

Abstract
Amino acid restriction is among promising potential cancer treatment strategies. However, cancer cells employ a multitude of mechanisms to mount resistance to amino acid restriction, which impede the latter’s clinical development. Here we show that MAPK signaling activation in asparagine‐restricted melanoma cells impairs GSK3‐β‐mediated c‐MYC degradation. In turn, elevated c‐MYC supports ATF4 translational induction by enhancing the expression of the amino acid transporter SLC7A5, increasing the uptake of essential amino acids, and the subsequent maintenance of mTORC1 activity in asparagine‐restricted melanoma cells. Blocking the MAPK‐c‐MYC‐SLC7A5 signaling axis cooperates with asparagine restriction to effectively suppress melanoma cell proliferation. This work reveals a previously unknown axis of cancer cell adaptation to asparagine restriction and informs mechanisms that may be targeted for enhanced therapeutic efficacy of asparagine limiting strategies.
Keywords: ATF4, c‐MYC, MAPK, melanoma, mTORC1
Subject Categories: Cancer, Metabolism
This study demonstrates that blocking MAPK cooperates with asparagine restriction to effectively suppress melanoma cell proliferation.
Introduction
Cancer cells are characterized by high proliferation rates, which entails sustained nutrient supply to support their amplified macromolecular biosynthesis (anabolism). Recognizing this, there has been a notable increase in efforts to evaluate the therapeutic value of nutrient restriction approaches. Due to toxicities associated with what could otherwise be a rational therapeutic approach, targeting of glucose metabolism has fallen short of translating into meaningful clinical interventions (Ganapathy‐Kanniappan & Geschwind, 2013).
Owing to enhanced protein synthesis, cancer cells also exhibit a heightened dependence on vascular amino acid availability (Kang, 2020). Mammalian cells express the enzymes needed to synthesize a subset of amino acid, referred to as non‐essential amino acids (NEAA). However, given their exorbitant biosynthetic needs, cancer cells still critically rely on vascular NEAA supply, prompting the evaluation of vascular amino acid restriction approaches (Kim et al, 2009; Egler et al, 2016; Maddocks et al, 2017; Gwinn et al, 2018). Moreover, some tumors either lack or express very low levels of enzymes required for NEAA biosynthesis, rendering them auxotrophic and dependent on vascular availability of these amino acids (Feun et al, 2008; Egler et al, 2016).
Asparagine synthetase (ASNS) is the cellular enzyme that catalyzes asparagine biosynthesis, which requires aspartate and glutamine, with the latter as the amido group donor. Recognizing a lack or extremely low expression of ASNS in acute lymphoblastic leukemia (ALL), asparagine hydrolyzing drug L‐asparaginase (L‐A’ase) received approval for clinical use in ALL (Egler et al, 2016). However, solid malignancies such as pancreatic cancer and melanoma, which also exhibit very low ASNS expression, have failed to show encouraging response to L‐A’ase (Lessner et al, 1980; Taylor et al, 2001; Bachet et al, 2015). Decrease in the levels of charged tRNAs consequent to depleted intracellular amino acid levels activate GCN2 kinase (Kilberg et al, 2009). Activated GCN2 phosphorylates and inactivates the translation initiation factor eukaryotic initiation factor 2α (eIF2α), an event that marks suppression of cap‐dependent translation, while enhancing the translation of mRNAs such as activating transcription factor 4 (ATF4) that harbor upstream open reading frames (uORF) in their 5′ untranslated region (UTR) (Dever et al, 1992; Harding et al, 2003). ATF4 promotes the expression of a large array of genes, including amino acid biosynthesis enzymes and amino acid transport proteins, which help orchestrate cellular adaptive response to amino acid restriction (Kilberg et al, 2009).
Previously, we showed that MAPK signaling is activated and critical for the induction of ATF4 and its target gene ASNS, which confers resistance to asparagine restriction in melanoma and pancreatic tumors (Pathria et al, 2019). Harboring basic helix‐loop‐helix and leucine zipper structural motifs, transcription factor c‐MYC is a key regulator of cell growth, proliferation, apoptosis, metabolism, and cellular stress response (Dang, 2012), and is a known downstream effector of the MAPK signaling pathway (Sears et al, 2000; Tsai et al, 2012). Here, we investigate the regulation and physiological role of c‐MYC in melanoma cell adaptation to asparagine restriction.
Results
Asparagine restriction upregulates c‐MYC expression
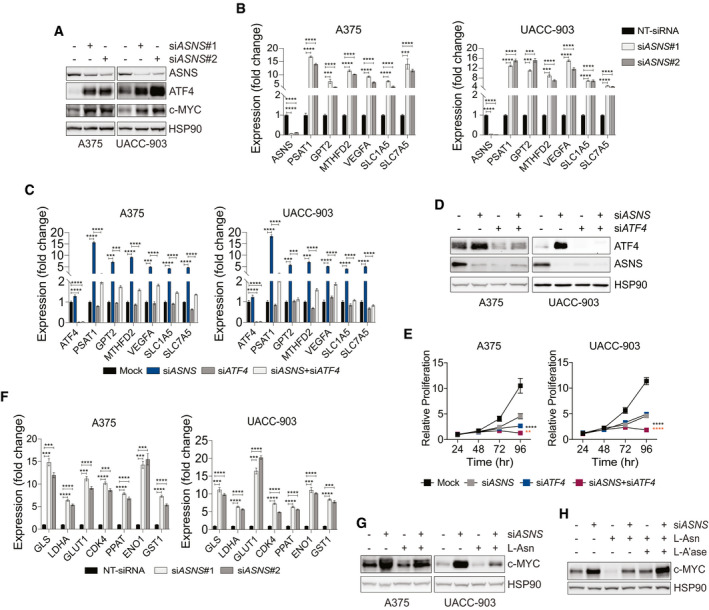
To understand the potential involvement of c‐MYC in cellular adaptive response to asparagine restriction, we first studied the effect of asparagine restriction on c‐MYC expression. ASNS suppression using short interfering RNA (siRNA) in A375 and UACC‐903 melanoma cell lines, while showing the induction of ATF4 (Pathria et al, 2019), also upregulated c‐MYC levels (Fig 1A). ASNS depletion induced the expression of ATF4 transcriptional target genes (PSAT1, phosphoserine aminotransferase 1; GPT2, glutamic‐pyruvic transaminase 2; MTHFD2, methylene tetrahydrofolate dehydrogenase 2; VEGFA, vascular endothelial growth factor A; SLC1A5, solute carrier family 1 member 5; and SLC7A5, solute carrier family 7 member 5), while ATF4 suppression effectively attenuated these changes and cooperated with ASNS knockdown to inhibit melanoma cell proliferation (Fig 1B–E). Increased expression of the established c‐MYC target genes (GLS, glutaminase; LDHA, lactate dehydrogenase A; GLUT1, glucose transporter 1; CDK4, cyclin‐dependent kinase 1; PPAT, phosphoribosyl pyrophosphate amidotransferase; ENO1, enolase 1; GST1, glutathione S‐transferase 1) in asparagine‐restricted melanoma cells underscored the functional integrity of upregulated c‐MYC (Fig 1F). siASNS‐mediated induction of c‐MYC was largely blocked in A375 cells cultured with L‐asparagine (L‐Asn) (Fig 1G), while the addition of asparagine hydrolyzing enzyme L‐A’ase to the growth medium reversed the effect of L‐Asn (Fig 1H). These results demonstrate that asparagine restriction in melanoma cells induces the expression of c‐MYC.
Figure 1. Asparagine restriction upregulates ATF4 and c‐MYC.
-
AImmunoblotting of c‐MYC and ATF4 in melanoma cells treated with NT‐siRNA (−) or siASNS (#1 or #2) for 72 h.
-
B, CReverse transcription with quantitative PCR (RT–qPCR) of the indicated transcripts in melanoma cells 36 h after treatment with siASNS (#1 or #2) (B), or siASNS, siATF4, or both (C).
-
DImmunoblotting of ATF4 and ASNS in melanoma cells 72 h after treatment with siASNS, siATF4, or both.
-
EProliferation of melanoma cells over indicated times after treatment with siASNS, siATF4, or both.
-
FRT–qPCR analysis of indicated transcripts in melanoma cells 36 h after treatment with siASNS (#1 or #2).
-
G, HImmunoblotting of c‐MYC in melanoma cells treated for 72 h with siASNS, L‐Asn, or both (G), or in A375 cells treated as indicated for 72 h (H).
Data information: Data are shown as the mean ± SD, n = 3 biological replicates. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Unpaired Student’s t‐test was used in (B), (C), and (F); two‐way ANOVA was utilized in (E). In (E), the statistical significance denoted in black and orange corresponds to the comparison of siASNS + siATF4 vs. siASNS and siATF4, respectively.
MAPK‐GSK3‐β signaling regulates c‐MYC expression upon asparagine restriction
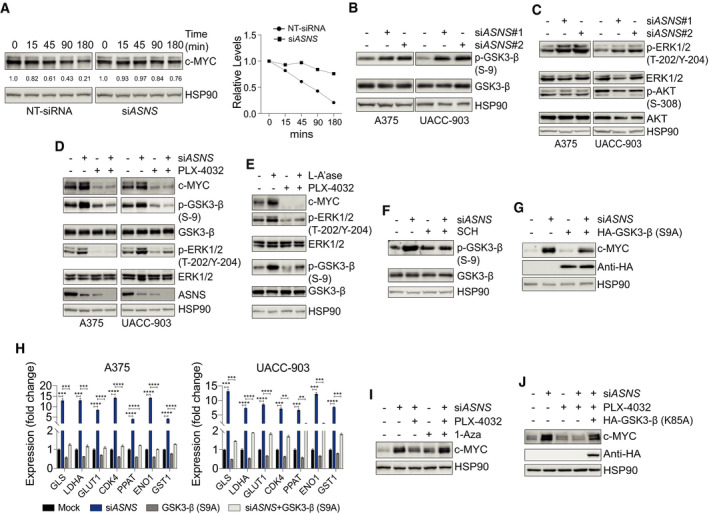
Just like a multitude of downstream processes regulated by c‐MYC, regulation of c‐MYC’s own expression and activity is quite complex and multi‐factorial (Stine et al, 2015). Previously, we showed that asparagine restriction triggers hyperactivation of MEK‐ERK (MAPK) signaling, a known positive modulator of c‐MYC expression and activity (Sears et al, 2000; Dang, 2012; Pathria et al, 2019). One mechanism of c‐MYC regulation by MAPK signaling involves phosphorylation of glycogen synthase kinase 3β (GSK3‐β) on Ser‐9, a post‐translational modification that impairs GSK3‐β activity (Ding et al, 2005; Tsai et al, 2012). Since an active GSK3‐β phosphorylates c‐MYC on Thr‐58, marking it for proteasomal degradation, activation of MAPK signaling in response to asparagine restriction is expected to stabilize c‐MYC (Sears et al, 2000; Tsai et al, 2012). To test this, A375 cells were transfected with a non‐targeting siRNA (NT‐siRNA) or siASNS and treated with the protein synthesis inhibitor cycloheximide for different timepoints. Inhibition of ASNS expression in A375 cells resulted in the stabilization of c‐MYC protein, as compared to the NT‐siRNA treatment (Fig 2A). To evaluate the potential role of altered GSK3‐β activity in the regulation of c‐MYC expression, we first studied the effect of asparagine restriction on GSK3‐β phosphorylation. ASNS suppression increased the phosphorylation of GSK3‐β on Ser‐9 (Fig 2B). Both MAPK and phosphatidylinositol 3‐kinase (PI3K)‐AKT signaling pathways phosphorylate GSK3‐β on Ser‐9 to suppress its activity (Tsai et al, 2012). Consistent with our prior observations (Pathria et al, 2019), ASNS suppression in melanoma cells induced ERK phosphorylation, while AKT phosphorylation remained unaffected (Fig 2C). Importantly, PLX‐4032, a BRAF inhibitor, which suppressed MAPK signaling, as illustrated by the decrease in p‐ERK1/2 levels, attenuated the enhancement of phospho‐(Ser‐9)‐GSK3‐β and c‐MYC levels in ASNS‐suppressed melanoma cells (Fig 2D). Similarly, treatment of ASNS‐depleted A375 cells cultured in L‐Asn‐containing media with L‐A’ase induced c‐MYC and phospho‐GSK3‐β levels, changes that were attenuated by concomitant BRAF inhibition (Fig 2E). Moreover, SCH772984, a specific ERK inhibitor, alleviated the increase in phospho‐GSK3‐β levels in ASNS‐suppressed A375 cells (Fig 2F). Notably, expression of a non‐phosphoryable (S9A) active mutant of GSK3‐β (hereafter referred to as GSK3‐β‐Active) ablated c‐MYC induction and blocked the upregulation of c‐MYC target genes in ASNS‐suppressed A375 cells (Fig 2G and H). The suppression of c‐MYC induction by PLX‐4032 in ASNS‐depleted A375 cells (Fig 2D) was antagonized by co‐treatment with a selective GSK3‐β inhibitor 1‐Azakenpaullone (1‐Aza; Fig 2I) Lastly, the expression of a kinase dead (K85A) mutant of GSK3‐β (Mariappan et al, 2008) rescued c‐MYC expression in ASNS‐silenced A375 cells treated with a BRAF inhibitor (Fig 2J). Together, these data demonstrate the central role of MAPK‐GSK3‐β signaling in the regulation of c‐MYC expression in asparagine‐deprived melanoma cells.
Figure 2. MAPK‐GSK3‐β signaling pathway is required for AAR signaling activation in asparagine‐restricted melanoma cells.
-
AImmunoblotting of c‐MYC in A375 cells treated with NT‐siRNA or siASNS for 24 h followed by cycloheximide treatment for indicated timepoints (left). Plot showing relative protein levels of c‐MYC (y‐axis) as a function of time lapsed (x‐axis) in the left panel (right).
-
B–DImmunoblotting of indicated proteins in melanoma cells 72 h after treatment with NT‐siRNA (−) or siASNS (#1 or #2) (B and C), or siASNS, PLX‐4032, or both (D).
-
EImmunoblotting of indicated proteins in A375 cells cultured in L‐Asn and treated with L‐A’ase, PLX‐4032, or both for 72 h.
-
FA375 cells were treated with siASNS, SCH772984, or both for 72 h followed by immunoblotting of phosphorylated and total GSK3‐β.
-
GA375 cells were treated with siASNS, HA‐GSK3‐β (S9A), or both for 72 h followed by immunoblotting of indicated proteins.
-
HRT–qPCR analysis of indicated transcripts in melanoma cells 36 h after treatment with siASNS, HA‐GSK3‐β (S9A), or both.
-
IA375 cells were treated as indicated for 72 h followed by immunoblotting of c‐MYC.
-
JImmunoblotting of c‐MYC and HA proteins in A375 cells treated as indicated for 72 h.
Data information: Data are shown as the mean ± SD, n = 3 biological replicates. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Unpaired Student’s t‐test was used in (H).
Source data are available online for this figure.
c‐MYC is essential for the induction of amino acid response signaling
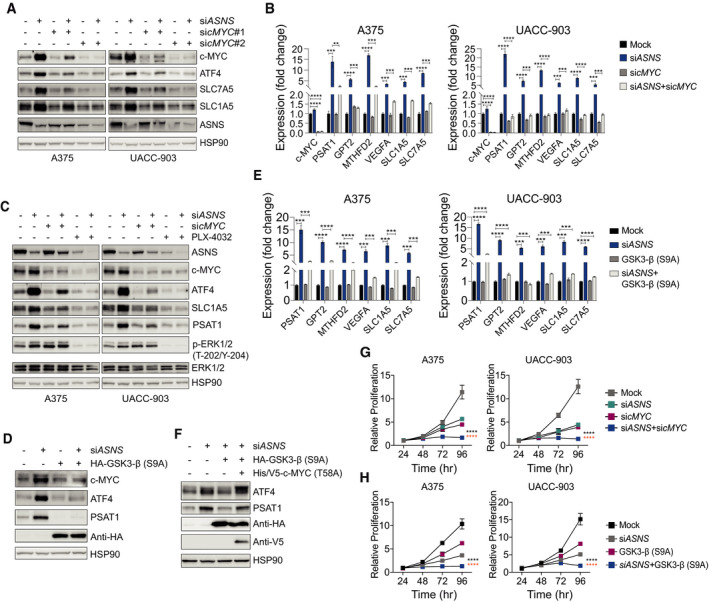
Induction of ATF4 and the associated transcriptional program (Fig 1A and B) marks the activation of the cellular adaptive response to amino acid restriction, referred to as amino acid response (AAR) signaling (Kilberg et al, 2009). To evaluate the role of c‐MYC in cellular adaptation to asparagine restriction, we first studied the effect of c‐MYC suppression on the levels of ATF4 and its key transcriptional targets. While ASNS suppression in melanoma cells, expectedly, enhanced the protein levels of ATF4 and the expression of ATF4 target genes, a concomitant c‐MYC suppression reversed these changes (Fig 3A and B). MAPK signaling inhibition, which suppresses c‐MYC induction (Fig 2D–F), similar to c‐MYC suppression (Fig 3A and B), abolished the induction of ATF4 and its target genes (Fig 3C). Moreover, the expression of GSK3‐β‐Active in A375 cells attenuated the induction of c‐MYC, ATF4, and ATF4 target genes associated with ASNS suppression (Fig 3D and E). As GSK3‐β‐mediated Thr‐58 phosphorylation of c‐MYC marks it for proteasomal degradation (Sears et al, 2000), next we asked whether the co‐expression of a non‐phosphoryable (T58A) c‐MYC mutant can rescue ATF4 induction in ASNS‐suppressed A375 cells expressing GSK3‐β‐Active. Indeed, (T58A) c‐MYC expression alleviated ATF4 suppression caused by the expression of GSK3‐β‐Active (Fig 3F). Suppressing the induction of ATF4 and the associated transcriptional program has previously been shown to promote the anti‐proliferative effect of asparagine restriction (Nakamura et al, 2018; Pathria et al, 2019). Accordingly, the attenuation of ATF4 induction via c‐MYC suppression or the expression of GSK3‐β‐Active led to a more effective inhibition of melanoma cell proliferation as compared to ASNS suppression alone (Fig 3G and H).
Figure 3. c‐MYC is critical for the induction of ATF4 in asparagine‐restricted melanoma cells.
-
AImmunoblotting of indicated proteins in melanoma cells treated with siASNS, sicMYC (#1 or #2), or a combination of siASNS and sicMYC (#1 or #2) for 72 h.
-
BRT–qPCR analysis of indicated transcripts in melanoma cells 36 h after treatment with siASNS, sicMYC, or both.
-
CImmunoblotting of indicated proteins in melanoma cells 72 h after treatment with siASNS, sicMYC, PLX‐4032, or a combination of siASNS and sicMYC, or siASNS and PLX‐4032.
-
DImmunoblotting of indicated proteins in A375 cells 72 h after treatment with siASNS, HA‐GSK3‐β (S9A), or both.
-
ERT–qPCR analysis of indicated transcripts in melanoma cells after 36 h treatment with siASNS, HA‐GSK3‐β (S9A), or both.
-
FA375 cells were treated as indicated for 72 h followed by immunoblotting of indicated proteins.
-
G, HProliferation of melanoma cells over indicated times after treatment with siASNS, sicMYC, or both (G), or siASNS, HA‐GSK3‐β (S9A), or both (H).
Data information: Data are shown as the mean ± SD, n = 3 biological replicates. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Unpaired Student’s t‐test was used in (B) and (E); two‐way ANOVA was utilized in (G) and (H). The statistical significance denoted in black and orange corresponds to the comparison of siASNS + sicMYC vs. siASNS and sicMYC, respectively (G), and the comparison of siASNS + GSK3‐β (S9A) vs. siASNS and GSK3‐β (S9A), respectively (H).
c‐MYC promotes essential amino acid uptake and mTORC1 activity to support AAR signaling
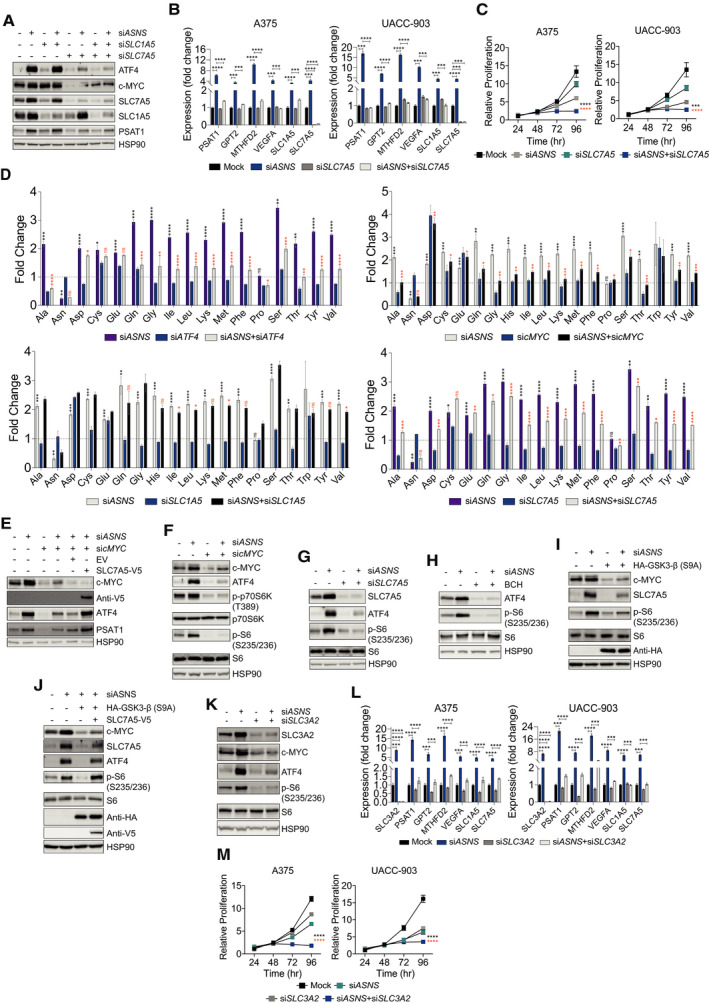
Along with growth factor signaling cues, essential amino acids (EAA) are critical for mTORC1 activity (Bar‐Peled & Sabatini, 2014). Ours and work of others have revealed the critical requirement of mTORC1 function for ATF4 translation and consequently the mediation of cellular response to amino acid restriction (Nofal et al, 2017; Pathria et al, 2018; Pathria et al, 2019). In addition to ATF4, c‐MYC directly promotes the transcription of the glutamine transporter SLC1A5 and an EAA transporter SLC7A5 (Stine et al, 2015; Yue et al, 2017) (Fig 3A and B). Therefore, we hypothesized that c‐MYC‐dependent upregulation of SLC1A5 and/or SLC7A5 and the ensuing EAA uptake is critical for the maintenance of mTORC1 activity and hence ATF4 translation. While suppression of SLC1A5 did not affect ATF4 induction in ASNS knocked‐down A375 cells, downregulation of SLC7A5 almost completely attenuated the induction of ATF4 and the associated transcriptional program (Fig 4A and B). Of note, SLC7A5 depletion negatively impacted basal c‐MYC expression (Fig 4A), consistent with the regulation of c‐MYC expression by mTORC1 in response to SLC7A5‐dependent EAA uptake (Yue et al, 2017). Along with the ability of SLC7A5 to alleviate ATF4‐associated adaptive signaling (Fig 4A and B), its suppression cooperated with ASNS depletion to impair melanoma cell proliferation (Fig 4C).
Figure 4. c‐MYC regulates essential amino acid uptake and mTORC1 activity to support AAR signaling.
-
AImmunoblotting of indicated proteins in A375 cells 72 h after treatment with siASNS, siSLC1A5, siSLC7A5, or a combination of siASNS and siSLC1A5, or siASNS and siSLC7A5.
-
BRT–qPCR analysis of indicated transcripts in melanoma cells after 36 h treatment with siASNS, siSLC7A5, or both.
-
CProliferation of melanoma cells over indicated times after treatment with siASNS, siSLC7A5, or both.
-
DGC‐MS‐based estimation of intracellular amino acid levels in A375 cells treated with siASNS, siATF4, or both (top left); siASNS, sicMYC, or both (top right); siASNS, siSLC1A5, or both (bottom left); or siASNS, siSLC7A5, or both (bottom right). The fold change is relative to NT‐siRNA‐treated A375 cells.
-
E–KImmunoblotting of indicated proteins in A375 cells 72 h after indicated treatment (E), treatment with siASNS, sicMYC, or both (F), siASNS, siSLC7A5, or both (G), siASNS, BCH, or both (H), siASNS, HA‐GSK3‐β (S9A), or both (I), siASNS or indicated combinations (J), or siASNS, siSLC3A2, or both (K).
-
LRT–qPCR analysis of indicated transcripts in melanoma cells after 36 h treatment with siASNS, siSLC3A2, or both.
-
MProliferation of melanoma cells over indicated times after treatment with siASNS, siSLC3A2, or both.
Data information: Data are shown as the mean ± SD, n = 3 biological replicates. ns: not significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Unpaired Student’s t‐test was used in (B), (D), and (L); two‐way ANOVA was utilized in (C) and (M). In (D), the statistical significance denoted in black and orange corresponds to the comparison of NT‐siRNA vs. siASNS and siASNS vs. siATF4, sicMYC, siSLC1A5, or siSLC7A5, respectively. Statistical significance shown only for the combination treatments reversing the effect of siASNS on the intracellular amino acid levels. In (D), siATF4 and siSLC7A5 graphs are part of the same experiment and sicMYC and siSLC1A5 graphs are part of the same experiment. The intracellular levels of histidine and tryptophan could not be measured in siASNS + siATF4 and siASNS + siSLC7A5 combination settings, and, therefore, not shown. The statistical significance denoted in black and orange corresponds to the comparison of siASNS + siSLC7A5 vs. siASNS and siSLC7A5, respectively (C), and the comparison of siASNS + siSLC3A2 vs. siASNS and siSLC3A2, respectively (M).
Gas chromatography‐mass spectrometry (GC‐MS)‐based analysis of A375 cells subjected to siASNS treatment revealed increased intracellular levels of virtually every amino acid, an effect that was reversed by ATF4 knockdown (Fig 4D, upper left). These observations were consistent with the ability of ATF4 to enhance amino acid uptake in cells deprived of a particular amino acid (Chen et al, 2014; Pathria et al, 2018). c‐MYC suppression, which overcomes ATF4 induction in siASNS‐treated A375 cells (Fig 3A), closely mimicked the effect of ATF4 silencing on amino acid uptake (Fig 4D, upper right). SLC1A5 knockdown, which failed to overcome ATF4 induction in ASNS‐suppressed A375 cells (Fig 4A), expectedly, did not have a major impact on the changes in the intracellular amino acid levels elicited by ASNS suppression (Fig 4D, lower left). On the other hand, SLC7A5 knockdown, which suppressed ATF4 induction in ASNS‐depleted A375 cells (Fig 4A), attenuated the increase in the intracellular amino acid levels associated with ASNS depletion (Fig 4D, lower right). Notably, sicMYC‐mediated attenuation of ATF4 and PSAT1 induction in ASNS‐depleted A375 cells was rescued by the overexpression of SLC7A5 (Fig 4E). While mTORC1 activity, as assessed by the phosphorylation of mTORC1 targets p70S6K and/or ribosomal S6 protein (S6), was maintained in ASNS‐depleted A375 cells, simultaneous suppression of either c‐MYC or SLC7A5, or chemical inhibition of SLC7A5 with the small‐molecule inhibitor BCH (Kim et al, 2008), effectively compromised mTORC1 activity (Fig 4F–H). Notably, consistent with its ability to suppress mTORC1 activity, BCH also suppressed ATF4 induction caused by ASNS depletion (Fig 4H). The expression of GSK3‐β‐Active in A375 cells, in accordance with the attenuation of c‐MYC induction in ASNS‐suppressed cells, also mitigated the induction of SLC7A5 and mTORC1 activity (Fig 4I). Conversely, ectopic expression of SLC7A5 rescued mTORC1 activity and restored ATF4 induction (Fig 4J). SLC7A5 membrane localization and transporter channel activity is dependent on its heterodimerization with solute carrier family 3 member 2 (SLC3A2) (Nicklin et al, 2009). ASNS suppression in A375 cells showed increased levels of SLC3A2, while depletion of SLC3A2 mimicked the effect of c‐MYC or SLC7A5 knockdown on ATF4 induction and mTORC1 activity and suppressed the upregulation of ATF4 target genes (Fig 4K and L). Lastly, similar to SLC7A5 knockdown, siSLC3A2 cooperated with ASNS suppression to effectively impair melanoma cell proliferation (Fig 4M). Collectively, these data demonstrate the important role of SLC7A5 in EAA uptake and concomitant mTORC1 activity for the maintenance of AAR signaling and the adaptation of melanoma cells to asparagine restriction (Fig 5).
Figure 5. The crosstalk between AAR and growth factor signaling pathways in melanoma cell resistance to asparagine restriction.
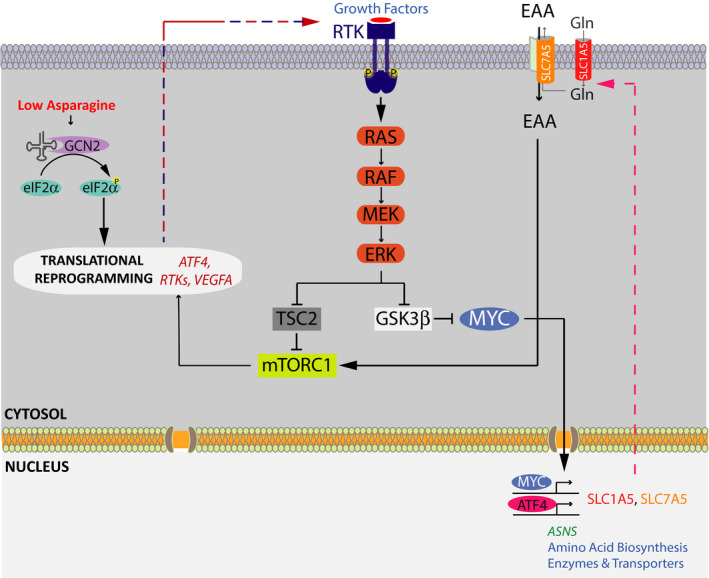
The model outlining the key elements of the AAR and growth factor signaling pathways working in concert to regulate melanoma cell adaptation to asparagine restriction. Asparagine restriction activates GCN2, which phosphorylates and inactivates eIF2α, leading to the suppression of cap‐dependent translation and the enhancement of cap‐independent translation. Among transcripts showing increased translation are ATF4, multiple RTKs, and VEGFA. ATF4 orchestrates an expansive adaptive program to amino acid/asparagine restriction via inducing the expression of a large array of amino acid biosynthetic enzymes and transport proteins. Activated in response to enhanced RTK expression, MAPK signaling promotes mTORC1 activity and the consequent ATF4 translation through a concerted regulation of two distinct mechanisms—(i) suppression of the tumor suppressor tuberous sclerosis 2 (TSC2) activity, a negative regulator of mTORC1 function, and (ii) alleviation of GSK3‐β‐mediated c‐MYC degradation, leading to the induction of c‐MYC transcriptional target SLC7A5. SLC7A5 in turn enhances the uptake of EAA, providing critical cues for mTORC1 activation.
Discussion
Cellular response to amino acid restriction is characterized by the activation of AAR signaling, a pathway that been well characterized (Kilberg et al, 2009). However, therapeutic targeting of AAR signaling has remained a challenge, primarily due to a lack of effective pharmacological inhibitors. In this study, we demonstrate the role of the MAPK‐GSK3‐β‐c‐MYC signaling axis in the uptake of EAA and its implications on the maintenance of mTORC1 activity, with the concomitant induction of ATF4 and the associated pro‐survival program in melanoma cells experiencing asparagine restriction. It is plausible that this pathway may also be relevant in other tumor types, where both c‐MYC and ATF4 are key players.
MAPK signaling along with the PI3K‐Akt signaling pathway negatively regulates GSK3‐β activity by catalyzing its phosphorylation on Ser‐9 residue (Sears et al, 2000; Tsai et al, 2012). This in turn relieves GSK3‐β mediated c‐MYC phosphorylation on Thr‐58, an event that primes c‐MYC for proteasomal degradation, thus stabilizing c‐MYC (Sears et al, 2000; Gregory et al, 2003). The physiological significance of these signaling relationships was shown in arginine deiminase‐treated (arginine‐depleted) melanoma cells, whereby c‐MYC induction enhances the expression of arginine synthesizing enzyme argininosuccinate synthetase, which confers resistance to arginine deiminase (Tsai et al, 2012). Although asparagine restriction did not promote the activity of the PI3K‐Akt pathway, and MAPK signaling inhibition was sufficient to suppress enhanced GSK3‐β Ser‐9 phosphorylation, it remains to be seen whether restriction of glutamine, which promotes the activity of PI3K‐Akt signaling in KRAS‐dependent manner, may regulate the GSK3‐β‐c‐MYC axis (Gwinn et al, 2018). While our studies have focused on the post‐translational regulation of c‐MYC levels by MAPK signaling, as an early response gene, c‐MYC may additionally be regulated by MAPK signaling through transcriptional modulation (Kerkhoff et al, 1998; Pathria et al, 2019). Future studies would allow to map the contribution of such regulation in the context of cellular ASNS depletion.
As a regulator of a wide array of cellular processes, c‐MYC controls an expansive gene expression program (Zeller et al, 2006). c‐MYC is an important regulator of cellular metabolism (Stine et al, 2015), promoting expression of the proteins involved in glucose transport, glycolysis, glutamine transport and assimilation, fatty acid and cholesterol biosynthesis, and nucleotide biosynthesis (Stine et al, 2015). In addition to its important role under steady‐state, c‐MYC also regulates cellular response to amino acid restriction. For example, c‐MYC was shown to promote the expression of several enzymes of the serine/glycine biosynthesis pathway (Sun et al, 2015). Although this study reported the upregulation of c‐MYC levels in response to glutamine restriction, the underlying mechanism remained unclear. Future investigations will determine whether the MAPK‐GSK3‐β‐c‐MYC axis, relevant to asparagine restriction, may also be pertinent to the restriction of other amino acids.
Although mTORC1 activation in response to amino acid/asparagine restriction may seem counter‐intuitive, mechanistic support for such regulation has been accumulating. Decrease in intracellular glutamine, asparagine, serine, or aspartate levels have been shown to enhance the uptake of EAA, which promote mTORC1 activity (Bar‐Peled & Sabatini, 2014; Chen et al, 2014; Pathria et al, 2018; Pathria et al, 2019). Notably, the upregulation of SLC7A5 and the associated uptake of EAA were shown to be critical for the maintenance of mTORC1 activity in response to decrease in cellular glutamine, serine, or aspartate levels (Chen et al, 2014; Pathria et al, 2018). Arginine uptake, mediated through p53‐dependent transcriptional upregulation of the arginine transporter SLC7A3, maintained mTORC1 activity in glutamine‐deprived cells (Lowman et al, 2019). Moreover, a leucine‐free diet in mice failed to suppress mTORC1 activity (Singh et al, 2011). Coupling of glutamine export with the uptake of EAA is an important mechanism for mTORC1 activation (Nicklin et al, 2009). Accordingly, glutamine uptake by SLC1A5 is critical for increased uptake of EAA and mTORC1 activity in melanoma cells subjected to LDHA inhibition, which suppressed intracellular serine and aspartate levels (Pathria et al, 2018). Notably, our data have established SLC1A5 dispensability for increased EAA uptake and mTORC1 activation in response to asparagine restriction. The effect of asparagine restriction on the uptake of other amino acids (Krall et al, 2016) may potentially influence this response, an avenue that needs to be further explored.
The role of growth factor signaling pathways in cancer cell adaptation to amino acid restriction is emerging (Tsai et al, 2012; Gwinn et al, 2018; Pathria et al, 2019). The critical requirement of the KRAS‐PI3K‐Akt‐NRF2 signaling axis in the transcriptional regulation of ATF4, which confers resistance to glutamine and asparagine restriction, was reported in NSCLC cells (Gwinn et al, 2018). In our studies, the receptor tyrosine kinase (RTK)‐MAPK‐mTORC1‐ATF4 signaling axis was found to be a synthetic vulnerability of asparagine‐restricted melanoma and pancreatic cancer cells (Pathria et al, 2019). Identifying the role of c‐MYC, downstream of the MAPK‐GSK3‐β signaling axis, in sustaining mTORC1‐mediated ATF4 induction, adds to our understanding of amino acid response signaling. The identification of SLC7A5 and the associated uptake of EAA as an integral component of the cellular adaptive response program to asparagine restriction provides the foundation for further investigations into the translational value of the MAPK‐GSK3‐β‐c‐MYC signaling axis and its potential role in cancer cell adaptation to the restriction of other amino acids.
Materials and Methods
Cell culture and reagents
Melanoma cell line A375 was obtained from ATCC. UACC‐903 melanoma cell line was obtained from the University of Arizona Cancer Center. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GE Healthcare Life Sciences, Marlborough, MA), supplemented with 5% fetal bovine serum and penicillin–streptomycin. Cells were grown at 37°C in a humidified atmosphere containing 5% carbon dioxide. L‐asparagine (used at 0.3 μM) and 1‐Azakenpaullone (used at 1 μM) were purchased from Sigma‐Aldrich (St. Louis, MO). PLX‐4032 (used at 200 nM), SCH‐772984 (used at 250 nM), cycloheximide (used at 100 μM), and BCH (used at 10 mM) were purchased from Selleckchem (Houston, TX). l‐asparaginase (used at 1 U/ml) was purchased from Prospec Protein Specialists (Rehevot, Israel). HA‐GSK3‐β (S9A) pcDNA3, HA‐GSK3‐β (K85A) pcDNA3, and pD40‐His/V5‐c‐MycT58A plasmids were purchased from Addgene (Watertown, MA). SLC7A5 (Homo sapiens) pLenti6.3/V5‐DEST plasmid was obtained from DNASU plasmid repository (Tempe, AZ).
Antibodies
The following antibodies were used: c‐MYC (D84C12) (dilution, 1:1,000) (Cat#5605), GSK3‐β (27C10) (dilution, 1:1,000) (Cat#9315), phospho‐(Ser‐9)‐GSK3‐β (dilution, 1:1,000) (Cat#9336), phospho‐(Ser‐308)‐Akt (244F9) (dilution, 1:1,000) (Cat#4056), Akt (dilution, 1:1,000) (Cat#9272), ATF4 (D4B8) (dilution, 1:1,000) (Cat#11815), SLC1A5 (ASCT2, V501) (dilution, 1:1,000) (Cat#5345), phospho‐(Ser235/236)‐S6 Ribosomal Protein (D68F8) (dilution, 1:1,000) (Cat#4858), S6 Ribosomal Protein (54D2) (dilution, 1:1,000) (Cat#2217), phospho‐(Thr389)‐p70S6 kinase (dilution, 1:1,000) (Cat#9205), p70S6 kinase (dilution, 1:1,000) (Cat#9202), phospho‐p44/42 Erk1/2 (Thr202/Tyr204) (dilution, 1:1,000) (Cat#9101), p44/42 Erk1/2 (dilution, 1:1,000) (Cat#9102), SLC7A5 or LAT1 (dilution, 1:1,000) (Cat#5347), and HRP‐conjugated anti‐mouse (dilution 1:10,000) (Cat#7076) and HRP‐conjugated anti‐rabbit (dilution, 1:10,000) (Cat#7074) antibodies from Cell Signaling Technology (Danvers, MA). PSAT1 (dilution, 1:1,000) (Cat#PA5‐22124), anti‐V5 (dilution, 1:1,000) (Cat#R960‐25) from Thermo‐Fisher Scientific (Waltham, MA). ASNS (G‐10) (dilution, 1:250) (Cat#sc‐365809), HSP90 (F‐8) (dilution, 1:5,000) (Cat#sc‐13119), anti‐HA (F‐7) (dilution, 1:1,000) (Cat#7392) from Santa Cruz Biotechnology, (Dallas, TX).
siRNA transfection
1 × 105 cells were seeded overnight (O/N) per well in 6‐well plates. Negative control (NT‐siRNA) or siRNA targeting the transcript of interest was transfected utilizing jetPRIME® transfection reagent, as per manufacturer’s instructions (Polyplus, Illkirch, France). Following siRNAs were used: siASNS#1 (SASI_Hs01_00116724), siASNS#2 (SASI_Hs01_00116721), siATF4 (SASI_Hs02_00332313), sicMYC#1 (SASI_Hs01_00222676), sicMYC#2 (SASI_Hs01_00222677), siSLC7A5 (SASI_Hs02_00335760), siSLC1A5 (SASI_Hs01_00162267), siSLC3A2 (SASI_Hs01_00063836), and the negative control siRNA (NT‐siRNA; SIC001) was purchased from Sigma‐Aldrich.
Immunoblotting
1–2 × 105 cells were seeded O/N per well in 6‐well plates. Following treatment, total protein was extracted in Laemmli buffer, fractionated by SDS polyacrylamide gels and transferred to PVDF membranes (Millipore, Burlington, MA). Following blocking with 5% non‐fat dry milk (BD Biosciences, San Jose, CA), the membranes were incubated with primary antibodies O/N at 4°C. Subsequently, 2 h incubation with HRP‐conjugated secondary antibodies was performed for the chemiluminescence reaction. The protein signal was visualized using the ChemiDoc imaging system (Bio‐Rad, Hercules, CA) according to the manufacturer’s instructions.
Cell proliferation
For cell proliferation, 0.3–0.5 × 105 cells were seeded O/N in triplicate in 6‐well plates. Following treatment, cells were trypsinized and the cell count was determined with Neubauer hemocytometer (Celeromics, Cambridge, UK).
qPCR analysis
1 × 105 cells were seeded O/N per well in 6‐well plates. Following 36 or 48 h of specified treatments, total RNA was extracted using RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized using oligo(dT) and random primers (AB Bioscience, Concord, MA), and qPCR analysis was performed with SYBR Green (Bio‐Rad). PrimerQuest tool (Coralville, IA) and Primer Bank (https://pga.mgh.harvard.edu/primerbank/) were used for primer design. β‐Actin was used as an internal control.
Sequence of the primers used:
SLC1A5‐F: CAGCCTTTCGCTCATACTCTAC
SLC1A5‐R: GTAAACCCACATCCTCCATCTC
SLC7A5‐F: CGATGGACTCCTGACCATAATC
SLC7A5‐R: GTGTCTGCCTTTCTTGTCTCT
SLC3A2‐F: TGAATGAGTTAGAGCCCGAGA
SLC3A2‐R: GTCTTCCGCCACCTTGATCTT
ASNS‐F: GTTCGTGCTTCAGTAGGTATGT
ASNS‐R: GGTGGCAGAGACAAGTAATAGG
GPT2‐F: AGAAACTCCCAACTGTCCTTAC
GPT2‐R: CCTCCTAGACTAGCTGACCTTAT
MYC‐F: GCTGTAGTAATTCCAGCGAGAG
MYC‐R: GAGTCGTAGTCGAGGTCATAGT
ATF4‐F: GTATGAGCCCAGAGTCCTATCT
ATF4‐R: CACATTGACGCTCCTGACTATC
PSAT1‐F: CAGGAAGGTGTGCTGACTATG
PSAT1‐R: CCCATGACGTAGATGCTGAAA
PHGDH‐F: GTCGCGGAGAGTTTGAGTATTT
PHGDH‐R: GGTGGCAGAGCGAACAATAA
VEGFA‐F: AGGGCAGAATCATCACGAAGT
VEGFA‐R: AGGGTCTCGATTGGATGGCA
GLS‐F: AGGGTCTGTTACCTAGCTTGG
GLS‐R: ACGTTCGCAATCCTGTAGATTT
LDHA‐F: ATGGCAACTCTAAAGGATCAGC
LDHA‐R: CCAACCCCAACAACTGTAATCT
GLUT1‐F: GGCCAAGAGTGTGCTAAAGAA
GLUT1‐R: ACAGCGTTGATGCCAGACAG
CDK4‐F: ATGGCTACCTCTCGATATGAGC
CDK4‐R: CATTGGGGACTCTCACACTCT
PPAT‐F: GATGGGAGTTCGGTGCCAA
PPAT‐R: CAACGAAGGGCTGACAATTTTC
ENO1‐F: AAAGCTGGTGCCGTTGAGAA
ENO1‐R: GGTTGTGGTAAACCTCTGCTC
GST1‐F: TCTGCCCTACTTGATTGATGGG
GST1‐R: TCCACACGAATCTTCTCCTCT
β‐Actin‐F: CATGTACGTTGCTATCCAGGC
β‐Actin‐R: CTCCTTAATGTCACGCACGAT
Statistical analysis
Statistical significance between two groups was assessed by the unpaired Student’s t‐test. Two‐way ANOVA was utilized to analyze cell proliferation at different time points. GraphPad Prism 8 software (GraphPad, La Jolla, CA) was used to perform all statistical calculations.
Author contributions
GP and ZAR conceived the study; GP, SV and DAS designed experiments; GP SV and DAS performed experiments; GP, SV, JY and DAS analyzed data; GP and ZAR wrote the manuscript and supervised various aspects of the study.
Conflict of interest
Z.A.R. is a co‐founder and serves as scientific advisor to Pangea Therapeutics. All other authors declare that they have no conflict of interest.
Supporting information
Review Process File
Source Data for Figure 2
Acknowledgements
We thank members of the Ronai laboratory for discussions. Support by NCI grants R35CA197465 (Z.A.R), P01 CA128814 (D.A.S., Z.A.R.), are gratefully acknowledged. The SBP Cancer Metabolism Core is supported by NCI Cancer Center grant P30 CA030199. We also thank support by the Hervey Family Support (G.P). We thank Olga Zagnitko for assistance in the processing and analysis of metabolic samples.
EMBO reports (2021) 22: e51436
Contributor Information
Gaurav Pathria, Email: gauravpathria@gmail.com.
Ze’ev A Ronai, Email: zeev@ronailab.net.
Data availability
No data were deposited in a public database. Inquiries for reagents used in this study should be addressed to Ze’ev Ronai (zeev@ronailab.net).
References
- Bachet JB, Gay F, Marechal R, Galais MP, Adenis A, Ms CD, Cros J, Demetter P, Svrcek M, Bardier‐Dupas A et al (2015) Asparagine synthetase expression and phase I study with L‐asparaginase encapsulated in red blood cells in patients with pancreatic adenocarcinoma. Pancreas 44: 1141–1147 [DOI] [PubMed] [Google Scholar]
- Bar‐Peled L, Sabatini DM (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol 24: 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W et al (2014) The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol 206: 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG (1992) Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene‐specific translational control of GCN4 in yeast. Cell 68: 585–596 [DOI] [PubMed] [Google Scholar]
- Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z et al (2005) Erk associates with and primes GSK‐3beta for its inactivation resulting in upregulation of beta‐catenin. Mol Cell 19: 159–170 [DOI] [PubMed] [Google Scholar]
- Egler RA, Ahuja SP, Matloub Y (2016) L‐asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother 7: 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, Savaraj N (2008) Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des 14: 1049–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy‐Kanniappan S, Geschwind JF (2013) Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR (2003) Phosphorylation by glycogen synthase kinase‐3 controls c‐myc proteolysis and subnuclear localization. J Biol Chem 278: 51606–51612 [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Lee AG, Briones‐Martin‐Del‐Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, Sweet‐Cordero EA (2018) Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L‐asparaginase. Cancer Cell 33: 91–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Kang JS (2020) Dietary restriction of amino acids for Cancer therapy. Nutr Metab (Lond) 17: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkhoff E, Houben R, Loffler S, Troppmair J, Lee JE, Rapp UR (1998) Regulation of c‐myc expression by Ras/Raf signalling. Oncogene 16: 211–216 [DOI] [PubMed] [Google Scholar]
- Kilberg MS, Shan J, Su N (2009) ATF4‐dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS, Cho SH, Chun HS, Lee SY, Endou H, Kanai Y, Kim DK (2008) BCH, an inhibitor of system L amino acid transporters, induces apoptosis in cancer cells. Biol Pharm Bull 31: 1096–1100 [DOI] [PubMed] [Google Scholar]
- Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, Gandour‐Edwards R, Chuang FY, Bold RJ, Kung HJ (2009) Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase‐independent apoptosis. Cancer Res 69: 700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7: 11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessner HE, Valenstein S, Kaplan R, DeSimone P, Yunis A (1980) Phase II study of L‐asparaginase in the treatment of pancreatic carcinoma. Cancer Treat Rep 64: 1359–1361 [PubMed] [Google Scholar]
- Lowman XH, Hanse EA, Yang Y, Ishak Gabra MB, Tran TQ, Li H, Kong M (2019) p53 promotes cancer cell adaptation to glutamine deprivation by upregulating Slc7a3 to increase arginine uptake. Cell Rep 26: 3051–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F et al (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544: 372–376 [DOI] [PubMed] [Google Scholar]
- Mariappan MM, Shetty M, Sataranatarajan K, Choudhury GG, Kasinath BS (2008) Glycogen synthase kinase 3beta is a novel regulator of high glucose‐ and high insulin‐induced extracellular matrix protein synthesis in renal proximal tubular epithelial cells. J Biol Chem 283: 30566–30575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C et al (2018) Inhibition of GCN2 sensitizes ASNS‐low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci USA 115: E7776–E7785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C et al (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofal M, Zhang K, Han S, Rabinowitz JD (2017) mTOR Inhibition restores amino acid balance in cells dependent on catabolism of extracellular protein. Mol Cell 67: 936–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Lee JS, Hasnis E, Tandoc K, Scott DA, Verma S, Feng Y, Larue L, Sahu AD, Topisirovic I et al (2019) Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat Cell Biol 21: 1590–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, Sahu AD, Ruppin E, Herlyn M, Osterman AL et al (2018) Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 37: e99735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR (2000) Multiple Ras‐dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Akcakanat A, Sharma C, Luyimbazi D, Naff KA, Meric‐Bernstam F (2011) The effect of leucine restriction on Akt/mTOR signaling in breast cancer cell lines in vitro and in vivo . Nutr Cancer 63: 264–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5: 1024–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Song L, Wan Q, Wu G, Li X, Wang Y, Wang J, Liu Z, Zhong X, He X et al (2015) cMyc‐mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res 25: 429–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW, Dorr RT, Fanta P, Hersh EM, Salmon SE (2001) A phase I and pharmacodynamic evaluation of polyethylene glycol‐conjugated L‐asparaginase in patients with advanced solid tumors. Cancer Chemother Pharmacol 47: 83–88 [DOI] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT (2012) Activation of Ras/PI3K/ERK pathway induces c‐Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 72: 2622–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue M, Jiang J, Gao P, Liu H, Qing G (2017) Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep 21: 3819–3832 [DOI] [PubMed] [Google Scholar]
- Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC et al (2006) Global mapping of c‐Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci USA 103: 17834–17839 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Source Data for Figure 2
Data Availability Statement
No data were deposited in a public database. Inquiries for reagents used in this study should be addressed to Ze’ev Ronai (zeev@ronailab.net).