. This increased gene expression of PDIA2 and ERO1A among DN subjects affirms the role of impaired redox folding and redox homeostasis by the release of reactive oxygen species by ERO1A in DN subjects.
Fig. 3.
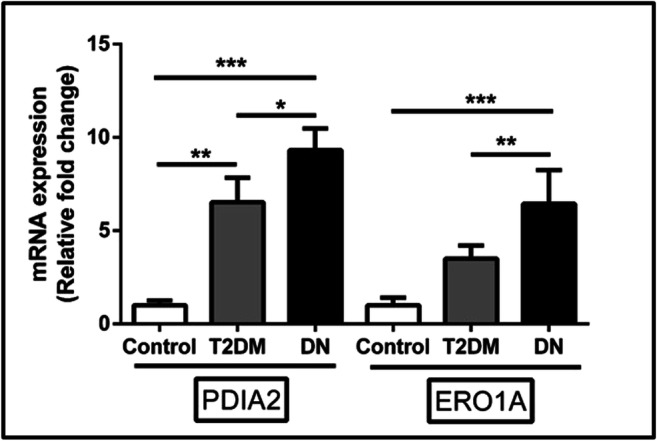
The levels of crosstalk genes such as PDIA2 and ERO1A in PBMCs of study subjects were measured using qRT-PCR analysis. A total of twenty-seven samples were analyzed for gene expression studies. Data are expressed as fold change over control and presented as mean ± SD of three independent experiments. Statistical analysis was performed by the Kruskal-Wallis test. *p < 0.05, **p < 0.01, and ***p < 0.001
Association between crosstalk markers with ER and oxidative stress markers
We further evaluated the association between the crosstalk markers and ER-OS markers in PBMCs of study subjects (Table 2). The levels of crosstalk markers PDIA2 and ERO1A individually correlated with the ER and OS signatures in DN subjects. Spearman’s correlation results revealed a fair/moderate degree of positive correlation of PDIA2 with PERK and p22pHox (r = 0.414; p = 0.016) and (r = 0.368; p = 0.021) respectively. In contrast, there was a fair degree of positive correlation of ERO1A with PERK (r = 0.432; p = 0.037) and a strong positive correlation with p22pHox (r = 0.814; p = 0.005). This signifies that the reactive oxygen species generated by ERO1A plays a pivotal role in overall cellular stress towards the pathogenesis of DN.
Table 2.
Spearman’s correlation coefficient of crosstalk markers (PDIA2 and ERO1A) with ER stress markers (PERK) and oxidative stress markers (p22pHox) among DN subjects
| Variables | PERK | p22pHox | ||
|---|---|---|---|---|
| r value | p value | r value | p value | |
| PDIA2 | 0.41 | 0.01 | 0.36 | 0.02 |
| ERO1A | 0.43 | 0.03 | 0.81 | 0.005 |
r and p values were calculated using Spearman’s correlation test at 95% confidence intervals
Effect of ER-OS-crosstalk markers in THP-1 cells under hyperglycemic environment
We further explored the mechanism by which the enzymes PDIA2 and ERO1A, mainly involved in redox protein folding, might involve in crosstalk between ERS and OS. In order to understand the molecular switch between the ERS and OS, we exposed THP-1 cells to hyperglycemic conditions and assessed the gene expression of ERS, OS, and crosstalk markers under different time points. Gene expression data revealed a progressive increase in the expression of the ER-resident protein GRP78 in THP-1 cells exposed to elevated glucose levels peaked around 6 h around 7.6-fold post treatment (Fig. 4). Interestingly, a significant decrease by about 3.1-fold in GRP78 was observed beyond 6 h high glucose treatment, but that was still elevated compared to control which was noticeable until the end period of the study. In contrast, though the level of the other OS marker TRPC-6 also increased by about 7.6-fold at 6 h, this remained relatively unchanged until 24 h after treatment. Similarly, the gene expression levels of PDIA2 and ERO1A were assessed and found to be elevated 3.0-fold at 3 h and remained constant from 9 to 24 h compared to the control group (Fig. 4). These results show that the crosstalk between ER and oxidative stress occurs at 3 h of glucose exposure in THP-1 cells resulting in the molecular switch between ER towards oxidative stress under hyperglycemic conditions.
Fig. 4.

a Effect of ER and oxidative stress markers in high glucose (33.3 mM) treated in THP-1 cells under different time points. b Effect of crosstalk markers in high glucose stimulated THP-1 cells under different time intervals and assessed the gene expression using qRT-PCR. Data are expressed as fold change over control and presented as mean ± SD of three separate experiments. Statistical analysis was performed using unpaired “t” test with the Welch corrections. *p < 0.05, **p < 0.01, and ***p < 0.001
Effect of high glucose exposure on eIF2α phosphorylation in THP-1 cells
To test the effect of high glucose exposure on eIF2α phosphorylation, we examined both total eIF2α and its phosphorylation at Ser 51 by immunoblot analysis. The eIF2α phosphorylation of the total cellular extract increased in 1 h of glucose exposure when compared to control cells (Fig. 5). At later time points, the reduction of eIF2α phosphorylation was observed by almost 3 folds in 24 h of glucose exposed cells.
Fig. 5.

Effect of high glucose (33.3 mM) on eIF2α phosphorylation by western blotting in THP-1 cells. Data are expressed as p-eIF2α(S51)/eIF2α ratio and presented as mean ± SD of three separate experiments. Statistical analysis was performed using by the Kruskal-Wallis test. *p < 0.05
Free radical generation in THP-1 cells under hyperglycemic conditions
Figure 6 demonstrates the reactive oxygen species levels in the THP-1 cells treated with glucose (33.3 mM) for various time intervals. Glucose exposure resulted in a time-dependent increase in levels of free radicals, confirmed by the right shift in comparison to untreated cells. We found that the mean fluorescent intensity increased up to 60% in high glucose–treated cells for 24 h.
Fig. 6.

Determination of reactive oxygen species generation via detection of DCFDA by flow cytometry. THP-1 cells were incubated with high glucose for different time points and stained with DCFDA. The fluorescence intensity was measured by flow cytometry. A shift to the right indicates increased reactive oxygen species levels (a). The mean fluorescence intensity (MIF) values are presented as means ± SD of three experiments (b). Statistical analysis was performed using by the Kruskal-Wallis test. *p < 0.05
Discussion
Oxidative stress is a major contributor to the pathogenesis of diabetes and its associated complications, and recent studies have shed light on ER stress in the pathogenesis of diabetes, insulin resistance, endothelial dysfunction, and inflammation in blood vessels (Basha et al. 2012; Flamment et al. 2012; Urano et al. 2000). Considering the importance of OS in mediating ERS in hyperglycemia and the development of DN, we first sought to assess relative gene expression levels of OS markers such as TXNIP and p22pHox in PBMCs of T2DM and DN. The results revealed elevated levels of both mediators in both T2DM and DN patients in comparison to healthy controls that were statistically significant. There is an increase in the level of oxidative stress markers such as TXNIP and p22pHox in DN subjects in the present study. In diabetes, NADPH oxidase 1-Nox1 and Nox2 are the major sources of reactive oxygen species, contributing to oxidative stress, endothelial dysfunction, and inflammation in blood vessels (Drummond et al. 2011). Here, we observed the NADPH oxidase subunit p22pHox to be significantly elevated in DN and T2DM subjects when compared to control groups. A recent finding from our laboratory reported that the level of p22pHox was increased in the onset of diabetes subjects and affirmed the pivotal role of oxidative stress in the pathogenesis of diabetes and associated complications (Sireesh et al. 2018). Thioredoxin-interacting protein is a protein that negatively regulates the redox protein, thioredoxin, which leads to an imbalance in cytoprotecting enzymes. The level of TXNIP was found to be significantly increased in prediabetes when compared to healthy control subjects (Gateva et al. 2019). Our results are in line with the previous findings which revealed the higher levels of oxidative stress in DN and T2DM subjects when compared to control subjects.
To ascertain the development of OS-mediated induction of ER stress, we also investigated gene expression analysis of five ER stress markers across the three groups. Our results on the gene expression of ER stress markers such as PERK, ATF6, IRE1, and GRP78 showed a significant increase in DN subjects compared to T2DM and control subjects. It has been demonstrated that there was an elevated level of MDA, GRP78, and CHOP in PBMCs of T2DM compared to control groups (Mozzini et al. 2015). Studies from our laboratory have shown that there were elevated levels of GRP78 and CHOP in ER stress-induced endothelial cells (Suganya et al. 2018). Our results are similar to the aforementioned reports and showed that the elevated levels of ER stress markers are associated with T2DM and DN subjects.
Since ER is the central organelle involved in protein folding, trafficking, and biogenesis, ER stress expectedly contributes towards many pathophysiological conditions arising from metabolic disturbances in the cell. ER stress and oxidative stress act in concert with many disease conditions, and therefore, we put forth a facile methodology to understand the crosstalk between ER-mediated reactive oxygen species generation and overall contribution towards oxidative stress via studying PDIA2 and ERO1A in PBMCs isolated from DN patients. Gene expression revealed that a progressive increase in PDIA2 and ERO1A in T2DM (6.53-fold) and DN (9.31-fold) was statistically significant compared to healthy control.
To the best of our knowledge, this is the first line of evidence we present that attempts characterizing the association between PDI and ERO1A in ongoing OS and ER cellular stress in DN subjects. Under chronic ER stress, there is an imbalance between protein folding and protein client load that favors the formation of non-native disulfide bridges between the cysteine residues by the PDI. This leads to protein aggregation in the ER, which results to the generation of reactive oxygen species by the ERO1A and depletes the level of glutathione in the ER lumen. Dysfunctional PDI and ERO1A imbalance the GSSG/GSH ratio in the ER lumen and contribute to enhanced oxidative stress. ER and mitochondria are physically and functionally connected through mitochondrial-associated membranes (MAMs) (Missiroli et al. 2018). ER stress also causes Ca2+ leak from the ER, travels through MAMs, and targets the complex III of electron transport chain in mitochondria, thus causing electron leak to molecular O2, thus aggravating the ongoing reactive oxygen species production contributing to overall oxidative stress in the cell (Pinton et al. 2008). However, under clinical settings, to the best of our knowledge, this is the first line of evidence we present that attempts characterizing the association between PDI and ERO1A in ongoing OS and ER cellular stress in DN subjects.
Compelling evidence demonstrates both PDI and ERO1A play a crucial role in many pathophysiological disease states, including neurodegenerative diseases, cancer, diabetes, and cardiovascular diseases (Parakh and Atkin 2015). In our present study, we observed a progressive increase in the expression of PDI in DN subjects when compared to T2DM and control subjects. This possibly suggests the accumulation of mis/unfolded proteins in the ER lumen in DN subjects. Over-expression of PDI was observed in a variety of cancers and has been suggested as a diagnostic marker to detect glial and breast cancers (Thongwatchara et al. 2011; Goplen et al. 2006). In the context of redox homeostasis, PDI is associated with NADPH complex subunits and raises reactive oxygen species levels in vascular smooth muscle cells. Only oxidized PDI triggers the production of reactive oxygen species, whereas reduced PDI ameliorates the production of reactive oxygen species (de A Paes et al. 2011). Also, reduced PDI helps to alleviate the misfolded protein load in the ER by stimulating endoplasmic reticulum-associated degradation (Tsai et al. 2001). In addition, the expression of ERO1A was increased in DN in comparison to T2DM and control subjects to suggest the reactive oxygen species-mediated elevation of ERO1A under hyperglycemic conditions that bolster oxidative stress. This has been evident that there were increased levels of oxidized ERO1 in microsomes of streptozotocin-induced diabetic rats (Nardai et al. 2005). This confirms that the PDI and ERO1 play a pivotal role in the crosstalk between ERS and OS under a hyperglycemic environment and accelerate the progression of DN. To further investigate the molecular mechanism behind the crosstalk, we put forth a mechanistic approach to understand the nexus between ER and the regulatory effect of PDI and ERO1A. The association of crosstalk markers with ERS and OS was evaluated using the Spearman's correlation. Interestingly, we found a positive correlation between PDIA2 and ERO1A with PERK and p22pHox. Our results shed light on the association between the PDIA2 and ERO1A with PERK and p22pHox towards enhanced oxidative stress. Our results further prove the relationship between the crosstalk markers with ERS and OS markers and provide a better understanding of the intracellular stress signaling cascade between ER and the cytosol that contributes to the progression of DN.
We exposed in vitro THP-1 cells under a hyperglycemic environment and assessed the gene expression of GRP78, TRPC6, PDIA2, and ERO1A at different time points. Accumulating evidence affirms the over-expression of pro-inflammatory cytokines (IL-1β and TNF-α) and oxidative stress in THP-1 cells under diabetic microenvironment (Aljada et al. 2002; Shanmugam et al. 2003). Interestingly, the upregulation of PDI was observed at different time points in azacytidine-induced THP-1 cells (Tang et al. 2013). In this present study, we found the level of the ER chaperone protein, GRP78, progressively increased and reached a peak at 6 h, after which it declined until 24 h, probably due to the initiation of the UPR. The oxidative stress marker was progressively increasing till 6 h and maintained the same level till 24 h. This is in agreement with the aforementioned report that oxidized PDI might regulate the ongoing reactive oxygen species production by targeting NADPH oxidase to maintain intracellular stress. From our data, it is tempting to speculate that the molecular switch between the ERS and OS appears to be turned on at 3 h where an increase in PDIA2 and ERO1A levels was observed at 3 h but which decreases after 3 h, though a constant yet elevated level was maintained when compared to untreated cells.
ER stress inducers such as hyperglycemia enhanced the production of reactive oxygen species and eventually induces cell damage by directly acting on lipid, protein, and DNA (Yu et al. 2006). Stimulation with high glucose is one of the factors that initiate diabetes development and its complications (Wu et al. 2014). The free radicals serve as upstream signal molecules of the high glucose–induced signaling pathway. Flow cytometry data demonstrated increased reactive oxygen species production in response to altered glucose levels in a time-dependent manner. This finding was also confirmed by another study where reactive oxygen species increased by high glucose exposure in human tumor cells (KB31, KBV1, A549, and DMS-53) (Seebacher et al. 2015).
ER stress induces the activation of unfolded protein response (UPR) pathways via the induction of PERK, which leads to the elevation of phosphorylated eIF2α, resulting in the promotion of a pro-adaptive signaling pathway by the inhibition of global protein synthesis and selective translation of ATF4 (Rozpedek et al. 2016). In parallel, ATF4 activates CHOP thereby induces expression of GADD34, which targets protein phosphatase 1 (PP1) to eIF2α for dephosphorylation and relief of translational inhibition (Novoa et al. 2001). In the present study, exposure to glucose for 1 h resulted in increased phosphorylation of eIF2α compared to control cells, while the long duration of exposure reduced the phosphorylation of eIF2α in a time-dependent manner. Our results are in agreement with the findings that high glucose exposure to renal proximal tubular cells induced the phosphorylation of eIF2α (Bao et al. 2019). Few studies underlined that eIF2α phosphorylation is necessary for regulating the expression of genes that maintain cellular function and limit oxidative stress, thus preventing oxidative damage and reducing ER stress (Back et al. 2009; Ju et al. 2017). The dephosphorylation of eIF2α was observed in 24 h exposure to glucose, possibly due to the recovery of the cells from a period of ER stress and to adapt to a higher level of ER folding charge. Also, the dephosphorylation of eIF2α enhances glucose tolerance and diminished hepatosteatosis in animals fed a high-fat diet (Oyadomari et al. 2008). Moreover, the ability of phosphorylated eIF2α to control cell survival or death depends on the type of stimuli and the specificity of the kinase that mediates the phosphorylation of eIF2α (Rajesh et al. 2015). These findings, together with our data, provide further evidence that eIF2α phosphorylation plays an essential role in the regulation of glucose metabolism.
To conclude, the ERS and OS markers were significantly increased in PBMCs of DN subjects when compared with T2DM and control subjects. The increase in crosstalk markers that play a pivotal role in the ER lumen also in DN compared to T2DM and control subjects symbolize protein misfolding and aggregation in the ER and aberrant reactive oxygen species generation due to ERO1 function. Correlation studies also confirm the association between the ERS and OS markers in diabetic complications. However, this is the first line of evidence to provide an insight into the crosstalk between ER and oxidative stress by clinical and mechanistic approach towards the progression of diabetic nephropathy among the South Indian population.
Supplementary Information
(DOCX 13 kb)
Acknowledgements
This study was financially supported by the SRM Institute of Science and Technology, Kattankulathur, Chennai, Tamil Nadu. The authors gratefully acknowledge the facilities provided by “SRM-DBT Partnership Platform for Contemporary Research Services and Skill Development in Advanced Life Sciences Technologies” (Grant No. BT/PR12987/INF/22/205/2015).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vijay Viswanathan, Email: drvijay@mvdiabetes.com.
Kunka Mohanram Ramkumar, Email: ramkumak@srmist.edu.in.
References
- Aljada A, Ghanim H, Dandona P. Translocation of p47phox and activation of NADPH oxidase in mononuclear cells. Methods Mol Biol. 2002;196:99–103. doi: 10.1385/1-59259-274-0:99. [DOI] [PubMed] [Google Scholar]
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Ao Y, Yi B, Batubayier J. High levels of glucose induce epithelial-mesenchymal transition in renal proximal tubular cells through PERK-eIF2alpha pathway. Chin Med J. 2019;132:868–872. doi: 10.1097/CM9.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha B, Samuel SM, Triggle CR, Ding H. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp Diabetes Res. 2012;2012:481840–481814. doi: 10.1155/2012/481840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuozzo JW, Kaiser CA. Competition between glutathione and protein thiols for disulphide-bond formation. Nat Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]
- Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13:681–696. doi: 10.1038/nrneph.2017.129. [DOI] [PubMed] [Google Scholar]
- de A Paes AM, Veríssimo-Filho S, Guimarães LL, Silva AC, Takiuti JT, Santos CX, Janiszewski M, Laurindo FR, Lopes LR. Protein disulfide isomerase redox-dependent association with p47(phox): evidence for an organizer role in leukocyte NADPH oxidase activation. J Leukoc Biol. 2011;90:799–810. doi: 10.1189/jlb.0610324. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamment M, Hajduch E, Ferre P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–390. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Gateva AT, Assyov YS, Velikova T, Kamenov ZA. Higher levels of thioredoxin interacting protein (TXNIP) in patients with prediabetes compared to obese normoglycemic subjects. Diabetes Metab Syndr. 2019;13:734–737. doi: 10.1016/j.dsx.2018.11.056. [DOI] [PubMed] [Google Scholar]
- Goplen D, Wang J, Enger PO, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006;66:9895–9902. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]
- Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- Hakim FA, Pflueger A. Role of oxidative stress in diabetic kidney disease. Med Sci Monit. 2010;16:RA37–RA48. [PubMed] [Google Scholar]
- Iwawaki T, Oikawa D. The role of the unfolded protein response in diabetes mellitus. Semin Immunopathol. 2013;35:333–350. doi: 10.1007/s00281-013-0369-5. [DOI] [PubMed] [Google Scholar]
- Ju SM, Jo YS, Jeon YM, Pae HO, Kang DG, Lee HS, Bae JS, Jeon BH. Phosphorylation of eIF2alpha suppresses cisplatin-induced p53 activation and apoptosis by attenuating oxidative stress via ATF4-mediated HO-1 expression in human renal proximal tubular cells. Int J Mol Med. 2017;40:1957–1964. doi: 10.3892/ijmm.2017.3181. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Kurz T, Brunk UT, Nilsson SE, Frennesson CI. What does the commonly used DCF test for oxidative stress really show? The Biochemical Journal. 2010;428:183–190. doi: 10.1042/BJ20100208. [DOI] [PubMed] [Google Scholar]
- Liu L, Li J. Communications between the endoplasmic reticulum and other organelles during abiotic stress response in plants. Front Plant Sci. 2019;10:749. doi: 10.3389/fpls.2019.00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M, Wieckowski MR, Giorgi C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018;9:329. doi: 10.1038/s41419-017-0027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes. 1983;32(Suppl 2):64–78. doi: 10.2337/diab.32.2.s64. [DOI] [PubMed] [Google Scholar]
- Mozzini C, Garbin U, Stranieri C, Pasini A, Solani E, Tinelli IA, Cominacini L, Fratta Pasini AM. Endoplasmic reticulum stress and Nrf2 repression in circulating cells of type 2 diabetic patients without the recommended glycemic goals. Free Radic Res. 2015;49:244–252. doi: 10.3109/10715762.2014.997229. [DOI] [PubMed] [Google Scholar]
- Nardai G, Stadler K, Papp E, Korcsmaros T, Jakus J, Csermely P. Diabetic changes in the redox status of the microsomal protein folding machinery. Biochem Biophys Res Commun. 2005;334:787–795. doi: 10.1016/j.bbrc.2005.06.172. [DOI] [PubMed] [Google Scholar]
- Needham PG, Guerriero CJ, Brodsky JL. Chaperoning endoplasmic reticulum-associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb Perspect Biol. 2019;11:a033928. doi: 10.1101/cshperspect.a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parakh S, Atkin JD. Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front Cell Dev Biol. 2015;3:30. doi: 10.3389/fcell.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh K, Krishnamoorthy J, Kazimierczak U, Tenkerian C, Papadakis AI, Wang S, Huang S, Koromilas AE. Phosphorylation of the translation initiation factor eIF2alpha at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 2015;6:e1591. doi: 10.1038/cddis.2014.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16:533–544. doi: 10.2174/1566524016666160523143937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebacher NA, Richardson DR, Jansson PJ. Glucose modulation induces reactive oxygen species and increases P-glycoprotein-mediated multidrug resistance to chemotherapeutics. Br J Pharmacol. 2015;172:2557–2572. doi: 10.1111/bph.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- Sireesh D, Dhamodharan U, Ezhilarasi K, Vijay V, Ramkumar KM. Association of NF-E2 related factor 2 (Nrf2) and inflammatory cytokines in recent onset type 2 diabetes mellitus. Sci Rep. 2018;8:5126. doi: 10.1038/s41598-018-22913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganya N, Mani KP, Sireesh D, Rajaguru P, Vairamani M, Suresh T, Suzuki T, Chatterjee S, Ramkumar KM. Establishment of pancreatic microenvironment model of ER stress: quercetin attenuates beta-cell apoptosis by invoking nitric oxide-cGMP signaling in endothelial cells. J Nutr Biochem. 2018;55:142–156. doi: 10.1016/j.jnutbio.2017.12.012. [DOI] [PubMed] [Google Scholar]
- Tang H, Tian E, Liu C, Wang Q, Deng H. Oxidative stress induces monocyte necrosis with enrichment of cell-bound albumin and overexpression of endoplasmic reticulum and mitochondrial chaperones. PLoS One. 2013;8:e59610. doi: 10.1371/journal.pone.0059610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongwatchara P, Promwikorn W, Srisomsap C, Chokchaichamnankit D, Boonyaphiphat P, Thongsuksai P. Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol Rep. 2011;26:185–191. doi: 10.3892/or.2011.1266. [DOI] [PubMed] [Google Scholar]
- Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, Post JA. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;5:381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- Wang MG, Fan RF, Li WH, Zhang D, Yang DB, Wang ZY, Wang L. Activation of PERK-eIF2alpha-ATF4-CHOP axis triggered by excessive ER stress contributes to lead-induced nephrotoxicity. Biochim Biophys Acta Mol Cell Res. 2019;1866:713–726. doi: 10.1016/j.bbamcr.2018.12.002. [DOI] [PubMed] [Google Scholar]
- Wu Y, Ding Y, Tanaka Y, Zhang W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci. 2014;11:1185–1200. doi: 10.7150/ijms.10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 13 kb)