

Abstract
Parkin promotes cell survival by removing damaged mitochondria via mitophagy. However, although some studies have suggested that Parkin induces cell death, the regulatory mechanism underlying the dual role of Parkin remains unknown. Herein, we report that mitochondrial ubiquitin ligase (MITOL/MARCH5) regulates Parkin‐mediated cell death through the FKBP38‐dependent dynamic translocation from the mitochondria to the ER during mitophagy. Mechanistically, MITOL mediates ubiquitination of Parkin at lysine 220 residue, which promotes its proteasomal degradation, and thereby fine‐tunes mitophagy by controlling the quantity of Parkin. Deletion of MITOL leads to accumulation of the phosphorylated active form of Parkin in the ER, resulting in FKBP38 degradation and enhanced cell death. Thus, we have shown that MITOL blocks Parkin‐induced cell death, at least partially, by protecting FKBP38 from Parkin. Our findings unveil the regulation of the dual function of Parkin and provide a novel perspective on the pathogenesis of PD.
Keywords: mitochondria, mitophagy, Parkin, MITOL/MARCH5, E3 ubiquitin ligase
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
This study reveals that MITOL is translocated from mitochondria to the ER during the late phase of mitophagy. At the ER, MITOL degrades Parkin, thereby stabilizing FKBP38, and thus promoting cell survival.
Introduction
Mutations in PARK2 (Parkin), a ubiquitin E3 ligase, and PARK6 (PINK1), a ubiquitin kinase, are associated with the familial form of Parkinson’s disease (PD; Kitada et al, 1998; Shimura et al, 2000). Recent studies have revealed that recruitment of Parkin from the cytosol to depolarized mitochondria is mediated by PINK1 to remove damaged mitochondria via autophagy (Narendra et al, 2008; Geisler et al, 2010; McLelland et al, 2014). PINK1 is constitutively degraded through the N‐end rule pathway (Yamano & Youle, 2013). However, when mitochondrial depolarization occurs, PINK1 is stabilized on the outer mitochondrial membrane, where it phosphorylates ubiquitin and Parkin; therefore, PINK1 is known as a parkin activator and recruiter (Geisler et al, 2010; Matsuda et al, 2010; Narendra et al, 2010; Vives‐Bauza et al, 2010; Kondapalli et al, 2012; Shiba‐Fukushima et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). Hence, it is widely believed that the PINK1–Parkin pathway has a cell‐protective function via mitophagy, and impaired mitophagy due to loss of function of PINK1 or Parkin is involved in the pathogenesis of PD.
On the other hand, widespread quantitative analysis of the ubiquitylome has led to the discovery of novel substrates for Parkin, such as cell survival‐related proteins, including FK506‐binding protein 38 (FKBP38; Sarraf et al, 2013; Rose et al, 2016). Some studies have indicated that Parkin induces cell death during mitophagy (Carroll et al, 2014; Zhang et al, 2014; Akabane et al, 2016), suggesting a dual role of Parkin in both cell survival and death signaling. However, the regulatory mechanism underlying the dual function of Parkin is unknown. In addition, although many studies have revealed the regulatory role of both the subcellular localization and ligase activity of Parkin in mitophagy, the quantitative aspects of Parkin regulation are still unknown.
FKBP38 is a membrane‐bound chaperon protein that mainly resides on the outer membrane of mitochondria. It is a tail‐anchored protein with a transmembrane domain at the carboxyl terminal. The known functions of FKBP38 include recruiting the anti‐apoptotic proteins Bcl‐2 and Bcl‐xL into the mitochondria to inhibit apoptosis (Shirane & Nakayama, 2003). Interestingly, it has been reported that FKBP38 escapes mitochondrial degradation by translocating from the mitochondria to the ER during mitophagy, thereby inhibiting apoptosis (Saita et al, 2013). Since FKBP38 can inhibit apoptosis even in the absence of mitochondria, it is considered to contribute to the inhibition of mitochondria‐dependent and ER‐dependent apoptosis.
Previously, we identified MITOL/MARCH5 and found that it specifically polyubiquitinates and degrades misfolded mutant SOD1 and expanded polyglutamine in a proteasome‐dependent manner to protect the cells (Yonashiro et al, 2009; Sugiura et al, 2011). With regard to its relationship with mitophagy, MITOL has been reported to influence the removal of mitochondria by controlling FUNDC1 in response to hypoxic stress (Liu et al, 2012). However, the function of MITOL in Parkin‐mediated mitophagy cannot be fully excluded. In this study, we found that MITOL not only binds to Parkin in a mitophagy induction‐dependent manner but also mediates the addition of K48‐linked ubiquitin chain to its lysine 220 residue, and thereby degrading Parkin, particularly, its phosphorylated active form. Moreover, we reported that MITOL translocates to the ER with FKBP38 in an FKBP38‐dependent manner and degrades Parkin to protect FKBP38 from Parkin for cell survival during the late phase of mitophagy. Our findings unveil the regulation of the dual role of Parkin and provide a novel perspective on the pathogenesis of Parkinson’s disease.
Results
MITOL mediates ubiquitination of Parkin in Parkin‐dependent mitophagy
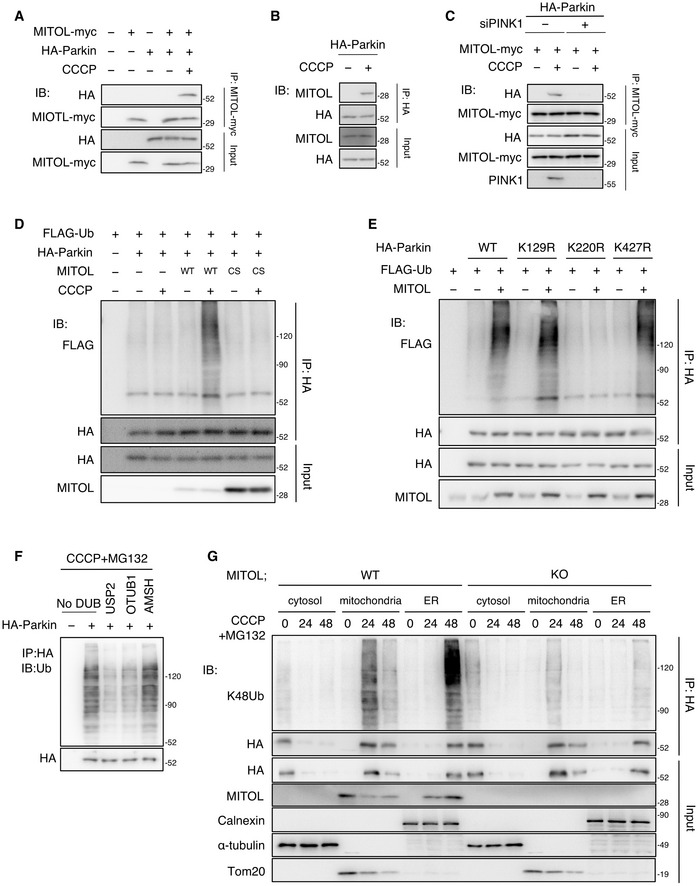
It is well known that Parkin is selectively recruited to mitochondria with low membrane potential that is induced by an uncoupler, carbonyl cyanide m‐chlorophenylhydrazone (CCCP; Narendra et al, 2008; Matsuda et al, 2010; Vives‐Bauza et al, 2010). To investigate the physiological aspects of the interaction between MITOL and Parkin, a co‐immunoprecipitation assay was first performed using HeLa cells with or without stable expression of HA‐Parkin, which demonstrated that Parkin was co‐immunoprecipitated with MITOL after CCCP treatment (Fig 1A). The interaction between endogenous MITOL and Parkin was also observed in a CCCP‐dependent manner (Figs 1, EV1 and 1, EV1). To determine which domain of MITOL is necessary for interaction with Parkin, three glutathione S‐transferase (GST)‐fused fragments of MITOL were generated, which are considered to be localized on the cytosolic surface of the outer mitochondrial membrane (Fig EV1B). In the GST pull‐down assay, the GST fragment containing the C‐terminal of MITOL was able to pull Parkin down from the lysate of the CCCP‐treated HeLa cells that stably expressed HA‐Parkin (Fig EV1B). In addition, in an immunoprecipitation experiment, MITOL mutants lacking the C‐terminal domain (ΔC) did not bind to Parkin after CCCP treatment (Fig EV1C). These results demonstrated that MITOL interacts with Parkin via its C‐terminal domain. To determine which domain of Parkin interacts with MITOL, GST‐fused Parkin fragments were generated (Fig EV1D). In the GST pull‐down assay, the GST‐fused Parkin RING2 domain was able to pull MITOL down (Fig EV1D). PINK1 is essential for the translocation of Parkin to depolarized mitochondria (Lazarou et al, 2012; Okatsu et al, 2012; Okatsu et al, 2013). In the PINK1 knockdown cells, the binding between MITOL and Parkin was decreased due to reduced recruitment of Parkin to the mitochondria (Fig 1C). Additionally, binding was reduced in the case of the pathogenic mutant Parkin K211N (Fig EV1E; Geisler et al, 2010; Matsuda et al, 2010), which is severely compromised the mitochondrial localization 8 h after CCCP treatment (Fig EV1F), indicating that Parkin is required for the mitochondrial recruitment to interact with MITOL. Thus, we conclude that MITOL binds to Parkin in a mitochondrial depolarization‐dependent manner and that the binding domains responsible for the interaction are present in the C‐terminal of MITOL and the RING2 domain of Parkin, respectively. To clarify the regulatory mechanism of Parkin by MITOL in detail, the ubiquitination of Parkin by MITOL was characterized. Upon CCCP treatment, overexpressed wild‐type (WT) MITOL in the HeLa cells stably expressing HA‐Parkin elevated the levels of ubiquitinated Parkin, compared with those of the control vectors or the MITOL CS (C65S, C68S) mutant lacking the E3 ligase activity (Fig 1D). On the other hand, as Parkin is also a ubiquitin ligase that is activated in a CCCP‐dependent manner, we tried to detect the ubiquitination of MITOL upon mitochondrial depolarization using immunoprecipitation assay. Interestingly, the Parkin‐dependent ubiquitination of MITOL was observed in the early phase of mitophagy consistent with a previous report (Koyano et al, 2019a); however, it could not be detected in the late stage (Fig EV2A). Next, to ascertain whether Parkin translocation to the mitochondria requires the ubiquitination of Parkin, we examined the ubiquitination of Parkin K211N mutant and found that MITOL‐mediated ubiquitination did not occur 8 h after CCCP treatment (Fig EV2B), suggesting that Parkin is ubiquitinated by MITOL after its translocation to the mitochondria. We used UbPred, an in silico tool for ubiquitination analysis (Radivojac et al, 2010), to identify the lysine residue(s) essential for the ubiquitination of Parkin by MITOL and predicted several lysine residues that were potential ubiquitin linkage sites. Based on this, three site‐specific KR mutants were prepared in which the candidate lysine residues of Parkin were mutated to arginine. Despite the overexpression of MITOL, the ubiquitination level of the K220R mutant was much lower than those of WT Parkin and other Parkin KR mutants (Fig 1E). The Lys48‐linked polyubiquitin chain is mainly responsible for the degradation of ubiquitinated proteins by the 26S proteasome, whereas Lys63‐linked polyubiquitination alters the activation and localization of substrates as well as the regulation of the binding proteins (Pickart & Fushman, 2004; Mukhopadhyay & Riezman, 2007). To determine the type of polyubiquitin chain, single KR ubiquitin mutants with a single point mutation (lysine to arginine) were generated. In the WT ubiquitin or the K63R ubiquitin mutant, Parkin ubiquitination was increased following the MITOL overexpression in a CCCP‐dependent manner, but it was not altered in the K48R ubiquitin mutant (Fig EV2C). Previously, to identify the type of polyubiquitin chain, we prepared a variety of ubiquitin mutants with a single intact K residue. Consistent with the results obtained using the KR mutants, the ubiquitination of Parkin by MITOL was confirmed in the R48K ubiquitin mutant‐transfected HeLa cells (Fig EV2D). Furthermore, we performed ubiquitin chain restriction assay (UbiCRest) to determine Parkin‐attached endogenous ubiquitin (Hospenthal et al, 2015). Parkin‐attached polyubiquitin chains were digested in the presence of the universal deubiquitinating enzyme USP2 and the K48‐polyubiquitin‐specific deubiquitinating enzyme OTUB1, whereas the K63‐polyubiquitin‐specific deubiquitinating enzyme AMSH did not alter the levels of the polyubiquitin chains in Parkin (Fig 1F). This indicates that K48 polyubiquitin linkages are attached to Parkin. We performed ubiquitination assay using K48‐linked polyubiquitin chain‐specific antibody and found that the K48‐linked ubiquitination in Parkin mediated by CCCP treatment was reduced in the mitochondrial and ER fraction due to MITOL deficiency (Fig 1G). Therefore, it was concluded that MITOL mediates the K48‐linked polyubiquitination of Parkin at the K220 residue.
Figure 1. MITOL mediated the K48‐linked polyubiquitination of Parkin at K220.
-
AMITOL interacts with Parkin under CCCP treatment. HeLa cells or HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 4 h. Lysates of cells were subjected to an IP assay with anti‐myc antibody and IB assay with the indicated antibodies.
-
BInteraction of endogenous MITOL and Parkin in HeLa cells stably expressing HA‐Parkin. HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for 12 h and subjected to an IP‐IB assay with the indicated antibodies.
-
CPINK1 knockdown decreases interaction between MITOL and Parkin. HeLa cells stably expressing HA‐Parkin were transfected with PINK1 siRNA or control siRNA and treated with DMSO or CCCP (10 μM) for 4 h. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP assay with anti‐myc antibody and IB assay with the indicated antibodies.
-
DMITOL overexpression enhances the ubiquitination of Parkin. HeLa cells or HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 8 h. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies. WT, wild‐type; CS, MITOL C65/67S mutant lacking ubiquitin ligase activity.
-
EMITOL fails to ubiquitinate the Parkin K220R mutant. HeLa cells were transfected with the indicated vectors and treated with CCCP (10 μM) for 8 h. MG132 (30 μM) was added 3 h after CCCP treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies and compared ubiquitination levels of WT, K129R, K220R, and K427R HA‐Parkin.
-
FParkin is ubiquitinated by K48‐polyubiquitin in mitophagy. HeLa cells stably expressing HA‐Parkin were treated with CCCP (10 μM) for 48 h together with MG132 (30 μM) for the final 45 h. Parkin was immunoprecipitated from cell lysates and then performed ubiquitin restriction assay with AMSH, OTUB1, and USP2 using UbiCRest kit. Lysates of cells were subjected to an IB assay with the indicated antibodies. DUBs, deubiquitinating enzyme.
-
GEndogenous MITOL enhances the K48‐polyubiquitin chains of Parkin. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were treated with CCCP (10 μM) for indicated time. MG132 (30 μM) was added 3 h after CCCP treatment. Lysates of cells were fractionated into cytosolic, mitochondrial, and ER fractions, and then subjected to an IP‐IB assay with the indicated antibodies. K48‐polyubiquitin chains were immunoblotted with K48‐specific ubiquitin antibody.
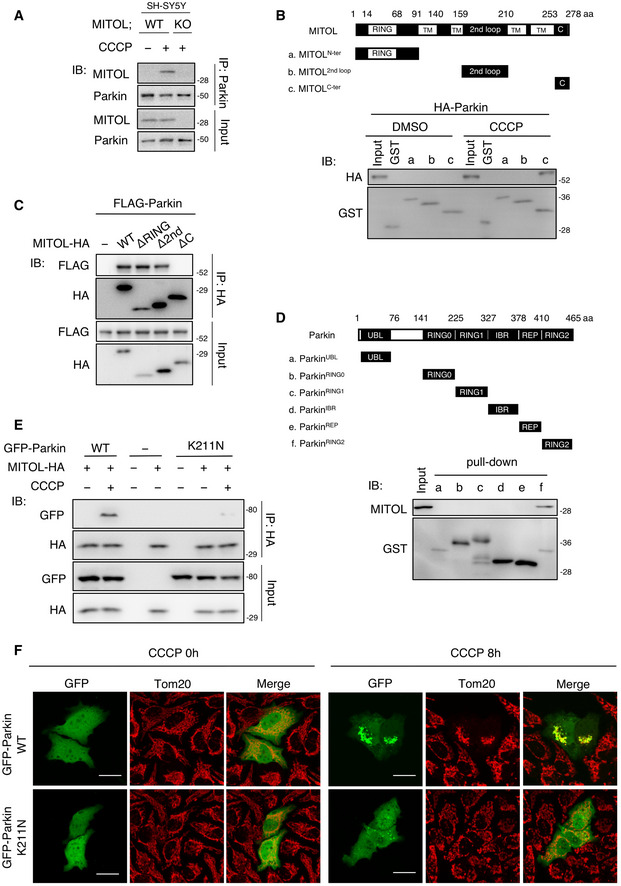
Figure EV1. The C‐terminal region of MITOL interacts with the RING2 domain of Parkin, related to Fig 1 .
-
AInteraction of endogenous MITOL and Parkin in SH‐SY5Y cells. SH‐SY5Y cells were treated with DMSO or CCCP (10 μM) for 12 h and subjected to an IP‐IB assay with the indicated antibodies.
-
BParkin interacts with C‐terminal region of MITOL. The schematic diagram shows the three truncated MITOL fragments that were fused to GST (Top). HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for 4 h. Lysates of cells were performed GST pull‐down assay and then subjected to an IB assay with anti‐HA or anti‐GST antibodies (Bottom). (a) N‐terminal RING domain (1–90 aa); (b) second loop domain (160–209 aa); (c) C‐terminal domain (254–278 aa); TM, transmembrane.
-
CThe C‐terminal deleted MITOL mutant failed to interact with Parkin. HeLa cells were transfected with the indicated vectors and treated with CCCP (10 μM) for 4 h. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies. ΔRING, 14–68 aa deletion. Δ2nd, 160–209 aa deletion. ΔC, 254–278 aa deletion.
-
DMITOL interacts with RING2 domain of Parkin. The schematic diagram shows the six truncated Parkin fragments that were fused to GST (Top). Mitochondrial fractions were isolated from HeLa cells and performed a GST pull‐down assay. IB assay was subjected with anti‐MITOL or anti‐GST antibodies (Bottom). (a) UBL domain (1–76 aa); (b) RING0 domain (141–224 aa); (c) RING1 domain (225–326 aa); (d) IBR domain (327–377 aa); (e) REP domain (378–409 aa); (f) RING2 domain (410–465 aa).
-
EParkin K211N fails to interact with MITOL. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 4 h. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies.
-
FParkin K211N mutant fails to translocate to the mitochondria in CCCP‐treated cells. HeLa cells were transfected with the indicated vectors and treated with CCCP (10 μM) for 8 h. Cells were immunostained with indicated antibody. Scale bar, 10 μm.
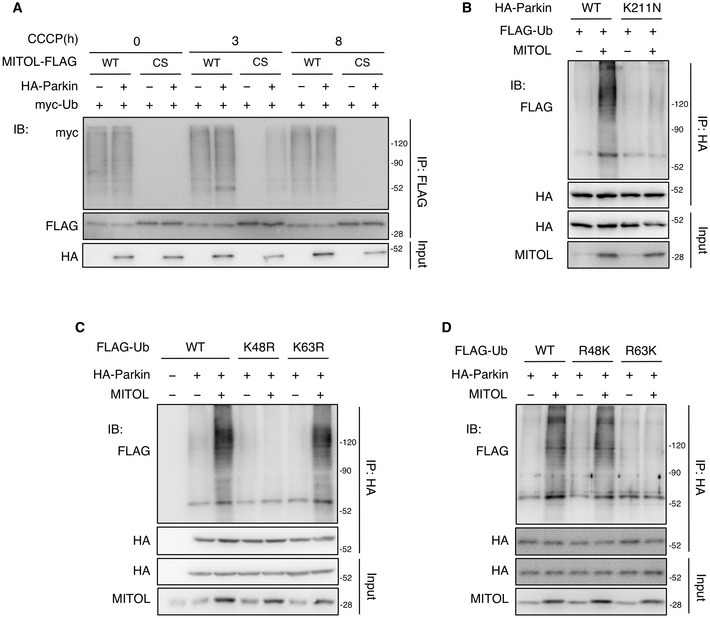
Figure EV2. MITOL mediated the K48‐linked polyubiquitination of Parkin in mitophagy, related to Fig 1 .
-
AUbiquitylation of MITOL occurs in the early phase but not the late phase of mitophagy. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for indicated times. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies. WT, wild‐type; CS, MITOL C65/67S mutant lacking ubiquitin ligase activity.
-
BMITOL fails to ubiquitinate the Parkin K211N mutant. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 8 h. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies and compared ubiquitination levels of WT and K211N HA‐Parkin. Parkin K211N mutant; impaired mitochondrial translocation.
-
C, DMITOL mediates K48‐linked polyubiquitination of Parkin. HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 8 h. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies, and compared the effect of various KR FLAG‐ubiquitin (Ub) mutants (C) or RK FLAG‐Ub mutants (D) on MITOL‐mediated Parkin ubiquitination. KR ubiquitin mutant, turn single lysine to arginine (K48R or K63R); RK ubiquitin mutant, turn all lysine to arginine and then put back single lysine (R48K or R63K).
MITOL regulates Parkin during mitophagy
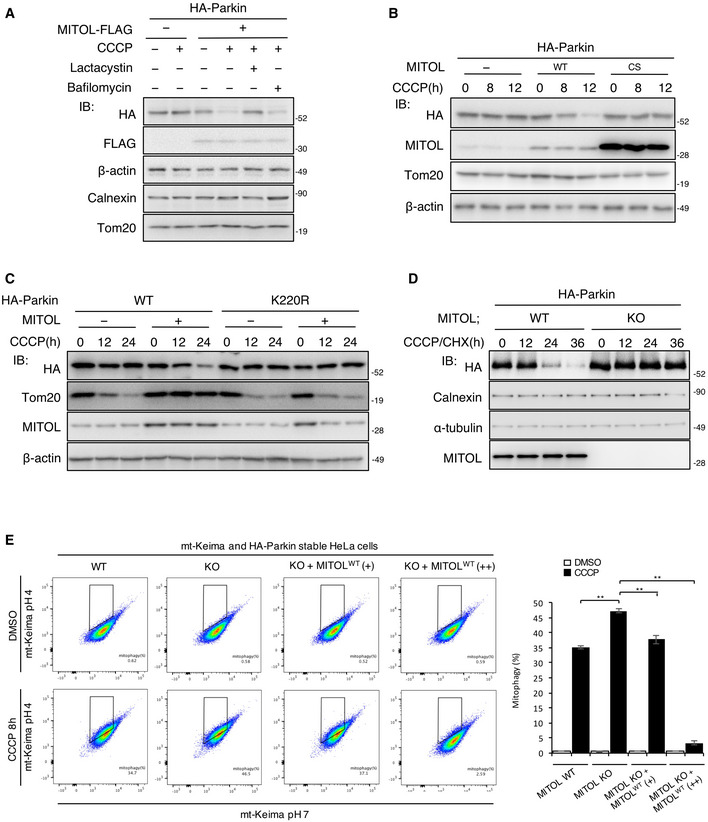
As MITOL promotes the addition of K48‐linked polyubiquitin to Parkin, we assumed that it induces proteasomal degradation of Parkin. To examine whether the degradation of Parkin depends on proteasomes or autophagy, we treated the cells with lactacystin, a proteasome‐specific inhibitor, or bafilomycin A1, an autophagic flux inhibitor. Lactacystin strongly inhibited the degradation of Parkin in the CCCP‐treated cells overexpressing MITOL (Fig 2A). Parkin degradation was also inhibited in the MITOL CS mutant and Parkin K220R mutant (Fig 2B and C). In contrast, MITOL overexpression downregulated the Parkin protein level in the mitochondrial fraction post CCCP treatment (Fig EV3A). Furthermore, cycloheximide (CHX) treatment reduced Parkin in CCCP‐treated MITOL WT but not MITOL KO cells (Figs 2D and EV3B). As it is known that Parkin nonspecifically ubiquitinates mitochondrial proteins during mitophagy (Koyano et al, 2019b), we confirmed the specificity of MITOL for its substrate using Mito‐GFP upon depolarization. While Parkin ubiquitinated Mito‐GFP in a mitophagy‐dependent manner, MITOL did not ubiquitinate it (Fig EV3C), indicating its substrate specificity during mitophagy. These findings suggest that MITOL ubiquitinates and subsequently degrades Parkin in a proteasome‐dependent manner. To understand whether the ubiquitination of Parkin by MITOL has a functional effect on mitophagy, we performed quantitative analysis of mitophagy using the mitochondria‐localized pH‐dependent fluorescent protein mt‐Keima (Katayama et al, 2011; Yamashita et al, 2016; Igarashi et al, 2020), which exhibits a short excitation in normal conditions (pH 8.0) and shifted to longer excitation wavelengths in mitophagy processes because of the acidic environment of the lysosome (pH 4.5). Flow cytometric analysis indicated that MITOL deletion significantly enhanced mitophagy, which was restored to normal levels by a low expression of MITOL. On the other hand, a high expression of MITOL strongly suppressed mitophagy through the intense degradation of Parkin by MITOL (Figs 2E and EV3E). Furthermore, MITOL overexpression failed to suppress the mitophagy induced by the Parkin K220R mutant (Fig EV3F), which exhibits normal translocation to mitochondria in mitophagy (Fig EV3D). Taken together, endogenous MITOL fine‐tunes mitophagy by controlling the quantity of Parkin on mitochondria.
Figure 2. MITOL regulates mitophagy through the degradation of Parkin.
-
AMITOL degrades Parkin through the ubiquitin–proteasome system. HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 8 h alone or either with bafilomycin A1 (10 μM) or lactacystin (10 μM) for the final 5 h. Lysates of cells were subjected to an IB assay with the indicated antibodies.
-
BOverexpression of MITOL CS mutant fails to degrade Parkin. HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for the indicated time. Lysates of cells were subjected to an IB assay with the indicated antibodies. MITOL CS mutant; lacking ubiquitin ligase activity.
-
CParkin K220R mutant is not regulated by MITOL in mitophagy. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for the indicated time. Lysates of cells were subjected to an IB assay with the indicated antibodies.
-
DEndogenous MITOL attenuates Parkin in both CHX‐ and CCCP‐treated conditions. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were treated with CCCP (10 μM) as indicated times. CHX (30 μM) was added 5 h after CCCP treatment. Lysates of cells were subjected to an IB assay with the indicated antibodies.
-
EMITOL regulates mitophagy. MITOL WT HeLa cells, MITOL KO HeLa cells, and KO HeLa cells transfected with MITOL WT plasmids 0.5 μg (+) and 5 μg (++) stably expressing HA‐Parkin and mt‐Keima were treated with CCCP (10 μM) for 8 h. Then, mKeima was measured at 488 (pH 7) and 561 (pH 4) nm lasers using Flow Cytometer. Percentages of mitophagy were calculated from 30,000 cells in each independent experiment. Data represent the mean ± SD of three independent experiments. For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01.
Figure EV3. MITOL degrades Parkin and slightly effect for mitophagy, related to Fig 2 .
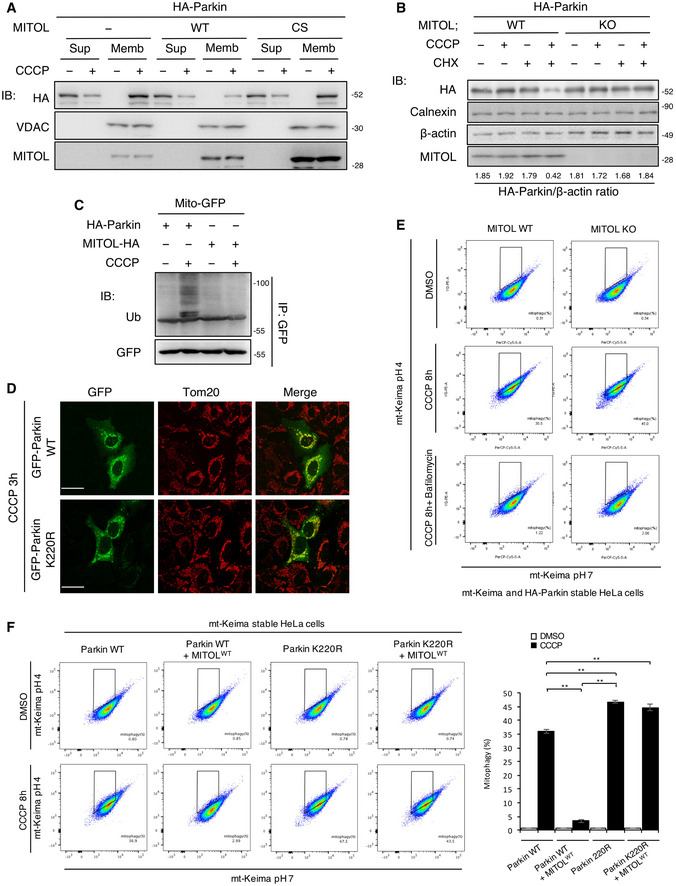
-
AMITOL overexpression degrades Parkin on mitochondrial membrane. HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 12 h. Lysates of cells were fractionated into mitochondria‐rich membrane fraction (Memb) and supernatant (Sup), and then subjected to an IB assay with the indicated antibodies. Loading was adjusted for approximately equal concentrations of VDAC in the membrane fraction (Memb).
-
BEndogenous MITOL attenuates mitophagy in CHX‐treated cells. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for 30 h. CHX (30 μM) was added 5 h after CCCP treatment. Lysates of cells were subjected to an IB assay with the indicated antibodies.
-
CMITOL has the specificity for the substrate. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 8 h. MG132 (30 μM) was added 3 h after each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies and compared ubiquitination levels of HA‐Parkin and MITOL‐HA to Mito‐GFP.
-
DParkin K220R mutant normally translocated to the mitochondria in CCCP‐treated cells. HeLa cells were transfected with indicated vectors and treated with CCCP (10 μM) for 3 h. Cells were immunostained with indicated antibody. Scale bar, 10 μm.
-
EMt‐Keima signal is responding to autophagy process. MITOL WT HeLa cells and MITOL KO HeLa cells expressing HA‐Parkin and mt‐Keima were treated with DMSO or CCCP (10 μM) for 8 h alone or either with bafilomycin A1 (10 μM). Then, mKeima was measured at 488 (pH 7) and 561 (pH 4) nm lasers using Flow Cytometer. Percentages of mitophagy were calculated from 30,000 cells.
-
FParkin K220R mutant induces mitophagy even in the overexpression of MITOL. HeLa cells stably expressing mt‐Keima were transfected with indicated vectors and treated with CCCP (10 μM) for 8 h. Then, mKeima was measured at 488 (pH 7) and 561 (pH 4) nm lasers using Flow Cytometer. Percentages of mitophagy were calculated from 30,000 cells in each independent experiment. Data represent the mean ± SD of three independent experiments. For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01.
MITOL translocates to the ER via FKBP38 in the later phase of mitophagy
Although MITOL is considered to localize in the mitochondria, the MITOL protein level was not reduced during the late phase of mitophagy when mitochondria were removed (Fig 3A). A previous study showed that the mitochondria‐localized protein FKBP38 translocates from the mitochondria to the ER to escape the degradation together with the mitochondria during mitophagy (Saita et al, 2013). Thus, it was speculated that MITOL might also exhibit similar dynamic translocation. To monitor endogenous MITOL localization during mitophagy, an EGFP‐MITOL knock‐in cell line was generated, and dynamic translocation from the mitochondria to the ER was observed in the later phase of mitophagy (Fig 3B and C). Consistently, immunostaining analysis revealed that MITOL was localized to the ER in the later phase of mitophagy, similar to FKBP38 (Fig EV4A), suggesting that MITOL translocates to the ER along with FKBP38. As expected, MITOL interacted with FKBP38 under both normal and mitochondrial depolarization conditions (Figs 3D and EV4B). As MITOL was bound to FKBP38, it was assumed that the translocation of MITOL to the ER might depend on FKBP38. Therefore, the effect of FKBP38 knockdown on MITOL localization was examined using siRNA in each organelle fraction. FKBP38 knockdown inhibited the translocation of MITOL to the ER and reduced MITOL expression under CCCP treatment conditions (Fig EV4C and D). To examine whether the ER‐localized MITOL was indeed translocated from the mitochondria and not due to its de novo synthesis in the ER, the photoconverting fluorescent tag protein Kikume Green‐Red (KikGR), which changes color from green to red following irradiation with ultraviolet rays (360–410 nm), was used. When MITOL‐KikGR‐transfected cells were exposed to ultraviolet rays before CCCP treatment, the MITOL‐KikGR synthesized before mitophagy displayed red fluorescence in the mitochondria (Fig 3E) as well as in the ER during the late phase of mitophagy, suggesting that MITOL was transported from the mitochondria to the ER during mitophagy (Fig 3E). These results show that MITOL translocates to the ER in an FKBP38‐dependent manner in the late phase of mitophagy.
Figure 3. MITOL translocates to the ER with FKBP38 in the late stage of mitophagy.
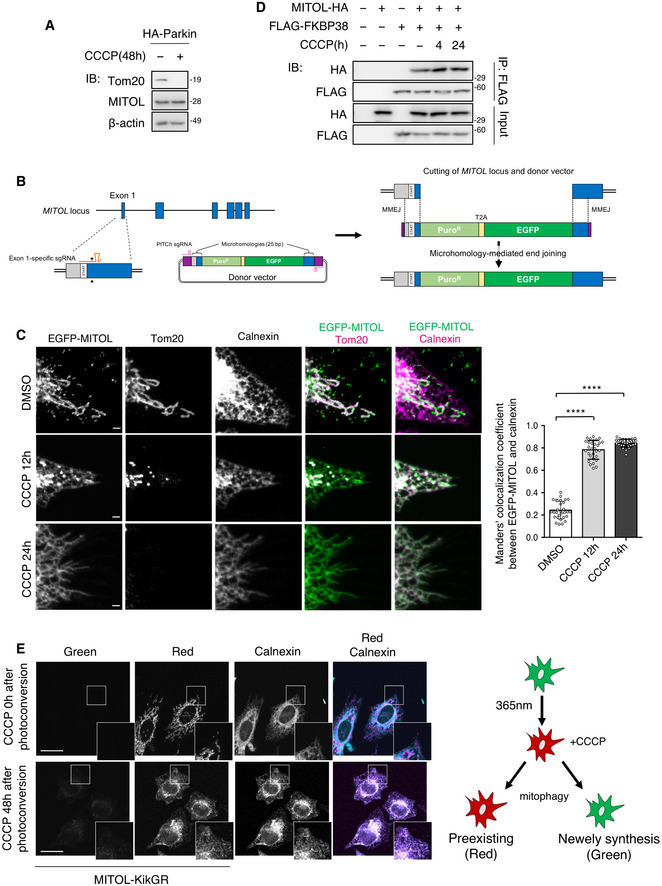
-
AMITOL was not degraded in mitophagy. HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for 48 h and subjected to an IB assay with the indicated antibodies.
-
BSchematic diagram of EGFP knock‐in for N‐terminal tagging. MITOL‐specific and PITCh‐specific sgRNAs expressed from pX330A‐MITOL/PITCh (not shown) individually target the MITOL exon 1 locus and the donor vector. This allows for both the cleavage of the genomic locus and the release of the EGFP‐containing cassette. MMEJ leads to the repair of double‐strand break via the insertion of the EGFP‐containing cassette, resulting in endogenously EGFP tagged MITOL.
-
CEndogenous MITOL translocates to the ER in later phase of mitophagy. EGFP‐MITOL knock‐in HeLa cells were transfected with HA‐Parkin and treated with DMSO or CCCP (10 μM) as indicated times. Cells were fixed, permeabilized, and subjected to immunofluorescence analysis with the indicated antibodies. Colocalization was quantified by Manders’s coefficient. Means ± SEM of more than 10 cells obtained from three independent experiments. For statistical analysis, a one‐way ANOVA with Tukey's multiple comparisons test was performed, ****P < 0.0001. Scale bar represents 1 μm.
-
DMITOL interacts with FKBP38. HeLa cells or HeLa cells stably expressing mCherry‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for indicated times. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies.
-
EER‐localized MITOL is transported from the mitochondria and is not newly synthesized protein. HeLa cells stably expressing HA‐Parkin were co‐transfected with vectors for MITOL‐KikGR and FLAG‐FKBP38. After ultraviolet light (365 nm) exposure to the whole plate, cells were incubated with or without CCCP (10 μM) for indicated time, and monitored for red and green KikGR fluorescence. Scale bar, 10 μm. Higher magnification images of the boxed regions are shown in the small panel. Right panel is schematic experimental model for KikGR in this study.
Figure EV4. MITOL translocates to the ER in an FKBP38‐dependent manner, related to Fig 3 .
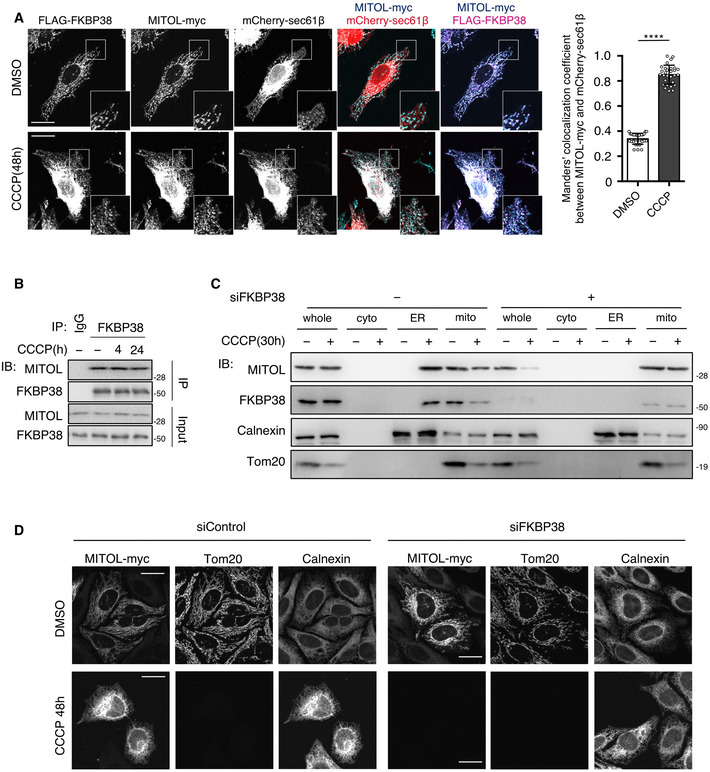
-
AMITOL translocates from the mitochondria to the ER in the late phase of mitophagy with FKBP38. HeLa cells stably expressing HA‐Parkin were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for 48 h. Cells were fixed, permeabilized, and subjected to immunofluorescence analysis with indicated antibodies. Colocalization was quantified by Manders’s coefficient. Means ± SEM of more than 10 cells obtained from three independent experiments. For statistical analysis, Student’s t‐tests were performed, ****P < 0.0001. Scale bar represents 10 μm. Higher magnification images of the boxed regions are shown in the small panel.
-
BInteraction of endogenous MITOL and FKBP38. HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for indicated times and subjected to an IP‐IB assay with the indicated antibodies.
-
C, DMITOL translocation depends on FKBP38. HeLa cells stably expressing HA‐Parkin were transfected with FKBP38 siRNA or control siRNA and treated with DMSO or CCCP (10 μM) for 30 h. Lysates of cells were fractioned into whole‐cell, cytosolic, ER, and mitochondrial fractions and then subjected to an IB assay with the indicated antibodies (C). MITOL KO HeLa cells stably expressing HA‐Parkin were transfected with MITOL‐myc and treated with DMSO or CCCP (10 μM) for 48 h. Cells were immunostained with indicated antibodies (D). Scale bar, 10 μm.
MITOL specifically degrades phosphorylated Parkin in mitophagy
Parkin and ubiquitin are phosphorylated by PINK1, which is stabilized on the outer membrane of the depolarized mitochondria. Subsequently, phosphorylated Parkin binds to phosphorylated ubiquitin to activate and induce mitophagy (Kondapalli et al, 2012; Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). However, the quantitative regulation between the active and inactive forms of Parkin in mitophagy remains largely unknown. Although endogenous MITOL mediates the K48‐linked polyubiquitination of Parkin under mitochondrial depolarization, the entire pool of Parkin did not change dramatically, suggesting the possibility that endogenous MITOL specifically recognizes the activated form of Parkin. We examined whether endogenous MITOL regulates the amount of phosphorylated Parkin and found continuous accumulation of phosphorylated Parkin in the MITOL KO cells after mitochondrial depolarization (Fig 4A). In MITOL KO cells, Parkin was bound to ubiquitin, confirming the accumulation of activated Parkin in MITOL KO (Fig 4B). To determine the significance of the translocation of MITOL to the ER, we examined the phosphorylated Parkin in the ER fractions in the later phase of mitophagy and found a dramatic increase in Ser65‐phosphorylated Parkin in the ER fraction of MITOL KO cells (Fig 4C). To examine whether the ER‐localized Parkin was synthesized de novo, we used the photoconverting fluorescent tag protein KikGR. KikGR‐Parkin exhibited red fluorescence in the ER at the late mitophagy phase, suggesting that Parkin was in the ER during this phase (Fig 4D). Parkin accumulation in the ER was observed in FKBP38‐knockdown cells (Fig EV5A), suggesting that Parkin accumulates in the ER in an FKBP38‐independent manner. Taken together, these data show that (i) endogenous MITOL also controls the degradation of Parkin, particularly phosphorylated Parkin, (ii) quantitative regulation of Parkin by endogenous MITOL affects mitophagy, and (iii) MITOL translocates to the ER in an FKBP38‐dependent manner in the late phase of mitophagy.
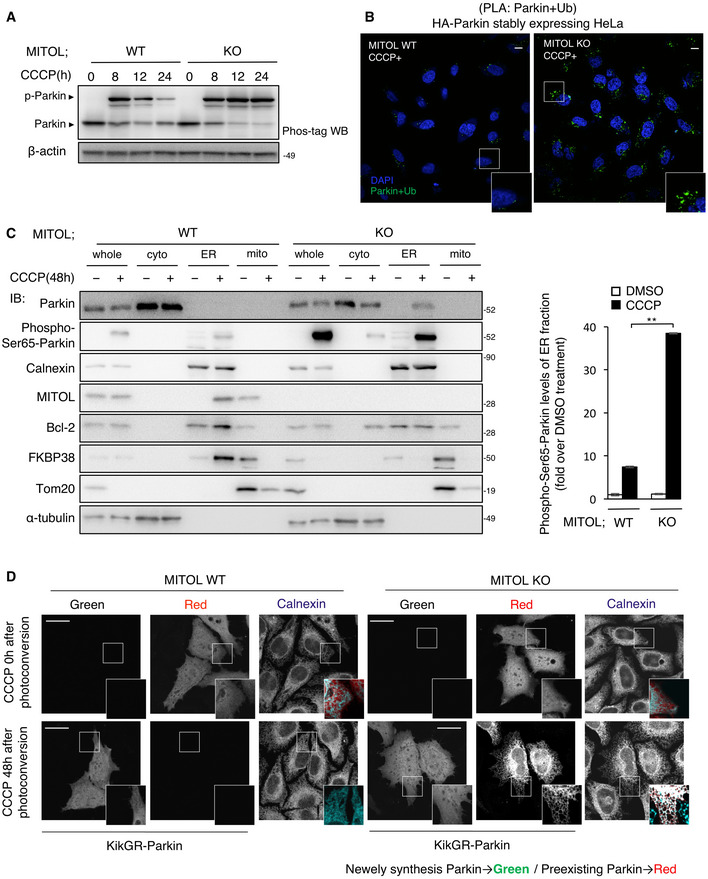
Figure 4. MITOL selectively recognizes phosphorylated form of Parkin.
-
APhosphorylated Parkin accumulates in MITOL KO cells. HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for the indicated time and subjected to IB assay with the indicated antibodies. Other cell lysates were subsequently separated on a Phos‐tag gel, followed by IB with anti‐Parkin antibodies (Phos‐tag). p‐Parkin, phosphorylated Parkin.
-
BAccumulation of activated Parkin in MITOL KO cells. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were treated with CCCP (10 μM) for 30 h and then performed Duolink in situ proximity ligation (PLA) assay demonstrating the interaction between Parkin and Ubiquitin. Scale bar, 5 μm. Higher magnification images of the boxed regions are shown in the small panel.
-
CPhosphorylated Parkin accumulates in the ER fraction in the absence of MITOL. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were treated with DMSO or CCCP (10 μM) for 48 h. Lysates of cells were fractionated into cytosolic, mitochondrial, and ER fractions, and then subjected to an IB assay with the indicated antibodies. Phospho‐Ser65‐Parkin is detected by using specific antibody. Right panels are the quantification of IB about Phospho‐Ser65‐Parkin protein levels of the ER fraction. The data represent the mean ± SD for three independent experiments. For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01.
-
DParkin is detected at the ER in the absence of MITOL. HeLa cells were transfected with vectors for KikGR‐Parkin. After ultraviolet light (365 nm) exposure to the whole plate, cells were incubated with or without CCCP (10 μM) for indicated time, and monitored for red and green KikGR fluorescence. Scale bar, 10 μm. Higher magnification images of the boxed regions are shown in the small panel.
Figure EV5. Parkin translocation to the ER does not depend on FKBP38 and ubiquitination of FKBP38 needs Parkin E3 activity, related to Figs 4 and 5 .
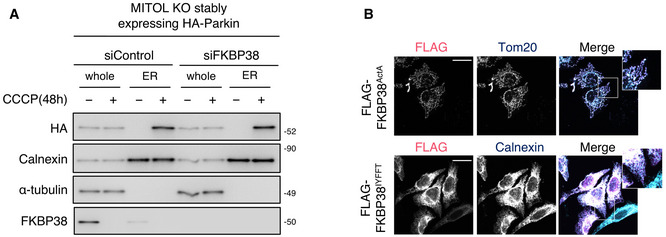
-
AParkin accumulation in the ER does not depend on FKBP38. MITOL KO HeLa cells stably expressing HA‐Parkin were transfected with FKBP38 siRNA or control siRNA and treated with CCCP (10 μM) for 48 h. Lysates of cells were fractionated into ER fractions and then subjected to IB assay with indicated antibodies.
-
BFKBP38IYFFT and FKBP38ActA are accurately localized in each targeting organelle. HeLa cells stably expressing HA‐Parkin were transfected with indicated vectors and immunostained with indicated antibodies. Scale bar, 10 μm. Higher magnification images of the boxed regions are shown in the small panel.
MITOL protects FKBP38 by degrading phosphorylated Parkin for cell survival during mitophagy
Parkin degrades the Pael receptor (Pael‐R) for the substrate in the ER (Imai et al, 2001), and based on this, we considered that the accumulation of Parkin in the MITOL KO cells might induce excessive degradation of its substrates in the ER. Therefore, we investigated the amount of FKBP38 in the MITOL KO cells and found that the amount of FKBP38 in the ER was markedly decreased by MITOL KO (Fig 4C). We examined whether FKBP38 is a specific substrate of Parkin by using phosphorylation‐defective Parkin mutant (S65A; Kane et al, 2014) and Parkin catalytic cysteine mutant (C431S; Lazarou et al, 2013), and found that Parkin polyubiquitinated FKBP38 at its K271/K273 residue and induced FKBP38 degradation during mitophagy (Fig 5A–C). Furthermore, to determine the relationship between the subcellular localization and Parkin‐induced degradation of FKBP38, we added either an ER targeting signal (IYFFT) or a mitochondria targeting signal (ActA) to FKBP38 (Zuchner et al, 2004; de Brito & Scorrano, 2008). In the MITOL KO cells, ER‐targeted FKBP38 was markedly degraded after CCCP treatment compared to WT cells (Figs EV5, 5). In contrast, the degradation of FKBP38 targeted in the mitochondria was not strongly enhanced by MITOL deficiency as mitochondrial degradation ensued during this time (Fig 5D). These data suggest that MITOL inhibits the degradation of FKBP38, especially in the ER via the degradation of activated Parkin in the later phase of mitophagy. Therefore, we considered that MITOL KO cells become vulnerable to cell death due to FKBP38 degradation. Analysis of cell death by propidium iodide (PI) staining indicated that the cell death in MITOL KO cells increased compared to the WT cells after CCCP treatment and this enhanced cell death was rescued by the overexpression of WT MITOL (Fig 5E). Moreover, the FKBP38 mutant K271/273R, lacking the essential lysine residues for the degradation modulated by Parkin, partly rescued the cell death in MITOL KO cells and CCCP‐treated Parkin K220R cells (Fig 5F and G), indicating that FKBP38 inhibits Parkin‐induced cell death at the ER during mitophagy. On the other hand, cell death in the FKBP38 K271/273R mutants was only partially rescued suggesting that activated Parkin targets not only FKBP38, but also other substrates in the ER. FKBP38 contributes to the localization of the anti‐apoptotic protein Bcl‐2 (Shirane & Nakayama, 2003). We hypothesized that the degradation of FKBP38 via Parkin causes mislocalization of Bcl‐2 in the MITOL KO cells. Thus, we analyzed the localization of Bcl‐2 between MITOL WT cells and MITOL KO cells during mitophagy. Consistent with previous reports (Saita et al, 2013), we found that Bcl‐2 translocated to the ER in the HA‐Parkin stably expressing MITOL WT cells in mitophagy while it was partially mislocalized to the cytosol in HA‐Parkin stably expressing MITOL KO cells (Fig 6A). Finally, we verified whether this cell death pathway requires caspase activation or not and for this, we treated the cells with the pan‐caspase inhibitors Z‐VAD‐FMK. Addition of Z‐VAD‐FMK to the MITOL KO cells prevented cell death in mitophagy (Fig 6B). Since Bcl‐2, which regulates the Ca2+ release from the ER to prevent excessive cell death (Lam et al, 1994), does not accurately localize at the ER in MITOL KO cells, we hypothesized that the mislocalization of Bcl‐2 caused cell death via calcium leak. Thus, we treated the MITOL KO cells with BAPTA‐AM, an intracellular calcium chelator and as expected, we observed that the cell death could be rescued in the MITOL KO cells (Fig 6C). Taken together, our findings demonstrated that MITOL inhibits Parkin‐induced cell death, at least partially, by protecting FKBP38 from activated Parkin at the ER during mitophagy.
Figure 5. MITOL protects FKBP38 by degrading phosphorylated Parkin for cell survival.
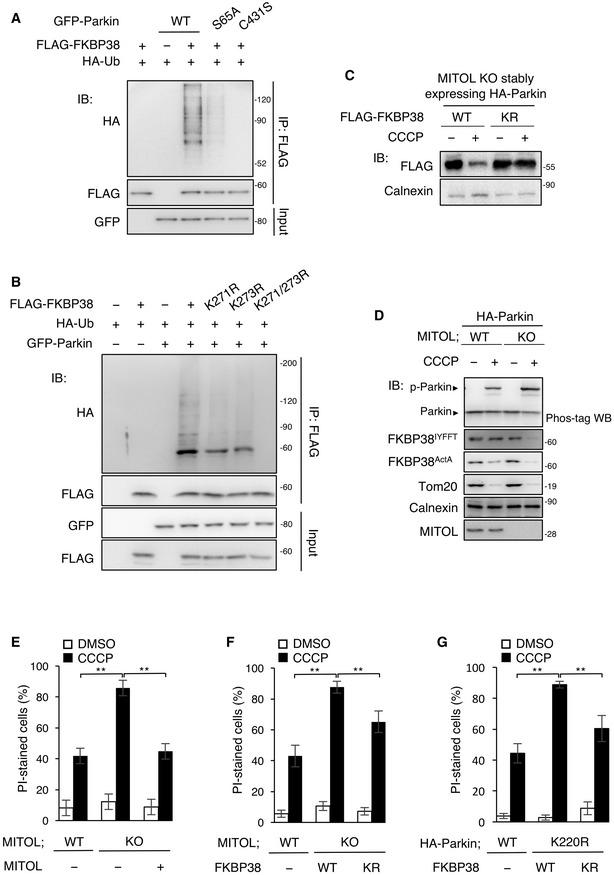
-
AUbiquitination of FKBP38 depends on Parkin E3 activity. HeLa cells were transfected with the indicated vectors and treated with DMSO or CCCP (10 μM) for indicated times. MG132 (30 μM) was added 1 h before each treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies. WT, wild‐type.
-
BParkin fails to ubiquitinate the FKBP38 K271/273R mutant. HeLa cells were transfected with the indicated vectors and treated with CCCP (10 μM) for 30 h. MG132 (30 μM) was added 5 h after CCCP treatment. Lysates of cells were subjected to an IP‐IB assay with the indicated antibodies and compared ubiquitination levels of WT, K271R, K273R, and K271/273R FLAG‐FKBP38.
-
COverexpression of WT FKBP38 is degraded in MITOL KO cells in mitophagy. MITOL KO HeLa cells stably expressing HA‐Parkin were transfected with WT or K271/273R mutant FLAG‐FKBP38 and treated with DMSO or CCCP (10 μM) for 24 h. Lysates of cells were subjected to an IB assay with the indicated antibodies.
-
DMITOL KO increases degradation of ER targeting FKBP38. HeLa cells stably expressing HA‐Parkin were transfected with mitochondrial targeting FLAG‐FKBP38 (FKBP38ActA) and ER targeting FLAG‐FKBP38 (FKBP38IYFFT), and treated with DMSO or CCCP (10 μM) for 24 h. Lysates of cells were subjected to an IB assay with FLAG antibodies for organelle targeting FKBP38 or indicated antibodies. Another cell lysates were subsequently separated on a Phos‐tag gel, followed by IB with anti‐Parkin antibodies (Phos‐tag).
-
E–GMITOL partly prevents CCCP‐induced cell death by reducing Parkin‐mediated degradation of FKBP38. WT or MITOL KO HeLa cells stably expressing HA‐Parkin and HeLa cells were transfected with the indicated vectors, and treated with DMSO or CCCP (10 μM) for 24 h. Cells were stained with propidium iodide (PI) for counting the percentage of dead cells using fluorescence microscopy. Data represent the mean ± SD of five independent experiments (>100 individual cells were counted). For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01. KR, FKBP38 K271/273R.
Figure 6. MITOL deletion causes mislocalization of Bcl‐2 and leads to excessive cell death via calcium leak in mitophagy.
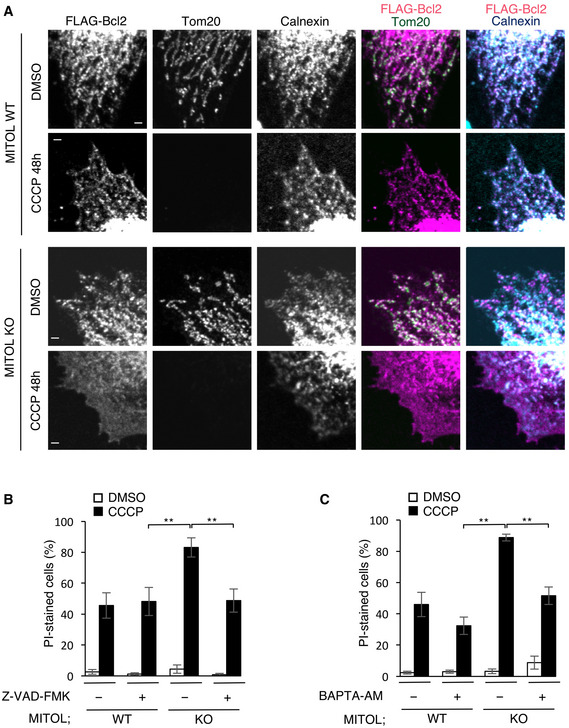
-
AMislocalization of Bcl‐2 is observed in MITOL KO cells in mitophagy. WT or MITOL KO HeLa cells stably expressing HA‐Parkin were transfected with FLAG‐Bcl2 and treated with DMSO or CCCP (10 μM) for the indicated times. Cells were immunostained with indicated antibody. Scale bar, 1 μm.
-
BMITOL prevents caspase‐dependent cell death in mitophagy. WT or MITOL KO HeLa cells stably expressing HA‐Parkin and HeLa cells were treated with DMSO or CCCP (10 μM) with or without Z‐VAD‐FMK (10 μM) for 24 h. Cells were stained with propidium iodide (PI) for counting fluorescence microscopy of the percentage of dead cells. Data represent the mean ± SD of three independent experiments (>100 individual cells were counted). For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01.
-
CCell death in mitophagy caused by MITOL deficiency depends on calcium leak. WT and MITOL KO HeLa cells stably expressing HA‐Parkin or HeLa cells were treated with DMSO or CCCP (10 μM) with or without BAPTA‐AM (10 μM) for 24 h. Cells were stained with propidium iodide (PI) for counting fluorescence microscopy of the percentage of dead cells. Data represent the mean ± SD of three independent experiments (>100 individual cells were counted). For statistical analysis, a one‐way ANOVA with Tukey post‐test was performed, **P < 0.01.
Discussion
There have been recent advancements in the analysis of PINK1–Parkin‐mediated mitophagy and its molecular mechanism has been elucidated in detail. It has also become clear that (i) PINK1 phosphorylates the S65 of ubiquitin during the lowering of the membrane potential in the mitochondria; (ii) this phosphorylated ubiquitin is a Parkin‐activating factor; and (iii) the conformation of Parkin changes based on its phosphorylation and binding with phosphorylated ubiquitin (Trempe et al, 2013). In vitro binding studies have revealed that MITOL binds to the RING2 domain of Parkin only when CCCP was added. Thus, it is considered that MITOL specifically binds to the activated Parkin that has already undergone conformational changes. We found that MITOL mediates ubiquitination of Parkin at the K220 residue and promotes the degradation of Parkin. As MITOL degrades phosphorylated Parkin rather than unphosphorylated form, it can be considered that although it is not certain whether MITOL‐mediated Parkin degradation is dependent on the structure of Parkin or not, MITOL can selectively recognize and degrade phosphorylated Parkin. Because the degradation of Parkin by endogenous MITOL is milder than that by overexpressed MITOL, the timing of degradation in endogenous MITOL is extremely slow. When Parkin is degraded at an early stage, mitophagy is strongly inhibited. This suggests that if the amount of phosphorylated Parkin does not surpass the threshold at the appropriate time, mitochondrial degradation might not occur. Based on this, we considered that endogenous MITOL mildly degrades phosphorylated Parkin at the appropriate timing to prevent any hindrance to quality control of the cells via Parkin. On the other hand, it has been recently reported that protein ubiquitination by MITOL is involved in Parkin recruitment and activation during the early phase of mitophagy (Koyano et al, 2019b), suggesting distinct roles of MITOL in the early and late phases. As mitophagy proceeded even when MITOL was completely depleted, it was concluded that although MITOL may be partially involved in Parkin recruitment, it is not essential for Parkin‐dependent mitophagy.
In this study, we found that MITOL translocated to the ER in the latter phase of mitophagy. A previous study revealed that the mechanism of the translocation of FKBP38 to the ER depends on the number of basic amino acids on the C‐terminal domain of the protein as high basicity in the C‐terminal domain disturbs the translocation to the ER (Saita et al, 2013). However, the C‐terminal domain of MITOL possesses seven basic amino acids, suggesting that MITOL cannot leave the mitochondria by itself. Indeed, FKBP38 knockdown abolished MITOL translocation to the ER, suggesting that MITOL is transported by FKBP38. In the present study, we were unable to clarify how MITOL, which is tightly integrated into the mitochondrial outer membrane, is translocated to the ER by FKBP38, or the mechanism underlying the translocation of Parkin. As Parkin is phosphorylated by PINK1 on the mitochondria, phosphorylated Parkin accumulated in the ER might pass through the mitochondria once. MITOL is known to be abundant in the membrane contact sites between the mitochondria and ER, where it regulates the substrate IRE1α (Sugiura et al, 2013; Takeda et al, 2019). However, as p97 reduces the contact sites between these two organelles, which are associated with mitophagy, translocation might not be mediated through the contact sites between ER and mitochondria (McLelland et al, 2018). Disruption of the mitochondrial outer membrane, which is triggered by the degradation of the substrates of Parkin, has been revealed to be essential for mitophagy (Yoshii et al, 2011; Wei et al, 2017). In a recent study, Koyano et al (2019a) reported that MITOL was translocated to the peroxisome in a Parkin ubiquitination‐dependent manner. We also found that the ubiquitination of MITOL by Parkin occurred in the early stage of mitophagy and thereafter disappeared in the late stage of mitophagy (Fig EV2). At present, it is not clear whether MITOL translocates to the ER via the peroxisome or directly, but it is considered that the loss of MITOL ubiquitination in the late stage of mitophagy might be key for this translocation.
Although previous studies using mass spectrometric analysis have suggested the possibility that the anti‐apoptotic protein FKBP38 is one of the substrates of Parkin (Sarraf et al, 2013; Rose et al, 2016), it has not been experimentally proven yet. In this study, we have demonstrated that Parkin interacts with, ubiquitinates and degrades FKBP38, indicating that FKBP38 is a physiological substrate of Parkin during mitophagy. In addition, we have shown that the accumulation of activated Parkin in MITOL KO cells induced the degradation of FKBP38, resulting in enhanced cell death. However, complete mitigation of cell death by the FKBP38 mutant, which could not be degraded by Parkin, was not completely achieved, and thus, there may be other anti‐apoptotic protein(s) that are ubiquitinated by Parkin in the ER. Further studies are required to clarify the cytotoxic pathway triggered by hyperactivation of Parkin in the ER. Our findings further uncover the physiological significance of Parkin by showing that Parkin performs mitochondrial quality control as well as actively induces cell death via the degradation of FKBP38 when the mitochondria are severely damaged, thereby maintaining cellular homeostasis.
On the other hand, during mitophagy, MITOL may respond to mitochondrial damage functionally or quantitatively, and regulate cell death through Parkin degradation. The studies conducted so far have shown that physiologically, Parkin dysfunction can induce cytotoxicity by allowing the accumulation of the substrate (Xu et al, 2002). This phenomenon has also been demonstrated in a familial loss‐of‐function mutant of PD (Dawson & Dawson, 2010). However, in patients with sporadic PD, Parkin is mostly functionally normal. Furthermore, in Parkin KO mice, the loss of dopaminergic neurons, which is a pathological hallmark of PD, was not observed (Goldberg et al, 2003; Itier et al, 2003; von Coelln et al, 2004; Perez & Palmiter, 2005). Taking these facts into consideration, it is possible that Parkin itself might partially contribute to the pathology of PD by inducing cell death and the disruption of the Parkin degradation by MITOL may cause excessive Parkin activation, thereby inducing cell death and causing sporadic PD. If this is the case, MITOL could be considered as a potential therapeutic target for PD.
Materials and Methods
Antibodies and reagents
Anti‐MITOL rabbit polyclonal antibody was produced as described previously. Anti‐β‐actin and anti‐FLAG‐M2 antibodies were purchased from Sigma. Anti‐Parkin and anti‐VDAC were purchased from Cell Signaling Technology. Anti‐Parkin (phospho‐Ser65) was purchased from UBIQUIGENT. Anti‐Tom20 and anti‐calnexin were purchased from Santa Cruz Biotechnology. Anti‐ubiquitin and anti‐GFP were purchased from MBL. Anti‐myc was obtained from Recenttec. Anti‐HA was obtained from Proteintech. Anti‐GST and BAPTA‐AM were purchased from Wako. Anti‐HSP60 was purchased from Enzo Life Sciences. Anti‐FKBP38 and anti‐K63‐chain were purchased from Abcam. Anti‐K48‐chain was purchased from Millipore. Carbonyl cyanide m‐chlorophenylhydrazone (CCCP) and chloroquine diphosphate were obtained from Wako. MG132, lactacystin, Z‐VAD‐FMK, and bafilomycin A1 were obtained from Peptide Institute. Cycloheximide (CHX) was purchased from Sigma.
DNA constructs
Mitochondrial ubiquitin ligase (MITOL), ubiquitin mutant expression vector, and Mito‐GFP were constructed as described previously (Sugiura et al, 2013). To generate stably expressing cells, HA‐Parkin and mCherry‐Parkin were subcloned into the pCX4 retrovirus vector (Akagi et al, 2000). The recombinant GST fusion proteins GST‐MITOL and GST‐Parkin were generated by subcloning into the pGEX‐3X vector. Point mutations of Parkin and FKBP38 were generated with the site‐directed mutagenesis kit (Stratagene). The expression plasmids for human Parkin and FKBP38 were generated by amplifying the corresponding cDNA by PCR and cloning it into pFLAG‐CMV2, or pEGFP‐C1 expression vectors. FLAG‐Bcl2 (Addgene #18003) and mCherry‐Sec61β (Addgene #49155) were purchased from Addgene. To generate localization‐restricted FKBP38, mitochondrial FKBP38 (FKBP38ActA) and ER FKBP38 (FKBP38IYFFT) were constructed as previously described (Zhu et al, 1996; Rojo et al, 2002). Complementary DNA was subcloned into the humanized‐codon monomeric KikGR vector (MBL). Plasmid pX330A‐MITOL/PITCh, an all‐in‐one CRISPR‐Cas9 vector for cutting the genomic MITOL exon 1 locus and the donor vector, was constructed using the pX330A‐1x2 (Addgene #58766) plasmid and pX330S‐2‐PITCh (Addgene #63670), in accordance with a previously described protocol (Sakuma et al, 2016). The oligonucleotides for the template of sgRNA targeting MITOL exon 1 are CRISPR‐hMITOL_sense (5'‐ caccGCCAAGCCCTACAGCAGATGC‐3') and CRISPR‐hMITOL_antisense (5'‐aaacGCATCTGCTGTAGGGCTTGGC‐3'). The donor vector, including PuroR, EGFP, and microhomologies, was constructed from pCRIS‐PITChv2‐FBL (Addgene #63672) using a standard PCR and a SLiCE cloning method (Motohashi, 2015).
Cell culture and transfection
HeLa cells and SH‐SY5Y cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. Cells were transfected with either Lipofectamine 3000 (Invitrogen) or RNAiMax (Invitrogen) according to the manufacture’s protocol. siPINK1 has been described previously (Villa et al, 2017). siMITOL was described previously (Yonashiro et al, 2009). Scrambled siRNA and siFKBP38 were purchased from Qiagen.
Generation of stable cell lines
Stable cell lines were generated using a retroviral expression system as previously described (Akagi et al, 2003). Briefly, Plat E cells were transfected with retroviral vectors using Lipofectamine LTX Reagent with PLUS Reagent (Invitrogen). After 48 h, the growth medium containing retrovirus was collected. HeLa cells were incubated with the collected virus‐containing medium with 8 μg/ml polybrene (Sigma) for 24 h.
MITOL KO Cells
The CRISPR Design Tool (http://www.genome‐engineering.org/crispr/) was used to select the genomic sequence target in hMITOL (5’‐caccgccaagccctacagcagatgc‐3’). Oligo pairs encoding 20‐nt guide sequences were annealed and ligated into the plasmid pX330. HeLa cells were transfected using Lipofectamine 3000 (Invitrogen), and KO clones were selected by serial dilution.
Generation of EGFP Knock‐in cell line
HeLa cells were plated in a 10‐cm dish and co‐transfected with 1.5 μg of the donor vector and 3 μg of pX330A‐MITOL/PITCh using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s instructions. After 72 h of transfection, cells were selected with 1 μg/ml puromycin for 7 days.
Subcellular fractionation
(Organelle fraction) HeLa cells or HeLa cells expressing HA‐Parkin were homogenized in homogenization buffer (HB; 5 mM HEPES pH 7.4, 0.5 mM EDTA, 250 mM mannitol). The homogenate was centrifuged at 800 g for 10 min at 4°C, and the resulting supernatant was further centrifuged at 8,000 g for 10 min at 4°C. The new pellet (crude mitochondrial fraction) was collected, and the supernatant was centrifuged further at 100,000 g for 1 h at 4°C. The resulting pellet (light membrane fraction) and supernatant (cytosolic fraction) were collected. The crude mitochondrial fraction was centrifuged at 100,000 g for 30 min at 4°C. The resulting mitochondrial and ER bands were isolated and resuspended in an isolation buffer (mitochondria‐rich membrane fraction). Cells were harvested and suspended in cell homogenization buffer. Cells were homogenized by 20 passages through a 27‐gauge needle with a 1‐ml syringe, on ice. The homogenates were separated into post‐nuclear supernatant by centrifugation at 1,000 g for 5 min at 4°C. The heavy membrane fraction was separated by spinning down supernatant from PNS (post‐nuclear supernatant) separation at 3,000 g for 10 min at 4°C. To wash the heavy membrane fraction, the pellet was resuspended in fresh buffer and spun back twice. The supernatant from the previous fractionation was spun down at 16,000 g for 15 min at 4°C to collect the light membrane fraction. The remaining supernatant was the cytosolic fraction (Sup). Light membranes and heavy membranes were combined to form the membrane fraction (Memb).
Immunoprecipitation
To examine the protein interaction, cells were solubilized with NP‐40 lysis buffer (1% NP‐40, 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5 mM EDTA, 10 mM NaF, and protease inhibitors) and centrifuged at 15,000 g for 20 min at 4°C. The supernatant was subjected to immunoprecipitation using the indicated antibodies. To evaluate ubiquitylation levels of protein, cells were solubilized with RIPA lysis buffer (0.1% SDS, 0.05% DOC, 1% Triton X‐100, 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, and protease inhibitors) and centrifuged at 15,000 g for 20 min at 4°C. The supernatant was sonicated 10 s and subjected to immunoprecipitation using the indicated antibodies.
Immunoblotting and Phos‐tag PAGE
Whole lysates were separated by SDS–PAGE and transferred to the PVDF membranes (Millipore). The blots were probed with the indicated antibodies, and protein bands on the blot were visualized using the enhanced chemiluminescence reagent (Millipore). Phos‐tag‐PAGE was performed using 8% SDS–PAGE minigels containing 10 μM Phos‐tag (Wako) in the presence of 100 μM MnCl2 following the manufacturer’s protocol.
GST pull‐down assay
To determine the specific domains required for the interaction between MITOL and Parkin, GST fusion deletion mutants were prepared as described previously (Yonashiro et al, 2006). Lysates from HeLa cells or HeLa cells stably expressing HA‐Parkin were incubated with the purified GST fusion proteins. Precipitates were analyzed by immunoblotting.
Morphological analysis by immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in phosphate‐buffered saline (PBS) for 15 min at room temperature, then washed twice with PBS, permeabilized with 0.2% Triton X‐100 in PBS for 5 min, then washed four times with PBS, and blocked with 1% bovine serum albumin in PBS, all at room temperature. For double staining, the cells were incubated with appropriate primary Abs for 1 h at room temperature, washed three times with PBS, and then incubated with appropriate secondary Abs for 30 min. The samples were washed as described above, mounted using Fluorescent Mounting Medium (Dako), and analyzed using an Olympus FV1000 and FV3000 confocal laser scanning microscope.
Photoconversion analysis
KikGR‐labeled proteins expressed in cells were photoconverted with a 365‐nm UV light for 5 min using the Benchtop UV Transilluminator, LM‐26 (2UV; Funakoshi).
In situ Proximity Ligation Assay
HeLa cells stably expressing HA‐Parkin were 4% paraformaldehyde in phosphate‐buffered saline (PBS) for 15 min at room temperature, then washed twice with PBS, permeabilized with 0.2% Triton X‐100 in PBS for 5 min, then washed four times with PBS, and blocked with 1% bovine serum albumin in PBS, all at room temperature. The Duolink in situ proximity ligation assay was carried out according to the manufacturer’s instructions (Olink Bioscience, Sigma) using the following primary antibodies, anti‐Parkin and anti‐ubiquitin, and analyzed using an Olympus Fluoview FV1000 laser scanning confocal microscope.
PI Staining
Plasma membrane integrity based on the exclusion of PI was measured as an index of dead cells. Cells were stained at 37°C for 10 min with PI (1 μM final concentration). After incubation, stained cells were scored as dead cells under a fluorescence microscope.
Mt‐Keima‐based mitophagy assay
For the detection of mitophagy, HeLa cells stably expressing mt‐Keima transiently transfected with the plasmid as indicated, and MITOL WT and KO HeLa cells stably expressing HA‐Parkin and mt‐Keima were treated with 10 μM CCCP for 8 h. For measurement of mKeima, a BD LSR II Flow Cytometer was used to detect the dual‐excitation ratiometric pH at 488 (pH 7) and 561 (pH 4) nm lasers with 695/40 and 575/26 nm emission filters. For each sample, 30,000 cells were counted and analyzed using FlowJo software (version 10.7.1).
Ubiquitin restriction assay
Ubiquitin restriction assay was performed using the UbiCRest kit (Boston Biochem) following the manufacturer’s instructions. Recombinant AMSH, OTUB1, and USP2 were used for the reaction. Parkin was immunoprecipitated from HA‐Parkin stably expressing cells treated with CCCP (10 μM, 3 h) as described in the above section. After washing with NP‐40 buffer three times, the beads were washed with reaction buffer for additional two times. The purified Parkin was then incubated with either with AMSH, OTUB1, Usp2, or buffer only (reaction buffer), in a 20 μl reaction, for 1 h at 37°C. The reaction was terminated by the addition of SDS loading buffer. The samples were separated by SDS–PAGE and analyzed by Western blotting with the indicated antibody.
Statistical analysis
Results are expressed as either mean ± SD or ± SEM. The obtained data were performed either two‐tailed Student’s t‐test or a one‐way ANOVA followed by post hoc Tukey test to determine significance.
Author contributions
IS, KT, NI, TT, SN, and SY conceived the project and designed the experimental strategy. IS, SN, KT, and NI performed the molecular biology experiments. S‐IY, TKa, TKo, YU, and YF provided material and technical assistance. IS and SY wrote the manuscript with contribution from RI.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Asei Hirai, Kanta Tsuda, and Tomohiro Maruyama for technical assistance and advice. We thank Drs. Toshifumi Fukuda and Nobuko Matsushita for useful discussions. This study was supported in part by the MEXT/JSPS KAKENHI [Grant Nos. 16K08246 (to SN and RI), and 15H01190, 17H04053, 18H04869, 20H03454, and 20H04911 (to SY)] and MEXT‐Supported Program for the Strategic Research Foundation at Private Universities [Grant No. S1411014 (to SN, RI, and SY), the Uehara Memorial Foundation, the Naito Foundation (to SN and SY)], the Takeda Science Foundation, the Sumitomo Foundation, the Cosmetology Research Foundation, the Ono Medical Research Foundation the Tokyo Biochemical Research Foundation and AMED under Grant Numbers JP17gm5010002, JP18gm5010002, JP19gm5010002, and JP20gm5010002 (to SY).
EMBO reports (2021) 22: e49097
Data availability
No primary data sets deposited in a public database.
References
- Akabane S, Matsuzaki K, Yamashita S, Arai K, Okatsu K, Kanki T, Matsuda N, Oka T (2016) Constitutive activation of PINK1 protein leads to proteasome‐mediated and non‐apoptotic cell death independently of mitochondrial autophagy. J Biol Chem 291: 16162–16174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Shishido T, Murata K, Hanafusa H (2000) v‐Crk activates the phosphoinositide 3‐kinase/AKT pathway in transformation. Proc Natl Acad Sci USA 97: 7290–7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Sasai K, Hanafusa H (2003) Refractory nature of normal human diploid fibroblasts with respect to oncogene‐mediated transformation. Proc Natl Acad Sci USA 100: 13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Carroll RG, Hollville E, Martin SJ (2014) Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl‐1. Cell Rep 9: 1538–1553 [DOI] [PubMed] [Google Scholar]
- von Coelln R, Dawson VL, Dawson TM (2004) Parkin‐associated Parkinson's disease. Cell Tissue Res 318: 175–184 [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2010) The role of parkin in familial and sporadic Parkinson's disease. Mov Disord 25(Suppl 1): S32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ et al (2003) Parkin‐deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628–43635 [DOI] [PubMed] [Google Scholar]
- Hospenthal MK, Mevissen TET, Komander D (2015) Deubiquitinase‐based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat Protoc 10: 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi R, Yamashita SI, Yamashita T, Inoue K, Fukuda T, Fukuchi T, Kanki T (2020) Gemcitabine induces Parkin‐independent mitophagy through mitochondrial‐resident E3 ligase MUL1‐mediated stabilization of PINK1. Sci Rep 10: 1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105: 891–902 [DOI] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen‐Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L et al (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12: 2277–2291 [DOI] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2: 120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Koyano F, Yamano K, Kosako H, Kimura Y, Kimura M, Fujiki Y, Tanaka K, Matsuda N (2019a) Parkin‐mediated ubiquitylation redistributes MITOL/March5 from mitochondria to peroxisomes. EMBO Rep 20: e47728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Yamano K, Kosako H, Tanaka K, Matsuda N (2019b) Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J Biol Chem 294: 10300–10314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW (1994) Evidence that BCL‐2 represses apoptosis by regulating endoplasmic reticulum‐associated Ca2+ fluxes. Proc Natl Acad Sci USA 91: 6569–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ (2013) PINK1 drives Parkin self‐association and HECT‐like E3 activity upstream of mitochondrial binding. J Cell Biol 200: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I et al (2018) Mfn2 ubiquitination by PINK1/parkin gates the p97‐dependent release of ER from mitochondria to drive mitophagy. eLife 7: e32866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi K (2015) A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site‐directed mutagenesis. BMC Biotechnol 15: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H (2007) Proteasome‐independent functions of ubiquitin in endocytosis and signaling. Science 315: 201–205 [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3: 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N (2013) A dimeric PINK1‐containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288: 36372–36384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD (2005) Parkin‐deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA 102: 2174–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Fushman D (2004) Polyubiquitin chains: polymeric protein signals. Curr Opin Cell Biol 8: 610–616 [DOI] [PubMed] [Google Scholar]
- Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM (2010) Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78: 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo M, Legros F, Chateau D, Lombes A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115: 1663–1674 [DOI] [PubMed] [Google Scholar]
- Rose CM, Isasa M, Ordureau A, Prado MA, Beausoleil SA, Jedrychowski MP, Finley DJ, Harper JW, Gygi SP (2016) Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst 3: 395–403.e394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saita S, Shirane M, Nakayama KI (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat Commun 4: 1410 [DOI] [PubMed] [Google Scholar]
- Sakuma T, Nakade S, Sakane Y, Suzuki KT, Yamamoto T (2016) MMEJ‐assisted gene knock‐in using TALENs and CRISPR‐Cas9 with the PITCh systems. Nat Protoc 11: 118–133 [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba‐Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1‐mediated phosphorylation of the Parkin ubiquitin‐like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2: 1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin‐protein ligase. Nat Genet 25: 302–305 [DOI] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl‐2 to mitochondria and inhibits apoptosis. Nat Cell Biol 5: 28–37 [DOI] [PubMed] [Google Scholar]
- Sugiura A, Yonashiro R, Fukuda T, Matsushita N, Nagashima S, Inatome R, Yanagi S (2011) A mitochondrial ubiquitin ligase MITOL controls cell toxicity of polyglutamine‐expanded protein. Mitochondrion 11: 139–146 [DOI] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N et al (2013) MITOL regulates endoplasmic reticulum‐mitochondria contacts via mitofusin2. Mol Cell 51: 20–34 [DOI] [PubMed] [Google Scholar]
- Takeda K, Nagashima S, Shiiba I, Uda A, Tokuyama T, Ito N, Fukuda T, Matsushita N, Ishido S, Iwawaki T et al (2019) MITOL prevents ER stress‐induced apoptosis by IRE1alpha ubiquitylation at ER‐mitochondria contact sites. EMBO J 38: e100999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al‐Abdul‐Wahid S, Krett J, Wong K, Kozlov G et al (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455 [DOI] [PubMed] [Google Scholar]
- Villa E, Proics E, Rubio‐Patino C, Obba S, Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragon L, Riley JS et al (2017) Parkin‐independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep 20: 2846–2859 [DOI] [PubMed] [Google Scholar]
- Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS et al (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238.e210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA (2002) Dopamine‐dependent neurotoxicity of alpha‐synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8: 600–606 [DOI] [PubMed] [Google Scholar]
- Yamano K, Youle RJ (2013) PINK1 is degraded through the N‐end rule pathway. Autophagy 9: 1758–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T, Yoshimori T, Sakai Y, Mihara K et al (2016) Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215: 649–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura‐Hoshino M, Sada K, Hotta H, Yamamura H et al (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25: 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Sugiura A, Miyachi M, Fukuda T, Matsushita N, Inatome R, Ogata Y, Suzuki T, Dohmae N, Yanagi S (2009) Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1‐induced ROS generation. Mol Biol Cell 20: 4254–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011) Parkin mediates proteasome‐dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee S, Peng Y, Bunker E, Giaime E, Shen J, Zhou Z, Liu X (2014) PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr Biol 24: 1854–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW (1996) Bcl‐2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15: 4130–4141 [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar‐Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J et al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet 36: 449–451 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
No primary data sets deposited in a public database.