

Abstract
Aims/Introduction
Micro‐ribonucleic acids (miRNAs) possess crucial functions in governing metabolisms associated with type 2 diabetes mellitus. This study aimed to investigate the role of miR‐23a‐3p in pyroptosis caused by nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3) inflammatory body activation, thereby reducing the occurrence of type 2 diabetes mellitus.
Materials and Methods
miR‐23a‐3p and NIMA‐related kinase 7 (NEK7) expression in type 2 diabetes mellitus patients and rat models was examined. Dual‐luciferase reporter gene experiments were used to verify the targeting relationship between miR‐23a‐3p and NEK7. Bone marrow‐derived macrophages were transfected with miR‐23a‐3p mimic, miR‐23a‐3p inhibitor or short hairpin NEK7 and were treated with a specific activator of NLRP3 inflammatory body (lipopolysaccharide + adenosine‐5′‐triphosphate) to evaluate expression of NEK7, miR‐23a‐3p, gasdermin D p30, pro‐caspase‐1 and caspase‐1 in cells, and interleukin‐1β and tumor necrosis factor‐α in supernatant. Type 2 diabetes mellitus rat models were used to observe the influences of miR‐23a‐3p, NEK7 and NLRP3 inflammatory body on pyroptosis and type 2 diabetes mellitus in vivo.
Results
NEK7 was overexpressed, whereas miR‐23a‐3p was underexpressed in patients and rat models with type 2 diabetes mellitus. NEK7 was a target gene of miR‐23a‐3p. After the addition of lipopolysaccharide + adenosine‐5′‐triphosphate in bone marrow‐derived macrophages, the expression of miR‐23a‐3p subsequently declined. Furthermore, the addition of lipopolysaccharide + adenosine‐5′‐triphosphate elevated NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1 and gasdermin D p30 expressions in bone marrow‐derived macrophages, and enhanced levels of interleukin‐1β and tumor necrosis factor‐α in the supernatant, accompanied with conspicuous cell pyroptosis, which was reversed after miR‐23a‐3p overexpression and NEK7 silencing. miR‐23a‐3p overexpression alleviated liver and kidney damage in type 2 diabetes mellitus rats, and reduced NLRP3‐induced pyroptosis.
Conclusions
Targeting NEK7 by miR‐23a‐3p could reduce NLRP3‐induced pyroptosis, and assuage liver and kidney injuries in type 2 diabetes mellitus rats.
Keywords: Micro‐ribonucleic acid‐23a‐3p, NOD‐, LRR‐ and pyrin domain‐containing protein 3, Type 2 diabetes mellitus
Targeting NIMA‐related kinase 7 by micro‐ribonucleic acid could reduce NOD‐, LRR‐ and pyrin domain‐containing protein 3‐induced pyroptosis, and assuage liver and kidney injuries in type 2 diabetes mellitus rats.
Introduction
The term, pyroptosis, is a Greek term that refers to fire/fever and falling, which is also called caspase 1‐dependent cell death in scientific terms 1 . It is a newly discovered pro‐inflammatory programmed cell death pathway in recent years, which can be initiated by diverse pathological stimuli, including stroke, heart failure and cancer 2 . On both endogenous and exogenous stimulations, apoptosis‐associated speck‐like protein containing a C‐terminal caspase recruitment domain drives pro‐caspase‐1 to form inflammatory bodies and further activates pro‐caspase‐1. Activated caspase‐1 stimulates the activation of downstream cytokines, such as interleukin (IL)‐1β and IL‐18 3 . In patients with type 2 diabetes mellitus, cellular pro‐inflammatory factor IL‐1β starts being expressed and elevated. Furthermore, IL‐1β is regarded as the ultimate pathway for autoimmune diabetes and type 2 diabetes mellitus. A decline in the quantity of functioning insulin‐producing β‐cells leads to the pathophysiological development of type 2 diabetes mellitus 4 . β‐Cell number grows when there are increased demands; for example, in obesity, the decline of the number will cause diabetes 5 . The pathophysiology of type 2 diabetes mellitus consists of two aspects: insulin resistance and a defect in β‐cells 6 . Recent studies have shown that nucleotide‐binding oligomerization (NOD)‐like receptor (NLR) family pyrin domain containing 3 (NLRP3) activators could induce reactive oxygen species to act as intermediates to trigger the activation of inflammasomes 7 .
It was reported that micro‐ribonucleic acids (miRNAs) play critical roles in the development of type 2 diabetes mellitus 8 . For instance, miR‐23a was found to be involved in the mediation of type 2 diabetes mellitus 9 . miR‐23a‐3p correlates to obesity and tumor necrosis factor‐α (TNF‐α)‐induced insulin resistance 10 . Meanwhile, a prior study revealed the decrease of serum level of miR‐23a in type 2 diabetes mellitus 11 . NIMA‐related kinase 7 (NEK7), a serine/threonine protein kinase, regulates proper spindle formation and cytokinesis production 12 . Furthermore, being a mitotic kinase, NEK7 is required for the activation of NLRP3 inflammasomes 13 . A prior study showed that NLRP3 inflammasomes activation was related to the development of type 2 diabetes mellitus 14 . However, the correlation between miR‐23a‐3p and NEK7 and the effect of NEK7 on type 2 diabetes mellitus still remains poorly understood. Therefore, the present study aimed to investigate whether miR‐23a‐3p modulates pyroptosis caused by NLRP3 inflammasomes in type 2 diabetes mellitus by targeting NEK7.
Methods
Sample collection from patients with type 2 diabetes mellitus
Samples were obtained from elderly patients with type 2 diabetes mellitus, who were outpatients and inpatients in The First Affiliated Hospital of Henan University, Kaifeng, China, from January 2018 to June 2019. There were a total of 30 patients (13 men and 17 women) aged 62–89 years (75.37 ± 7.99 years). All patients met with the 2019 World Health Organization criteria for diabetes diagnosis. They did not receive any prior treatment with hypoglycemic drugs. Patients with severe heart, liver and kidney diseases, chronic complications of diabetes, acute and chronic infections, recent surgeries and trauma, cancer malignancies, autoimmune diseases, and other endocrine system diseases were excluded. Another 30 cases of healthy individuals who underwent physical examination in The First Affiliated Hospital of Henan University during the same period were selected as controls, including 16 men and 14 women. They were aged 56–85 years (68.43 ± 9.93 years). People with type 2 diabetes mellitus, atherosclerosis and gout were excluded for study selection. There was no significant difference in both sex and age between the two groups (both P > 0.05). The clinical data of all participants were complete and are listed in Table 1.
Table 1.
Clinical data for type 2 diabetes mellitus patients
| Control | Diabetes | |
|---|---|---|
| Age (years) | 75.37 ± 7.99 | 68.43 ± 9.93 |
| Weight (kg) | 65.2 ± 3.6 | 74.3 ± 6.4 |
| BMI | 24.1 ± 0.2 | 26.2 ± 0.8 |
| Blood glucose (mmol/L) | 5.4 | 9.6 |
| Glycosylated hemoglobin | 4.3% | 8.6% |
BMI, body mass index.
This study was approved by the ethics committee of The First Affiliated Hospital of Henan University, and all patients submitted signed informed consent forms. All operations involving animal experiments met with the ethical requirements and were approved by the Animal Ethics Committee of The First Affiliated Hospital of Henan University. The animals received humane care according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Isolation of rat bone marrow‐derived macrophage cells
Bone marrow‐derived macrophages (BMDMs) were collected from the leg bones of rats. The extracted bone marrow was centrifuged. Ammonium–chloride–potassium lysing buffer was used to lyse red blood cells, which were then centrifuged in Dulbecco’s modified Eagle’s medium. Rat mononuclear cells were isolated by density gradient centrifugation and differentiated into macrophages. Once the cells grew adequately, phorbol myristate acetate was added to induce cells to differentiate into macrophages for the subsequent experiments. The cell density was adjusted to 1 × 106 cells/mL, and cells seeded into a petri dish or a plate were left to stimulate with 100 ng/mL phorbol myristate acetate for 72 h. The cell adherence, deformation and extension of pseudopods were observed under an inverted microscope, suggesting transformation of cells into macrophages that can be used for experiments.
Plasmid transfection
BMDMs were seeded in a six‐well plate (4 × 105 cells/well). When cell confluence reached 80–90%, plasmids (the final concentration of 50 nmol/L) were transfected for 48 h according to the instructions of lipofectamine 2000 (11668‐019; Invitrogen, New York, NY, USA). BMDMs were grouped into: (i) the control group: macrophages were co‐cultured with phosphate‐buffered saline for 48 h; (ii) the positive control group (lipopolysaccharide [LPS] + adenosine‐5′‐triphosphate [ATP]): macrophages were co‐cultured with LPS (1 μg/mL; S1732; Beyotime Biotechnology, Shanghai, China) for 6 h, and then co‐cultured with ATP (5 mol/L) for another 30 min; (iii) the miR‐23a‐3p group (LPS [L]‐mimic negative control [NC] and L‐miR‐23a‐3p mimic [MI]): macrophages were transfected with miR‐23a‐3p mimic or the corresponding NC for 48 h, co‐cultured with LPS (1 μg/mL) for 6 h, and then co‐cultured with ATP (5 mol/L) for 30 min; (iv) the miR‐23a‐3p inhibitor group (L‐inhibitor NC and L‐miR‐23a‐3p inhibitor [IN]): macrophages were transfected with miR‐23a‐3p inhibitor or corresponding NC for 48 h, and then co‐cultured with LPS and ATP; (v) the NEK7 silenced group (L‐short hairpin RNA NC [sh‐NC] and L‐sh‐NEK7): macrophages were transfected with sh‐NEK7 plasmid or corresponding NC for 48 h, and then co‐cultured with LPS and ATP; and (vi) the co‐transfection group (L‐miR‐23a‐3p inhibitor + sh‐NC and L‐miR‐23a‐3p inhibitor + sh‐NEK7): macrophages were transfected with miR‐23a‐3p inhibitor, further transfected with sh‐NEK7 plasmid or corresponding NC for 48 h, and then co‐cultured with LPS and ATP.
Reverse transcription quantitative polymerase chain reaction
The total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and the RiboPure™ Blood Kit (Ambion, Austin, TX, USA). Reverse transcription was carried out using a reverse transcription kit (RR047A; Takara, Tokyo, Japan) to obtain complementary deoxyribonucleic acid (cDNA) for messenger RNA detection. A miRNA First Strand cDNA Synthesis (Tailing Reaction) kit (B532451‐0020; Shanghai Sangon Biotech, Shanghai, China) was used to obtain cDNA by reverse transcription for miRNA detection. The samples were subjected to reverse transcription quantitative polymerase chain reaction (RT–qPCR) in a real‐time quantitative PCR instrument (ABI 7500; ABI, Foster City, CA, USA). The miRNA universal negative primer and the U6 internal reference upstream primer were provided by the miRNA First Strand cDNA Synthesis (Tailing Reaction) kit. Other primers were synthesized by Shanghai Sangon Biotech (Table 2). To normalize qPCR detection, U6 was used as the internal control for the detection of miR‐23a‐3p in cells, and cel‐miR‐39 for the detection of serum miR‐23a‐3p. Glyceraldehyde 3‐phosphate dehydrogenase was the internal control for messenger RNA. The equation method was used to calculate the relative expression of the product.
Table 2.
Primer sequences of reverse transcription quantitative polymerase chain reactions
| Gene | Primer sequences |
|---|---|
| miR‐23a‐3p | F: 5′‐TGCGTGCGA TCACATTGCCAGGGA‐3′ |
| R: 5′‐TACGAAGGGTCCGAACAC‐3′ | |
| NEK7 | F: 5′‐CCGTTACTCAGTTCCAGCCA‐3′ |
| R: 5′‐CTACCGGCACTCCATCCAAG‐3′ | |
| U6 | F: 5′‐CTCGCTTCGGCAGCACA‐3′ |
| R: 5′‐AACGCTTCACGAATTTGCGT‐3′ | |
| GAPDH | F: 5′‐TTCACCACCATGGAGAAGGC‐3′ |
| R: 5′‐GGCATGGACTGTGGTCATGA‐3′ | |
| Cel‐miR‐39 | F: GGTCACCGGGTGTAAATCAGCTTG |
Cel‐miR‐39, nematode microRNA 39; F, forward; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; miR, micro‐ribonucleic acid; NEK7, NIMA‐related kinase 7; R, reverse; U6, small nuclear RNA U6.
Detection of cytokines by enzyme‐linked immunosorbent assay
A double antibody sandwich enzyme‐linked immunosorbent assay kit (eBioscience, San Diego, CA, USA) was used to detect the levels of TNF‐α and IL‐1β in the peripheral blood and cell culture supernatant according to the instructions provided by the kit. A plate reader (BIO‐RAD, Hercules, CA, USA) was utilized to measure the absorbance (A) value at 450 nm.
Dual‐luciferase reporter experiment
The NEK7 3′untranslated region (UTR) dual‐luciferase reporter gene plasmids and mutant plasmids with mutations at the binding site with miR‐23a‐3p were constructed; namely, PmirGLO‐NEK7‐wild type and PmirGLO‐NEK7‐mutant type. Reporter plasmids were co‐transfected with miR‐23a‐3p mimic and NC plasmid into HEK293T cells (American Type Culture Collection, Manassas, VA, USA). The Dual‐Luciferase® Reporter Assay System (E1910; Promega, Madison, WI, USA) was adopted to detect luciferase activity. The activity of the Renilla luciferase was used as an internal reference, and the ratio of firefly luciferase activity‐to‐Renilla luciferase activity was expressed as the relative luciferase activity.
Western blot analysis
The total protein of tissues or cells was extracted by using phenylmethylsulfonyl fluoride and protease inhibitors. The supernatant was extracted after tissues or cells were allowed to lyse with radio‐immunoprecipitation assay lysis buffer (P0013B; Beyotime Biotechnology) at 4°C for 15 min, and centrifuged at 7,546.5 g for 15 min. The protein concentration was then measured using a bicinchoninic acid kit (23227; Thermo Fisher Scientific Inc., Waltham, MA, USA). Proteins were transferred onto a polyvinylidene fluoride membrane after polyacrylamide gel electrophoresis, and the membrane was blocked with 5% bovine serum albumin at room temperature for 1 h. The membrane was later incubated with primary anti‐rabbit antibodies from Abcam (Cambridge, UK) to NEK7 (ab133514), NEK6 (ab133494), NEK9 (ab235052), NLRP3 (ab214185), caspase‐1 p10 (ab179515), IL‐1β (ab9722), β‐actin (ab8226) and GSDMD (ab209845), and primary anti‐rabbit caspase‐1 antibody (#4199; CST, Framingham, MA, USA) overnight. The diluted horseradish peroxidase‐labeled goat anti‐rabbit immunoglobulin G (ab205718; Abcam) was added and incubated with the membrane at room temperature for 1.5 h. After incubation, the developer reagent (NCI4106; Pierce, Rockford, IL, USA) was added for development. ImageJ 1.48u software (Bio‐Rad) was used for protein quantitative analysis.
Establishment of a rat model of type 2 diabetes mellitus 15
In this substudy, 88 male Wistar rats (weighing 180–220 g, aged 4–6 weeks) were selected. The rats were housed in specific pathogen‐free cages, and the surrounding environment was comprised of sterile, dry, constant temperature conditions, with water that had been disinfected with in high temperature conditions and food that had been disinfected with ultraviolet light. Eight Wistar rats were randomly selected as the normal control, and the remaining rats were designated as type 2 diabetes mellitus models. Type 2 diabetes mellitus rats were fed with a high‐fat diet (containing 2% cholesterol, 0.2% propylthiouracil, 0.3% sodium cholate, 7.5% lard, and 90% basic feed) for 9 weeks. Afterwards, rats were intraperitoneally injected with streptozotocin (S0130; Sigma, Santa Clara, CA, USA), which was diluted with citric acid buffer to a 1% streptozotocin solution first, at 60 mg/kg. Rats in the normal control group were given the same amount of citric acid buffer for two consecutive weeks. The fasting blood glucose of rats was measured by obtaining blood samples from the tail vein on the seventh day after injection. Rats with a random blood glucose >16.7 mmol/L were classified as type 2 diabetes mellitus rats. The metabolic parameters of normal rats and type 2 diabetes mellitus rats are listed in Table S1. Type 2 diabetes mellitus rats were injected with shRNA NEK7, miR‐23a‐3p agomir (miR‐23a‐3p) and miR‐23a‐3p antagomir (miR‐23a‐3p IN), and corresponding NCs (20 μmol/L in 5 μL of saline) through the tail vein, or stimulated with 5 μL of ATP (NLRP3 activator). Small interfering RNA, stimulants or inhibitors were injected into the model rats daily for three consecutive days, and controls were disrupted for two consecutive days to allow sufficient overexpression or inhibition before sample collection or further behavioral testing. All rats underwent oral gavage continuously for 4 weeks. During the intervention period, normal rats continued to be fed with common diet, whereas type 2 diabetes mellitus rats continued to be fed with a high‐sugar and high‐fat diet. All rats had free access to water. After four consecutive weeks of oral gavage, all rats were deprived of food, but not water, overnight, anesthetized by intraperitoneal injection of 2% sodium pentobarbital, followed by the collection of blood from the carotid artery. The blood was allowed to stand for 15 min, and was subsequently centrifuged. A blood glucose meter was used to measure blood glucose by the paper strip method, and rat liver and kidney tissues were collected to measure various indicators.
Glucose tolerance test
Rats were allowed to fast overnight and were given 50% glucose solution (2 g/kg) by oral gavage. Blood was collected by the tail incision method. A glucometer was used to measure blood glucose levels at oral glucose tolerance test (OGTT)‐0 min, OGTT‐15 min, OGTT‐30 min, OGTT‐60 min and OGTT‐120 min. The area under the glucose curve was calculated according to the trapezoidal formula.
Insulin tolerance test
Rats were intraperitoneally injected with normal short‐acting insulin (0.75 U/kg body mass) after 6 days of fasting, and blood samples were taken from the tail vein to determine blood glucose levels at 0, 30, 60, 90, 120, 150 and 180 min after injection to evaluate the presence of insulin resistance in type 2 diabetes mellitus rats.
Biochemical analysis
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured according to the kit instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). After rats underwent fasting for 12 h, glucose tolerance and insulin tolerance were measured using Roche Blood Glucose Test Strips (Roche Diagnostics Limited Company, Mannheim, Germany) at 0, 15, 30, 60 and 120 min after glucose stimulation.
Hematoxylin–eosin staining of rat tissue
Tissue specimens of rats were fixed in 4% paraformaldehyde solution for 30–50 min, dehydrated, cleared, immersed in wax, embedded and sectioned. The tissue sections were flattened and loaded onto a glass slide, dried in a 45°C incubator, dewaxed, and then washed with descending concentrations of alcohol and distilled water for 5 min each. Shortly after, the sections were left to stain with hematoxylin for 5 min, differentiated with 1% hydrochloric acid ethanol for 3 s, and were left to stain with 5% eosin for approximately 3 min. The sections were dehydrated, cleared and mounted for observation under a microscope.
Insulin immunohistochemistry
After the preparation of paraffin‐embedded pancreatic sections, the endogenous peroxidase activity was blocked to prevent non‐specific binding. The sections were then incubated with mouse monoclonal primary insulin Ab‐6 (INS04 + INS05) antibodies at a concentration of 0.5–1 μg/mL for 30 min at room temperature. After buffer washing, samples were incubated with biotinylated goat anti‐polyvalent secondary antibodies for 10 min. Samples were cultured with streptavidin peroxidase for 10 min according to the manuals of the insulin Ab‐6 kit (Lab Vision Corporation‐Thermo Fisher Scientific). The samples were cultured with Di Amino benzidine Plus chromogen and substrate for 5–15 min, counterstained with Mayer’s hematoxylin, and cover slipped for examination under a light microscope.
Statistical analysis
All measurement data are shown as the mean ± standard deviation and analyzed by SPSS 21.0 software (IBM, Armonk, NY, USA), with P < 0.05 as a level of statistically significance. If data conformed to normal distribution and homogeneity of variance, an unpaired t‐test was used to compare unpaired data between two groups. Data comparisons among multiple groups were carried out using a one‐way analysis of variance (anova) and Tukey’s post‐hoc test. Data comparisons between groups at different time points were carried out using repeated measures of anova and the Bonferroni post‐hoc test. The correlation between NEK7 and miR‐23a‐3p expression was analyzed using Pearson’s correlation coefficient.
Results
miR‐23a‐3p is lowly expressed and NEK7 is highly expressed in type 2 diabetes mellitus patients and rats
RT–qPCR results showed that miR‐23a‐3p expression was downregulated and NEK7 was highly expressed (Figure 1a,b) in type 2 diabetes mellitus patients, when compared with that of healthy individuals’ blood. In addition, patients were arranged into the low‐expression group and the high‐expression group (50% being the cut‐off value) based on the expression of miR‐23a‐3p. The fasting blood glucose and insulin levels in the high‐expression group were significantly lower than those in the low‐expression group (Figure 1c,d). A Pearson correlation analysis showed a negative correlation between miR‐23a‐3p and NEK7 expression (Figure 1e). RT–qPCR results showed that miR‐23a‐3p expression was lower and NEK7 expression was higher (Figure 1f,g) in the serum of rats with type 2 diabetes mellitus than in control rats. Western blot analysis showed increased expression of NEK7 in liver and kidney tissues of type 2 diabetes mellitus rats, whereas RT–qPCR showed low expression of miR‐23a‐3p (Figure 1h,i), compared with that of the control rats.
Figure 1.
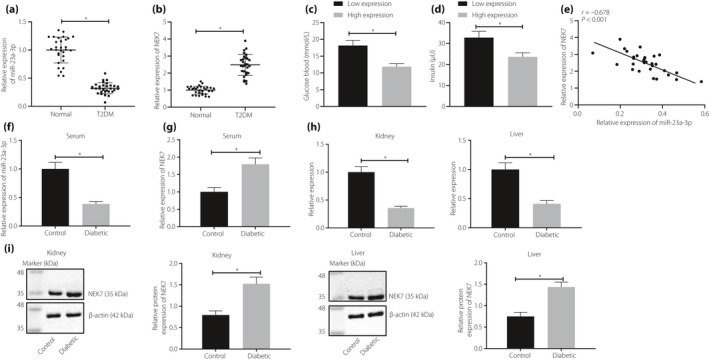
Micro‐ribonucleic acid (miR)‐23a‐3p is downregulated, but NIMA‐related kinase 7 (NEK7) is upregulated in patients and rats with type 2 diabetes mellitus (T2DM). (a) miR‐23a‐3p and (b) NEK7 expressions detected in blood samples from patients with type 2 diabetes mellitus and healthy people (30 participants each) detected using reverse transcription quantitative polymerase chain reaction (RT–qPCR). (c) Oral glucose tolerance test and (d) insulin tolerance test results of patients with high or low expression of miR‐23a‐3p. (e) Pearson’s correlation analysis of correlation between miR‐23a‐3p and NEK7. The expression levels of (f) miR‐23a‐3p and (g) NEK7 in serum of type 2 diabetes mellitus and control rats detected by RT–qPCR (n = 8). (h) miR‐23a‐3p expression in the liver and kidneys of type 2 diabetes mellitus rats and control rats detected by RT–qPCR (n = 8). (i) NEK7 in the liver and kidneys from type 2 diabetes mellitus rats and control rats detected by western blot analysis. *P < 0.05. Measurement data were expressed by the mean ± standard deviation. Comparison between two groups was carried out by an unpaired t‐test. The correlation between NEK7 and miR‐23a‐3p was analyzed using Pearson’s correlation coefficient.
miR‐23a‐3p inhibits expression of NLRP3 inflammatory bodies by targeting NEK7
TargetScan database (http://www.targetscan.org/vert_72/) was first used to predict the presence of a targeted binding site between miR‐23a‐3p and NEK7 3′UTR. Dual‐luciferase reporter gene assay showed that miR‐23a‐3p and wild‐type NEK7 3′UTR co‐transfection indicated a significant reduction of luciferase activity, whereas miR‐23a‐3p and mutant‐type NEK7 3′UTR co‐transfection showed no significant difference in luciferase activity (Figure 2a,b). Based on RT–qPCR, after transfection with miR‐23a‐3p mimic, miR‐23a‐3p expression was significantly increased, and NEK7 and NLRP3 expression was decreased, which was contrary to after the transfection with miR‐23a‐3p inhibitor (Figure 2c,d). The co‐immunoprecipitation results (Figure 2e) showed that the number of complexes formed by NEK7 and NLRP3 was increased in diabetic rats, suggesting that NEK7 activated NLRP3 in a protein‐binding‐dependent manner under diabetic conditions.
Figure 2.
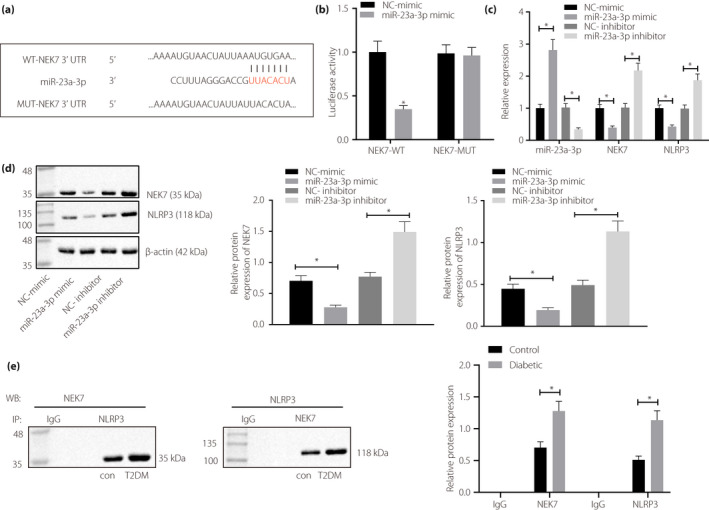
Micro‐ribonucleic acid (miR)‐23a‐3p negatively targets NIMA‐related kinase 7 (NEK7) to suppress the activation of nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3) inflammatory bodies. (a) TargetScan software was used to predict the binding of miR‐23a‐3p on NEK7, and to identify the site of miR‐23a‐3p binding to NEK7 by sequence comparison. (b) Dual‐luciferase reporter gene assay was used to validate the binding of miR‐23a‐3p to the 3′untranslated region of NEK7 in HEK‐293T cells. (c) The expression of miR‐23a‐3p, NLRP3 and NEK7 was detected by reverse transcription quantitative polymerase chain reaction after miR‐23a‐3p was either overexpressed or inhibited in BMDM cells isolated from normal rats. (d) A Western blot analysis was carried out to detect the expression of NEK7 and NLRP3 in BMDM cells isolated from normal rats after overexpression or inhibition of miR‐23a‐3p. (e) Co‐immunoprecipitation was carried out to detect the binding of NEK7 and NLRP3 in kidney tissues of diabetic rats (8 rats in each group). *P < 0.05. Measurement data are presented by the mean ± standard deviation. Comparison between two groups was carried out by an unpaired t‐test. Data comparison among multiple groups was carried out using a one‐way anova and Tukey’s post‐hoc test. IgG, immunoglobulin G; NC, negative control; T2DM, type 2 diabetes mellitus.
miR‐23a‐3p inhibits pyroptosis caused by activation of NEK7/NLRP3 inflammatory bodies
After the addition of LPS + ATP, miR‐23a‐3p expression weakened, whereas NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1 and GSDMD p30 (pyroptosis in innate immunity is caused by cleavage of a common substrate protein, GSDMD, by the activated cysteine proteases caspase‐1 and caspase‐4/5/11 16 ) had elevated levels in BMDM, and IL‐1β and TNF‐α levels in the supernatant were enhanced, accompanied with conspicuous cell pyroptosis (the morphology of the cells expanded and a large number of empty vesicles appeared), which was reversed after additional miR‐23a‐3p mimic treatment (Figure 3a‐f). Furthermore, in miR‐23a‐3p inhibitor‐transfected BMDM treated with LPS + ATP, miR‐23a‐3p expression was reduced; however, IL‐1β and TNF‐α levels in the supernatant and NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1 and GSDMD p30 expression in cells were increased, along with aggravated cell pyroptosis (the morphology of the cells expanded and a large number of empty vesicles appeared; Figures 3e‐j). After NEK7 was silenced in BMDM, which was then added with LPS + ATP, the levels of IL‐1β and TNF‐α in the supernatant decreased, and expression of NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1 and GSDMD p30 in cells was also diminished with reduced cell pyroptosis, whereas the expression of miR‐23a‐3p still remained unchanged (Figure 3e‐j). In BMDMs treated with both LPS + ATP and miR‐23a‐3p inhibitor, NEK3 silencing reduced the levels of IL‐1β and TNF‐α in the supernatant, the expression of NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1, and GSDMD p30, as well as pyroptosis in cells; on the contrary, the expression of miR‐23a‐3p remained unchanged (Figure 3e‐j).
Figure 3.
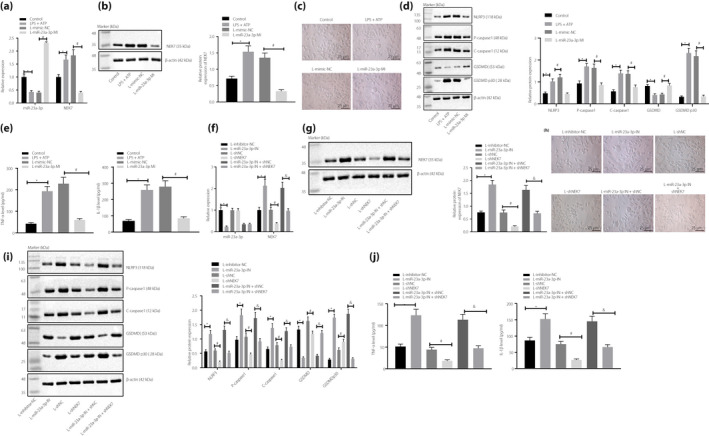
Micro‐ribonucleic acid (miR)‐23a‐3p suppresses pyroptosis by inactivating NIMA‐related kinase 7 (NEK7)/nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3) inflammatory bodies. (a) Reverse transcription quantitative polymerase chain reaction (RT–qPCR) was used to detect miR‐23a‐3p and NEK7 expressions in cells after treatment with miR‐23a‐3p mimic and lipopolysaccharide (LPS) + adenosine‐5′‐triphosphate (ATP). (b) Expression of NEK7 protein in cells after treatment with miR‐23a‐3p mimic and LPS + ATP detected by western blot analysis. (c) Cellular morphology observed by microscope (×400). (d) Expression of NLRP3, pro‐caspase‐1, cle‐caspase‐1, gasdermin D (GSDMD) and GSDMD p30 in cells after treatment with miR‐23a‐3p mimic and LPS + ATP detected by western blot analysis. (e) The levels of interleukin (IL)‐1β and tumor necrosis factor (TNF)‐α in the supernatant after treatment with miR‐23a‐3p mimic and LPS + ATP detected by enzyme‐linked immunosorbent assay. (f) A RT–qPCR was used to detect miR‐23a‐3p and NEK7 expression in cells after treatment with short hairpin (sh)‐NEK7, miR‐23a‐3p inhibitor and LPS + ATP. (g) Expression of NEK7 protein in cells after treatment with sh‐NEK7, miR‐23a‐3p inhibitor and LPS + ATP detected by western blot analysis. (h) Morphology observed by microscope in cells after treatment with sh‐NEK7, miR‐23a‐3p inhibitor and LPS + ATP (×400). (i) Expression of NLRP3, pro‐caspase‐1, cle‐caspase‐1 and GSDMD p30 in cells after treatment with sh‐NEK7, miR‐23a‐3p inhibitor and LPS + ATP detected by western blot analysis. (j) The levels of IL‐1β and TNF‐α in the supernatant after treatment with sh‐NEK7, miR‐23a‐3p inhibitor and LPS + ATP detected by enzyme‐linked immunosorbent assay. Values were presented as the mean ± standard error of the mean, and compared by a one‐way analysis of variance. *P < 0.05; # P < 0.05; & P < 0.05. The measurement data was expressed by the mean ± standard deviation, and the comparison of data among multiple groups was carried out by a one‐way anova. Tukey’s was used for post‐hoc tests.
miR‐23a‐3p inhibits the occurrence of type 2 diabetes mellitus by inhibiting pyroptosis in vivo
RT–qPCR results showed that miR‐23a‐3p expression was reduced, in contrast to that of the normal rats, whereas NEK7 expression was elevated in BMDMs of rats with type 2 diabetes mellitus (Figure 4a). In type 2 diabetes mellitus rats, miR‐23a‐3p was downregulated, whereas NEK7 was upregulated (Figure 4b,c); liver weight, liver‐to‐bodyweight ratio, serum ALT and AST levels (Figure 4d,e), and insulin and glucose tolerance (Figure 4f) were significantly elevated when compared with that of normal rats. Meanwhile in type 2 diabetes mellitus rats, IL‐1β and TNF‐α both showed increased expression (Figure 4g); rat kidney and liver presented with pathological changes (Figure 4h), and the expression of NLRP3, pro‐caspase‐1 and cle‐caspase‐1 was enhanced (Figure 4i) compared with that of normal rats. These changes were negated by injection with miR‐23a‐3p agomir (Figure 4a‐h). In the presence of miR‐23a‐3p agomir, ATP treatment increased liver weight, liver‐to‐bodyweight ratio, serum ALT and AST levels, insulin tolerance and glucose tolerance, IL‐1β and TNF‐α levels, pathological changes of rat kidneys and livers, and NLRP3, pro‐caspase‐1 and cle‐caspase‐1 expression (Figure 4d‐i). In addition, insulin immunohistochemistry results showed that insulin levels were diminished in type 2 diabetes mellitus rats, compared with that of the normal rats, which was restored by additional miR‐23a‐3p agomir treatment. However, in the presence of miR‐23a‐3p agomir, the addition of ATP decreased insulin levels in type 2 diabetes mellitus rats (Figure 4j).
Figure 4.
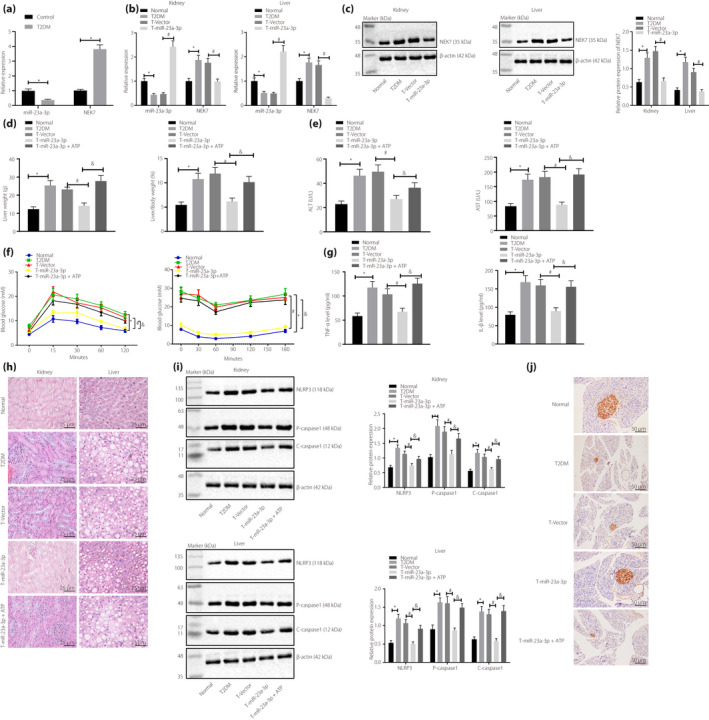
Micro‐ribonucleic acid (miR)‐23a‐3p upregulation results in inhibition of type 2 diabetes mellitus (T2DM) occurrence in rats by suppressing pyroptosis. Normal rats were used as controls, and type 2 diabetes mellitus rats were either injected or not injected with vectors, miR‐23a‐3p agomir or miR‐23a‐3p agomir + adenosine‐5′‐triphosphate (ATP). Reverse transcription quantitative polymerase chain reaction was carried out to detect (a) miR‐23a‐3p and (b) NEK7 expression in the liver and kidney tissues of type 2 diabetes mellitus (T2DM) rats. (c) Western blot analysis was carried out to detect NEK7 expression in liver and kidney tissues of type 2 diabetes mellitus rats. (d) Liver weight and liver‐to‐bodyweight measurements of type 2 diabetes mellitus rats. (e) Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in type 2 diabetes mellitus rats. (f) Detection of insulin tolerance and glucose tolerance in type 2 diabetes mellitus rats. (g) Enzyme‐linked immunosorbent assay detection of serum interleukin (IL)‐1β and tumor necrosis factor (TNF)‐α levels in type 2 diabetes mellitus rats. (h) Hematoxylin–eosin staining to observe the pathological changes of rat kidney and liver tissues of type 2 diabetes mellitus rats (×400). (i) Western blot analysis was carried out to detect NIMA‐related kinase 7 (NEK7), nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3), pro‐caspase‐1, cle‐caspase‐1 and c‐caspase‐1, expression in type 2 diabetes mellitus rats (n = 8). (j) Pancreatic insulin abundance measured by insulin immunohistochemistry (×200). *P < 0.05; # P < 0.05; & P < 0.05. Measurement data were expressed by the mean ± standard deviation. Data comparisons among multiple groups were performed using the one‐way anova and Tukey’s post‐hoc test. Data comparisons between groups at different time points were carried out using repeated measures analysis of variance and post‐hoc testing was carried out using the Bonferroni post‐hoc test.
miR‐23a‐3p targets NEK7 to inhibit NLRP3 activation‐caused pyroptosis and to relieve type 2 diabetes mellitus in rats
miR‐23a‐3p antagomir treatment triggered the reduction of miR‐23a‐3p expression and the increase of NEK7 expression in type 2 diabetes mellitus rats (Figure 5a,b), along with the elevation of liver weight and liver‐to‐bodyweight ratio, serum ALT and AST levels, insulin tolerance and glucose tolerance (Figure 5c‐e), kidney and liver pathological changes, serum IL‐1β and TNF‐α levels (Figure 5f‐h), and NLRP3, pro‐caspase‐1 and cle‐caspase‐1 expression (Figure 5i). These results were contrary to that of NEK7‐silenced type 2 diabetes mellitus rats (Figures 5a‐i). Furthermore, insulin immunohistochemistry showed that the insulin levels in type 2 diabetes mellitus rats were reduced after treatment with miR‐23a‐3p antagomir, but were enhanced after silencing NEK7 (Figure 5g). Co‐treatment with miR‐23a‐3p antagomir and sh‐NEK7 restored the effects of the single treatment of miR‐23a‐3p antagomir or sh‐NEK7 in type 2 diabetes mellitus rats (Figures 5a‐i).
Figure 5.
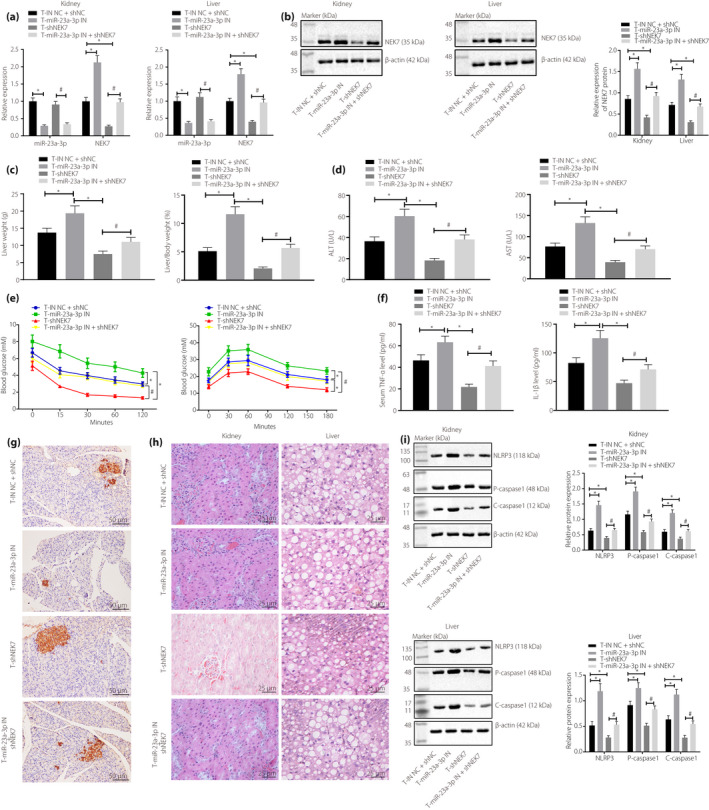
Micro‐ribonucleic acid (miR)‐23a‐3p targets NIMA‐related kinase 7 (NEK7) to suppress pyroptosis caused by nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3) inflammation body activation and to attenuate type 2 diabetes mellitus in rats. Type 2 diabetes mellitus rats were injected with inhibitor negative control (NC) + short hairpin (sh)‐NC, miR‐23a‐3p antagomir, sh‐NEK7 or miR‐23a‐3p antagomir + sh‐NEK7. (a) A reverse transcription quantitative polymerase chain reaction was carried out to detect miR‐23a‐3p and NEK7 levels in the liver and kidney tissues of type 2 diabetes mellitus rats. (b) A Western blot analysis was carried out to detect NEK7 expression in the liver and kidney tissues of type 2 diabetes mellitus rats. (c) Liver weight and liver‐to‐bodyweight ratio measurements in type 2 diabetes mellitus rats. (d) Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in type 2 diabetes mellitus rats. (e) Detection of insulin tolerance and glucose tolerance in type 2 diabetes mellitus rats. (f) Enzyme‐linked immunosorbent assay detection of the levels of interleukin (IL)‐1β and tumor necrosis factor (TNF)‐α in serum. (g) Insulin levels determined by insulin immunohistochemistry experiment (×200). (h) Hematoxylin–eosin staining to observe the pathological changes of rat kidney and liver tissues (×400). (i) Western blot analysis detection of expression of NEK7, NLRP3, pro‐caspase‐1, cle‐caspase‐1 and c‐caspase‐1 (n = 8). *P < 0.05; # P < 0.05. Measurement data are expressed by the mean ± standard deviation. Data comparisons among multiple groups were carried out using the one‐way anova and Tukey’s post‐hoc test. Data comparisons between groups at different time points were carried out using repeated measures analysis of variance; post‐hoc testing was carried out using the Bonferroni post‐hoc test.
Discussion
Type 2 diabetes mellitus is a growing worldwide healthcare burden, and is tightly associated with the widespread prevalence of obesity 17 . It is the consequence of disrupted insulin secretion and resistance, and can also be described as an imbalance of carbohydrate, protein and lipid metabolisms 17 . Over the recent decades of investigations, the understanding of type 2 diabetes mellitus has been developing and making progress expeditiously. However, it is a complicated chronic condition that demands perpetual healthcare, together with the necessity of patient self‐care for monitoring aberrant glucose level fluctuations, both making it a disorder that generates a burden for healthcare. Therefore, studies aiming to develop and improve more effective and safer treatments are at of importance.
miRNAs are short non‐coding RNAs (17–24 nucleotides) that bind to the 3′UTRs of targeted messenger RNAs to modulate gene expression by translational suppression or degradation 10 . miRNAs have been discovered to regulate metabolisms correlated with type 2 diabetes mellitus 18 , including the insulin pathway and glucose homeostasis, emphasizing their feasibility as therapeutic targets in treating obesity and metabolic disorders. The plasma of patients with cardiovascular diseases has been observed to carry inhibited expression of miR‐23 19 , and some research has shown that miR‐23 plays a role in glucose metabolism in dog muscles 20 . In the current study, we identified that the expression of miR‐23a‐3p and NEK7 was negatively correlated in patients and rat models with type 2 diabetes mellitus; more specifically, miR‐23a‐3p is downregulated, whereas NEK7 is upregulated. With the prediction functionality of TargetScan software in predicting the presence of a targeted binding site between miR‐23a‐3p and NEK7 3′UTR, we have proved that miR‐23a‐3p negatively regulates NEK7 expression by binding to its 3′UTR region. This negative regulation affects the expression of NLRP3 inflammatory bodies, which is also consistent with another finding that NEK7 regulates the activation of NLRP3 inflammatory bodies 21 .
Recent studies have shed light on novel forms of programmed cell death, such as necroptosis and pyroptosis. Pyroptosis is a pro‐inflammatory form of programmed cell death that depends on the enzymatic function of inflammatory proteases that are part of cysteine‐dependent aspartate‐specific caspase family 22 . Pyroptosis has been mostly reported to occur in phagocytes of the myeloid lineage, including neutrophils, macrophages and dendritic cells, epithelial cells, endothelial cells, and neurons. One underlying potential mechanism is that the aforementioned cells show higher expression levels of inflammatory caspases that could promote pyroptosis, such as caspase‐1 22 . Inflammasomes are multimeric protein structures that govern inflammatory reactions and pyroptosis as part of a host’s defenses to prevent microbial invasion 23 . The sensors of inflammasomes are closely related to apoptosis‐associated speck‐like protein, which contains an apoptosis‐associated speck‐like protein containing a C‐terminal caspase recruitment domain, could trigger inflammatory caspase‐1 to allow the secretion of inflammatory cytokines and initiate programmed cell death to enable the host with the defensive responses in fighting pathogens 23 . The capability of NLRs to construct inflammasomes was originally attributed to the NLF‐family pyrin domain‐containing 1 (NLRP1) 24 , 25 . It has also been reported that the increased production of reactive oxygen species and thiol oxidation could stimulate NEK7‐dependent NLRP3 activation, and that a decrease in NADH level and mitochondrial reactive oxygen species level results in the dysregulation of cellular glycolytic flux that would further promote NLRP3 inflammasomes and pyroptosis 26 , 27 . In addition, we found that miR‐23a‐3p inhibits pyroptosis through the inactivation of NEK7/NLRP3 inflammatory bodies, which have been verified by overexpressing miR‐23a‐3p, depleting miR‐23a‐3p or depleting NEK7 experiment, as shown.
In brief, we have provided evidence that miR‐23a‐3p could target NEK7 to further inhibit the activation of NLRP3, which suppressed pyroptosis. Hence, the symptoms of type 2 diabetes mellitus in rats could be mitigated (Figure 6). These results gave us implications that the miR‐23a‐3p–NEK7–NLRP3 axis could be utilized as a potential therapeutic target in the alleviation or treatment of type 2 diabetes mellitus in the future.
Figure 6.
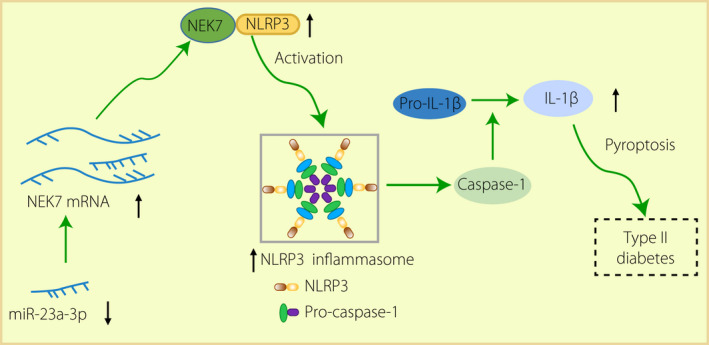
The mechanism of Micro‐ribonucleic acid (miR)‐23a‐3p in type 2 diabetes mellitus. In type 2 diabetes mellitus rats, miR‐23a‐3p was downregulated and NIMA‐related kinase 7 (NEK7) was upregulated to activate pyroptosis caused by nucleotide‐binding oligomerization‐like receptor family pyrin domain containing 3 (NLRP3) inflammatory body activation, thus inducing the development of type 2 diabetes mellitus. IL, interleukin; mRNA, messenger ribonucleic acid.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Metabolic parameters of normal rats and type 2 diabetes mellitus rats.
Acknowledgments
This study was supported by Key Scientific Research Project Plan of Colleges and Universities of Education Department in Henan Province (No. 17A320022); Education Department of Henan Province (No. 192102310052).
J Diabetes Investig 2021; 12: 334–345
Contributor Information
Hongye Chang, Email: 287757199@qq.com.
Hongjuan Chang, Email: changhj0812@126.com.
References
- 1. Cookson BT, Brennan MA. Pro‐inflammatory programmed cell death. Trends Microbiol 2001; 9: 113–114. [DOI] [PubMed] [Google Scholar]
- 2. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 2005; 73: 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Donath MY, Ehses JA, Maedler K, et al. Mechanisms of beta‐cell death in type 2 diabetes. Diabetes 2005; 54(Suppl 2): S108–S113. [DOI] [PubMed] [Google Scholar]
- 5. Bonner‐Weir S. Perspective postnatal pancreatic beta cell growth. Endocrinology 2000; 141: 1926–1929. [DOI] [PubMed] [Google Scholar]
- 6. Taylor R, Al‐Mrabeh A, Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol 2019; 7: 726–736. [DOI] [PubMed] [Google Scholar]
- 7. Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol 2014; 5: 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Improta Caria AC, Nonaka CKV, Pereira CS, et al. Exercise training‐induced changes in MicroRNAs: beneficial regulatory effects in hypertension, type 2 diabetes, and obesity. Int J Mol Sci 2018; 19: 3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Candia P, Spinetti G, Specchia C, et al. A unique plasma microRNA profile defines type 2 diabetes progression. PLoS One 2017; 12: e0188980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lozano‐Bartolome J, Llaurado G, Portero‐Otin M, et al. Altered expression of miR‐181a‐5p and miR‐23a‐3p is associated with obesity and TNFalpha‐Induced insulin resistance. J Clin Endocrinol Metab 2018; 103: 1447–1458. [DOI] [PubMed] [Google Scholar]
- 11. Yang Z, Chen H, Si H, et al. Serum miR‐23a, a potential biomarker for diagnosis of pre‐diabetes and type 2 diabetes. Acta Diabetol 2014; 51: 823–831. [DOI] [PubMed] [Google Scholar]
- 12. Byrne M, Nasir N, Basmadjian C, et al. Nek7 conformational flexibility and inhibitor binding probed through protein engineering of the R‐spine. Biochem J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharif H, Wang L, Wang WL, et al. Structural mechanism for NEK7‐licensed activation of NLRP3 inflammasome. Nature 2019; 570: 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rovira‐Llopis S, Apostolova N, Banuls C, et al. Mitochondria, the NLRP3 inflammasome, and sirtuins in type 2 diabetes: new therapeutic targets. Antioxid Redox Signal 2018; 29: 749–791. [DOI] [PubMed] [Google Scholar]
- 15. Yu CY, Yang CY, Rui ZL. MicroRNA‐125b‐5p improves pancreatic beta‐cell function through inhibiting JNK signaling pathway by targeting DACT1 in mice with type 2 diabetes mellitus. Life Sci 2019; 224: 67–75. [DOI] [PubMed] [Google Scholar]
- 16. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 17. DeFronzo RA, Ferrannini E, Groop L, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers 2015; 1: 15019. [DOI] [PubMed] [Google Scholar]
- 18. Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol 2012; 13: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han H, Qu G, Han C, et al. MiR‐34a, miR‐21 and miR‐23a as potential biomarkers for coronary artery disease: a pilot microarray study and confirmation in a 32 patient cohort. Exp Mol Med 2015; 47: e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herrera Uribe J, Vitger AD, Ritz C, et al. Physical training and weight loss in dogs lead to transcriptional changes in genes involved in the glucose‐transport pathway in muscle and adipose tissues. Vet J 2016; 208: 22–27. [DOI] [PubMed] [Google Scholar]
- 21. He Y, Zeng MY, Yang D, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016; 530: 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol 2016; 26: R568–R572. [DOI] [PubMed] [Google Scholar]
- 23. Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol 2017; 29: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bertin J, Nir WJ, Fischer CM, et al. Human CARD4 protein is a novel CED‐4/Apaf‐1 cell death family member that activates NF‐kappaB. J Biol Chem 1999; 274: 12955–12958. [DOI] [PubMed] [Google Scholar]
- 25. Inohara N, Koseki T, del Peso L, et al. Nod1, an Apaf‐1‐like activator of caspase‐9 and nuclear factor‐kappaB. J Biol Chem 1999; 274: 14560–14567. [DOI] [PubMed] [Google Scholar]
- 26. Gross CJ, Mishra R, Schneider KS, et al. K(+) Efflux‐independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 2016; 45: 761–773. [DOI] [PubMed] [Google Scholar]
- 27. Sanman LE, Qian Y, Eisele NA, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 2016; 5: e13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Metabolic parameters of normal rats and type 2 diabetes mellitus rats.