

Abstract
The intra‐erythrocyte stage of P. falciparum relies primarily on glycolysis to generate adenosine triphosphate (ATP) and the energy required to support growth and reproduction. Lactic acid, a metabolic byproduct of glycolysis, is potentially toxic as it lowers the pH inside the parasite. Plasmodium falciparum formate–nitrite transporter (PfFNT), a 34‐kDa transmembrane protein, has been identified as a novel drug target as it exports lactate from inside the parasite to the surrounding parasitophorous vacuole within the erythrocyte cytosol. The structure and detailed molecular mechanism of this membrane protein are not yet available. Here we present structures of PfFNT in the absence and presence of the functional inhibitor MMV007839 at resolutions of 2.56 Å and 2.78 Å using single‐particle cryo‐electron microscopy. Genetic analysis and transport assay indicate that PfFNT is able to transfer lactate across the membrane. Combined, our data suggest a stepwise displacement mechanism for substrate transport. The PfFNT membrane protein is capable of picking up lactate ions from the parasite’s cytosol, converting them to lactic acids and then exporting these acids into the extracellular space.
Keywords: cryo‐EM, Malaria, membrane protein, PfFNT, Plasmodium falciparum transporter
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Structural and genetic analyses of the P. falciparum transporter PfFNT suggest a stepwise displacement mechanism for substrate transport. This membrane protein is capable of acquiring lactate ions from the parasite’s cytosol, converting them into lactic acids and then exporting these acid molecules into the extracellular space.
Introduction
Malaria is an extremely devastating disease as it infects more than 200 million people with nearly half a million deaths worldwide each year (WHO, 2018). It appears that more than 90% of pregnant women and children with Plasmodium falciparum infection reside in sub‐Saharan Africa (WHO, 2019). Resistance to several classes of antimalarial drugs has developed over the past century, e.g., 4‐aminoquinolines such as chloroquine and dihydrofolate reductase inhibitors such as pyrimethamine and cycloguanil. More recently, resistance to highly effective artemisinin‐combination therapies has emerged in southeast Asia (Blasco et al, 2017). The lethality of malaria combined with its propensity to develop drug resistance indicates that identification of novel classes of antimalarials should be a high global health priority.
Glucose is the major energy source for the malaria parasite (McKee et al, 1946). The abundance of glucose in the mammalian host’s blood provides a favorable environment for the survival of the asexual intra‐erythrocytic form of P. falciparum, which primarily relies on anaerobic glycolysis to fulfill its energy requirements (MacRae et al, 2013). Within the parasite, glucose is metabolically converted to pyruvate and the waste product lactate in order to generate adenosine triphosphate (ATP). Both glucose influx and lactate efflux are essential for this process and maintaining intracellular homeostasis. It has thus been proposed that this pathway is a potential target for a novel class of antimalarial drugs (Woodrow et al, 1999; Joet et al, 2003; Marchetti et al, 2015; Wu et al, 2015). A potential barrier to this is the evolutionary conservation of glucose transporters and glycolytic enzymes between the parasite and human host.
The P. falciparum membrane protein PfFNT, which belongs to the formate/nitrite transporter (FNT) family, is responsible for mediating the efflux of the waste product lactate from the intra‐erythrocytic malaria parasite (Marchetti et al, 2015; Wu et al, 2015). PfFNT represents a novel, valid drug target because the human genome does not encode this family of membrane proteins (Golldack et al, 2017; Hapuarachchi et al, 2017). Using the Malaria Box (Spangenberg et al, 2013; Van Voorhis et al, 2016) to screen for potent PfFNT inhibitors, two promising small molecule compounds, MMV007839 and MMV000972, have been identified (Wu et al, 2015; Golldack et al, 2017; Hapuarachchi et al, 2017). These compounds inhibit the function of the PfFNT transporter, leading to the accumulation of intracellular lactate which in turn kills the malaria parasite (Wu et al, 2015; Golldack et al, 2017; Hapuarachchi et al, 2017). PfFNT is localized at the parasite’s plasma membrane and functions via a substrate/proton symport mechanism (Marchetti et al, 2015; Wu et al, 2015; Golldack et al, 2017; Hapuarachchi et al, 2017). Although PfFNT was also thought to exist in the membrane of the digestive vacuole, a study using GFP‐fused PfFNT indicated that this membrane protein is only confined to the plasma membrane (Wu et al, 2015). In addition to exporting lactate, this transporter mediates efflux of various monocarboxylates, including formate, acetate, pyruvate, and propionate (Marchetti et al, 2015). To elucidate the molecular mechanism of substrate translocation across the plasma membrane via the PfFNT transporter, we obtained structural information of this membrane protein both in the absence and presence of the transporter’s inhibitor MMV007839. We here present structures of apo‐PfFNT and PfFNT bound with MMV007839 using single‐particle cryo‐electron microscopy (cryo‐EM) to resolutions of 2.56 Å and 2.78 Å. Coupled with genetic analysis and transport assay, these structures suggest that PfFNT contains transient binding sites for lactate and lactic acid. This channel sequentially shuttles lactate ions and converts these ions into lactic acids via a stepwise displacement mechanism. Our work provides molecular insights into the mechanism of lactate extrusion that involves substrate binding and displacement, as well as proton transfer via the PfFNT membrane protein to facilitate substrate transport across the membrane.
Results
Structure of apo‐PfFNT
To obtain the structural information of PfFNT, we cloned the full‐length (309 amino acids) P. falciparum PfFNT transporter into pcDNA3.1‐N‐DYK. This PfFNT protein was expressed in HEK293 cells and purified using a Strep‐Tactin affinity column. We then reconstituted purified PfFNT into lipidic nanodiscs and solved the structure of the 34‐kDa transporter using single‐particle cryo‐EM. The three‐dimensional reconstitution of the PfFNT transporter led to a cryo‐EM map at a nominal resolution of 2.56 Å (Table EV1, Appendix Figs S1 and S2), which enabled us to build a model of this membrane protein. The final structural model of this apo‐PfFNT transporter includes residues 12–292.
PfFNT appears to be pentameric in oligomerization (Fig 1A–E) with most of the secondary structures embedded in the membrane. Each protomer consists of six transmembrane (TM) helices (TM1‐TM6) (Fig 1F). Two juxtamembrane helices (JM1 and JM2), approximately parallel to and on opposite sides of the parasite plasma membrane, are located at the N‐terminus and space between TM3 and TM4, respectively. In addition, an extracellular helix (α1) is observed to connect TM1 and TM2. Both the N‐ and C‐termini of PfFNT localize at the cytoplasmic side (Fig 1D and F), consistent with the known structures of bacterial FNT‐family proteins (Wang et al, 2009; Waight et al, 2010; Lu et al, 2011; Czyzewski & Wang, 2012; Lu et al, 2012b). The N‐terminal end of each protomer contains a small β‐strand (β1), whereas the C‐terminal end possesses a small α‐helix (α2). These two secondary structural features protrude into the cytoplasm and form the cytoplasmic domain (Fig 1F).
Figure 1. Cryo‐EM structure of the Plasmodium falciparum PfFNT transporter.
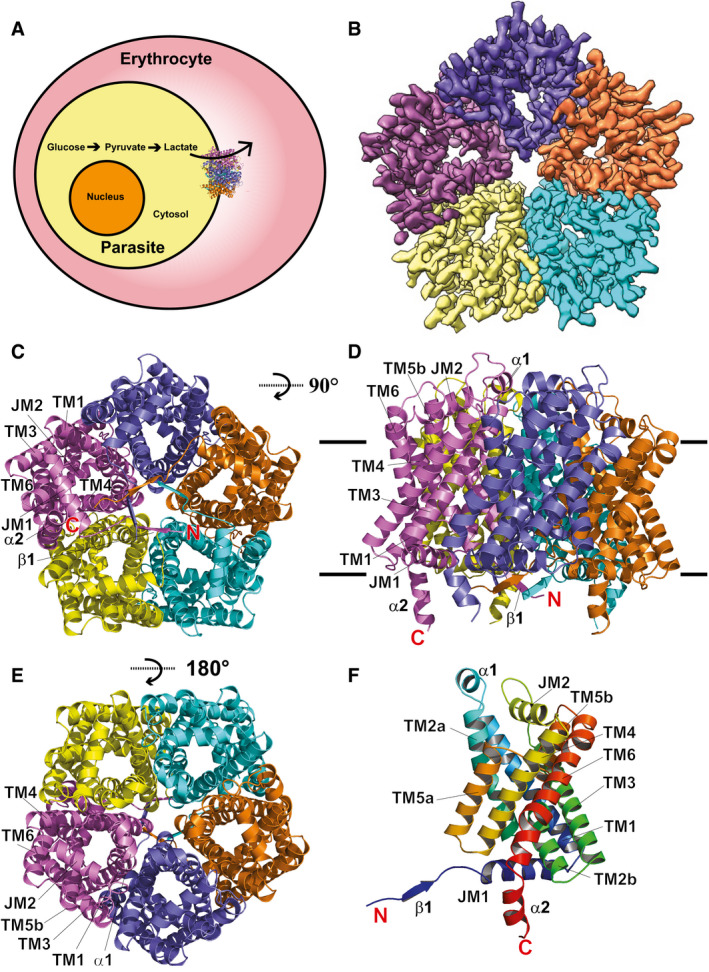
- The intra‐erythrocytic state of P. falciparum illustrating the localization of PfFNT within the parasite’s plasma membrane.
- Cryo‐EM density map of pentameric PfFNT viewed from the parasite’s cytoplasm. The five protomers of pentameric PfFNT are colored yellow, cyan, orange, slate, and purple.
- Ribbon diagram of the 2.56‐Å resolution structure of pentameric PfFNT viewed from the parasite’s cytoplasm. The five protomers of pentameric PfFNT are colored yellow, cyan, orange, slate, and purple.
- Ribbon diagram of pentameric PfFNT viewed from the parasite’s membrane plane. The five protomers of pentameric PfFNT are colored yellow, cyan, orange, slate, and purple.
- Ribbon diagram of pentameric PfFNT viewed from the extracellular side of the parasite. The five protomers of pentameric PfFNT are colored yellow, cyan, orange, slate, and purple.
- Structure of a protomer of PfFNT. This ribbon diagram is colored using a rainbow gradient from the N‐terminus (blue) to the C‐terminus (red).
The TMs, JMs, α‐helices, and β‐strands are designated numerically from the N‐ to C‐termini: β1 (13–16), JM1 (20‐35), TM1 (41–68), α1 (71–77), TM2 (a (80–100) and b (105–118)), TM3 (123–150), JM2 (164–179), TM4 (182–206), TM5 (a (210–226) and b (231–244)), TM6 (250–280), and α2 (282–292). Of the six TMs, TM2 and TM5 are broken into two segments that are connected by extended loops, L2 and L5, within the membrane, respectively. Both L2 and L5 contain four amino acids. The orientations of these two flexible loops are roughly parallel to the plane of the lipid membrane. The six TMs are arranged as three helical pairs, which form an inverted antiparallel topology (Appendix Fig S3). Interestingly, some of the amino acids within the N‐terminal half of each protomer (e.g., K35, D103, and N108) mirror the seemingly related C‐terminal half residues (e.g., K177, E229, and N234). The pentameric protein has a pseudo‐fivefold symmetrical architecture throughout with each protomer within the pentamer similar in conformation. Superimposition of these five protomers results in a root‐mean‐square deviation (r.m.s.d.) of 0.4–0.5 Å between backbone atoms, indicating that their secondary structures are very similar. However, a detailed inspection indicates that the side‐chain orientations of many residues are quite distinct, suggesting that these five PfFNT molecules may represent various conformational states of the protein. Viewed in parallel to the membrane, the pentamer is about 50 Å tall and 75 Å wide. Additional densities, corresponding to the belt formed by nanodisks, were found to encircle the transmembrane region of the pentameric PfFNT transporter (Appendix Fig S1).
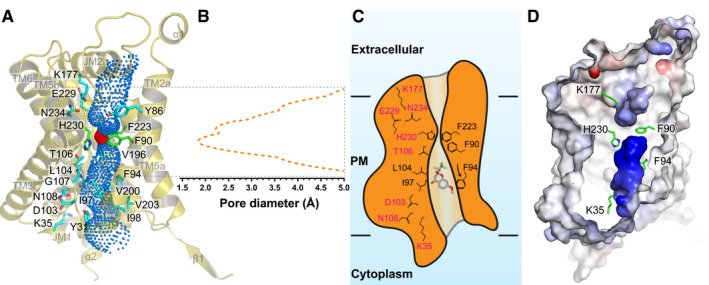
The overall secondary structure at the transmembrane region of PfFNT is conserved relative to other known structures of bacterial membrane proteins belonging to the FNT family, including Escherichia coli FocA (Wang et al, 2009), Vibrio cholerae FocA (Waight et al, 2010), Salmonella typhimurium FocA (Lu et al, 2011), Salmonella typhimurium NirC (Lu et al, 2012b), and Clostridium difficile HSC (Czyzewski & Wang, 2012). Pairwise superimpositions of the structure of PfFNT to those of its bacterial homologs give rise to r.m.s.d. of 1.6–1.9 Å (Appendix Fig S4). Consistent with its bacterial counterparts, each PfFNT protomer forms a central channel starting from the cytoplasmic side up to the extracellular space (Fig 2A–D). There are at least 20 residues that line the wall of the central channel, including seven hydrophobic, five aromatic, three polar, three positively charged, and two negatively charged amino acids. Many of these are conserved residues, such as Y31, K35, V54, Y86, F90, F94, I97, I98, D103, L104, T106, G107, N108, K177, V200, V203, F223, E229, H230, and N234, which may be important for the function of this membrane protein (Fig 2A and Appendix Fig S5). Since PfFNT shuttles lactate across the membrane, it is somewhat surprising that this central channel is quite narrow and constitutes multiple constriction sites. TM1, TM2, TM4, and TM5 are responsible for creating this central channel in the middle of each monomer. Calculation of the channel diameter along the axis perpendicular to the membrane plane indicates that the narrowest region of the channel can be as small as 1.8 Å (protomer A) and even the widest portion is < 5.0 Å (protomer E) (Fig 2B and Appendix Fig S6), indicating that this central channel is quite narrow in comparison with those of the related bacterial FNT proteins. This observation does not appear to agree with a previous proposition that eukaryotic FNTs possess wider channels that allow larger monocarboxylates to pass through (Wiechert et al, 2017; Erler et al, 2018; Helmstetter et al, 2019). In the middle of the membrane, the constriction sites of the channel line with amino acids V54, F90, F94, I97, L104, V220, F223, and H230 (Fig 2A and B and Appendix Fig S6). These residues roughly create two constriction sites of the central channel. It appears that residues F94, I97, and L104 make up the first constriction site located right above the membrane surface at the cytoplasmic side, whereas residues F90, F223, and H230 contribute to the second constriction site situated closer to the extracellular surface. Presumably, these are critical residues needed to facilitate the export of lactate across the parasite’s plasma membrane.
Figure 2. Structure of the central channel in the apo‐PfFNT protomer.
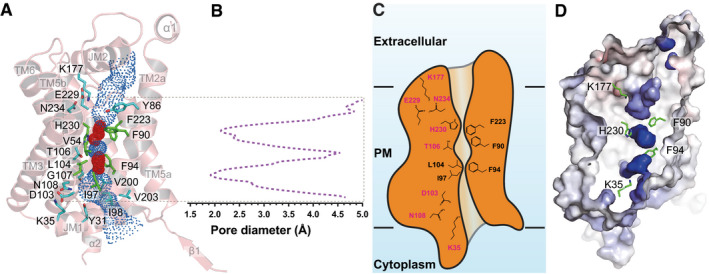
- The central channel in the apo‐PfFNT protomer viewed from the parasite’s membrane plane. The channel, calculated by HOLE (Smart et al, 1996), is indicated by blue dots. The two constriction sites are colored red. Residues forming the constriction sites are in green sticks. Residues that line the rest of the channel are in cyan sticks.
- Representative figure of the calculated diameters of an apo‐PfFNT protomer.
- A cartoon of the central channel formed within a PfFNT protomer. The channel contains two constriction sites in this conformational state. Residues forming these constrictions and the K35‐D103‐N108 and K177‐E229‐N234 triads are illustrated as sticks.
- Surface representation of the inside of the channel formed by a PfFNT protomer (red, negative −15 kT/e; blue, positive +15 kT/e). The five residues K35, F90, F94, K177, and H230, which are subjected for mutagenesis studies, are in green sticks.
Structure of the PfFNT‐inhibitor complex
Recently, a Malaria Box chemical library (Spangenberg et al, 2013; Van Voorhis et al, 2016) was used to screen for novel inhibitors that perturb the growth of the P. falciparum parasite. A new class of fluoroalkyl vinylogous acids that potently block PfFNT and kill cultured parasites was identified in these studies (Golldack et al, 2017; Hapuarachchi et al, 2017) which provided the first evidence that PfFNT is a druggable antimalarial target. To elucidate how these novel PfFNT inhibitors hinder the function of the transporter, we incubated a 2 μM PfFNT‐nanodisc sample with 10 μM MMV007839 (Fig 3A) for two hours to form the PfFNT‐MMV007839 complex and then solved the cryo‐EM structure of this PfFNT‐inhibitor complex at a resolution of 2.78 Å (Fig 3, Table EV1, Appendix Figs S7 and S8).
Figure 3. Cryo‐EM structure of the PfFNT‐MMV007839 complex.
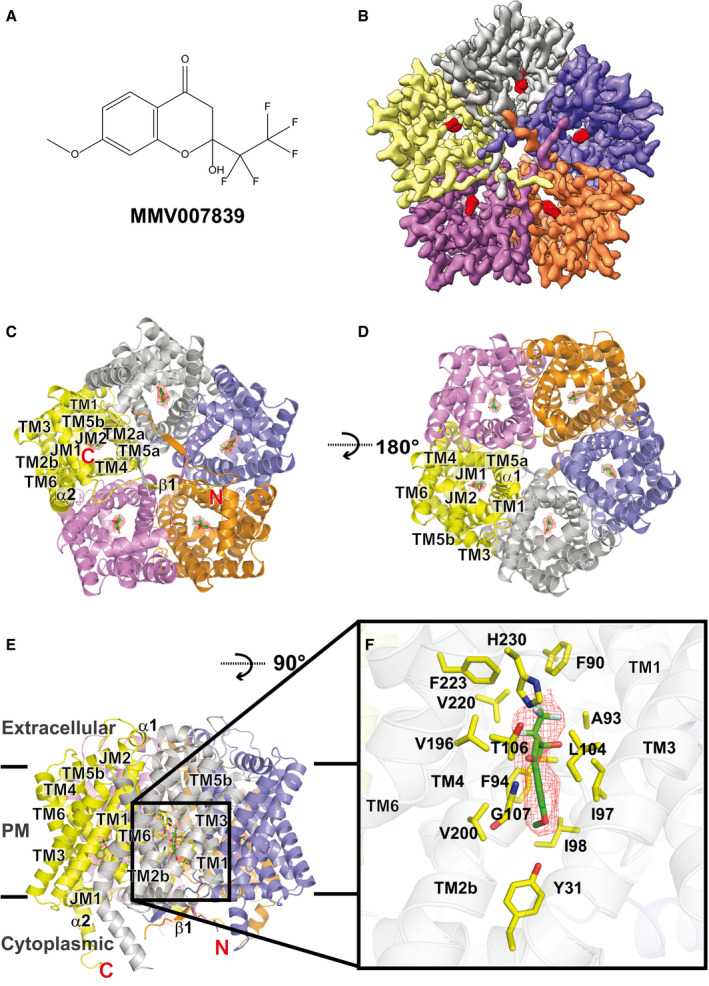
- Chemical structure of MMV007839. The chemical name of MMV007839 is 2‐hydroxy‐7‐methoxy‐2‐(perfluoroethyl)chroman‐4‐one, and its chemical formula is C12H9F5O4.
- Cryo‐EM density map of pentameric PfFNT viewed from the parasite’s cytoplasm. Densities of the five bound MMV007839 within the pentamer are colored red. The five protomers of pentameric PfFNT are colored yellow, slate, orange, purple, and gray.
- Ribbon diagram of the 2.78‐Å resolution structure of pentameric PfFNT‐MMV007839 viewed from the parasite’s cytoplasm. The five protomers of pentameric PfFNT are colored yellow, slate, orange, purple, and gray.
- Ribbon diagram of pentameric PfFNT‐MMV007839 viewed from the extracellular side of the parasite. The five protomers of pentameric PfFNT are colored yellow, slate, orange, purple, and gray.
- Ribbon diagram of pentameric PfFNT‐MMV007839 viewed from the parasite’s membrane plane. The five protomers of pentameric PfFNT are colored yellow, slate, orange, purple, and gray. Densities of the five bound MMV007839 are in red meshes.
- The MMV007839 of PfFNT. The bound MMV007839 is colored green. Density of the bound MMV007839 is in red mesh. Residues involved in forming the inhibitor‐binding site are colored yellow.
Surprisingly, the overall structure of pentameric PfFNT‐MMV007839 (Fig 3B–D) resembles that of the apo‐PfFNT pentamer, with a r.m.s.d. of 0.59 Å between backbone atoms, suggesting that the secondary structure of PfFNT does not significantly change after inhibitor binding. Within the pentamer, the five β1 strands develop a ring architecture situated at the center of the pentamer (Fig 3B and C), leaving the five α2 helices near the edge of the pentamer to mimic five leg‐like features (Fig 3E). Single‐particle 2D class images reveal that the overall shape of the pentameric molecule bears resemblance to a jellyfish (Appendix Fig S7).
Our high quality cryo‐EM map allows us to unambiguously depict the location of bound MMV007839. The bound inhibitor localized right next to G107 and is closely attached to this residue (Fig 3F). It appears that the backbone nitrogen of G107 forms a hydrogen bond with an oxygen atom from the carbonyl group of the MMV007839 inhibitor. Apparently, the space between G107 and MMV007839 is so tight that there is no room to accommodate an amino acid side chain in this region. This observation is in good agreement with the previous finding that P. falciparum carrying a G107S mutation elevates resistance to the MMV007839 inhibitor (Golldack et al, 2017; Hapuarachchi et al, 2017), probably via van der Waals repulsion between this mutated residue and the inhibitor. The MMV007839 binding mode is also consistent with the predicted model of PfFNT‐inhibitor interactions (Golldack et al, 2017). Residues Y31, F90, A93, F94, I97, I98, L104, T106, G107, V196, V200, V220, F223, and H230, which are within 4.5 Å from MMV007839, form the inhibitor‐binding site (Fig 3F). Surprisingly, most of these residues are hydrophobic in nature. Therefore, the primary association between PfFNT and the inhibitor should be via hydrophobic interaction. In addition, the two nitrogen atoms of the imidazole ring of H230 are 4.6 and 4.7 Å away from the oxygen atoms of the hydroxyl and carbonyl groups of MMV007839, respectively. We postulate that these two nitrogen atoms participate in electrostatic contributions to secure inhibitor binding.
Although the secondary structure of PfFNT‐MMV007839 does not alter much in comparison with that of apo‐PfFNT, the conformation of the central channel formed by each PfFNT protomer is quite distinct in the presence of this inhibitor. We observed that this channel is wider in diameter after inhibitor binding. The narrowest and widest regions of the channel were measured to be 1.8 Å (protomer D) and 5.3 Å (protomer E), respectively (Fig 4A–D and Appendix Fig S9). There is a noticeable constriction site, which is located immediately above the bound MMV007839 molecule in relation to the extracellular surface of the plasma membrane. F90, F223, and H230 are responsible for creating this constriction (Fig 4A–D and Appendix Fig S9). These three residues have been previously predicted to form a constriction based on the crystal structures of bacterial FNT transporters (Golldack et al, 2017). According to the structural information, these residues may be very important for transport function.
Figure 4. Structure of the central channel in the PfFNT‐MMV007839 protomer.
- The central channel in the PfFNT‐MMV007839 protomer viewed from the parasite’s membrane plane. The channel, calculated by HOLE (Smart et al, 1996), is indicated by blue dots. There is one constriction site (colored red) formed in the central channel. Residues forming the constriction are in green sticks. Residues that line the rest of the channel are in cyan sticks.
- Representative figure of the calculated diameters of a PfFNT‐MMV007839 protomer.
- A cartoon of the central channel formed within a PfFNT protomer. The channel contains one constriction site in this conformational state. Residues forming the constriction and the K35‐D103‐N108 and K177‐E229‐N234 triads are illustrated as sticks. Residues F94, I97, and L104, which form the first constriction site in the apo‐PfFNT structure, are also included in the figure.
- Surface representation of the inside of the channel formed by a PfFNT protomer (red, negative −15 kT/e; blue, positive +15 kT/e). The five residues K35, F90, F94, K177, and H230, which are subjected for mutagenesis studies, are in green sticks.
In comparison with the apo‐PfFNT and PfFNT‐MMV007839 structures, the side chains of several amino acids that participate in forming the channel have significantly shifted their location and orientation to accommodate for inhibitor binding. These residues include Y31, F90, F94, I97, I98, L104, T106, V196, V200, F223, and H230, in which many of them are involved in forming the binding site (Fig 3F and Appendix Fig S10). In addition, G107 is found to shift the backbone location to expand the size of the pocket for housing the MMV007839 molecule. This observation indeed highlights the flexibility of the side chains of these binding residues, which are able to adjust their orientation and position to facilitate and accommodate the function of the PfFNT transporter.
Mutations on the PfFNT transporter
Based on the information obtained from cryo‐EM structures of apo‐PfFNT and PfFNT‐MMV007839, there are four distinct clusters of amino acids which may be important for the function of this transporter. It has been proposed that FNT transporters translocate substrate via a substrate/proton symport mechanism (Lu et al, 2012b; Wu et al, 2015). As mentioned earlier, the N‐ and C‐terminal halves of PfFNT are structurally related and form an inverted antiparallel topology. The two halves of the transporter contain two triads, K35‐D103‐N108 and K177‐E229‐N234, located near the cytoplasmic side and extracellular surface, respectively. These two triads are roughly mirror reflections of each other and may be functionally important. In addition, there are two constrictions within the central channel. The first constriction consists of residues F94, I97, and L104, whereas the composition of the second constriction includes residues F90, F223, and H230. To determine whether these clusters of residues are important for the function of PfFNT, we mutated the positively charged and aromatic residues K35, F90, F94, K177, and H230 into alanines, individually. We also converted H230 into an asparagine. We reconstituted the wild‐type and mutant transporters into liposomes. We then used an osmotic method coupled with stopped‐flow to measure influx of lactate into these reconstituted proteoliposomes (Fig 5 and Appendix Fig S11). Upon addition of lactate into the extravesicular buffer, the size of proteoliposomes shrinks rapidly because of the osmotic effect. The rate of lactate influx was measured by the rate of recovery of the volume of proteoliposomes (Fig 5A). The results showed that the K35A, F94A, and K177A mutants generated exponential reswelling curves with the rates of 8.3 × 10−3 ± 0.7 × 10−3 s‐1, 8.6 × 10−3 ± 0.2 × 10−3 s‐1 and 7.2 × 10−3 ± 0.5 × 10−3 s‐1, respectively (Fig 5B). These reswelling rates are faster than that of wild‐type (5.7 × 10−3 ± 0.2 × 10−3 s‐1), suggesting that a mutation on these residues enhances the transport of lactate across the membrane via PfFNT. In contrast, the time course curves for both the H230A and H230N mutants lacked any reswelling phase, posing that the H230A and H230N mutants are not capable of shuttling lactate into the proteoliposomes and the transport function of these two mutant proteins are completely abolished (Fig 5B). For the F90A mutant, there was an exponential reswelling with the swelling rate of 2.7 × 10−3 ± 0.2 × 10−3 s‐1. In comparison with that of the wild‐type transporter, the rate of lactate transport for the F90A mutant appears to be approximately twofold slower (Fig 5B).
Figure 5. Relative rates for transport of lactic acid via PfFNT.
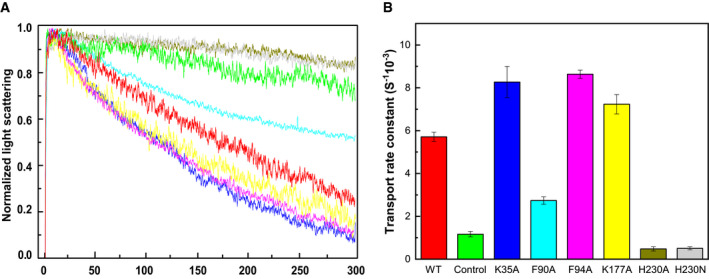
- Stopped‐flow assay measuring relative rates of lactic acid transport into reconstituted vesicles containing wild‐type or mutant transporter (green, control with empty vesicles; red, wild‐type; blue, K35A; cyan, F90A; magenta, F94A; yellow, K177A; olive green, H230A; and gray, H230N).
- Relative rates of lactic acid transport based on reswelling rates of proteoliposomes. Each data point is an average of five different light scattering data sets. Error bars depict standard deviation for these five light scattering data. All of the transport rates from the mutant transporters are significantly different compare from that of wild‐type PfFNT (K35A, P = 2.8 × 10−3; F90A, P = 4.4 × 10−7; F94A, P = 4.9 × 10−6; K177A, P = 1.2 × 10−3; H230A, P = 4.5 × 10−7; H230N, P = 2.0 × 10−6; Student’s t‐test) (green, control with empty vesicles; red, wild‐type; blue, K35A; cyan, F90A; magenta, F94A; yellow, K177A; olive green, H230A; and gray, H230N).
Discussion
Our cryo‐EM data indicate that the overall secondary structure of PfFNT does not change much before and after inhibitor binding. This result indeed agrees with the idea that FNTs are not expected to undergo major conformational changes during substrate transport (Lu et al, 2012b; Wu et al, 2015). Therefore, it is most likely that the transport process requires a large degree of flexibility of residue side chains within the central channel. This can be readily achieved by the rearrangement of side‐chain orientations of residues that line the wall of the channel. Thus, the residues forming the constriction sites are sufficiently flexible to quickly rearrange for substrate translocation. This is in line with previous suggestions that the side chains of residues at the constrictions are flexible enough to facilitate the passage of substrates (Waight et al, 2010; Lu et al, 2012a; Wiechert et al, 2017). Indeed, the aromatic residue F94, which forms one of the constrictions in the middle of the channel, significantly changes its side‐chain location when compared with the structures of the five PfFNT apo‐protomers (Appendix Fig S12). In addition, the side chains of residues Y31, K35, F90, I98, D103, L104, T106, N108, K177, F223, E229, N234, and H230 are found to significantly shift their positions compared with the five protomers of the apo‐PfFNT structure (Appendix Fig S12). In comparison with the five PfFNT‐inhibitor protomers, the positively charged amino acid H230, which contributes to form the central channel, alters its side‐chain orientation. Additionally, the side chains of residues Y31, K35, F90, I98, D103, L104, T106, N108, K177, F223, E229, and N234 are observed to switch their relative positions when compared with the conformations of these five protomers (Appendix Fig S13). As there are multiple constrictions within the channel, we propose that the residues lining the channel are highly flexible that can quickly rearrange to sequentially contract and relax the channel for propelling the movement of substrate across the membrane.
As the pKa of lactic acid is 3.86, this compound should be in the form of lactate ion in the cytoplasm of P. falciparum, where the cytoplasmic pH is approximately 7.15 (Kuhn et al, 2007). In view of the structures of PfFNT, the first half of the central channel formed in each protomer, starting from the cytoplasmic portion and up to the position of the histidine H230 residue, is positively charged (Figs 2D and 4D). However, the second half of this central channel spanning the midway and up to the extracellular surface of the parasite is quite neutral in charge (Figs 2D and 4D). This observation suggests that the more stable form of the substrate should be in the deprotonated state as a lactate ion inside the first half of the channel, whereas this substrate should be in the form of lactic acid in the second half of this channel. Thus, we propose that PfFNT shuttles lactate ions from the parasite’s cytoplasm to the midway of the central channel. These ions will be converted to lactic acids, probably via proton transfer from H230, and then shuttled across the second half of the channel to the parasite’s extracellular surface (Fig 6). This proposed mechanism is in good agreement with results from a study using radiolabeled substrate that the transport mechanism should be involved in a direct one‐step protonation of weak acid substrates (Wiechert & Beitz, 2017). Another study using radiolabeled lactate also indicated that the PFT‐type proteins should use the proton gradient to neutralize the weak acid anion within the central channel to facilitate transport (Bader & Beitz, 2020).
Figure 6. Proposed mechanism for substrate transport.
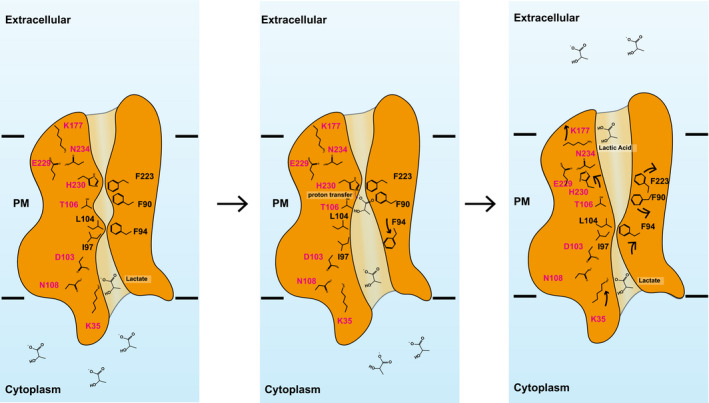
Based on the structures of apo‐PfFNT and PfFNT‐MMV007839, we propose that the PfFNT membrane protein is capable of picking up lactate ions from the parasite’s cytosol, converting them to lactic acids and then exporting these acids into the extracellular space via a sequential displacement mechanism. We believe that residues lining the central channel are highly flexible and can quickly rearrange sequentially to contract and relax the channel for propelling the export of substrate across the membrane. This schematic diagram indicates that PfFNT shuttles lactate ions from the parasite’s cytoplasm to the midway of the central channel. These ions will be converted to lactic acids, via proton transfer from H230, and then shuttled across the second half of the channel to the parasite’s extracellular surface. Residue K35 contributes to form a transient binding site to bind lactate ion nearby to the cytoplasmic surface of the parasite. As soon as the second lactate ion arrives from the parasite’s cytoplasm, the first lactate will be displaced and continue to propagate to the midway of the channel, where H230 is responsible for donating a proton to convert this lactate ion into lactic acid and open the constriction for further propagation. Residue K177 participates in forming a transient binding site, which binds lactic acid nearby the extracellular surface. A second ion enters this site will cause displacement and extrusion of lactic acid out of the parasite.
Our mutational data suggest that a mutation of the conserved residue K35 to an alanine facilitates the transport of lactate across the membrane, posing a possibility that K35 may contribute to form a transient binding site for lactate ion nearby to the cytoplasmic surface of the parasite. When there is only one lactate in the channel, this ion may be bound at this binding site. As soon as the second lactate ion arrives from the parasite’s cytoplasm, the first lactate may be displaced and released so that it will continue to propagate forward along the channel (Fig 6). Therefore, it is likely that PfFNT may utilize a displacement mechanism to extrude lactate ions within the first half of the central channel and deliver these ions to the midway of the central channel.
We observed that a mutation of H230 to an alanine or asparagine completely abolishes the transport function of PfFNT, suggesting that this residue is indispensable for substrate export. The residue is located in the middle of this elongated channel. Coupled with F90 and F223, H230 forms the second constriction. This constriction probably will need to open in order to allow for substrate translocation. We believe that H230 is responsible for donating a proton to convert the lactate ion into lactic acid when lactate arrives at this gate. It appears that proton transfer may be coupled with the opening of this constriction, which permits the lactic acid molecule to further propagate along the channel. Thus, PfFNT may export the neutral lactic acid in the second half of the central channel to eliminate this toxic compound from the parasite. It is likely that F90 and F223 may provide with important van der Waals interactions, allowing the substrate to specifically contact H230 to facilitate the proton transfer process. This idea indeed is in good agreement with our mutagenesis study that a mutation of F90 to an alanine partially abrogates the transport function of this membrane protein (Fig 5).
Interestingly, a mutation of the conserved lysine K177 nearby the extracellular surface of the parasite to an alanine also enhances the activity of lactate transport. Similar to K35, K177 may participate in forming a transient binding site, which stabilizes the lactic acid position. A second ion enters this site may cause the displacement of lactic acid to the opposite side via van der Waal repulsion (Fig 6).
Materials and Methods
Expression and purification of PfFNT
The codon‐optimized DNA of full‐length PfFNT from Plasmodium falciparum 3D7 was synthesized and cloned into pcDNA3.1‐N‐DYK (GenScript) in frame with a thrombin cleavage site and Twin‐Strep‐tag at the C‐terminus. The resulting plasmid was confirmed by the Sanger method of DNA sequencing.
The human embryonic kidney Expi293F (Thermo Fisher) cells were cultured in Expi 293TM Expression Medium (Gibco) at 37°C supplemented with 8% CO2. The PfFNT protein was expressed using a transient expression system with the following procedures. The purified pcDNA3.1‐N‐DYK plasmid expressing full‐length PfFNT was mixed with cationic liposomes (Transfection reagent I, Avanti Polar lipid) at a 1:10 (DNA:liposome) (w/w) ratio in Opti‐MEM™ I reduced‐serum medium (Gibco) and incubated at room temperature for 15 min. The resulting lipoplexes were added to cells (cell density of 2.5–3 × 106 cells/ml) at a final DNA concentration of 1 mg/l. To boost protein expression, valproic acid (Sigma) was added at a final concentration of 3 mM after 18–24 h and allowed to proceed for total 72 h.
Cells were collected and resuspended in lysis buffer (20 mM HEPES‐NaOH (pH 7.5) and 150 mM NaCl) and disrupted with a French pressure cell. The membrane fraction was collected and washed once with the lysis buffer. The membrane protein was then solubilized in 1% (w/v) n‐dodecyl‐β‐D‐maltoside (DDM) and 0.1% cholesteryl hemisuccinate (CHS) for 3 h at 4°C. Insoluble material was removed by ultracentrifugation at 100,000 g. The extracted protein was applied to a Strep‐Tactin affinity column (IBA Lifesciences) and washed twice with 15 column volumes of lysis buffer supplemented with 0.05% DDM and 0.005% CHS. The PfFNT protein was eluted by adding 2.5 mM desthiobiotin. The purity of the PfFNT protein (> 90%) was judged using SDS–PAGE stained with Coomassie Brilliant Blue. The purified protein was then concentrated to a final concentration of 20 mg/ml.
Nanodisc preparation
To reconstitute purified PfFNT into nanodiscs, the purified membrane protein was mixed with the scaffold protein MSP1E3D1 and E. coli polar lipid extract at a molar ratio of 1:2.5:70 for 15 min at room temperature. 0.8 mg/ml pre‐washed Bio‐beads (Bio‐Rad) was subsequently added, and the mixture was incubated for 1 h on ice followed by overnight incubation at 4°C. The protein–nanodisc solution was filtered through 0.22‐µm nitrocellulose‐filter tubes to remove the Bio‐beads and further purified using a Superose 6 column (GE Healthcare) equilibrated with buffer containing 20 mM Tris–HCl, pH 7.5, and 100 mM NaCl.
Electron microscopy sample preparation
The PfFNT‐nanodisc sample was concentrated to 0.7 mg/ml (3 μM). A 2.5 μl sample was applied to glow‐discharged holey carbon grids (Quantifoil Cu R1.2/1.3, 300 mesh), blotted for 15 s, and then plunge‐frozen in liquid ethane using a Vitrobot (Thermo Fisher). The grids were transferred into cartridges. For high‐resolution data collection, the sample grids were loaded into a Titan Krios cryo‐electron microscope operated at 300 kV equipped with Gatan BioQuantum imaging filter (GIF) and a K3 summit direct electron detector (Gatan). The micrographs were recorded using SerialEM (Mastronarde, 2005) multi‐shot method over nine neighboring holes (3 × 3) with counting mode at nominal × 81,000 magnification corresponding to a calibrated pixel size of 1.08 Å (super‐resolution, 0.54 Å/pixel) and a defocus range of −1 to −2.5 μm. To remove inelastically scattered electrons, the slit width was set to 20 eV. Each micrograph was exposed for 2 s with a total specimen dose of 29.2 e‐/Å2 and 40 frames were captured per specimen area.
For the PfFNT‐MMV007839 inhibitor complex, a 2 μM PfFNT sample was incubated with 10 μM MMV007839 (MolPort) for 2 h to form the PfFNT‐MMV007839 complex. The procedures for making cryo‐EM grids and high‐resolution cryo‐EM data collection were the same as those of apo‐PfFNT. Each micrograph was exposed for 2.8 s with a total specimen dose of 41.0 e‐/Å2 and 56 frames were captured per specimen area.
Cryo‐EM data processing
The micrographs of PfFNT were aligned by using patch‐based motion correction for beam induced motion using cryoSPARC (Punjani et al, 2017). The contrast transfer function (CTF) parameters of the micrographs were determined using Patch CTF (Zhang, 2016). After manual inspection and sorting to discard poor micrographs, ∼2,000 particles of PfFNT were manually picked to generate templates for automatic picking. Initially, 2,949,036 particles were selected after autopicking in cryoSPARC (Punjani et al, 2017). Several iterative rounds of two‐dimensional (2D) classifications were carried to remove false picks and classes with unclear features. The resulting 228,441 particles were used to generate a reference free ab initio three‐dimensional (3D) reconstruction with C1 symmetry. After two rounds of heterogeneous refinement, 85,976 particles were chosen for further processing with non‐uniform and local CTF refinement (Zhang, 2016). The local focused refinement with a soft mask covering the PfFNT protein area was done using cryoSPARC (Punjani et al, 2017), resulting in a 2.86 Å global resolution map based on the gold‐standard Fourier shell correlation (FSC 0.143). The cryo‐EM map was then improved using the RESOLVE density modification program (Terwilliger et al, 2020) implemented in PHENIX (Adams et al, 2002) to a final resolution of 2.56 Å (gold‐standard FSC 0.143).
For PfFNT‐MMV007839, 6,808,391 particles were initially selected from autopicking in cryoSPARC (Punjani et al, 2017). After several rounds of 2D classifications, 492,146 particles were selected for three additional rounds of 3D heterogeneous refinement. The resulting 141,224 particles were chosen for further processing with non‐uniform and local CTF refinement (Zhang, 2016). A soft mask covering the PfFNT protein area was used for the final local focused refinement, resulting in a 3.04 Å global resolution map based on the gold‐standard Fourier shell correlation (FSC 0.143). The cryo‐EM map was then improved using the RESOLVE density modification program (Terwilliger et al, 2020) implemented in PHENIX (Adams et al, 2002) to a final resolution of 2.78 Å (gold‐standard FSC 0.143).
Model building and refinement
Model building of apo‐PfFNT was based on the 2.86 Å cryo‐EM map. The homology modeling structure of PfFNT generated by the SWISS‐MODEL server (Waterhouse et al, 2018) based on the atomic coordinates of the HSC transporter (Czyzewski & Wang, 2012) (pdb id: 3TDP) was fit into the density map using Chimera (Pettersen et al, 2004). The subsequent model rebuilding was performed using Coot (Emsley & Cowtan, 2004). Structure refinements were performed using the phenix.real_space_refine program (Afonine et al, 2018) from the PHENIX suite (Adams et al, 2002). The final atomic model was evaluated using MolProbity (Chen et al, 2010). The statistics associated with data collection, 3D reconstruction, and model refinement are included in Table EV1.
The PfFNT‐MMV007839 structural model was built based on the apo‐PfFNT structure. A 3D conformer of the inhibitor MMV007839 (Pub Chem SID:89270032) was processed using phenix.elbow implemented in PHENIX (Adams et al, 2002). Structural refinements were done using the same approach as above (Table EV1).
Stopped‐flow light scattering assay
The purified wild‐type PfFNT and mutant K35A, F90A, F94A, K177A, and H230A proteins were reconstituted into liposomes made of E. coli polar lipid and egg yolk phosphatidylcholine (Avanti Polar Lipids) in a molar ratio of 3:1. In brief, each protein was mixed with unilamellar liposomes in 20 mM HEPES‐NaOH (pH 7.0) and 0.2% DDM at a protein‐to‐lipid ratio of 1:50 (w:w). The liposome–protein mixture was then incubated for 1 h at room temperature under gentle agitation. Subsequently, this mixture was diluted stepwise three times within 45 min. The final concentration of DDM should be below the critical micelle concentration of ~0.008%. The detergent molecules were then removed through extensively dialysis against 20 mM HEPES‐NaOH (pH 7.0) buffer. The protein samples were completely incorporated into liposomes as judged by 15% SDS–PAGE stained with Coomassie Blue. Control liposomes were prepared using the same procedure without the addition of protein. Before measurement, the control liposomes and various proteoliposomes were extruded through a 200‐nm pore‐size NanoSizer MINI Liposome Extruder (T&T Scientific).
The procedures of the light scattering experiments were similar to those for the formate transporter FocA (Wang et al, 2009) and glycerol‐conducting channel GlpF (Fu et al, 2000). To measure the lactate permeability and identify key residues involved in lactic acid transport, the proteoliposome samples were rapidly mixed with 100 mM sodium lactate and 20 mM HEPES‐NaOH (pH 6.0). The change in vesicle size was detected by recording the light scattering signal at 450 nm wavelength. The experiments were performed at 25°C on a stopped‐flow apparatus (Hi‐Tech Scientific) connected to a spectrofluorimeter (LS55; PerkinElmer). The change in light scattering was fitted by two exponentials using the equation Y = [a(1 ‐ e‐kt) ‐ b](e‐µt) + c. In this equation, a, b, and c are constants, whereas t and Y represent time and signal of light scattering. The first time constant (k) corresponds to the rapid water efflux caused by osmosis. The second time constant (µ) corresponds to the relative rates of reswelling due to the influx of lactate. The data were analyzed using Origin (OriginLab Corporation).
Author contributions
ML, C‐CS, and EWY designed experiments. ML made the construct. ML and C‐CS expressed and purified the PfFNT membrane protein. ML and C‐CS performed cryo‐EM and light scattering experiments. EWY wrote the manuscript with input from ML, C‐CS, and JWK.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Review Process File
Acknowledgements
We thank Dr. Philip A. Klenotic for proofreading the manuscript. This work was supported by NIH Grants R01AI145069 (E.W.Y.), R01AI130131 (J.W.K.), and U19AI129326 (J.W.K.). We are grateful to the Cryo‐Electron Microscopy Core at the CWRU School of Medicine and Dr. Kunpeng Li for access to the sample preparation and Cryo‐EM instrumentation.
EMBO Reports (2021) 22: e51628
Data availability
The cryo‐EM maps of the apo‐PfFNT and PfFNT‐MMV007839 were deposited in the EMDB under ID codes EMD‐21354 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD‐21354) and EMD‐21355 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD‐21355), respectively. Atomic coordinates have been deposited in the PDB with accession codes 6VQQ (https://www.rcsb.org/structure/6VQQ) for apo‐PfFNT and 6VQR (https://www.rcsb.org/structure/6VQR) for PfFNT‐MMV007839. Raw cryo‐EM data are available on request.
References
- Adams PD, Grosse‐Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58: 1948–1954 [DOI] [PubMed] [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader A, Beitz E (2020) Transmembrane facilitation of lactate/H(+) instead of lactic acid is not a question of semantics but of cell viability. Membranes 10: 236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco B, Leroy D, Fidock DA (2017) Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat Med 23: 917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzewski BK, Wang DN (2012) Identification and characterization of a bacterial hydrosulphide ion channel. Nature 483: 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Erler H, Ren B, Gupta N, Beitz E (2018) The intracellular parasite Toxoplasma gondii harbors three druggable FNT‐type formate and l‐lactate transporters in the plasma membrane. J Biol Chem 293: 17622–17630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu DX, Libson A, Miercke LJW, Weitzman C, Nollert P, Krucinski J, Stroud RM (2000) Structure of a glycerol‐conducting channel and the basis for its selectivity. Science 290: 481–486 [DOI] [PubMed] [Google Scholar]
- Golldack A, Henke B, Bergmann B, Wiechert M, Erler H, Blancke Soares A, Spielmann T, Beitz E (2017) Substrate‐analogous inhibitors exert antimalarial action by targeting the Plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog 13: e1006172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapuarachchi SV, Cobbold SA, Shafik SH, Dennis AS, McConville MJ, Martin RE, Kirk K, Lehane AM (2017) The malaria parasite's lactate transporter PfFNT Is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog 13: e1006180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter F, Arnold P, Höger B, Petersen LM, Beitz E (2019) Formate‐nitrite transporters carrying nonprotonatable amide amino acids instead of a central histidine maintain pH‐dependent transport. J Biol Chem 294: 623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joet T, Eckstein‐Ludwig U, Morin C, Krishna S (2003) Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci USA 100: 7476–7479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn Y, Rohrbach P, Lanzer M (2007) Quantitative pH measurements in Plasmodium falciparum‐infected erythrocytes using pHluorin. Cell Microbiol 9: 1004–1013 [DOI] [PubMed] [Google Scholar]
- Lu W, Du J, Wacker T, Gerbig‐Smentek E, Andrade SL, Einsle O (2011) pH‐dependent gating in a FocA formate channel. Science 332: 352–354 [DOI] [PubMed] [Google Scholar]
- Lu W, Du J, Schwarzer NJ, Gerbig‐Smentek E, Einsle O, Andrade SL (2012a) The formate channel FocA exports the products of mixed‐acid fermentation. Proc Natl Acad Sci USA 109: 13254–13259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Schwarzer NJ, Du J, Gerbig‐Smentek E, Andrade SL, Einsle O (2012b) Structural and functional characterization of the nitrite channel NirC from Salmonella typhimurium. Proc Natl Acad Sci USA 109: 18395–18400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae JI, Dixon MW, Dearnley MK, Chua HH, Chambers JM, Kenny S, Bottova I, Tilley L, McConville MJ (2013) Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum . BMC Biol 11: 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti RV, Lehane AM, Shafik SH, Winterberg M, Martin RE, Kirk K (2015) A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum . Nat Commun 6: 6721 [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- McKee RW, Ormsbee RA, Anfinsen CB, Geiman QM, Ball EG (1946) Studies on malarial parasites: Vi. the chemistry and metabolism of normal and parasitized (P. Knowlesi) monkey blood. J Exp Med 84: 569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP (1996) HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph Model 14: 354–360 [DOI] [PubMed] [Google Scholar]
- Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TN, Willis P (2013) The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8: e62906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Ludtke SJ, Read RJ, Adams PD, Afonine PV (2020) Improvement of cryo‐EM maps by density modification. Nat Methods 17: 923–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Voorhis WC, Adams JH, Adelfio R, Ahyong V, Akabas MH, Alano P, Alday A, Aleman Resto Y, Alsibaee A, Alzualde A et al (2016) Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog 12: e1005763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waight AB, Love J, Wang DN (2010) Structure and mechanism of a pentameric formate channel. Nat Struct Mol Biol 17: 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Wang J, Cheng C, Huang W, Lu P, Xu YN, Wang P, Yan N, Shi Y (2009) Structure of the formate transporter FocA reveals a pentameric aquaporin‐like channel. Nature 462: 467–472 [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2018) World Malaria Report 2018
- WHO (2019) World Malaria Report 2019
- Wiechert M, Beitz E (2017) Mechanism of formate‐nitrite transporters by dielectric shift of substrate acidity. EMBO J 36: 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechert M, Erler H, Golldack A, Beitz E (2017) A widened substrate selectivity filter of eukaryotic formate‐nitrite transporters enables high‐level lactate conductance. Febs J 284: 2663–2673 [DOI] [PubMed] [Google Scholar]
- Woodrow CJ, Penny JI, Krishna S (1999) Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J Biol Chem 274: 7272–7277 [DOI] [PubMed] [Google Scholar]
- Wu B, Rambow J, Bock S, Holm‐Bertelsen J, Wiechert M, Soares AB, Spielmann T, Beitz E (2015) Identity of a Plasmodium lactate/H(+) symporter structurally unrelated to human transporters. Nat Commun 6: 6284 [DOI] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: Real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Review Process File
Data Availability Statement
The cryo‐EM maps of the apo‐PfFNT and PfFNT‐MMV007839 were deposited in the EMDB under ID codes EMD‐21354 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD‐21354) and EMD‐21355 (https://www.ebi.ac.uk/pdbe/entry/emdb/EMD‐21355), respectively. Atomic coordinates have been deposited in the PDB with accession codes 6VQQ (https://www.rcsb.org/structure/6VQQ) for apo‐PfFNT and 6VQR (https://www.rcsb.org/structure/6VQR) for PfFNT‐MMV007839. Raw cryo‐EM data are available on request.