

Abstract
Pre‐mRNAs from thousands of eukaryotic genes can be non‐canonically spliced to generate circular RNAs (circRNAs) that have covalently linked ends. Most mature circular RNAs are expressed at low levels, but some have known physiological functions and/or accumulate to higher levels than their associated linear mRNAs. These observations have sparked great interest into this class of previously underappreciated RNAs and prompted the development of new experimental approaches to study them, especially methods to measure or modulate circular RNA expression levels. Nonetheless, each of these approaches has caveats and potential pitfalls that must be controlled for when designing experiments and interpreting results. Here, we provide practical advice, tips, and suggested guidelines for performing robust identification, validation, and functional characterization of circular RNAs. Beyond promoting rigor and reproducibility, these suggestions should help bring clarity to the field, especially how circular RNAs function and whether these transcripts may sponge microRNAs/proteins or serve as templates for translation.
Keywords: backsplicing, circRNA, microRNA sponge, RNase R, translation
Subject Categories: Methods & Resources, RNA Biology
This review provides advice and guidelines for performing robust identification, validation, and characterization of circular RNAs and discusses how to experimentally assess specific functions of circRNAs.

Glossary
- 4sU
4‐thiouridine
- cDNA
Complementary DNA
- CDR1
Cerebellar degeneration related protein 1
- CDR1as
CDR1 antisense RNA
- circRNA
Circular RNA
- ciRS‐7
Circular RNA sponge for miR‐7
- CRISPR
Clustered regularly interspaced short palindromic repeats
- DNA
Deoxyribonucleic acid
- eIF4E
Eukaryotic translation initiation factor 4E
- HEK293
Human embryonic kidney 293 cells
- IRES
Internal ribosome entry site
- m7G
7‐methylguanosine
- MCS
Multicloning site
- miR‐7
MicroRNA‐7
- miRNA
MicroRNA
- MRE
MicroRNA recognition element
- mRNA
Messenger RNA
- nt
Nucleotide
- oligo(dT)
Oligodeoxythymidylic acid
- poly(A)
Polyadenosine
- pre‐mRNA
Precursor messenger RNA
- RNA
Ribonucleic acid
- RNA‐seq
RNA sequencing
- RNase
Ribonuclease
- rRNA
Ribosomal RNA
- RT
Reverse transcriptase
- RT–PCR
Reverse transcription–polymerase chain reaction
- shRNA
Short hairpin RNA
- siRNA
Small interfering RNA
- snRNA
Small nuclear RNA
- tRNA
Transfer RNA
Introduction
Most eukaryotic genes contain intronic sequences that must be removed from nascent pre‐mRNAs by the spliceosome in a highly regulated fashion (reviewed in Nilsen & Graveley, 2010; Fica & Nagai, 2017; Shi, 2017). Many introns and exons can be alternatively included or excluded, which allows a variety of stable, mature transcripts to be produced from a single gene. Besides generating linear RNAs, many protein‐coding genes and some non‐coding RNA genes can produce covalently closed circular RNAs (circRNAs) when a splice donor (5′ splice site) is joined to an upstream splice acceptor (3′ splice site) via a form of alternative splicing called backsplicing (Fig 1A) (reviewed in Wilusz, 2018; Kristensen et al, 2019; Patop et al, 2019; Chen, 2020; Xiao et al, 2020). The first circular RNAs in eukaryotic cells were identified in the early 1990s (Nigro et al, 1991; Cocquerelle et al, 1992; Capel et al, 1993), and the field has exploded in recent years as high‐throughput sequencing projects have identified tens of thousands of previously missed circular transcripts (Salzman et al, 2012; Jeck et al, 2013; Memczak et al, 2013). Some circular RNAs, e.g., CDR1as/ciRS‐7, are highly conserved, accumulate to levels that exceed that of their cognate linear mRNA, and have clear molecular functions (Hansen et al, 2011; Hansen et al, 2013; Memczak et al, 2013; Piwecka et al, 2017). Other circular RNAs have been associated with developmental or disease processes (reviewed in Patop & Kadener, 2017; Knupp & Miura, 2018). It is thus perhaps not surprising that there has been great excitement and many groups entering the field, with over 7,000 new manuscripts that contain the keyword “circRNA” indexed on PubMed since 2012. Here, we aim to provide practical advice, tips, and suggested guidelines for identifying, validating, and characterizing circular RNAs. We break down some of the computational and experimental approaches that are often used in the field, stressing best practices and ways to avoid common pitfalls. Our goal is to help investigators ensure their circular RNA experiments are performed rigorously as well as aid the community in their efforts to critically assess articles in this rapidly growing field.
Figure 1.
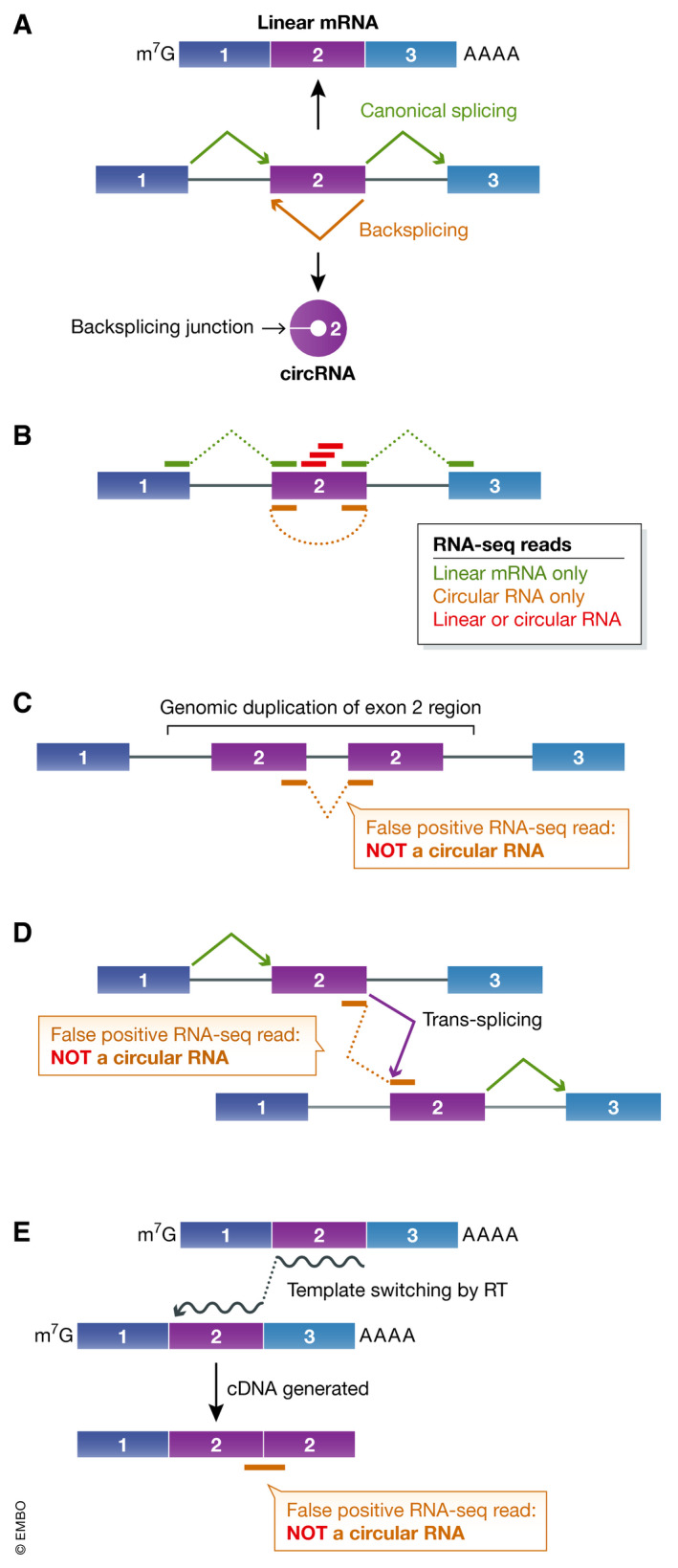
Annotation of circular RNAs using RNA‐seq reads that span backsplicing junctions.
(A) Splice sites can be joined in a linear order by the pre‐mRNA splicing machinery to generate a canonical linear mRNA that is also capped and polyadenylated (top). Alternatively, a pre‐mRNA can be subjected to backsplicing, which yields a circular RNA whose ends are covalently linked (bottom). (B) High‐throughput RNA‐seq reads can be used to annotate RNA isoforms generated from a gene locus. In this example, red sequencing reads do not span a splicing junction and cannot distinguish linear vs. circular RNA isoforms, whereas the green reads are consistent with production of the canonically spliced linear mRNA. The orange read suggests the existence of a transcript that has the end of exon 2 joined to the beginning of exon 2. This is consistent with a backsplicing event that results in production of a circular RNA from exon 2 (as in (A)). (C–E) However, the orange sequencing read may not be derived from a circular RNA backsplicing junction and thus may represent a false positive. (C) If exon 2 is duplicated in the genomic DNA, transcription of the gene results in a linear RNA that has the apparent backsplicing junction. (D) If pre‐mRNAs derived from the gene are subjected to a trans‐splicing event, a linear RNA that has the apparent backsplicing junction is produced. (E) During cDNA synthesis, reverse transcriptase (RT) can dissociate from a template RNA and resume extension from a second template, which can also result in a false‐positive backsplicing junction.
RNA‐seq datasets provide some, but not all of the critical data that are needed to support the existence of a circular RNA
Most circular RNAs have been identified using high‐throughput RNA‐seq followed by computationally finding spliced reads that span backsplicing junctions (reviewed in Szabo & Salzman, 2016) (orange read in Fig 1B). Circular RNAs do not end in poly(A) tails so they are depleted (but not completely absent) from RNA‐seq libraries that were generated using an oligo(dT) affinity purification step. Ribosomal RNA (rRNA) depletion should instead be used for library preparation, although differences in the efficiencies of commercially available rRNA depletion kits have been noted (Zinshteyn et al, 2020). Reads spanning backsplicing junctions typically represent a tiny amount (< 1%) of the overall sequencing data, but, as one would expect, biochemically enriching for circular RNAs (discussed further below) or increasing the length of RNA‐seq reads can help with their identification. A number of computational algorithms have been developed to identify backsplicing reads (reviewed in Szabo & Salzman, 2016), but there are three important caveats that must be considered regardless of the annotation approach one selects.
First, sequencing reads that appear to span a backsplicing junction may not, in fact, be derived from a circular RNA. Rearrangements in the genomic DNA (e.g., those that result in a tandem duplication of exons) (Fig 1C) or trans‐splicing events, in which the spliceosome joins together exons from two independent pre‐mRNAs (Fig 1D), each can yield sequencing reads that resemble backsplicing junctions, but are derived from linear RNAs (Chuang et al, 2018). In addition, reverse transcriptase can switch RNA templates during cDNA synthesis (Kulpa et al, 1997), creating junctions that do not naturally exist in cellular RNAs (Fig 1E). Therefore, before focusing in detail on any particular circular RNA candidate, it is good to check whether it is flanked by canonical splice sites and resistant to digestion by the exonuclease RNase R (discussed below).
Second, circular RNA detection algorithms yield divergent results, in part because of differences in how they identify reads spanning backsplicing junctions. Some algorithms are guided by genome annotations, while others make de novo predictions (reviewed in Szabo & Salzman, 2016). Regardless of the algorithm, there are often many false positives (10–20%) in the set of circular RNAs predicted, and these false positives may sometimes be as high as 45% (Hansen et al, 2016; Zeng et al, 2017; Hansen, 2018). When the same RNA‐seq dataset was analyzed using five different algorithms, the most highly abundant circular RNAs were generally predicted by all algorithms but ~ 40% of all putative circular RNAs were predicted with only a single approach (Hansen et al, 2016). Some of these transcripts may represent true circular RNAs, but these results underscore the importance of using at least two algorithms for annotating circular RNAs (Hansen, 2018). In our group, we typically have used CIRCexplorer2 (Zhang et al, 2016a) and CIRI2 (Gao et al, 2018) for annotation, and only used the intersecting set of circular RNA predictions for downstream analyses. It should be noted that circular RNAs that are listed in databases like circBase (Glazar et al, 2014) or assayed by commercially available microarrays were not all annotated by the same approach (or even a stringent approach), so appropriate caution should always be applied.
Third, RNA‐seq reads are typically short (< 250 bp) and often do not capture the entire circular RNA sequence (Fig 1B). This leaves the internal structure of the transcript ambiguous (Fig 2). It has often been assumed that all introns are removed from mature multi‐exonic circular RNAs (e.g., this is how circular RNAs are annotated in circBase). However, there are now many examples of alternative splicing events in circular RNAs, including retained introns or inclusion of exons that are never observed in the cognate linear mRNA (Li et al, 2015; Gao et al, 2016; Zhang et al, 2016a; preprint: Rahimi et al, 2019). Follow‐up sequencing of RT–PCR products is thus critical for defining the exact sequence of a circular RNA as well as determining if there may, in fact, be multiple distinct circular RNAs that share the same backsplicing junction (Fig 2). It is also now possible to use high‐throughput long‐read sequencing platforms, e.g., Oxford Nanopore, to define full‐length circular RNA sequences (preprint: Rahimi et al, 2019). For clarity and to promote reproducibility, the full sequences of all investigated circular RNAs should be experimentally determined and provided in all published works, e.g., in supplemental material.
Figure 2.
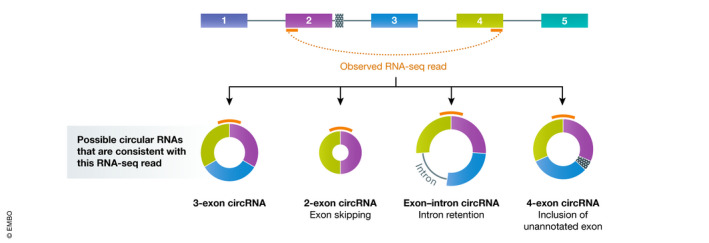
RNA‐seq reads that span backsplicing junctions often do not reveal the internal structure of the circular RNA
An example RNA‐seq read (orange) that connects the end of exon 4 to the beginning of exon 2. This may be derived from a circular RNA that contains the three annotated exons or from a circular RNA that has been alternatively spliced. Follow‐up analyses, e.g., using RT–PCR or Northern blots, are required to determine which circular RNA isoform(s) are, in fact, present in cells.
There is not yet a broadly accepted consensus for how to name circular RNAs, but the HUGO Gene Nomenclature Committee has suggested they be named circ[gene symbol]‐n, where n is an iterative five‐digit number (with the first identified circular RNA for a given gene being 00001) (Seal et al, 2020).
RNase R digests many, but not all linear RNAs
Once a set of circular RNA candidates has been identified, the next critical step is to validate that the transcripts have covalently linked ends and are resistant to digestion by RNA exonucleases. RNase R is commonly used for this purpose as it is a highly processive 3′–5′ exonuclease that contains helicase activity and can digest many linear RNAs (Jeck et al, 2013; Hossain et al, 2016; Zhang et al, 2016c). However, it is important to recognize that RNase R often does not digest all linear RNAs, especially those with structured 3′ ends, such as histone mRNAs, snRNAs, and tRNAs (Panda et al, 2017; Xiao & Wilusz, 2019) (Fig 3). This is because RNase R only binds to transcripts that have a 3′ single‐stranded tail of at least seven nucleotides (Vincent & Deutscher, 2006). Therefore, to better deplete RNAs with structured 3′ ends, poly(A) polymerase (e.g., from Escherichia coli) can be used in an in vitro reaction to add poly(A) tails to the 3′ ends of purified RNAs. This is then followed by either an oligo(dT) pulldown step (Panda et al, 2017) or incubation with RNase R (Xiao & Wilusz, 2019). Recent work has found that RNase R also abruptly stalls at internal G‐quadruplex structures, thereby only partially degrading linear RNAs that contain them (Xiao & Wilusz, 2019). G‐quadruplex structures are stabilized by K+ cations, which are present in commonly used RNase R reaction buffers, but not by cations with smaller ionic radius such as Li+. We showed that replacing K+ with Li+ in the reaction buffer was sufficient to enable RNase R to proceed through these sequences and fully degrade the linear RNAs (Xiao & Wilusz, 2019). Therefore, to obtain higher purity circular RNAs, we now suggest treating purified RNA samples with poly(A) polymerase followed by RNase R digestion in the presence of Li+ cations (Fig 3). Note, however, that this protocol still does not remove all linear RNAs. We have further noticed differences in digestion efficiencies across commercial lots of RNase R, so one should always verify that the reaction conditions used in an experiment lead to efficient digestion of housekeeping linear mRNAs. In addition, some circular RNAs (especially larger ones) have been found to be sensitive to RNase R digestion, especially when high RNase R concentrations are used, and this may be due to nicking by contaminating endonucleases.
Figure 3.
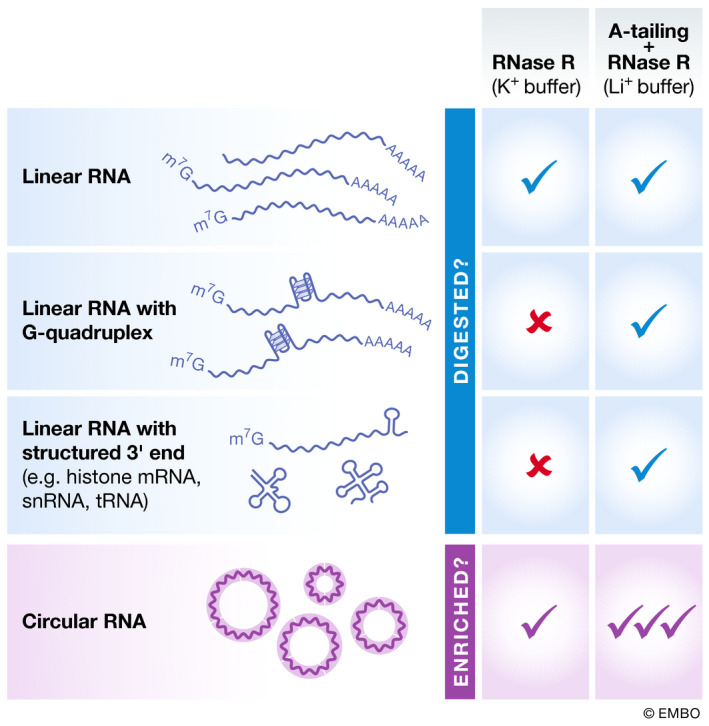
Differences in the sets of transcripts digested by RNase R depend on the assay conditions
RNase R digestions are typically performed in a K+ containing buffer, but this approach fails to remove many linear RNAs that contain G‐quadruplexes (G4) or have highly structured 3′ ends. By treating purified RNA samples with poly(A) polymerase followed by RNase R digestion in the presence of a Li+ containing buffer, linear RNAs are more efficiently removed. This allows circular RNAs to be more highly enriched.
Multiple complementary approaches need to be used to validate circular RNA expression
To confirm the expression of individual circular RNAs of interest, the easiest approach is RT–PCR using primers that amplify across the backsplicing junction (Panda & Gorospe, 2018). Unlike standard “convergent” primer pairs that face toward each other on the linear RNA (as well as on the corresponding cDNA and genomic DNA) (Fig 4A, green and purple primers), one must instead use “divergent” primers that face away from each other (Fig 4A, orange primers). These primers thus will not amplify cDNA derived from a linear RNA, but should instead be specific for the circular RNA due to its unique topology. Oligo(dT) should be avoided for cDNA synthesis (we instead typically use random hexamers) and, if desired, primers can be designed immediately next to one another so that the entire circular RNA sequence is amplified. Regardless of the primers selected, the amplified RT–PCR products should be sequenced to verify on‐target amplification and that the expected backsplicing junction is indeed used. For additional tips on performing and designing quantitative RT–PCR experiments, we recommend the Minimum Information for Publication of Quantitative Real‐Time PCR Experiments (MIQE) guidelines (Bustin et al, 2009; Udvardi et al, 2008).
Figure 4.
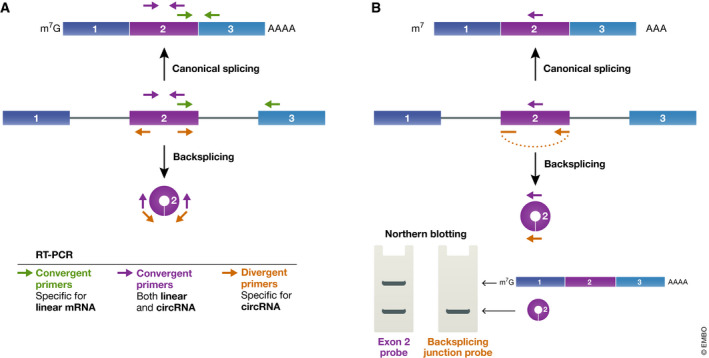
RT–PCR and Northern blotting to validate circular RNA expression
(A) By designing different primer pairs, RT–PCR can be used to quantify expression of the linear mRNA (green primers), the circular RNA (orange primers), or both transcripts (purple primers) derived from a gene. (B) Likewise, antisense oligonucleotides that are complementary to the middle of the exon (purple probe) or the backsplicing junction (orange probe) can be used as probes to visualize gene outputs using a Northern blot.
Just like when analyzing RNA‐seq data, it is important to remember that some RT–PCR products that appear to span a backsplicing junction may not be derived from a circular RNA (Fig 1C–E). Potential sources of artifacts again need to be ruled out. Potential DNA rearrangements can be dismissed by verifying that the divergent primers do not amplify genomic DNA, but keep in mind that the genomic DNA amplicon may be very long due to introns. Likewise, trans‐splicing events are fairly easy to rule out, as they generally result in linear RNAs that should become depleted after RNase R digestion. Artifacts due to template switching during cDNA synthesis are harder to control for, although comparing the results from two distinct reverse transcriptases may be useful as the enzymes are unlikely to jump at the same sequences. It is further important to check whether the predicted backsplicing junction is at consensus splice sites as well as validate circular RNAs of interest through orthologous approaches that do not involve reverse transcriptase.
In our laboratory, we prefer to use Northern blotting to validate circular RNA expression as this approach is both quantitative and independent of reverse transcriptase (Liang et al, 2017; Tatomer et al, 2017; Fig 4B). A probe complementary to the exon of interest can be used to confirm and quantify the relative amounts of linear vs. circular RNAs that accumulate from a given gene. Importantly, Northerns also allow one to determine if there are multiple different RNA isoforms (linear or circular) being generated, and the inclusion of an RNase R treatment can help one better distinguish linear from circular RNAs. One must nonetheless rule out cross‐hybridization artifacts and verify that multiple independent probes detect the transcripts. Keep in mind that a short (~ 20 nt) oligonucleotide probe complementary to the backsplicing junction should only detect the circular RNA (Fig 4B, orange probe) and thus such a probe provides no information as to whether the gene may produce linear mRNAs at significantly higher levels. We thus recommend that, at minimum, two Northern blots are shown: one that was hybridized with a probe complementary to the middle of the exon (detecting linear and circular RNAs, purple probe in Fig 4B) and a second that was hybridized with a probe complementary to the backsplicing junction (detecting only circular RNAs) (Fig 4B). Circular RNAs should migrate aberrantly (compared to linear RNAs of the same sequence) in polyacrylamide gels, although we find that they run fairly true to size in formaldehyde agarose gels. It must be stressed that the main drawback of Northerns is that they are much less sensitive than RT–PCR, so only the most abundant circular RNAs can typically be detected by this approach. RNase protection assays (Carey et al, 2013) may provide increased sensitivity compared to Northerns, but care needs to be taken when designing an RNA probe and interpreting the results. As an additional approach to prove circularity, RNase H, which cleaves RNA‐DNA hybrids, can be used (Starke et al, 2015). Cleavage of a circular RNA should result in a single linear fragment, whereas cleavage of a linear RNA should result in two fragments.
Regardless of the technique used to determine circular RNA levels, a key point to recognize is that steady‐state transcript levels are almost always what is being measured. Steady‐state RNA levels are determined by the interplay of RNA synthesis and degradation. Thus, one may be tempted to say that abundant circular RNAs arise from host genes that prefer backsplicing over canonical linear splicing, but 4‐thiouridine (4sU) labeling of nascent RNAs has shown that the efficiency of circular RNA production is generally very low (Zhang et al, 2016b). Hence, most circular RNAs accumulate to low levels because they are rarely produced. Circular RNA decay is nonetheless also very slow, and this can enable some circular RNAs to reach high levels that exceed that of their cognate linear mRNA.
Over‐expression of circular RNAs often also leads to production of undesired transcripts
Once a circular RNA of interest has been validated, a next obvious question to ask is what happens to cells when its expression is altered. The presence of inverted repeat sequences (e.g., Alu sequences) in the flanking introns can drive backsplicing reactions (Dubin et al, 1995; Jeck et al, 2013; Liang & Wilusz, 2014; Zhang et al, 2014), and thus, it is possible to over‐express circular RNAs using plasmids or viral vectors. These constructs typically consist of a promoter that drives expression of the exon(s) that circularize along with their immediate flanking sequences (Fig 5). The lack of flanking exons limits the types of undesired linear RNAs that can be produced. However, most circular RNA over‐expression constructs still backsplice at low efficiency and generate many undesired transcripts, including unspliced RNAs, concatamers, or trans‐spliced RNAs (Ho‐Xuan et al, 2020; Fig 5). These undesired RNAs all potentially limit the utility of these constructs for defining circular RNA functions. It is thus critical that Northern blots be performed to adequately characterize the outputs of any over‐expression system (Tatomer et al, 2017) and that such results be clearly presented in published works. We strongly recommend against using RT–PCR for this purpose as prominent by‐product RNAs can easily be missed, especially if only a single set of primers that amplify across the backsplicing junction is used.
Figure 5.
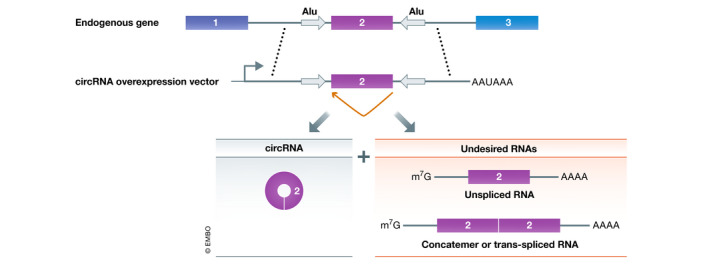
Over‐expression constructs can generate circular RNAs as well as other transcripts
Backsplicing can be induced when inverted repeats (gray arrows) in the flanking introns base pair to one another. To generate circular RNA over‐expression constructs, the exon that circularizes along with the immediate flanking sequences is often cloned. This can result in backsplicing (orange) and the production of the circular RNA, but a number of linear transcripts that are not desired are also often produced from these vectors.
We suggest the use of the Drosophila Laccase2 or human ZKSCAN1 flanking intronic sequences for over‐expressing circular RNAs, as these natural intronic sequences have inverted repeat sequences and have been shown to produce more circular RNA than constructs containing artificial intronic repeats (Kramer et al, 2015). Plasmids that have easy‐to‐use restriction sites between the introns are available (pcDNA3.1(+) Laccase2 MCS Exon and pcDNA3.1(+) ZKSCAN1 MCS Exon, respectively), and a variety of single exon circular RNAs (ranging in size from 300 to 1,500 nucleotides) have been produced in cells using this approach (Kramer et al, 2015; Meganck et al, 2018; Garikipati et al, 2019). It should be noted that these plasmids backsplice more efficiently than our originally described over‐expression plasmids (e.g., pcDNA3.1(+) CircRNA Mini Vector) (Liang & Wilusz, 2014), but they nonetheless still generate undesired transcripts besides the circular RNA of interest. For example, recent work has shown that the ZKSCAN1 introns can result in low levels of trans‐spliced RNAs that can be translated into protein (Ho‐Xuan et al, 2020). Therefore, to test whether any observed phenotype is truly due to over‐expression of a circular RNA, one should test constructs that lack one or both of the intronic repeats (as this should eliminate circular RNA generation from the construct) and confirm that the phenotype has been lost.
Beyond using plasmids or viral vectors to over‐express circular RNAs, it is becoming increasingly common to also test the effect of transfecting into cells purified circular RNAs that were made in vitro. A number of approaches have been described, including ones based on ribozymes (Ford & Ares, 1994; Wesselhoeft et al, 2018; Litke & Jaffrey, 2019), splint ligation (Moore, 1999), and chemical methods (Petkovic & Muller, 2015; Müller & Appel, 2017). There are currently conflicting data as to whether circular RNAs made in vitro trigger innate immune responses when added to cells (Chen et al, 2017; Wesselhoeft et al, 2018; Wesselhoeft et al, 2019; Chen et al, 2019b). The discrepancy may be due to differences in circular RNA sequence/nucleotide modification status or how efficiently reaction by‐products/impurities were removed by purification procedures. Immune responses thus need to be kept in mind, but ideally one should be able to show similar phenotypes regardless of whether the circular RNA was over‐expressed from a plasmid/viral vector or made in vitro and subsequently transfected.
Knocking down (or out) a circular RNA can have unintended effects on the cognate linear mRNA
The flip side to an over‐expression experiment is, of course, to deplete or fully knock out the transcript of interest, but this task is challenging when trying to manipulate circular RNAs. The sequence of the mature circular RNA often fully overlaps with that of its cognate linear mRNA, leaving the backsplicing junction as the only distinguishing (and thus targetable) feature in the mature circular RNA (Fig 1A). Small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) complementary to the backsplicing junction have been successfully used to deplete some endogenous circular RNAs (Zheng et al, 2016; Legnini et al, 2017; Chen et al, 2019a; Pamudurti et al, 2020; Suenkel et al, 2020). However, such an approach sometimes also results in reduced levels of the cognate linear mRNA, likely due to miRNA‐type silencing effects from pairing of the seed sequence to the mRNA (Bartel, 2018). This can make it impossible to determine whether an observed phenotype is due to depletion of the circular RNA, the linear mRNA, or a combination of both transcripts. Recent work from Pamudurti and colleagues described a number of methods to computationally determine si/shRNA off‐target effects, including ways to look for downregulated mRNAs that have sequence complementary to the si/shRNA (Pamudurti et al, 2020).
Because the backsplicing junction is the only unique, targetable feature within a mature circular RNA, a second si/shRNA is often used that has the region of complementarity shifted by a few nucleotides. This is an okay, but not ideal solution. Stronger genetic evidence for circular RNA functionality comes from knockdown‐rescue experiments. Here, the endogenous circular RNA is depleted with an si/shRNA, and one then tests whether the phenotypes can be rescued by a construct that expresses the circular RNA (with an altered backsplicing junction to make it resistant to the si/shRNA). Besides si/shRNAs, emerging work indicates that CRISPR/Cas13 systems can be targeted to backsplicing junctions to deplete circular RNAs (Li et al, 2020; Zhang et al, 2021), but some caution may need to be applied as some Cas13 enzymes are known to cause significant collateral damage and cleavage of bystander RNAs (Zhang et al, 2018).
Making a circular RNA knockout is feasible (Piwecka et al, 2017; Xia et al, 2018) but has significant challenges, especially if the goal is to prove that the circular RNA and not the overlapping, cognate linear mRNA is responsible for a phenotype. CDR1as/ciRS‐7 has been knocked out in mice by using CRISPR/Cas9 to remove the entire exon that circularizes, and this resulted in impaired sensorimotor gating (Piwecka et al, 2017). For this particular locus, the cognate linear transcript is very lowly expressed and the protein‐coding CDR1 gene on the opposite strand seems to never be transcribed. The observed brain phenotypes thus are most likely due to removal of the CDR1as/ciRS‐7 circular RNA, but one must keep in mind that additional transcripts besides the circular RNA have been removed and it remains unknown if they contribute in any way to the phenotype. It is thus not ideal to generate knockouts by removing the entire exon(s) that are backspliced, and a cleaner approach may instead be to prevent circular RNA production by removing one or both of the complementary repeats in the flanking introns that drive backsplicing (Zheng et al, 2016; Zhang et al, 2016b; Xia et al, 2018; Yoshimoto et al, 2020). This approach is, however, technically challenging as some circular RNAs are flanked by no repeats (and thus there is nothing obvious to delete) or many repeats, some of which may compensate when one repeat is deleted to result in residual expression of the circular RNA. One must, of course, keep in mind that deletion of an intronic repeat may affect linear mRNA levels or alternative splicing events. Therefore, demonstrating that any observed phenotype is rescued by re‐expressing the circular RNA remains the gold standard.
Most circular RNAs are not highly expressed, so stoichiometry must be considered before proposing a sponging model
The CDR1as/ciRS‐7 circular RNA has been studied in most detail and has driven great interest in the field as it represents a striking example of a functional circular RNA. CDR1as/ciRS‐7 is highly expressed in the brain and harbors > 60 evolutionarily conserved binding sites (also known as microRNA recognition elements, MREs) for miR‐7, which suggests it has the potential to sequester (or sponge) this microRNA and prevent miR‐7 from regulating expression of its target mRNAs (Hansen et al, 2013; Memczak et al, 2013). These results have motivated countless other groups to examine whether additional circular RNAs may function as microRNA sponges, also known as competing endogenous RNAs (ceRNAs) (Ebert et al, 2007; Salmena et al, 2011). A few additional circular RNAs (e.g., mouse Sry) do indeed have many microRNA binding sites (Hansen et al, 2013), but computational evidence indicates that the vast majority of circular RNAs are not enriched in microRNA binding sites (Guo et al, 2014; Enuka et al, 2016). There have nonetheless been hundreds of published reports claiming that many different endogenous circular RNAs function as microRNA sponges. Here, we discuss why such dichotomous conclusions have been reached and suggest that most reported circular RNAs likely are not microRNA sponges.
The ability of a microRNA to interact with a target transcript (regardless if it is linear or circular) is tightly linked to the relative abundance of that target in the overall pool of cellular RNAs (Fig 6). As the expression of one target mRNA or circular RNA increases, it theoretically can bind and titrate the microRNA away from other transcripts that have target sites, thereby de‐repressing the expression of these other transcripts (Ebert et al, 2007; Salmena et al, 2011; Thomson & Dinger, 2016). However, quantitative measurements have shown that, at minimum, thousands of new microRNA target sites are typically needed in order to induce significant changes in the set of mRNAs that are bound by a given microRNA (Bosson et al, 2014; Denzler et al, 2014; Jens & Rajewsky, 2015; Denzler et al, 2016). Most reported circular RNAs have a single predicted target site (or a few at most) for a given microRNA (Fig 6, left), which suggests the transcript must be present in thousands of copies per cell for any functional effect on microRNA targeting to likely be observed. Alternatively, the circular RNA must be present at a very high concentration in a specific subcellular domain for localized regulation of a microRNA to occur.
Figure 6.
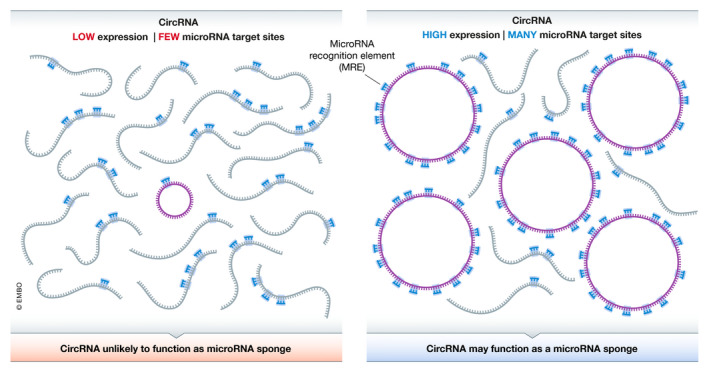
Stoichiometry dictates whether a circular RNA can function as a microRNA sponge
(Left) A circular RNA that is expressed at low levels and has a single microRNA recognition element (MRE) is unlikely to out‐compete other cellular transcripts for binding to that microRNA. (Right) Only when a circular RNA is highly expressed and has many MREs, does it have the potential to titrate away the microRNA and serve as a sponge.
Focusing again on CDR1as/ciRS‐7, this is one of the top expressed circular RNAs and is orders of magnitude more abundant than the average circular RNA. Yet, CDR1as/ciRS‐7 is still only present at ~ 200–300 copies per HEK293 cell (Memczak et al, 2013). The presence of > 60 miR‐7 binding sites per CDR1as/ciRS‐7 transcript allows it to potentially bind thousands of miR‐7 molecules per cell, which is consistent with a potential sponge function (Fig 6, right). Nonetheless, it is now clear that CDR1as/ciRS‐7 functions in a more nuanced way (Piwecka et al, 2017). The sponging model predicts that the expression of miR‐7 target genes should decrease when CDR1as/ciRS‐7 is knocked out (as miR‐7 can now more efficiently bind those other targets), but the opposite phenotype was, in fact, observed. This is because miR‐7 levels are reduced ~ 2‐fold when CDR1as/ciRS‐7 is knocked out in mice (Piwecka et al, 2017). CDR1as/ciRS‐7 thus likely does not function as a standard microRNA sponge, but instead appears to function as part of a larger post‐transcriptional regulatory network that protects miR‐7 from degradation and/or controls its temporal and spatial localization (Piwecka et al, 2017; Kleaveland et al, 2018).
CDR1as/ciRS‐7 has further been described as a putative oncogene and microRNA sponge in various cancers, but recent in situ hybridization experiments have revealed that this circular RNA is often only expressed in stromal cells, not in the cancer cells themselves (Kristensen et al, 2020). Therefore, the observed correlations between expression of CDR1as/ciRS‐7 and specific cancer‐associated mRNAs are not due to a sponging function. Kristensen et al (2020) nicely showed that these correlations can instead be explained by different cancer‐to‐stromal cell ratios in the tumor specimens that were examined.
In the vast majority of studies that have claimed microRNA sponging roles for other circular RNAs, the stoichiometry of the endogenous circular RNA has not been adequately considered (Fig 6). Instead, the microRNA and circular RNA are often over‐expressed to non‐physiological levels and the outputs of reporter genes or cherry‐picked endogenous genes are then examined. Such experiments represent logical initial steps that can clarify whether one should further consider a microRNA sponging model. However, they should be seen as just the beginning and recent work from Kristensen et al (2020) underscores why one should always be cautious when interpreting correlations between circular RNA and mRNA expression. One needs to test whether similar sponging effects are observed when expressing a mutated circular RNA that lacks the microRNA binding sites. More importantly, one needs to accurately measure endogenous circular RNA and microRNA levels, e.g., using digital droplet PCR and a standard curve, as well as determine transcriptome‐wide effects on the expression of predicted targets of that microRNA after modulating circular RNA levels. If a circular RNA is present at 1 copy or less per cell, it near certainly has minimal or no effect on microRNA targeting and the transcriptome‐wide results should confirm that this is indeed the case. The same logic holds true when considering whether a circular RNA may serve as a sponge for other molecules. For a circular RNA to sponge a protein, the sum of the circular RNA copy number and the number of protein binding sites on that circular RNA must be high relative to the concentration of the protein (Schreiner et al, 2020). In addition, the circular RNA and protein must be localized in the same subcellular compartment, e.g., as determined by RNA fluorescence in situ hybridization and immunofluorescence. If a circular RNA is not present at high stoichiometry or in the proper location, a sponging model is highly unlikely.
Endogenous circular RNAs may be translated, but obtaining convincing evidence remains challenging
In eukaryotic cells, initiation of protein synthesis typically requires eIF4E to first recognize the mRNA 5′ 7‐methylguanosine (m7G) cap, as this step enables recruitment of additional initiation factors and the small (40S) subunit of the ribosome (reviewed in Jackson et al, 2010). Circular RNAs lack a cap so alternate initiation mechanisms are required for ribosome recruitment. It has long been known that artificial circular RNAs can be translated when a viral internal ribosome entry site (IRES) is present (Chen & Sarnow, 1995). This has made it very tempting to speculate that endogenous circular RNAs may likewise have IRES elements and be translated to yield proteins that have critical cellular functions, e.g., during stress. However, there is still ongoing debate as to whether endogenous circular RNAs are, in fact, translated (Guo et al, 2014; Legnini et al, 2017; Pamudurti et al, 2017; Tatomer & Wilusz, 2017; Yang et al, 2017; Stagsted et al, 2019; van Heesch et al, 2019; Ho‐Xuan et al, 2020; Weigelt et al, 2020).
One of the strongest and most obvious pieces of evidence for translation of a circular RNA is detection of the encoded protein product by mass spectrometry. However, this task is difficult as (i) most circular RNAs are expressed at low levels, (ii) IRES‐driven translation mechanisms are typically not efficient (often only 1–10% as efficient as cap‐dependent initiation (Merrick, 2004)), and (iii) only those peptides that span the backsplicing junction are truly informative. Such junction spanning peptides have been found for some circular RNAs, but an open question is how many of these peptides represent false positives, especially when very large proteomics datasets have been analyzed. The same caveats hold true for ribosome profiling experiments (reviewed in Ingolia et al, 2019) where one looks for ribosome‐protected fragments that span backsplicing junctions. Such fragments are rare but, in some cases, they can be detected. The question now is whether they represent true translation events or false positives that come from regions of RNA that are protected from nuclease digestion by non‐ribosomal proteins. Further complicating matters, standard quality control metrics (Fields et al, 2015) that significantly increase the robustness of ribosome profiling data cannot be applied to circular RNAs. For example, one cannot confirm 3‐nucleotide periodicity of ribosome‐protected fragments on circular RNAs as it is impossible to determine whether a fragment is derived from a linear or circular RNA when the backsplicing junction is not present in the sequencing read.
Ribosome profiling or mass spectrometry can thus be informative starting points, but follow‐up analyses are critical to support any claim that a circular RNA is translated. Antibodies should be generated to detect endogenous circular RNA‐derived proteins and polysome profiling of the endogenous transcripts can be especially informative. Note that a puromycin treatment control must be run in parallel to confirm that co‐sedimentation of the circular RNA with polysomes is lost when translation is terminated. One can try to over‐express or deplete the circular RNA and look for corresponding changes in translational output, but one must remember all the caveats that we discussed earlier regarding these approaches. In particular, recent work from the Meister laboratory (Ho‐Xuan et al, 2020) should be considered a must read for anyone planning to use circular RNA over‐expression plasmids to study translation. In that manuscript, the authors describe a set of controls that are absolutely critical for determining whether the circular RNA or undesired RNAs made from a plasmid are the transcripts, in fact, being translated. Without these controls, the data are not interpretable. If a circular RNA is indeed translated, it likely contains an IRES element that should show activity using well‐established bicistronic reporters, but one must again rule out common experimental artifacts (e.g., cryptic promoters or splicing events) in these assays (Thompson, 2012). In total, we want to stress the importance of looking at the translational outputs of endogenous loci and using properly controlled follow‐up experiments to confirm any findings.
Concluding remarks
Circular RNAs were once considered to be rare errors in pre‐mRNA splicing, but it is now clear that these transcripts are much more widespread than previously appreciated. This has led to a tremendous influx of new studies into how these transcripts are made, regulated, and function (reviewed in Wilusz, 2018; Kristensen et al, 2019; Patop et al, 2019; Chen, 2020; Xiao et al, 2020). Here, we have highlighted how every step in studying circular RNAs (from initial identification to functional characterization) is inherently complicated, in large part due to the fact that mature circular RNA sequences overlap with that of their cognate linear mRNA. It is thus critical that appropriate experimental controls be included and that the caveats of each method are recognized. We have stressed the use of multiple complementary approaches at each step to ensure that results are robust, reproducible, and correctly interpreted. Suggestions and tips from our own experience have been provided, but this review is not intended to be exhaustive and other methodologies can be equally valid (e.g., also see Li et al, 2018; Tsitsipatis & Gorospe, 2021).
Going forward, it is important to recognize that only a very small subset of circular RNAs identified by RNA‐seq experiments have so far been validated or studied in sufficient detail, so many important questions about circular RNAs remain to be addressed (see Box 1 “In need of answers”). In particular, more work is needed to address how many circular RNAs are important for cellular physiology and to clarify if some of these transcripts simply represent noise or splicing errors. These efforts will be challenging and will require continued improvements to current methodologies, but they should reveal unprecedented insights into how circular RNAs act at the molecular level to impact normal development and disease processes.
Box 1. “In need of answers”.
What criteria should be used to confidently identify only true circular RNAs from RNA‐seq datasets?
Biochemical methods enable enrichment of circular RNAs but contaminating linear RNAs inevitably remain. Can approaches be further improved to enable the clean isolation of only circular RNAs from a pool of cellular total RNA?
Plasmids and viral vectors used to over‐express circular RNAs often generate undesired transcripts. Can improved constructs be developed that allow only the circular RNA of interest to be produced?
How many circular RNAs result in cellular phenotypes when over‐expressed or knocked down?
How many circular RNAs truly function to sponge microRNAs or RNA binding proteins?
How many endogenous circular RNAs are translated at significant levels? How is the ribosome assembled on these transcripts and what are the molecular functions of the encoded proteins?
What determines whether circular RNAs made in vitro induce an innate immune response?
Can circular RNAs be used as disease biomarkers or as therapeutic modalities?
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank all members of the Wilusz laboratory for helpful discussions and suggestions. Research on circular RNAs in our laboratory is supported by National Institutes of Health grants R35‐GM119735 and R01‐NS099371.
EMBO Reports (2021) 22: e52072
See the Glossary for abbreviations used in this article.
References
- Bartel DP (2018) Metazoan microRNAs. Cell 173: 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA (2014) Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56: 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL et al (2009) The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55: 611–622 [DOI] [PubMed] [Google Scholar]
- Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell‐Badge R (1993) Circular transcripts of the testis‐determining gene Sry in adult mouse testis. Cell 73: 1019–1030 [DOI] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, Smale ST (2013) The RNase protection assay. Cold Spring Harb Protoc 10.1101/pdb.prot071910 [DOI] [PubMed] [Google Scholar]
- Chen CY, Sarnow P (1995) Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268: 415–417 [DOI] [PubMed] [Google Scholar]
- Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, Iwasaki A, Chang HY (2017) Sensing self and foreign circular RNAs by intron identity. Mol Cell 67: 228–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Huang V, Xu X, Livingstone J, Soares F, Jeon J, Zeng Y, Hua JT, Petricca J, Guo H et al (2019a) Widespread and functional RNA circularization in localized prostate cancer. Cell 176: 831–843 [DOI] [PubMed] [Google Scholar]
- Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B et al (2019b) N6‐methyladenosine modification controls circular RNA immunity. Mol Cell 76: 96–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL (2020) The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol 21: 475–490 [DOI] [PubMed] [Google Scholar]
- Chuang TJ, Chen YJ, Chen CY, Mai TL, Wang YD, Yeh CS, Yang MY, Hsiao YT, Chang TH, Kuo TC et al (2018) Integrative transcriptome sequencing reveals extensive alternative trans‐splicing and cis‐backsplicing in human cells. Nucleic Acids Res 46: 3671–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerelle C, Daubersies P, Majerus MA, Kerckaert JP, Bailleul B (1992) Splicing with inverted order of exons occurs proximal to large introns. EMBO J 11: 1095–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M (2014) Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54: 766–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M (2016) Impact of microRNA levels, target‐site complementarity, and cooperativity on competing endogenous RNA‐regulated gene expression. Mol Cell 64: 565–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin RA, Kazmi MA, Ostrer H (1995) Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 167: 245–248 [DOI] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4: 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enuka Y, Lauriola M, Feldman ME, Sas‐Chen A, Ulitsky I, Yarden Y (2016) Circular RNAs are long‐lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 44: 1370–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fica SM, Nagai K (2017) Cryo‐electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat Struct Mol Biol 24: 791–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AP, Rodriguez EH, Jovanovic M, Stern‐Ginossar N, Haas BJ, Mertins P, Raychowdhury R, Hacohen N, Carr SA, Ingolia NT et al (2015) A regression‐based analysis of ribosome‐profiling data reveals a conserved complexity to mammalian translation. Mol Cell 60: 816–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Ares M Jr (1994) Synthesis of circular RNA in bacteria and yeast using RNA cyclase ribozymes derived from a group I intron of phage T4. Proc Natl Acad Sci USA 91: 3117–3121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wang J, Zheng Y, Zhang J, Chen S, Zhao F (2016) Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun 7: 12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhang J, Zhao F (2018) Circular RNA identification based on multiple seed matching. Brief Bioinform 19: 803–810 [DOI] [PubMed] [Google Scholar]
- Garikipati VNS, Verma SK, Cheng Z, Liang D, Truongcao MM, Cimini M, Yue Y, Huang G, Wang C, Benedict C et al (2019) Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF‐A axis. Nat Commun 10: 4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazar P, Papavasileiou P, Rajewsky N (2014) circBase: a database for circular RNAs. RNA 20: 1666–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Agarwal V, Guo H, Bartel DP (2014) Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15: 409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J (2011) miRNA‐dependent gene silencing involving Ago2‐mediated cleavage of a circular antisense RNA. EMBO J 30: 4414–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388 [DOI] [PubMed] [Google Scholar]
- Hansen TB, Veno MT, Damgaard CK, Kjems J (2016) Comparison of circular RNA prediction tools. Nucleic Acids Res 44: e58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB (2018) Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol 6: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, Witte F, Schneider‐Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL et al (2019) The translational landscape of the human heart. Cell 178: 242–260 [DOI] [PubMed] [Google Scholar]
- Hossain ST, Malhotra A, Deutscher MP (2016) How RNase R degrades structured RNA: role of the helicase activity and the S1 domain. J Biol Chem 291: 7877–7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho‐Xuan H, Glažar P, Latini C, Heizler K, Haase J, Hett R, Anders M, Weichmann F, Bruckmann A, Van den Berg D et al (2020) Comprehensive analysis of translation from overexpressed circular RNAs reveals pervasive translation from linear transcripts. Nucleic Acids Res 48: 10368–10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Hussmann JA, Weissman JS (2019) Ribosome profiling: global views of translation. Cold Spring Harb Perspect Biol 11: a032698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jens M, Rajewsky N (2015) Competition between target sites of regulators shapes post‐transcriptional gene regulation. Nat Rev Genet 16: 113–126 [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, Bartel DP (2018) A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174: 350–362.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupp D, Miura P (2018) CircRNA accumulation: a new hallmark of aging? Mech Ageing Dev 173: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, Wilusz JE (2015) Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev 29: 2168–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J (2019) The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20: 675–691 [DOI] [PubMed] [Google Scholar]
- Kristensen LS, Ebbesen KK, Sokol M, Jakobsen T, Korsgaard U, Eriksen AC, Hansen TB, Kjems J, Hager H (2020) Spatial expression analyses of the putative oncogene ciRS‐7 in cancer reshape the microRNA sponge theory. Nature Commun 11: 4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulpa D, Topping R, Telesnitsky A (1997) Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer‐associated reverse transcriptase errors. EMBO J 16: 856–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M et al (2017) Circ‐ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66: 22–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L et al (2015) Exon‐intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22: 256–264 [DOI] [PubMed] [Google Scholar]
- Li X, Yang L, Chen LL (2018) The biogenesis, functions, and challenges of circular RNAs. Mol Cell 71: 428–442 [DOI] [PubMed] [Google Scholar]
- Li S, Li X, Xue W, Zhang L, Yang LZ, Cao SM, Lei YN, Liu CX, Guo SK, Shan L et al (2020) Screening for functional circular RNAs using the CRISPR‐Cas13 system. Nat Methods 18: 51–59 [DOI] [PubMed] [Google Scholar]
- Liang D, Wilusz JE (2014) Short intronic repeat sequences facilitate circular RNA production. Genes Dev 28: 2233–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Tatomer DC, Luo Z, Wu H, Yang L, Chen LL, Cherry S, Wilusz JE (2017) The output of protein‐coding genes shifts to circular RNAs when the Pre‐mRNA processing machinery is limiting. Mol Cell 68: 940–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litke JL, Jaffrey SR (2019) Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat Biotechnol 37: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meganck RM, Borchardt EK, Castellanos Rivera RM, Scalabrino ML, Wilusz JE, Marzluff WF, Asokan A (2018) Tissue‐dependent expression and translation of circular RNAs with recombinant AAV vectors in vivo . Mol Ther Nucleic Acids 13: 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338 [DOI] [PubMed] [Google Scholar]
- Merrick WC (2004) Cap‐dependent and cap‐independent translation in eukaryotic systems. Gene 332: 1–11 [DOI] [PubMed] [Google Scholar]
- Moore MJ (1999) Joining RNA molecules with T4 DNA ligase. Methods Mol Biol 118: 11–19 [DOI] [PubMed] [Google Scholar]
- Müller S, Appel B (2017) In vitro circularization of RNA. RNA Biol 14: 1018–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B (1991) Scrambled exons. Cell 64: 607–613 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamudurti NR, Bartok O, Jens M, Ashwal‐Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez‐Hernandez D, Ramberger E et al (2017) Translation of CircRNAs. Mol Cell 66: 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamudurti NR, Patop IL, Krishnamoorthy A, Ashwal‐Fluss R, Bartok O, Kadener S (2020) An in vivo strategy for knockdown of circular RNAs. Cell Discov 6: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, De S, Grammatikakis I, Munk R, Yang X, Piao Y, Dudekula DB, Abdelmohsen K, Gorospe M (2017) High‐purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res 45: e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Gorospe M (2018) Detection and analysis of circular RNAs by RT‐PCR. Bio Protoc 8: e2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patop IL, Kadener S (2017) circRNAs in cancer. Curr Opin Genet Dev 48: 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patop IL, Wust S, Kadener S (2019) Past, present, and future of circRNAs. EMBO J 38: e100836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovic S, Muller S (2015) RNA circularization strategies in vivo and in vitro . Nucleic Acids Res 43: 2454–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwecka M, Glazar P, Hernandez‐Miranda LR, Memczak S, Wolf SA, Rybak‐Wolf A, Filipchyk A, Klironomos F, Cerda Jara CA, Fenske P et al (2017) Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357: eaam8526 [DOI] [PubMed] [Google Scholar]
- Rahimi K, Venø MT, Dupont DM, Kjems J (2019) Nanopore sequencing of full‐length circRNAs in human and mouse brains reveals circRNA‐specific exon usage and intron retention. bioRxiv 10.1101/567164 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner S, Didio A, Hung LH, Bindereif A (2020) Design and application of circular RNAs with protein‐sponge function. Nucleic Acids Res 48: 12326–12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RL, Chen LL, Griffiths‐Jones S, Lowe TM, Mathews MB, O'Reilly D, Pierce AJ, Stadler PF, Ulitsky I, Wolin SL et al (2020) A guide to naming human non‐coding RNA genes. EMBO J 39: e103777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y (2017) Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat Rev Mol Cell Biol 18: 655–670 [DOI] [PubMed] [Google Scholar]
- Stagsted LV, Nielsen KM, Daugaard I, Hansen TB (2019) Noncoding AUG circRNAs constitute an abundant and conserved subclass of circles. Life Sci Alliance 2: e201900398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung LH, Bindereif A (2015) Exon circularization requires canonical splice signals. Cell Rep 10: 103–111 [DOI] [PubMed] [Google Scholar]
- Suenkel C, Cavalli D, Massalini S, Calegari F, Rajewsky N (2020) A highly conserved circular RNA is required to keep neural cells in a progenitor state in the mammalian brain. Cell Rep 30: 2170–2179 [DOI] [PubMed] [Google Scholar]
- Szabo L, Salzman J (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17: 679–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Liang D, Wilusz JE (2017) Inducible expression of eukaryotic circular RNAs from plasmids. Methods Mol Biol 1648: 143–154 [DOI] [PubMed] [Google Scholar]
- Tatomer DC, Wilusz JE (2017) An unchartered journey for ribosomes: circumnavigating circular RNAs to produce proteins. Mol Cell 66: 1–2 [DOI] [PubMed] [Google Scholar]
- Thompson SR (2012) So you want to know if your message has an IRES? Wiley Interdiscip Rev RNA 3: 697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson DW, Dinger ME (2016) Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17: 272–283 [DOI] [PubMed] [Google Scholar]
- Tsitsipatis D, Gorospe M (2021) Practical guide for circular RNA analysis: steps, tips, and resources. Wiley Interdiscip Rev RNA 12: e1633 [DOI] [PubMed] [Google Scholar]
- Udvardi MK, Czechowski T, Scheible WR (2008) Eleven golden rules of quantitative RT‐PCR. Plant Cell 20: 1736–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent HA, Deutscher MP (2006) Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem 281: 29769–29775 [DOI] [PubMed] [Google Scholar]
- Weigelt CM, Sehgal R, Tain LS, Cheng J, Eßer J, Pahl A, Dieterich C, Grönke S, Partridge L (2020) An insulin‐sensitive circular RNA that regulates lifespan in Drosophila . Mol Cell 79: 268–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselhoeft RA, Kowalski PS, Anderson DG (2018) Engineering circular RNA for potent and stable translation in eukaryotic cells. Nature Commun 9: 2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselhoeft RA, Kowalski PS, Parker‐Hale FC, Huang Y, Bisaria N, Anderson DG (2019) RNA circularization diminishes immunogenicity and can extend translation duration in vivo . Mol Cell 74: 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE (2018) A 360 degrees view of circular RNAs: From biogenesis to functions. Wiley Interdiscip Rev RNA 9: e1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Wang S, Ye B, Du Y, Li C, Xiong Z, Qu Y, Fan Z (2018) A circular RNA protects dormant hematopoietic stem cells from DNA sensor cGAS‐mediated exhaustion. Immunity 48: 688–701 [DOI] [PubMed] [Google Scholar]
- Xiao MS, Wilusz JE (2019) An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G‐quadruplexes or structured 3' ends. Nucleic Acids Res 47: 8755–8769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MS, Ai Y, Wilusz JE (2020) Biogenesis and functions of circular RNAs come into focus. Trends Cell Biol 30: 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen L, Wang Y et al (2017) Extensive translation of circular RNAs driven by N6‐methyladenosine. Cell Res 27: 626–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto R, Rahimi K, Hansen TB, Kjems J, Mayeda A (2020) Biosynthesis of circular RNA ciRS‐7/CDR1as is mediated by mammalian‐wide interspersed repeats. iScience 23: 101345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Lin W, Guo M, Zou Q (2017) A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 13: e1005420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L (2014) Complementary sequence‐mediated exon circularization. Cell 159: 134–147 [DOI] [PubMed] [Google Scholar]
- Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, Chen LL, Yang L (2016a) Diverse alternative back‐splicing and alternative splicing landscape of circular RNAs. Genome Res 26: 1277–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Li X, Zhang J, Chen S, Zhang JL, Yang L, Chen LL (2016b) The biogenesis of nascent circular RNAs. Cell Rep 15: 611–624 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang L, Chen LL (2016c) Characterization of circular RNAs. Methods Mol Biol 1402: 215–227 [DOI] [PubMed] [Google Scholar]
- Zhang C, Konermann S, Brideau NJ, Lotfy P, Wu X, Novick SJ, Strutzenberg T, Griffin PR, Hsu PD, Lyumkis D (2018) Structural basis for the RNA‐guided ribonuclease activity of CRISPR‐Cas13d. Cell 175: 212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nguyen TM, Zhang X‐O, Wang L, Phan T, Clohessy JG, Pandolfi PP (2021) Optimized RNA‐targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol 22: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, Wangen JR, Hua B, Green R (2020) Nuclease‐mediated depletion biases in ribosome footprint profiling libraries. RNA 26: 1481–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G et al (2016) Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nature Commun 7: 11215 [DOI] [PMC free article] [PubMed] [Google Scholar]