

Abstract

Multidrug resistant Gram-negative bacterial infections are an increasing public health threat due to rapidly rising resistance toward β-lactam antibiotics. The hydrolytic enzymes called β-lactamases are responsible for a large proportion of the resistance phenotype. β-Lactamase inhibitors (BLIs) can be administered in combination with β-lactam antibiotics to negate the action of the β-lactamases, thereby restoring activity of the β-lactam. Newly developed BLIs offer some advantage over older BLIs in terms of enzymatic spectrum but are limited to the intravenous route of administration. Reported here is a novel, orally bioavailable diazabicyclooctane (DBO) β-lactamase inhibitor. This new DBO, ETX1317, contains an endocyclic carbon–carbon double bond and a fluoroacetate activating group and exhibits broad spectrum activity against class A, C, and D serine β-lactamases. The ester prodrug of ETX1317, ETX0282, is orally bioavailable and, in combination with cefpodoxime proxetil, is currently in development as an oral therapy for multidrug resistant and carbapenem-resistant Enterobacterales infections.
Introduction
The global threat of antibacterial resistance is growing rapidly, and immediate worldwide attention is warranted to ensure new antibiotics become available to treat resistant infections.1 Without effective antibiotics, surgeries, organ transplants, and cancer chemotherapy are also at risk. Of particular concern are infections caused by multidrug resistant (MDR) Gram-negative bacteria. Recently the WHO2 and the CDC3 highlighted “priority pathogens” that pose the greatest threat to humanity from an infection point of view. Included as critical are carbapenem-resistant Acinetobacter baumannii, carbapenem-resistant Pseudomonas aeruginosa, and carbapenem-resistant, extended-spectrum β-lactamase- (ESBL-) producing Enterobacterales, for which new treatment options are urgently needed.
Since the introduction of penicillin in the 1940s, β-lactams have become the safest and most effective class of antibiotics to cure infections, especially those caused by Gram-negative bacteria.4 Unfortunately, many β-lactams have lost their activity over the years as bacteria relentlessly evolve to become resistant to these agents. One of the main resistance mechanisms in contemporary Gram-negative clinical isolates is mediated by the expression of multiple β-lactamases, enzymes made by the bacteria that hydrolyze and destroy the β-lactams.4 One approach to counter the action of β-lactamases is to combine a β-lactamase inhibitor (BLI) with the β-lactam to protect it from degradation and restore its antibacterial activity. The only approved oral BLI is clavulanic acid, which became available to the healthcare community in 1984.5 Its spectrum of activity is unfortunately limited to the Ambler class A β-lactamases,6 minimizing its clinical utility against contemporary isolates. Recently, three new BLIs with greater spectra of inhibition were approved: avibactam,7 vaborbactam, and relebactam.8 Unfortunately, none of these are suitable for oral dosing and they all need to be administered in a hospital setting, resulting in a high cost burden on the healthcare system. A potent, broad-spectrum, safe, and orally available BLI would therefore represent a welcome addition to the armamentarium against MDR Gram-negative infections. Specifically, an oral β-lactam-BLI combination therapy for urinary tract infections (UTIs) remains an important unmet medical need9,10 because the prevalence of ESBL-producing and carbapenem-resistant Enterobacterales (CRE) is increasing at an alarming rate.11
This work describes the discovery and characterization of ETX0282, an orally available BLI with a broad spectrum of activity against class A, C, and D serine β-lactamases. The combination of this novel BLI with a β-lactam has the potential to become an effective oral treatment option against MDR Gram-negative infections.
Results and Discussion
Design and Testing
The diazabicyclooctane (DBO) scaffold is the basis for several previously developed serine β-lactamase inhibitors such as avibactam and durlobactam (Figure 1).12,13 The high polarity and low pKa of these compounds are well-suited to intravenous administration but lead to low oral bioavailability. We therefore investigated a prodrug approach to improve oral absorption. Our effort was focused on replacing the sulfate activating group of the endocyclic double bond series of DBOs. This starting point had several advantages. (1) We possessed a deep understanding of the SAR of β-lactamase inhibitors. (2) These compounds have a covalent mechanism of inhibition. (3) We had available advanced intermediates from well-established syntheses. The very high polarity (log D < 0) and low pKa of avibactam and durlobactam are beneficial for their antibacterial activity, urinary excretion, and intravenous route of administration but result in minimal oral bioavailability (F < 10% 14). We therefore turned our attention to the corresponding prodrugs to increase oral bioavailability, an approach that should deliver high oral absorption and release the active BLI upon liver-mediated metabolism. While it seemed possible to prepare a sulfate prodrug of avibactam (as exemplified by ARX-179615), we decided to focus our efforts on the endocyclic double bond (EDB) series and replace the sulfate activating group by an acetate for several reasons. First, the tetrahydropyridine core in the EDB series is more reactive than the piperidine core,13 which is critical to achieve potent inhibition of a broad spectrum of serine β-lactamases. Second, the carboxylic acid group and its ester prodrugs are among the best-studied moieties in drug discovery to enhance oral absorption of poorly permeable compounds.16 The acetate also presents more opportunities for SAR exploration compared to sulfate prodrugs, which are considered to be unstable and potentially toxic alkylating agents.17 Several recent disclosures have also described oral BLIs from the DBO class (EBL-2253) and from the cyclic boronate class (VNRX-714518 and QPX772819).
Figure 1.
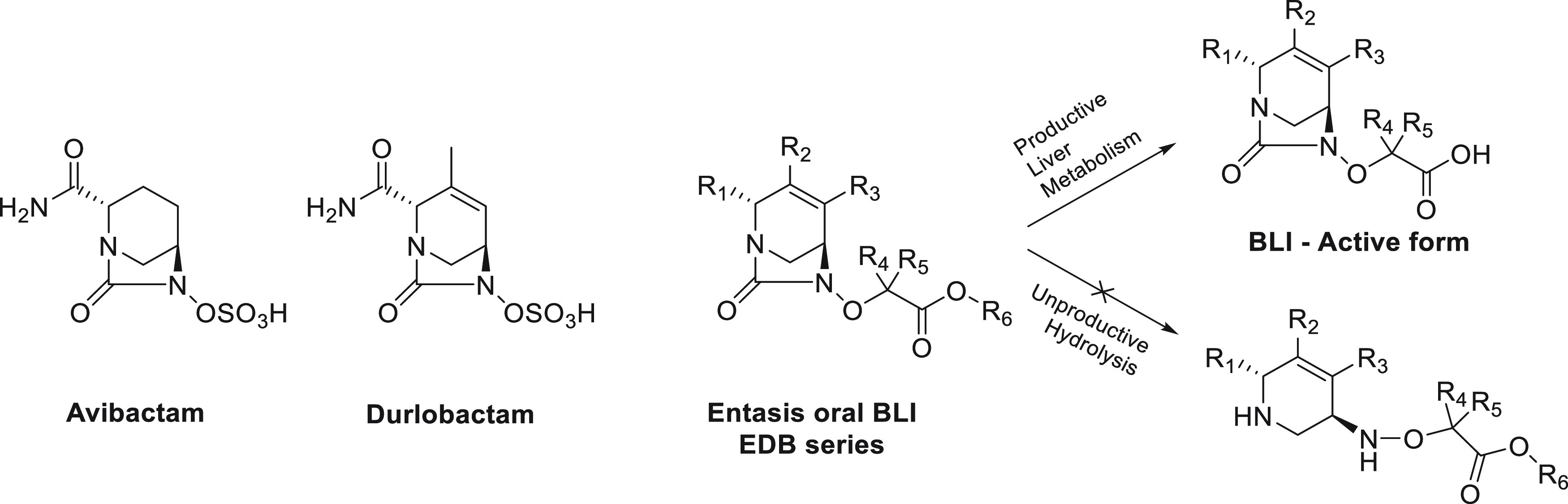
Structures of diazabicyclooctane β-lactamase inhibitors.
This program was prosecuted in two steps: (1) the discovery of a new BLI, the active ingredient that will circulate in the bloodstream and be responsible for the pharmacodynamic effect; (2) its corresponding prodrug, to be dosed orally, absorbed, and metabolized to yield the BLI. The majority of the medicinal chemistry design around R1, R2, and R3 was informed by our experience with the discovery of durlobactam (Figure 1). It was important to keep the BLI to a relatively low molecular weight (MW < 450) to ensure favorable bacterial permeation and provide inhibition of the largest number of β-lactamase enzymes possible. On the basis of the SAR generated during the durlobactam project,13 a combination of a hydrogen bond donor and acceptor was required at R1, while small alkyl groups at R2 and R3 were favored. Attention was also paid to the pKa of the carboxylic acid to optimize the interaction with the β-lactamase conserved KTG region, the part of the β-lactam substrate binding pocket that recognizes their carboxylic acid or sulfate group.20 We also sought to maintain the required low log D. This was achieved by varying the substitution at the R4 and R5 positions (Figure 1).
The new analogs were tested for inhibition of representative enzymes from each class of serine β-lactamases. Due to the time-dependent covalent inhibition by these compounds, we used the IC50 measured at 10 and 60 min of incubation to drive the SAR exploration.21 IC50 values were used as it allowed comparison of the potencies of compounds when some showed time-dependent inhibition and others did not. This was followed by evaluation of their ability (at a fixed concentration of 4 μg/mL) to restore the antibacterial potency of the β-lactam cefpodoxime against two resistant clinical strains of Escherichia coli and Klebsiella pneumoniae, as measured by the minimal inhibitory concentration (MIC) required to prevent bacterial growth. A combination MIC of less than 1 μg/mL was chosen as favorable criteria for progression as it would be similar to the potency observed for avibactam and durlobactam. Their intrinsic antibacterial activity was also evaluated as other DBOs in this series have showed activity on their own.13
After optimizing the active form of the BLI (Figure 1), we turned our attention to the ester prodrugs and their metabolic and pharmacokinetic profiles. Four parameters were taken into consideration to achieve the desired profile: (1) high stability to spontaneous hydrolysis in aqueous medium (pH 7.4 buffer, unproductive hydrolysis, Figure 1), (2) stability to intestinal esterases in vitro for stability during absorption, (3) rapid conversion to the active form by liver esterases in vitro (productive metabolism of the ester to the acid, Figure 1), and (4) high oral bioavailability in rats. Different linear and branched alkyl esters were prepared and subjected to this screening cascade to identify an orally bioavailable BLI with microbiological and DMPK profiles that could be matched with an oral β-lactam.
Structure–Activity Relationship of Analogs
The initial SAR exploration started from the R2-methyl hydroxyurea core. Sulfate replacement by acetic acid gave compound 1, which was less potent overall than avibactam or durlobactam (Table 1). Since the pKa of the acidic moiety and chemical reactivity of the urea carbonyl are critical for potency, the difluoroacetate analog 2 was prepared. The addition of the electronegative fluorine atoms was predicted to reduce the pKa of the acid to favor binding into the electropositive KTG binding subpocket and increase the electrophilicity of the urea carbonyl to generate a more reactive covalent inhibitor. As expected, analog 2 showed significantly improved potency against β-lactamases, with all IC50 values less than 15 nM at 60 min. Unfortunately, its antimicrobial activity was not measurable due to rapid decomposition under the conditions of the MIC assay. In order to mitigate this stability issue, we replaced one of the fluorine atoms by a methyl group (compound 3). This compound showed weak inhibition of the class D OXA-48 β-lactamase (IC50 = 22 μM at 60 min), likely due to a conformational constraint around the acetate side chain. The monofluoroacetic acid analogs 4 and 5 were potent β-lactamase inhibitors, with the (R)-diastereomer being the more potent. Compound 4 was equipotent with difluoroacetate 2 but without the instability under MIC measurement conditions. The compound had favorable MICs in combination with cefpodoxime (0.06 μg/mL vs E. coli ARC3627 and 0.25 μg/mL vs K. pneumoniae ARC561) with a greater than 128-fold reduction compared to the MIC with no BLI. Compound 4 also showed measurable intrinsic antibacterial activity against both strains. The (S)-diastereomer 5 showed slightly lower potency against the class A TEM-1 and class C AmpC β-lactamases and significantly lower antibacterial activity. The unexpected higher MIC values for 5 compared to 4 is not fully understood at this time, but we hypothesize that the unique stereochemistry of the fluoroacetate side chain could influence bacterial permeation and efflux. A cyclopropyl group at R2 (6) gave similar potency as a methyl group (5).
Table 1.
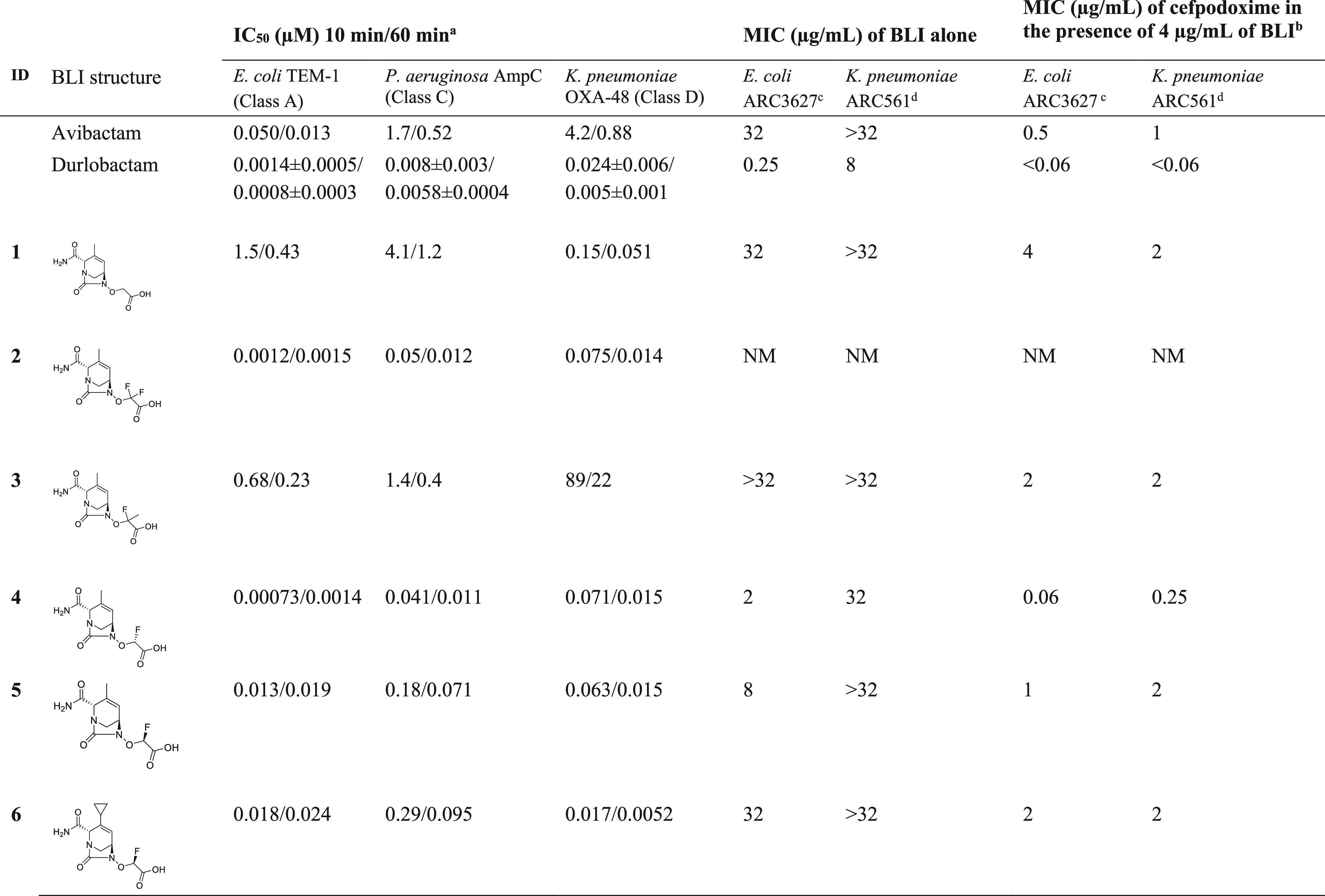
N = 1 determination except N = 3 for durlobactam, for which the average ± standard deviation is shown.
MIC of cefpodoxime in the absence of a BLI: ARC3627 > 64 μg/mL and ARC561 = 32 μg/mL.
E. coli (OXA-1, AmpC, CTX-M-15, TEM-1).
K. pneumoniae (OKP-6, AmpC, OXA-2, SHV-18) ATCC700603.
NM: nonmeasurable.
In an attempt to improve the activity of compound 5, we explored substitutions at the R1 position (Table 2). The sulfonamide- (7) and sulfonyl urea- (11) containing analogs displayed the best antibacterial activity (MIC ≤ 1 μg/mL), but lost potency against OXA-48. The MIC improvement may, in part, be explained by the increased polar surface area compared to the R1-primary amide (from TPSA = 113.2 Å2 for 5 to TPSA = 167.7 Å2 for 7 and TPSA = 179.8 Å2 for 11) and additional hydrogen bond donors and acceptors, leading to better bacterial uptake through porins and lower efflux.22 These two analogs also showed the lowest intrinsic MICs of the set against the E. coli strain while inactive against the K. pneumonia strain (MIC > 32 μg/mL). Introduction of ionizable side chains was not explored due to their likely reduction of oral absorption.
Table 2. β-Lactamase Inhibition and Antibacterial Activity in Combination with Cefpodoxime of R1-Substituted Inhibitors.

N = 1 determination.
MIC of cefpodoxime in the absence of a BLI: ARC3627 > 64 μg/mL and ARC561 = 32 μg/mL
E. coli (OXA-1, AmpC, CTX-M-15, TEM-1).
K. pneumoniae (OKP-6; AmpC; OXA-2; SHV-18) ATCC700603.
The SAR around the acetate side chain was explored on the R3-methyl scaffold (Table 3). The des-fluoro and both monofluoro acetic acid analogs all showed similar antibacterial activity in combination with cefpodoxime (MIC ≤ 0.25 μg/mL, Table 3). Compound 12 had low potency against AmpC (IC50 = 4.6 μM at 60 min). Unexpectedly, the position of the methyl group on the core (R2 vs R3) had a very significant effect on the activity of the monofluoro diastereomers, particularly with respect to the MICs. Compounds 4 and 5 (R2-methyl series, Table 1) showed a large difference in antibacterial potency, whereas 13 and 14 (R3-methyl series, Table 3) were much closer in biological activity (alone or in combination with cefpodoxime). This result clearly shows that the antibacterial activity from such structurally similar compounds will remain hard to predict, even with the most advanced computational chemistry and biology tools available.
Table 3. β-Lactamase Inhibition and Antibacterial Activity in Combination with Cefpodoxime of R3-methyl-Substituted Inhibitors.

N = 1 determination except N = 3 for 13, for which the average ± standard deviation is shown.
MIC of cefpodoxime in the absence of a BLI: ARC3627 > 64 μg/mL and ARC561 = 32 μg/mL.
E. coli (OXA-1, AmpC, CTX-M-15, TEM-1)
K. pneumoniae (OKP-6, AmpC, OXA-2, SHV-18), ATCC700603.
The best analogs from each series were selected (4, 11, 13, and 14) for testing prodrugs. Several esters were prepared and profiled in vitro to assess their aqueous stability and their stability to intestinal and liver esterases and in vivo to measure their oral bioavailability (Table 4). The goal was to identify an orally available DBO acetate that would be stable enough to reach the liver and subsequently to be efficiently hydrolyzed by carboxylesterases23 to unmask the active DBO acid prior to tissue distribution. Carboxylesterase CES-1 and CES-2 are the predominant isoforms found in human liver and intestinal tissues, respectively. In addition to tissue expression level differences, these isoforms also demonstrate different substrate affinity SAR.24 Targeted conversion in the liver during first-pass absorption and metabolism was a strategy we explored based upon the success of other approved and marketed prodrugs which have utilized conversion by liver (CES-1) carboxylesterase to maximize the production of active drug. Interestingly prodrug conversion in the intestinal enterocyte by CES-2 has shown mixed results with multiple mechanisms often required for optimum conversion and transfer of active drug to systemic circulation.24,25
Table 4. Stability Data and Rat Oral Bioavailability of Ester Prodrugs.

Not detected.
Linear and branched alkyl esters of the R2-methyl series had relatively low bioavailability, with the best bioavailability of 45% in rats obtained with ethyl ester 15. Isopropyl ester 16 had similar bioavailability despite having greater aqueous stability as well as greater stability to intestinal esterases (t1/2 > 2 h). Notably, the proxetil side chain rendered the ester 17 very unstable despite being the prodrug of choice for the marketed oral β-lactam cefpodoxime. R2-methyl analogs with a modified R1 had poor oral bioavailability, as exemplified by compound 19 (F = 4%). In contrast, esters of the R3-methyl series (20–23) had high oral bioavailabilities of 71–98%, which was independent of the chirality at R4/R5. Within the R3-methyl series, compound 23 showed markedly better stability than closely related analogs 20, 21, and 22. While we do not fully understand the reasons for this remarkable feature, we hypothesize that a unique conformation and orbital overlap are adopted by compound 23. That conformation could translate into a differentiated steric environment around the ester carbonyl and could influence the electrophilicity of the sp2 carbon.
At the end of this SAR exploration, compound 23 (ETX0282) was selected for further profiling based on its high oral bioavailability in rats, efficient conversion in vitro to ETX1317 in human liver S9, and the ability of the corresponding active BLI acid 13 (ETX1317) to restore the antibacterial activity of β-lactams.
In Vitro Profiling of ETX1317
The spectrum of class A, C, and D β-lactamase inhibition by ETX1317 was evaluated by measuring its ability to protect the β-lactamase substrate nitrocefin from degradation using previously described methods.13 ETX1317 shows potent inhibition of all serine β-lactamases tested with kinact/Ki acylation rate constants greater than 1.2 × 104 M–1 s–1 for class A and C enzymes and greater than 6.8 × 102 M–1 s–1 for class D enzymes (Table 5). ETX1317 not onlyis more potent than avibactam but also broadens the activity to include the large family of class D OXA enzymes.
Table 5. Kinetic Values of β-Lactamase and Penicillin-Binding Protein Inhibition by ETX1317a.
| β-lactamase | ETX1317 kinact/Ki (M–1 s–1) | durlobactam kinact/Ki (M–1 s–1) | avibactam kinact/Ki (M–1 s–1) |
|---|---|---|---|
| Class A | |||
| CTX-M-14 | (2.0 ± 0.2) × 106 | (2.2 ±0.3) × 106 | (1.81 ± 0.09) × 105 |
| CTX-M-15 | (4.8 ± 0.4) × 106 | (7 ± 2) × 106 | 8 × 105 |
| KPC-2 | (4.9 ± 0.2) × 104 | (9.3 ± 0.6) × 105 | 6 × 103 |
| SHV-5 | (3.2 ± 0.1) × 106 | (6.4 ± 0.5) × 106 | 1 × 105 |
| TEM-1 | NV | (1.4 ± 0.6) × 107 | 4 × 105 |
| Class C | |||
| P. aeruginosa AmpC | (1.20 ± 0.04) × 104 | (9 ± 5) × 105 | 3 × 103 |
| E. cloacae P99 | (3.9 ± 0.2) × 104 | (2.3 ± 0.4) × 106 | 8 × 103 |
| Class D | |||
| OXA-10 | (6.8 ± 0.3) × 102 | (9 ± 2) × 103 | 7 × 101 |
| OXA-23 | (1.54 ± 0.06) × 103 | (5.1 ± 0.2) × 103 | 1 × 102 |
| OXA-24 | (4.6 ± 0.2) × 103 | (9 ± 2) × 103 | 8 × 101 |
| OXA-48 | (5.3 ± 0.2) × 104 | (8 ± 2) × 105 | 5 × 103 |
| Penicillin-Binding Protein | |||
| E. coli PBP2 | (9.3 ± 0.7) × 103 | (1.7 ± 0.3) × 104 | 240 ± 40 |
NV: no value because the inhibitor was in rapid equilibrium. Ki = (3.4 ± 0.4) × 10–4 μM.
Such a broad spectrum of activity is necessary for potent activity against current MDR Gram-negative clinical isolates, which very often bear a wide variety of β-lactamases, often in combination.11 As expected based on its structure and mode of inhibition (covalent binding to active site serine), ETX1317 is not active against class B β-lactamases, as these enzymes use a metal ion in the active site to hydrolyze β-lactams. Like durlobactam,13 ETX1317 additionally inhibits E. coli penicillin-binding protein 2 (PBP2), with a kinact/Ki acylation rate constant close to 104 M–1 s–1, a value very similar to that of durlobactam and ∼40 times more potent than avibactam.
The crystal structure of the ETX1317 and CTX-M-14 (class A β-lactamase) complex was solved at 1.28 Å resolution with a refined Rwork/Rfree of 0.1427/0.1625, offering a clear depiction of the inhibitor’s postreaction binding mode (Figure 2A). ETX1317 retains many of the chemical characteristics of other BLIs in the DBO class, including avibactam and durlobactam, and therefore their binding poses are very similar (Figure 2B).26
Figure 2.

ETX1317 acyl-enzyme complex with CTX-M-14 β-lactamase. (A) Complex crystal structure of ETX1317 with CTX-M-14 β-lactamase determined at 1.28 Å resolution (PDB code 6VHS). An unbiased Fo – Fc map, shown in green, is contoured at 3σ. The ligand and protein are shown in purple and blue, respectively. Hydrogen bonds between the ligand and protein are depicted as black dashed lines. The catalytic water is shown as a red sphere. (B) Superimposition of ETX1317 with CTX-M-14 (purple/blue) and avibactam with CTX-M-14 (yellow/orange, PDB code 6MZ2, showing only the major conformation of avibactam).
The cyclic urea of the diazabicyclooctane core reacts with the catalytic serine, Ser70, forming a carbamoyl acyl-enzyme covalent linkage. Here, the carbonyl oxygen occupies the oxyanion hole formed by the backbone amide groups of Ser237 and Ser70. Following ring opening, ETX1317 adopts a half chair conformation due to the planarity of the C–C double bond, allowing the methyl substituent to project upward and form hydrophobic interactions with Tyr105. In contrast, avibactam lacks a C–C double bond and methyl group. Consequently, avibactam adopts a full chair conformation, and Tyr105 adopts an alternative conformation where it moves closer to the piperidine ring. Like avibactam, the carboxamide moiety of ETX1317 forms extensive hydrogen bonding interactions with Asn132 of the SXN motif, Asn104, and the backbone carbonyl of Ser237. The carboxamide and hydroxylamine substituents adopt a pseudoaxial orientation similar to the one observed for other DBO/β-lactamase cocrystal structures. This unique conformation in the active site has been hypothesized to facilitate the recyclization of the urea and release of the active inhibitor from the enzyme.27 Indeed, ETX1317 was shown to recyclize and be released intact from β-lactamases as detected by acylation exchange from KPC-3 to OXA-48.28
Consistent with all known β-lactamase substrates and inhibitors, ETX1317 has an anionic activating group, in this case a fluoroacetate group, that interacts with the highly conserved KTG motif. The carboxylate of the fluoroacetate group forms hydrogen bonds with Thr235 and Ser237, while the fluoride atom projects toward Lys234, Thr216, and Thr235 and is within hydrogen bonding distance with Thr235 (2.7 Å), in a similar manner to the third oxygen of the avibactam sulfate. This important interaction is further facilitated by a hydrogen bond between Ser130 and the NH group of the hydroxylamine side chain, which helps orient the flexible N–O side chain toward the electropositive KTG subpocket. The unique conformation of the monofluoroacetate activating group could explain the differences in potency between ETX1317 and its diastereomer (14), especially against class A β-lactamases.
ETX1317 displaces six water molecules from the active site upon binding, compared with the apo CTX-M-14 structure (PDB code 4UA6).29 Interestingly, the catalytic water molecule remains undisturbed and lies just 3 Å from the CTX-M-14-ETX1317 carbamate carbon. Since ETX1317 remains covalently bound, this suggests that ETX1317 possesses intrinsic hydrolytic stability and/or perturbs the protonation state of the catalytic Glu166, as has been suggested for avibactam.26,27
The potency and spectrum of β-lactamase inhibition by ETX1317 supported further antibacterial profiling in combination with β-lactams against relevant Gram-negative clinical isolates. Approved penicillins and cephalosporins administered orally were evaluated, using not only MIC as a criterion but also oral bioavailability, approved dose levels, and unbound plasma exposures. Cefpodoxime (CPD) emerged as the lead β-lactam candidate.30 Its approved use as an orally bioavailable proxetil ester prodrug to treat UTI infections was also aligned with our potential target product profile. A fixed 1:2 ratio of cefpodoxime/ETX1317 was utilized as the testing paradigm for antimicrobial susceptibility. This was found to give MIC values consistent with the outcome of in vivo antibacterial efficacy experiments.31 The clinical isolates used in this study (Table 6) were all highly resistant to CPD (MIC > 64 μg/mL), due to the expression of multiple β-lactamases, including ESBLs and carbapenemases. This panel did not contain any strains with class B metallo-β-lactamases, as DBOs do not inhibit those. ETX1317 itself showed various levels of antibacterial activity (MIC from 0.25 to 64 μg/mL) due to the inhibition of PBP2. The addition of ETX1317 to CPD restored its activity to ≤0.5 μg/mL across all Enterobacterales isolates tested, including the CRE strains (identified by “a” in Table 6). These results demonstrate that inhibition by ETX1317 of a broad spectrum of β-lactamases translates into potent antibacterial activity in combination with CPD against MDR Enterobacterales clinical isolates.
Table 6. MIC Values of ETX1317, Cefpodoxime, and the Cefpodoxime/ETX1317 (1:2) Combination against a Set of MDR Enterobacterales Clinical Isolates.
| β-lactamase genes encoded |
MIC (μg/mL) |
||||||
|---|---|---|---|---|---|---|---|
| species | strain identifier | class A | class C | class D | ETX1317 | CPD | CPD:ETX1317b |
| E. coli | ARC2687 | CTX-M-14 | AmpC | 0.5 | >64 | 0.25 | |
| ARC6059 | LAP-2 | AmpC, CMY-2 | 16 | >64 | 0.125 | ||
| ARC6064 | CTX-M-15, TEM-40 | AmpC | 8 | >64 | 0.5 | ||
| ARC6077 | CTX-M-15 | AmpC | OXA-1 | 4 | >64 | 0.125 | |
| ARC6078 | AmpC, CMY-2 | 0.25 | >64 | 0.25 | |||
| K. pneumoniae | ARC6082 | CTX-M-2, SHV-11, TEM-1 | OXA-9 | 64 | >64 | 0.125 | |
| ARC6088a | CTX-M-15, SHV-11, TEM-1 | OXA-1, OXA-48 | 16 | >64 | 0.125 | ||
| ARC6098 | CTX-M-15, SHV-1 | DHA-1 | OXA-1 | 16 | >64 | 0.125 | |
| ARC6107a | CTX-M-15, SHV-1, TEM-1 | OXA-1, OXA-244 | 4 | >64 | 0.25 | ||
| K. oxytoca | ARC5389a | OXY-1, KPC-2, PER-2 | 16 | >64 | 0.5 | ||
| C. freundii | ARC3518a | TEM-1, KPC-2 | AmpC | OXA-1 | 0.25 | >64 | 0.13 |
| E. cloacae | ARC6049 | CTX-M-15, TEM-1 | AmpC | OXA-1 | 1 | >64 | 0.125 |
| ARC6055 | SHV-5, TEM-1 | ACT variant | 1 | >64 | 0.125 | ||
CRE (carbapenem-resistant Enterobacterales) strains express β-lactamases capable of hydrolyzing carbapenems (highlighted with bold font).
MIC value shown is that of the CPD component of the combination; CPD = cefpodoxime; CPD:ETX1317 = cefpodoxime titrated with ETX1317 in a 1:2 ratio; C. freundii = Citrobacter freundii; E. cloacae = Enterobacter cloacae. All MIC values are made of at least three replicates.
In Vivo Profiling of Oral Prodrug ETX0282
The intravenous (iv) and oral (po) pharmacokinetics of ETX1317 and its isopropyl ester prodrug ETX0282 were determined in rats (N = 3/arm). Pharmacokinetic parameters are summarized in Table 7. Moderate clearance (CL) and a low volume of distribution (Vdss) resulted in a short half-life of 0.4 h which is consistent with other members of the DBO class.13 Nearly 60% of the administered iv dose of ETX1317 was eliminated in the urine as unchanged drug suggesting renal excretion was the predominant clearance mechanism. Oral administration of ETX0282 at a 10 mg/kg equivalent dose of ETX1317 resulted in high exposure of ETX1317 (Cmax = 5.8 μg/mL and AUC = 7.0 μg·h/mL) with no circulating concentrations of ETX0282 observed suggesting rapid conversion of ETX0282 to ETX1317 and high bioavailability (F) in this species. The metabolism of ETX0282 was studied in vitro and in vivo. In addition to ester prodrug cleavage of ETX0282 to yield ETX1317, hydrolytic cleavage of the DBO core was observed to yield the diamine metabolite of both ETX1317 and ETX0282 (Figure S1, Supporting Information). The diamine metabolite of ETX1317 was synthesized and used as a standard to quantify the exposure of the metabolite in definitive toxicology studies. We also looked for evidence of N–O bond cleavage to yield the monofluoro acetate and have not observed these metabolites or potentially associated structures in both in vitro and in vivo plasma and liver tissue samples. Rat protein binding of ETX1317 was low with a mean fraction unbound of 0.91 across a concentration range of 5–100 μM (Table 7). Protein binding of ETX0282 was not determined due to its limited relevance.
Table 7. Pharmacokinetic Values of ETX0282/ETX1317 in Ratsa.
| compd | dose/route (mg/kg) | Cmax (μg/mL) | AUC (μg·h/mL) | T1/2 (h) | Vdss (L/kg) | CL (mL min–1 kg–1) | renal CL (mL min–1 kg–1) | F (%) | PPB (% unbound) |
|---|---|---|---|---|---|---|---|---|---|
| ETX1317 | 10/iv | 17.5 ± 2.0 | 7.19 ± 0.34 | 0.4 ± 0.02 | 0.70 ± 0.01 | 23.1 ± 1.1 | 13.4 ± 0.3 | N/A | 91 ± 7 |
| ETX0282 | 11.6/pob | 5.8 ± 0.16 | 7.04 ± 0.62 | 1.1 ± 0.3 | N/A | N/A | N/A | 98 | ND |
Cmax, peak concentration; AUC, area under the curve; T1/2, apparent half-life based upon linear regression of semilog plot of concentration–time data; CL, clearance; Vdss, volume of distribution; F, bioavailability = (AUCpo/AUCiv) × 100; PPB, plasma protein binding, mean % unbound of 5–100 μM.
Equivalent to 10 mg/kg ETX1317 dose.
In support of determining the PK/PD of the combination of ETX1317 and cefpodoxime, the ability of ETX0282 to restore the activity of cefpodoxime proxetil in vivo upon oral dosing was evaluated in a neutropenic mouse thigh infection model,32 using the E. coli MDR clinical isolate ARC2687. While the ultimate clinical indication for the combination of ETX0282 and cefpodoxime proxetil (CPDP) is for the treatment of complicated urinary tract infections (cUTI), the murine neutropenic thigh model is the standard model utilized in exposure-efficacy PK/PD analysis of tissue and nontissue based infections.33 Achievement of PK/PD exposure end points determined in this model has been shown to correlate to clinical success.34 The isolate ARC2687 is resistant to fluoroquinolones (MIC > 4 μg/mL for levofloxacin) and cephalosporins (MIC > 64 μg/mL for cefpodoxime) but susceptible to meropenem (MIC = 0.03 μg/mL) and the CPD:ETX1317 combination (MIC = 0.25 μg/mL). For all arms, three CD-1 female mice were inoculated in each thigh resulting in 2 data points per animal. At 24 h, the vehicle control showed a large increase in colony-forming unit (CFU) count compared to the initial inoculum (+4.26 log(CFU/g)) confirming the virulence of this isolate (Figure 3). As expected based on its MIC, CPDP alone (po, q6h) was not efficacious, whereas meropenem was (−1.46 log(CFU/g) compared to pretreatment). Upon oral dosing of the combination of CPDP and ETX0282 every 6 h, a significant decrease in CFU count was observed for all three doses of the BLI. A reduction of 1.11 log(CFU/g) was obtained with a dose of 50 mg/kg and 100 mg/kg (po, q6h) of CPDP and ETX0282, respectively.
Figure 3.

Oral efficacy of the ETX0282/CPDP combination in the neutropenic mouse thigh infection model against the MDR isolate E. coli ARC2687 (AmpC, CTX-M-14). The strain is levofloxacin-resistant (MIC > 4 μg/mL), cefpodoxime-resistant (MIC > 64 μg/mL), meropenem-sensitive (MIC = 0.03 μg/mL), and cefpodoxime/ETX1317-sensitive (MIC = 0.25 μg/mL). Meropenem was dosed subcutaneously (sc, q6h).
The oral efficacy of this combination has also been demonstrated in a murine ascending pyelonephritis (cUTI) infection model where cidal activity was observed at much lower doses, consistent with high urinary excretion of both ETX1317 and CPD.35
The safety of ETX0282 was investigated with a 14-day toxicology study under Good Laboratory Practice (GLP) in rats and dogs following daily oral administration. The doses of 500 mg kg–1 day–1 and 400 mg kg–1 day–1 were established as the NOAELs (no observed adverse effect level) for rats and dogs, respectively. These results support continuing development of ETX0282. Phase 1 clinical trials in healthy volunteers showed that ETX0282 was generally well-tolerated, either alone or in combination with cefpodoxime proxetil, with no serious adverse events reported (ClinicalTrials.gov identifier: NCT03491748).
Chemical Synthesis
The ester and carboxylic acid analogs were all synthesized in a similar manner from the corresponding substituted, enantiopure hydroxyureas (Scheme 1).36 The esters were obtained by alkylation of the hydroxy group with the appropriate bromoacetates. When a chiral bromoacetate was utilized, the corresponding chiral ester was obtained with complete inversion of stereochemistry at R4/R5. The corresponding acids were isolated by saponification of the esters using lithium hydroxide. Under these basic conditions, special attention was paid to the double bond migration into conjugation with the R1 amide group and the opening of the urea that would subsequently give the corresponding diamine. Indeed, some of these reactions required careful temperature control and reaction times using UPLC/MS monitoring.
Scheme 1. General Synthesis of Diazabicyclooctane Esters and Carboxylic Acids.

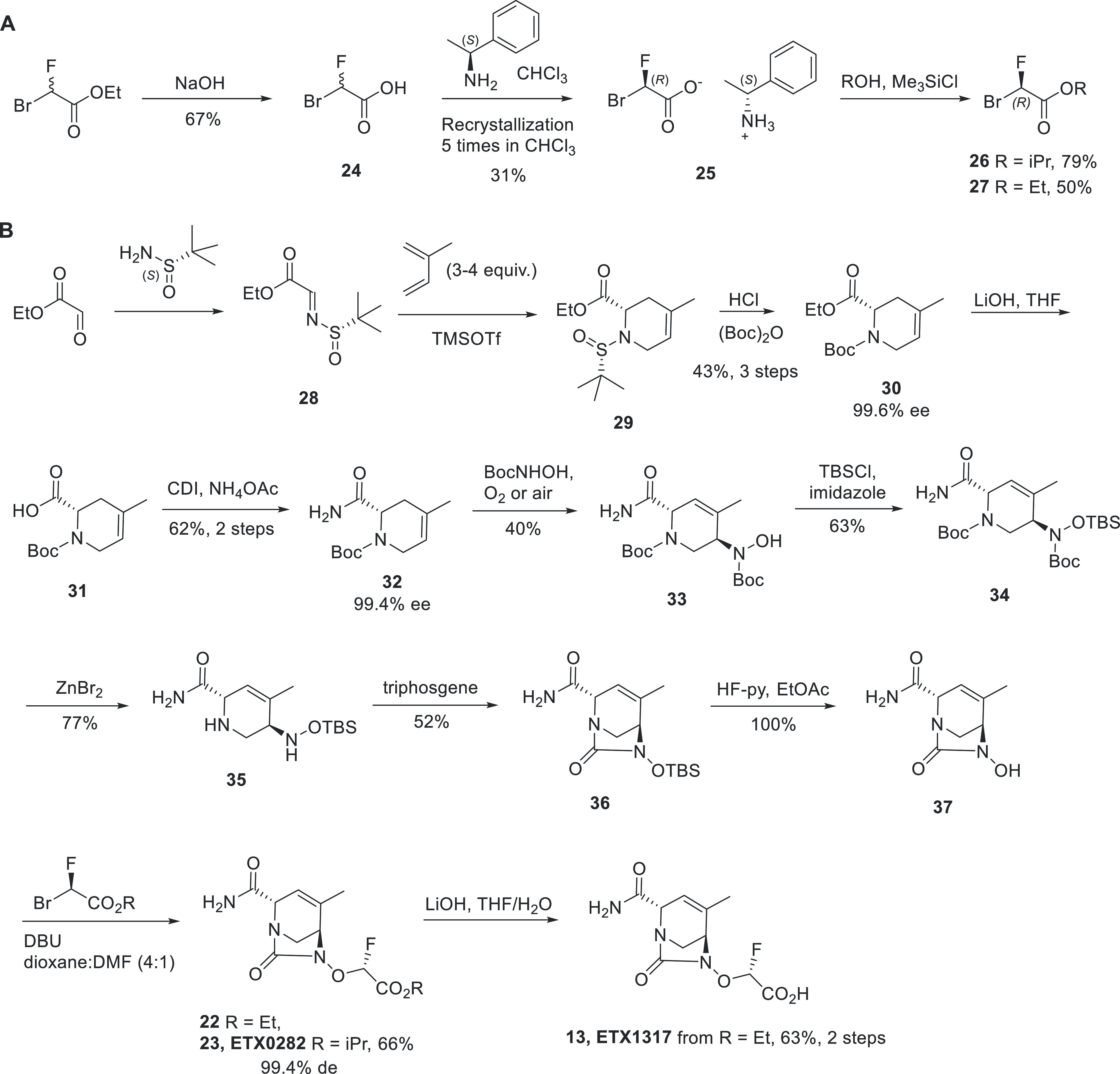
The gram-scale synthesis of ETX0282 and ETX1317 is presented below (Scheme 2). The chiral bromofluoroacetates 26 and 27 were first prepared in three steps from racemic ethyl bromofluoroacetate by saponification, followed by chiral salt resolution using (S)-1-phenylethanamine. The chiral salt was then re-esterified using isopropanol for 26 and ethanol for 27 to give the (R)-bromofluoroacetates in 79% and 50% yield, respectively. The key intermediate to the synthesis of ETX0282 and ETX1317 was the hydroxyurea 37, which was prepared in 10 steps from commercially available material.36 Ethyl 2-oxoacetate and (S)-2-methylpropane-2-sulfinamide were condensed to afford the chiral imine 28. An aza-Diels–Alder reaction with isoprene, followed by deprotection of the tert-butylsulfinyl and subsequent Boc protection, gave compound 30 in 43% yield for three steps and in 99.6% ee. Saponification of the ester followed by amide coupling afforded compound 32 in 62% yield, 99.4% ee. At this stage, many strategies were developed to install the hydroxylamine functionality in a regio and stereoselective manner. While the SN2 reaction on a chiral alcohol intermediate gave the right product, we identified the nitroso ene reaction37 as the most efficient transformation. As hypothesized from the concerted nature of its mechanism and the orientation of the primary amide and Boc group, the fully regio- and stereoselective nitroso ene reaction of alkene 32 with N-Boc-hydroxylamine gave compound 33 in a single step in 40% yield. This chiral intermediate was then protected with a TBS group, and Boc removal using zinc bromide gave compound 35. Cyclization of the diamine with triphosgene provided the corresponding cyclic urea, and the TBS was deprotected with HF-pyridine to give key intermediate 37. ETX0282 and 22 were prepared from the hydroxyurea 37 by alkylation with intermediates 26 and 27, respectively. ETX1317 was prepared in 63% yield from 22 by saponification with lithium hydroxide.
Scheme 2. Synthesis of ETX0282 and ETX1317: (A) Preparation of the Chiral Bromofluoroacetates and (B) Stereoselective Synthesis of ETX1317 and Its Ester Prodrug ETX0282.
Conclusions
We describe the discovery of ETX1317, a novel broad-spectrum inhibitor of class A, C, and D serine β-lactamases. ETX1317 retains the β-lactamase and antibacterial potencies of durlobactam, and the ester prodrug ETX0282 provides high oral bioavailability. Cefpodoxime proxetil is an orally bioavailable cephalosporin approved for treatment of a variety of bacterial infections. Its clinical utility is currently limited by β-lactamase-mediated resistance. In vitro, ETX1317 restores cefpodoxime’s antimicrobial activity against a variety of pathogens, including Enterobacterales resistant to fluoroquinolones, cephalosporins, and carbapenems. The combination of their orally bioavailable prodrugs, ETX0282 and cefpodoxime proxetil, respectively, was efficacious in murine infection models and had favorable tolerability in preclinical safety studies. ETX0282CPDP, the combination of ETX0282 and cefpodoxime proxetil, is currently being developed for the treatment of infections caused by Enterobacterales, including multidrug-resistant (MDR) and carbapenem-resistant Enterobacterales (CRE). By providing the option of an effective course of oral antibiotic treatment, ETX0282CPDP has the potential to benefit patients as well as the healthcare systems by reducing the risk of nosocomial infections and avoiding the healthcare costs associated with hospitalizations. Moreover, if potent, orally available β-lactams with activity against nonfermenter Gram-negative species (Pseudomonas aeruginosa and Acinetobacter baumannii, for example) become available, ETX0282 could be an oral BLI partner to treat infections due to these highly resistant and problematic pathogens.
Experimental Section
Chemical Synthesis: General Methods, Reagents, and Materials
All of the solvents and reagents used were obtained commercially and used as received unless noted otherwise. 1H and 13C NMR spectra were recorded in CDCl3, D2O, or DMSO-d6 solutions at 300 K using a Bruker Ultrashield 300 MHz instrument. Chemical shifts are reported as parts per million relative to tetramethylsilane (TMS) (0.00) for 1H and 13C NMR. Silica gel chromatographies were performed on ISCO Combiflash Companion Instruments using ISCO RediSep Flash Cartridges (particle size, 35–70 μm) or Silicycle SilicaSep Flash Cartridges (particle size, 40–63 μm). Reverse phase chromatography was performed on ISCO Combiflash Rf 200 Instruments using RediSep High Performance Gold C18 and C18Aq columns. Preparative reverse phase HPLC was carried out using a Gilson instrument. When not indicated, compound intermediates and reagents were purchased from chemical supply houses. All electrospray ionization mass spectra (ESI-MS) were recorded via reverse phase UPLC–MS with a Waters Acquity UPLC instrument with diode array and electrospray ionization detectors, a UPLC HSS T3, 2.1 mm × 30 mm, 1.8 μm column, and a gradient of 2–98% acetonitrile in water with 0.1% formic acid over 2.0 min at 1 mL/min. Injection volume was 1 μL, and the column temperature was 30 °C. Detection was based on electrospray ionization (ESI) in positive and negative polarity using a Waters ZQ mass spectrometer (Milford, MA, USA), diode-array UV detector from 210 to 400 nm, and evaporative light scattering detector (Cedex 75, Sedere, Alfortville, France). Purities of final compounds were determined using a Waters Acquity UPLC instrument with diode array detector, a UPLC ACE Excel 2 C18-AR 50 mm × 4.6 mm, 2 μm column, and a gradient of 5–95% acetonitrile in water with 0.1% formic acid over 6.0 min at 1 mL/min. Injection volume was 1 μL, and the column temperature was 30 °C. All compounds were isolated with ≥95% purity unless otherwise noted.
Ethyl (S,E)-2-((tert-Butylsulfinyl)imino)acetate (28)
To a solution of ethyl 2-oxoacetate (66.0 mL, 321 mmol, 50% in toluene) in DCM (1 L) at 0 °C were added (S)-2-methylpropane-2-sulfinamide (30.0 g, 248 mmol) and molecular sieves (4 Å, 500 g). The resulting solution was stirred at room temperature for 18 h. Molecular sieves were removed by filtration. Filtrate was concentrated by distillation under vacuum to give a crude product, which was purified by flash silica chromatography (0% to 5% EtOAc in petroleum ether) to give a colorless oil, 45.0 g, 88%. 1H NMR (400 MHz, CDCl3-d) δ: 1.28 (s, 9H), 1.39 (t, 3H, J = 12 Hz), 4.38 (q, 2H, J = 12 Hz), 8.01 (s, 1H).
Ethyl (S)-1-((S)-tert-Butylsulfinyl)-4-methyl-1,2,3,6-tetrahydropyridine-2-carboxylate (29)
To a solution of (S,E)-ethyl 2-(tert-butylsulfinylimino)acetate (28, 50.0 g, 244 mmol) in DCM (600 mL) at −78 °C was added isoprene (97.2 mL, 972 mmol), followed by addition of TMSOTf (97.4 mL, 417 mmol). The resulting solution was stirred at −78 °C for 3 h and quenched slowly at −78 °C with phosphate buffer solution (pH = 7.4, 1 L). After warming to room temperature, the mixture was extracted with DCM (3 × 500 mL). The combined organic extracts were washed with water (2 × 500 mL) and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to afford 60.0 g of crude product as a brown oil. The product was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ: 1.08(s, 9H), 1.18 (t, 3H, J = 12 Hz), 1.55 (m, 2H), 1.64 (s, 3H), 2.15 (m, 2H), 3.55 (m, 1H), 4.11 (dq, 2H, J = 2, 4 Hz), 5.37 (m, 1H). MS (ESI+) [M + H]+ = 274 (C13H23NO3S).
1-(tert-Butyl) 2-Ethyl (S)-4-methyl-3,6-dihydropyridine-1,2(2H)-dicarboxylate (30)
To a solution of the crude (S)-ethyl 1-((S)-tert-butylsulfinyl)-4-methyl-1,2,3,6-tetrahydropyridine-2-carboxylate (29, 100 g, 366 mmol) in MeOH (1.00 L) at 0 °C was added hydrogen chloride (100 mL, 4 M in dioxane, 400 mmol). The resulting solution was stirred at room temperature for 18 h. MeOH and HCl/dioxane were removed by distillation under vacuum to give a crude product, which was dissolved in water (1.00 L) and extracted with EtOAc (3 × 500 mL). The pH of the aqueous solution was adjusted to 7 with solid NaHCO3. The aqueous was extracted with EtOAc until LCMS showed no product detected. The organic phases were combined and dried over Na2SO4, filtered, and concentrated to afford crude product (30.0 g, 177 mmol) as a light-yellow oil. The oil was dissolved in THF (500 mL) and cooled by ice–water bath. To the cooled solution was added a solution of sodium bicarbonate (22.3 g, 266 mmol) in water (500 mL), followed by di-tert-butyl dicarbonate (57.8 g, 266 mmol). The resulting solution was stirred at room temperature for 18 h. The two layers were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and evaporated. Crude product was purified by flash silica chromatography (0–30% EtOAc in PE) to afford the title compound 47.5 g, 43% yield, 99.6% ee from 28. 1H NMR (400 MHz, DMSO-d6) δ: 1.12 (t, 3H), 1.40 (m, 9H), 1.63 (s, 3H), 2.34 (m, 2H), 3.58 (m, 1H), 3.87 (m, 1H), 4.08 (m, 2H), 4.80 (m, 1H), 5.34 (m, 1H). MS (ESI+) [M + Na]+ = 292 (C14H23NO4Na).
tert-Butyl (S)-2-cCarbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (31)
To a solution of 1-(tert-butyl) 2-ethyl (S)-4-methyl-3,6-dihydropyridine-1,2(2H)-dicarboxylate (30, 47.5 g, 176 mmol) in THF (1000 mL) and water (500 mL) at 0 °C was added dropwise lithium hydroxide (1 M, 440 mL, 440 mmol). The reaction mixture was warmed to room temperature and stirred for 16 h. Solvent was removed; residue was diluted with water. The pH of the solution was adjusted to ∼3 with HCl (1 N) solution. The mixture was extracted with EtOAc (3 × 300 mL). Organic layers were combined, washed with water and brine, dried over MgSO4, filtered, and concentrated to give a colorless oil (40.3 g). 1H NMR (300 MHz, DMSO-d6) δ: 1.38 (m, 9H), 1.64 (s, 3H), 2.36 (m, 2H), 3.60 (m, 1H), 3.89 (m, 1H), 4.75 (m, 1H), 5.35 (dd, 1H, J = 3, 15 Hz), 12.73 (s, 1H). MS (ESI+) [M + Na]+ = 264 (C12H19NO4Na).
tert-Butyl (S)-2-Carbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (32)
To a solution of (S)-1-(tert-butoxycarbonyl)-4-methyl-1,2,3,6-tetrahydropyridine-2-carboxylic acid (31, 40.3 g, 167 mmol) in THF (500 mL) at 0 °C was added N,N′-carbonyldiimidazole (32.5 g, 201 mmol) in portions. The crude was stirred at 0 °C for 5 h. Then ammonium acetate (38.2 g, 503 mmol) was added. The reaction was stirred at room temperature for an additional 18 h, quenched with water, and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, filtered, and concentrated. Crude product was purified by flash silica chromatography (0%–30% EtOAc in PE) to give a white solid, 25.0 g, 62% yield, 99.4% ee. 1H NMR (400 MHz, DMSO-d6) δ: 1.41 (s, 9H), 1.66 (s, 3H), 2.35 (s, 2H), 3.84 (m, 2H), 4.66 (m, 1H), 5.35 (m, 1H), 6.96 (s, 1H), 7.19 (s, 1H). MS (ESI+) [M + Na]+ = 263 (C12H20N2O3Na).
tert-Butyl (3R,6S)-3-((tert-Butoxycarbonyl)(hydroxy)amino)-6-carbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (33)
To a solution of tert-butyl (S)-2-carbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (32, 25.0 g, 104 mmol) in DCM (250 mL) were added BocNHOH (70.6 g, 531 mmol), CuCl (6.10 g, 62.5 mmol), and pyridine (107 mL, 1.30 mmol). The resulting solution was stirred at room temperature for 44 h under oxygen. The solids were removed by filtration. The filtrate was washed with water (6 × 500 mL) and brine, dried over Na2SO4, filtered, and concentrated. The crude product was purified by flash silica chromatography (0%–50% EtOAc in PE) to give the title compound as a white solid, 40% yield. Starting material was recovered (10 g). The same procedure was repeated three times to afford 15.0 g of product in total. 1H NMR (400 MHz, DMSO-d6) δ: 1.34–1.42 (m, 18H), 1.68 (s, 3H), 3.31 (dd, J = 4.4, 13.6 Hz, 1H), 3.92 (dd, J = 13.6, 48.8 Hz, 1H), 4.21 (d, J = 78 Hz, 1H), 4.61 (d, J = 72.4 Hz, 1H), 5.69 (d, J = 42 Hz, 1H), 6.98 (s, 1H), 7.40 (s, 1H), 8.77 (d, J = 62.4 Hz, 1H). MS (ESI+) [M + Na]+ = 394 (C17H29N3O6Na).
tert-Butyl (3R,6S)-3-((tert-Butoxycarbonyl)((tert-butyldimethylsilyl)oxy)amino)-6-carbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (34)
To a solution of tert-butyl (3R,6S)-3-((tert-butoxycarbonyl)(hydroxy)amino)-6-carbamoyl-4-methyl-3,6-dihydropyridine-1(2H)-carboxylate (33, 12.0 g, 32.3 mmol) in DCM (96 mL) at 0 ± 5 °C was added imidazole (4.40 g, 64.6 mmol). The resulting solution was stirred at room temperature for 10 min, and then TBS-Cl (4.80 g, 32.3 mmol) in DCM (10 mL) was added dropwise. The reaction mixture was stirred at 0 °C for an additional 18 h, washed with water and brine, dried over Na2SO4, filtered, and concentrated. The crude product was purified by flash silica chromatography (0%–20% EtOAc in PE) to afford the title compound as a white solid, 10.0 g, 63%. 1H NMR (300 MHz, DMSO-d6) δ: 0.08–0.12 (m, 6H), 0.84 (s, 9H), 1.31–1.40 (m, 18H), 1.75 (s, 3H), 3.20–3.29 (m, 1H), 4.00–4.15 (m, 2H), 4.59 (d, J = 25.8 Hz, 1H), 5.69 (d, J = 13.8 Hz, 1H), 7.01 (d, J = 15.3 Hz, 1H), 7.49 (s, 1H). MS (ESI+) [M + H]+: 486 (C23H43N3O6Si).
(2S,5R)-5-(((tert-Butyldimethylsilyl)oxy)amino)-4-methyl-1,2,5,6-tetrahydropyridine-2-carboxamide (35)
To a solution of tert-butyl (3R,6S)-3-[tert-butoxycarbonyl-[tert-butyl(dimethyl)silyl]oxy-amino]-6-carbamoyl-4-methyl-3,6-dihydro-2H-pyridine-1-carboxylate (34, 21.7 g, 44.7 mmol) in DCM (250 mL) at 0 °C was added zinc bromide (40.2 g, 179 mmol). The resulting suspension was allowed to warm to room temperature and stir ∼66 h. The reaction mixture was cooled by ice–water bath, to which a slurry of NaHCO3 (38.23 g, 10 equiv) in water (300 mL) was added. The resulting mixture was stirred for 1 h. Solid was removed by filtration and washed 3–4 times with DCM until no product was detected from the rinsing solution. The two layers from the filtrate were separated. The aqueous layer was extracted with DCM three times (until no product was detected from aqueous layer). The combined DCM solution was concentrated to remove most of the solvent. The residue was partially dissolved in 10% MeOH in DCM and was loaded onto a short silica gel pad and eluted with 10% MeOH in DCM. The filtrate was evaporated and dried under vacuum to give a yellow foam solid (crude 9.9 g, 77%). 1H NMR (300 MHz, CDCl3-d1) δ: 0.00 (s, 6H), 0.80 (s, 9H), 1.71–1.72 (m, 3H), 2.55 (dd, J = 13.8 Hz, 3.0 Hz, 1H), 2.54 (dd, J = 3.0, 10.8, 1H), 3.12 (dd, J = 1.8, 13.8 1H), 2.91 (s, 1H), 3.69 (d, J = 1.2 Hz, 1H), 4.95 (bs, 1H), 5.50 (s, 1H), 5.92 (d, J = 1.5 Hz, 1H), 7.15 (s, 1H). MS (ESI+) [M + H]+: 286 (C13H27N3O2Si).
(2S,5R)-6-((tert-Butyldimethylsilyl)oxy)-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-ene-2-carboxamide (36)
To a clear solution of (3R,6S)-3-[[tert-butyl(dimethyl)silyl]oxyamino]-4-methyl-1,2,3,6-tetrahydropyridine-6-carboxamide (35, 7.66 g, 26.8 mmol) in MeCN (150 mL) and DCM (200 mL) at 0 °C was added N,N′-diisopropylethylamine (19.1 mL, 107 mmol) followed by a solution of triphosgene (2.71 g, 9.12 mmol) in MeCN (50 mL) dropwise (2 mL/hour by a syringe pump). After addition, the solution was allowed to warm to room temperature and stirred overnight. The reaction mixture was concentrated to dryness. The resulting residue was diluted with EtOAc and washed with brine. The aqueous layer was extracted with EtOAc. The combined organic extracts were dried over MgSO4, filtered, and concentrated. Crude product was purified by silica gel chromatography (0%–100% EtOAc/hexane) to give the title compound as a white solid 4.36 g, 52%. 1H NMR (300 MHz, DMSO-d6) δ: 0.12 (s, 3H), 0.14 (s, 3H), 0.90 (s, 9H), 1.78 (s, 3H), 3.19 (m, 2H), 3.57 (s, 1H), 4.12 (q, J = 2.1 Hz, 1H), 5.47 (s, 1H), 7.32 (s, 1H), 7.51 (s, 1H). MS (ESI+) [M + H]+: 312 (C14H25N3O3Si).
(2S,5R)-6-Hydroxy-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-ene-2-carboxamide (37)
To a solution of (2S,5R)-5-(((tert-butyldimethylsilyl)oxy)amino)-4-methyl-1,2,5,6-tetrahydropyridine-2-carboxamide (36, 214 mg, 0.690 mmol) in ethyl acetate (5 mL) at 0 °C was added HF–pyridine (0.070 mL, 2.75 mmol). The reaction mixture was warmed to room temperature and stirred for 2 h. The reaction mixture was concentrated to afford title compound as a tan solid, 135 mg (100%). 1H NMR (300 MHz, DMSO-d6) δ: 1.79 (m, 3H), 3.16 (m, 2H), 3.54 (m, 1H), 4.05–4.09 (m, 1H), 5.41–5.43 (m, 1H), 7.27 (s, 1H), 7.49 (s, 1H), 9.60 (s, 1H). MS (ESI+) [M + H]+: 198(C8H11N3O3).
2-Bromo-2-fluoroacetic Acid (24)
To a 50 L reactor at 0–5 °C was charged a solution of ethyl 2-bromo-2-fluoroacetate (3.5 kg) in tetrahydrofuran (7 L, 2 V) and a solution of sodium hydroxide (830 g) in water (7 L, 2 V) dropwise over 1 h. The resulting solution was stirred at 0–5 °C for 1 h. HCl (160 mL) was added dropwise at 0–5 °C. Water and tetrahydrofuran were removed by concentration under vacuum. The residue was suspended in tetrahydrofuran (35 L, 10 V), and conc HCl (1.57 L, 1.0 equiv) was added dropwise. Anhydrous sodium sulfate was added, and the resulting mixture was stirred for 2 h. The solid was filtered off and washed with THF (1 L × 2). The filtrate was concentrated under vacuum to give 2-bromo-2-fluoroacetic acid (2.2 kg) as a yellow oil, which was combined with a previous batch made by the same method (940 g, purity, 72%) and distilled in vacuum (65–70 °C, 100 Pa) to give 2-bromo-2-fluoroacetic acid (2.55 kg, total yield 67%) as a colorless oil. 1H NMR (400 MHz, CDCl3-d) δ: 6.66 (d, 1H, J = 68 Hz), 11.15 (s, 1H).
(S)-1-Phenylethan-1-amine (R)-2-Bromo-2-fluoroacetate (25)
To a 10 L reactor at 0–5 °C was charged a solution of 2-bromo-2-fluoroacetic acid (24, 2.0 kg) in 1 L of chloroform (1 V), to which a solution of (S)-1-phenylethanamine (1.4 kg) in 1 L of chloroform (1 V) was added dropwise. The mixture was stirred at room temperature overnight, and the resulting white solid was collected by filtration to give a salt of (S)-1-phenylethanamine 2-bromo-2-fluoroacetate (2.5 kg; ee, 6%), which was charged into a 10 L reactor, followed by addition of chloroform (5 L, 2 V). The resulting mixture was stirred for 2 h at 50 °C (solid was partially dissolved in chloroform), cooled to 0 °C, and allowed to stand for 2 h. Solid was collected by filtration and washed with cooled chloroform (500 mL, 0.2 V). The recrystallization procedure was repeated 4 times to afford 1.09 kg (97% ee) of the title compound as a white solid with an overall yield of 31% (2 steps). 1H NMR (400 MHz, CD3OD-d4) δ: 1.65 (d, J = 6.8 Hz, 3H), 4.47 (q, J = 6.8 Hz, 1H), 6.51 (d, JH–F = 53.2 Hz, 1H), 7.40–7.50 (m, 5H).
Pentan-3-yl (R)-2-Bromo-2-fluoroacetate (26)
To a 2 L reactor at room temperature was charged (S)-1-phenylethanamine (R)-2-bromo-2-fluoroacetate (25, 450 g), dichloromethane (900 mL, 2 V), and iPrOH (2.0 equiv). Chlorotrimethylsilane (1.12 L) was added slowly, and a white precipitate was formed. The resulting mixture was stirred at room temperature overnight. White precipitate was filtered off, and the filter cake was washed with hexane (450 mL, 1 V). The combined filtrate was washed with water (3 × 100 mL). Organic solution was dried with anhydrous sodium sulfate, filtered, concentrated under vacuum. Residue was distilled (54–60 °C, 100 Pa) to give the title compound as a colorless oil (290 g, 79% yield, 95% purity, 97.2% ee). 1H NMR (400 MHz, CDCl3-d) δ: 1.32 (m, 6H), 5.17 (m, 1H), 6.53 (d, 1H, J = 51.2 Hz).
Ethyl (R)-2-Bromo-2-fluoroacetate (27)
Into a 50 mL three-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, were placed (1R)-1-phenylethan-1-amine, (2S)-2-bromo-2-fluoroacetic acid (25, 30.0 g, 108 mmol), and ethanol (34.7 g, 755 mmol). This was followed by the addition of chlorotrimethylsilane (82.0 g, 755 mmol) dropwise with stirring at room temperature. The resulting solution was stirred for 4 h at room temperature, then quenched by the addition of 10 mL of water/ice. The resulting solution was extracted 3 times with 20 mL of petroleum ether (30–60 °C), and the organic layers were combined. The resulting mixture was washed 2 times with 20 mL of brine. The mixture was dried over anhydrous sodium sulfate, filtered, and concentrated. The residue was applied onto a silica gel column with petroleum ether (30–60 °C). This resulted in 10.0 g (50%) of the title compound as a colorless oil. 1H NMR (300 MHz, CDCl3-d) δ:1.38 (t, 3H, J = 6 Hz), 4.38 (q, 2H, J = 6 Hz), 6.58 (d, 1H, J = 51 Hz).
(2R)-Isopropyl 2-(((2S,5R)-2-Carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl)oxy)-2-fluoroacetate (23, ETX0282)
To a solution of (2S,5R)-6-hydroxy-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-ene-2-carboxamide (37, 582 mg, 2.95 mmol) in 1,4-dioxane (16 mL) and DMF (2 mL) was added isopropyl (2R)-2-bromo-2-fluoroacetate (26, 881 mg, 4.43 mmol). The reaction mixture was cooled to 0 °C, and DBU (0.440 mL, 2.95 mmol) was added dropwise. The reaction mixture was stirred for 10 min, then diluted with ethyl acetate and washed with 1:1 brine water twice. The organics were dried over magnesium sulfate, filtered, and concentrated. Silica gel chromatography (0%–90% ethyl acetate/hexanes) afforded a white foam. The foam was dissolved in 1:1 acetonitrile:water, frozen, and lyophilized to afford a white solid, 614 mg, 66% yield, 99.4% de. HPLC purity: 98% (220 nm). Optical rotation: −3.71 (acetone, c = 1.25 g/dL). MS (ESI+) [M + H]+: 316 (C13H18FN3O5). HRMS (ESI+): (m/z) calculated for C13H19FN3O5 [M + H]+ 316.1303, found 316.1313. 1H NMR (300 MHz, DMSO-d6) δ: 1.24 (m, 6H), 1.82 (t, 3H, J = 1.79 Hz), 3.21 (m, 1H), 3.33 (m, 1H), 3.95 (d, 1H, J = 2.1 Hz), 4.22 (m, 1H), 5.01 (m, 1H), 5.52 (m, 1H), 6.15–6.33 (d, 1H, J = 55.8 Hz), 7.32 (s, 1H), 7.55 (s, 1H). 13C NMR (300 MHz, DMSO-d6) δ: 21.62, 21.76, 23.00, 46.49, 62.56, 64.16, 70.66, 104.37, 107.49, 120.98, 138.69, 162.63, 163.07, 169.45, 169.90.
Ethyl (2R)-2-[[(2S,5R)-2-Carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl]oxy]-2-fluoro-acetate (22)
The title compound was prepared from (37, 215 mg, 1.09 mmol) and ethyl (2R)-2-bromo-2-fluoroacetate (27, 342.9 mg, 1.85 mmol) according to the procedure for 23, ETX0282. HPLC purity: 96% (diode array detection). MS (ESI+) [M + H]+: 302 (C12H16FN3O5). HRMS (ESI+): (m/z) calculated for C12H17FN3O5 [M + H]+ 302.1147, found 302.1151. 1H NMR (300 MHz, DMSO-d6) δ: 1.21 (t, 3H, J = 7.2 Hz), 1.82 (m, 3H), 3.19 (m, 1H), 3.29 (m, 1H), 3.95 (d, 1H, J = 1.8 Hz), 4.23 (m, 1H), 4.24 (m, 2H), 5.52 (m, 1H), 6.19–6.37 (d, 1H, J = 56.1 Hz), 7.35 (br, 1H), 7.58 (br, 1H).
(2R)-2-(((2S,5R)-2-Carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl)oxy)-2-fluoroacetic Acid Lithium Salt (13, ETX1317)
To a solution of (2R)-ethyl 2-(((2S,5R)-2-carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl)oxy)-2-fluoroacetate (22, 107 mg, 0.36 mmol) in THF (3 mL) and water (1 mL) at 0 °C was added 1 M lithium hydroxide (0.360 mL, 0.360 mmol). The reaction mixture was stirred at 0 °C for 10 min. Another 0.2 equiv of lithium hydroxide was added. After 10 min, the reaction mixture was adjusted to pH = 7 with 0.5 N HCl. The THF was evaporated, and the remaining aqueous portion was frozen and lyophilized to afford a pale yellow solid. Reverse phase HPLC (YMC Carotenoid C30, 19 mm × 150 mm, 5 μm coupled with Synergi Polar RP 21.2 mm × 100 mm, 4 μm, 0%–40% acetonitrile in water, 20 mL/min, 15 min) afforded the title compound as a white solid after lyophilization, 64.6 mg, 63%. HPLC purity: 96% (220 nm). Optical rotation: −3.64 (water, c = 1.23 g/dL). MS (ESI+) [M + H]+: 274 (C10H12FN3O5). HRMS (ESI+): (m/z) calculated for C10H13FN3O5 [M + H]+ 274.0834, found 274.0823. 1H NMR (300 MHz, DMSO-d6) δ: 1.83 (m, 3H), 3.21 (m, 2H), 3.91 (d, 1H, J = 2.7 Hz), 4.16 (m, 1H), 5.19–5.41 (d, 1H, J = 65.4 Hz), 5.44 (m, 1H), 7.26 (br, 1H), 7.52 (br, 1H). 13C NMR (300 MHz, DMSO-d6) δ: 22.34, 46.94, 62.64, 63.59, 106.43, 109.62, 117.77, 140.66, 169.01, 169.37, 171.20, 173.39.
Enzyme Inhibition Assays
The methods used to measure inhibition of β-lactamases were the same as those described in the supplement to Durand-Reville et al.13 Reaction progress curves at 490 nm for hydrolysis of the chromogenic β-lactamase substrate nitrocefin (SynGene, Bangalore, India) at 100 μM were measured for a range of inhibitor concentrations in 0.1 M sodium phosphate (pH 7.0), 10 mM NaHCO3, and 0.005% Triton X-100 at ambient temperature using a Spectramax absorbance plate reader (Molecular Devices, Sunnyvale, CA). The set of 12 progress curves were fit globally to a second-order kinetic model of enzyme inactivation. The 10 min and 60 min time points on the set of best-fit curves were used to calculate the IC50 by nonlinear regression to the Hill equation: % inhibition = 100[I]n/(IC50n + [I]n), where [I] is the inhibitor concentration and n is the Hill coefficient. The second-order inactivation rate constant kinact/Ki was calculated from the set of progress curves using Global Kinetic Explorer (Kintek) as previously described.13
Inhibition of E. coli PBP2 was measured according to the method published in Shapiro et al.38 A set of 2-fold serial dilutions of each inhibitor from 61.44 to 0.06 and 0 μM were prepared from a fresh solution of the compound dissolved in assay buffer consisting of 0.1 M sodium phosphate (pH 6.0) and 0.01% Triton X-100. Inhibitor solution (2 μL) and 2 μL of 90 nM 5-TAMRA-ampicillin were added to the wells of a low-volume, shallow-well, black polystyrene, 384-well microplate (Corning). Reactions were initiated by addition of 2 μL of 300 nM E. coli PBP2. Reactions were followed by the change in fluorescence anisotropy. The second-order inactivation rate constant kinact/Ki was calculated from the set of 12 progress curves using Global Kinetic Explorer.
Antimicrobial Susceptibility Testing
The broth microdilution susceptibility testing was conducted according to CLSI39 using CAMHB. Durlobactam and avibactam were synthesized at Entasis Therapeutics. Cefpodoxime (catalog no. J66225) was purchased from Alfa Aesar. The minimal inhibitory concentration (MIC) of cefpodoxime combined with ETX1317 against MDR Enterobacterales clinical isolates was tested by titrating 2-fold dilutions of the combination in a fixed 1:2 weight ratio.
Crystallization Experiments and Structure Determination
CTX-M-14 was purified as previously described.40 Apo crystals of CTX-M-14 were grown in a hanging drop apparatus consisting of a 1:1 mixture of 20 mg·mL–1 CTX-M-14 and 1.2 M potassium phosphate, pH 7.9, at 20 °C. Apo crystals were transferred into a crystallization solution containing 3 mM ETX1317, for 15 min, then cryoprotected with 30% sucrose and 1.8 M potassium phosphate (pH 7.9) and flash frozen in liquid nitrogen. Data were collected on the SERCAT-22ID beamline at the Advanced Photon Source (APS) in Argonne, IL, and processed with iMOSFLM.41 The CCP4 versions of Scala and REFMAC were used for scaling and refinement.42,43 All model building was performed with COOT44 and Figure 2A and Figure 2B were generated with PyMol (Schrödinger, LLC).
In Vitro DMPK Experiments
Liver and intestinal S9 subcellular fractions (Xenotech) from rat and human tissue were diluted to a protein concentration of 0.8 mg/mL in 100 mM potassium phosphate buffer, pH 7.4, and preincubated in a 37 °C water bath for 5 min prior to addition of 10 μM (final) of compound of interest. Serial aliquots were removed at 0, 2, 5, 10, 20, 40, and 60 min and quenched in acetonitrile with internal standard prior to LC/MS/MS30 to determine concentrations of prodrug. First order degradation half-lives were determined from the slope of the log–linear plots of the depletion data.
Buffer stability (potassium phosphate buffer, pH 7.4) was evaluated using the same protocol but without adding the S9 subcellular fractions.
In Vivo Pharmacokinetics and Pharmacology Experiments
All in vivo procedures were completed in compliance with the Animal Welfare Act Regulations (9 CFR 3) under Entasis-reviewed IACUC protocols and under the supervision of a site attending veterinarian.
Intravenous pharmacokinetics of ETX1317 were investigated in jugular vein-cannulated male Sprague Dawley rats (Charles River Laboratories, 180–200 g, n = 3/route) at a dose of 10 mg/kg formulated in 0.9% saline. Oral pharmacokinetics of ETX0282 were evaluated at a 10 mg/kg equivalent dose of ETX1317 formulated in 25:75 PEG 400/WFI. The pH of each formulation was verified to fall within a range of 4–7, and compounds were confirmed to be in solution prior to dosing. Potency was verified by LC/MS/MS. Dose volumes were 5 and 10 mL/kg for ETX1317 (iv) and ETX0282 (po), respectively. Blood samples to be processed for plasma using K2EDTA as an anticoagulant were taken at 0.08, 0.17, 0.25, 0.5, 1, 2, 4, 8 h (iv) and 0.25, 0.5, 1, 2, 4, 8, and 24 h (po) postdose in rats. Blood samples collected into 0.5 mL of BD microtainers (Becton, Dickinson and Company) containing 1.0 mg of K2EDTA were centrifuged in a microfuge for 10 min. Plasma was transferred to 96-well cryotubes and stored at −80 °C prior to analysis.
In vivo neutropenic infection models in the mouse were conducted as previously described.32,45 All procedures were performed to Entasis-approved IACUC policies and guidelines as well as OLAW standards. Briefly, female CD-1 mice (N = 3 per arm, 20–22 g, Charles River Laboratories) were acclimated for 5 days prior to start of study. Animals were housed 3 per cage with free access to food and water. Mice were rendered neutropenic via two doses of cyclophosphamide on days −4 and −1 with 150 mg/kg and 100 mg/kg delivered intraperitoneally in a dose volume of 10 mL/kg, respectively. E. coli isolate ARC2687 was prepared for infection from an overnight plate culture. A portion of the plate was resuspended in sterile saline and adjusted to an OD of 0.1 at 625 nm. The adjusted bacterial suspension was further diluted to target an infecting inoculum of approximately 5.0 × 106 to 1.0 × 107 CFU/mouse. The actual inoculum size varied between 5.5 × 106 and 1.6 × 107 CFU/thigh and was administered via intramuscular injection of 100 μL. Plate counts of the inoculum were performed to confirm inoculum concentration. Treatment was initiated 2 h after bacterial challenge. ETX0282 and CPDP were formulated in 0.5% HPMC/0.1% Tween 80 suspension. Meropenem was dissolved in water for injection. The in vivo efficacy study used meropenem dosed at 600 mg/kg q6h subcutaneously as positive control. For dose arms utilizing a combination of ETX0282 and CPDP, both agents were reconstituted in the same vehicle. All dose concentrations were adjusted to deliver targeted mg/kg doses within a dose volume of 10 mL/kg. Formulation potency was verified by LC/MS/MS.30 ETX0282 and CPDP were orally administered via q6h dose intervals in order to achieve targeted unbound exposures. At 24 h after initiation of therapy, animals were euthanized, and thighs (2 separate samples/animal) were aseptically collected and homogenized in 1 mL of sterile saline. Bacterial colony enumeration of tissue homogenate was performed by serial dilution on tryptic soy agar (TSA) plates, which were incubated overnight at 35 °C prior to colony-forming unit (CFU) counting.
Acknowledgments
The authors acknowledge Lise Gauthier for assistance with analytical chemistry and Helen McGuire, Brendan Chen, Ye Wu, and Fei Zhou with synthesis. The authors thank Sharon Tentarelli at AstraZeneca R&D Boston for HRMS data. They thank Pharmaron for synthesis and scale-up and Neosome for pharmacology support. They thank Vinayak Hosagrahara and Minli Zhang for their input in DMPK studies. The authors thank the staff members of the Advanced Photon Source of Argonne National Laboratory, particularly those at SER-CAT for their assistance with X-ray diffraction data collection. The use of beamlines at SER-CAT was supported by its member institutions, and equipment grants (Grants S10_RR25528 and S10_RR028976) from the NIH. The authors thank Trent Kemp for assistance with CTX-M-14 crystallization.
Glossary
Abbreviations Used
- BOC
tert-butyloxycarbonyl
- CDC
Centers for Disease Control and Prevention
- DMPK
drug metabolism and pharmacokinetics
- IACUC
Institutional Animal Care and Use Committee
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- OLAW
Office of Laboratory Animal Welfare
- PE
petroleum ether
- SAR
structure–activity relationship
- TBS
tert-butyldimethylsilyl
- TPSA
topological polar surface area
- UPLC
ultraperformance liquid chromatography
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00579.
Synthesis and characterization (NMR, LC/MS, HPLC, HRMS) of intermediates and final compounds, synthetic schemes (Schemes S1 and S2), crystallographic data collection and refinement statistics (Table S1), LC/MS/MS conditions (Table S2), and metabolism pathways (Figure S1) (PDF)
Molecular formula strings and some data (CSV)
Accession Codes
The coordinates and structure factors of ETX1317 acyl-enzyme complex with CTX-M-14 β-lactamase (PDB code 6VHS) have been deposited with the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
∥ J.Z.: Johnson & Johnson Pharmaceutical Research & Development, Welsh & McKean Road, Spring House, PA 19477, U.S.
Author Present Address
⊥ X.W.: Novartis Institutes for BioMedical Research, Inc., 250 Massachusetts Avenue, Cambridge, MA 02139, U.S.
Author Present Address
# T.L.M.-D.: AbbVie, 1 North Waukegan Road, North Chicago, IL 60064, U.S.
Author Present Address
● J.A.C.R.: Pfizer, Inc., 1 Portland Street, Cambridge, MA 02139, U.S.
Author Present Address
∇ R.A.G.: 230 Westmont Avenue, Lavallette, NJ 08735, U.S.
Author Present Address
∞ J.C.V.: Novartis Institutes for BioMedical Research, Inc., 220 Massachusetts Avenue, Cambridge, MA 02139, U.S.
Author Present Address
◇ S.D.L.: Boehringer Ingelheim International GmbH, 900 Ridgebury Road, Ridgefield, CT 06877, U.S.
This research program was partially supported by Cooperative Agreement IDSEP160030 from ASPR/BARDA and by awards from Wellcome Trust and Germany’s Federal Ministry of Education and Research (BMBF), as administrated by CARB-X. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response, other funders, or CARB-X.
The authors declare the following competing financial interest(s): All authors, except Michael D. Sacco and Yu Chen, are current or former employees of Entasis Therapeutics or AstraZeneca and may own stock or stock options from these companies.
Supplementary Material
References
- Antibacterial Agents in Clinical Development: an Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Global Priority List of Antibiotic-Resistant Bacteria to guide Research, Discovery and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention, U.S. Department of Health and Human Services: Atlanta, GA, 2019. [Google Scholar]
- Bush K.; Bradford P. A. β-lactams and β-lactamase inhibitors: an overview. Cold Spring Harbor Perspect. Med. 2016, 6, a025247. 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading C.; Cole M. Clavulanic acid: a β-lactamase-inhibiting β-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. 10.1128/AAC.11.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M.; Papp-Wallace K. M.; Bonomo R. A. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. 10.1128/AAC.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley M. Ceftazidime-avibactam: A review in the treatment of serious Gram-negative bacterial Infections. Drugs 2018, 78, 675–692. 10.1007/s40265-018-0902-x. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G.; Lawrence C. K.; Adam H.; Schweizer F.; Zelenitsky S.; Zhanel M.; Lagace-Wiens P. R. S.; Walkty A.; Denisuik A.; Golden A.; Gin A. S.; Hoban D. J.; Lynch J. P. 3rd; Karlowsky J. A. Imipenem-relebactam and meropenem-vaborbactam: two novel carbapenem-β-lactamase inhibitor combinations. Drugs 2018, 78, 65–98. 10.1007/s40265-017-0851-9. [DOI] [PubMed] [Google Scholar]
- Zowawi H. M.; Harris P. N.; Roberts M. J.; Tambyah P. A.; Schembri M. A.; Pezzani M. D.; Williamson D. A.; Paterson D. L. The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat. Rev. Urol. 2015, 12, 570–584. 10.1038/nrurol.2015.199. [DOI] [PubMed] [Google Scholar]
- Bush K. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int. J. Antimicrob. Agents 2015, 46, 483–493. 10.1016/j.ijantimicag.2015.08.011. [DOI] [PubMed] [Google Scholar]
- Bush K.; Bradford P. A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33, e00047–19. 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman K. Diazabicyclooctanes (DBOs): a potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. 10.1016/j.mib.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Durand-Reville T. F.; Guler S.; Comita-Prevoir J.; Chen B.; Bifulco N.; Huynh H.; Lahiri S.; Shapiro A. B.; McLeod S. M.; Carter N. M.; Moussa S. H.; Velez-Vega C.; Olivier N. B.; McLaughlin R.; Gao N.; Thresher J.; Palmer T.; Andrews B.; Giacobbe R. A.; Newman J. V.; Ehmann D. E.; de Jonge B.; O’Donnell J.; Mueller J. P.; Tommasi R. A.; Miller A. A. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, e17104 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- Merdjan H.; Rangaraju M.; Tarral A. Safety and pharmacokinetics of single and multiple ascending doses of avibactam alone and in combination with ceftazidime in healthy male volunteers: results of two randomized, placebo-controlled studies. Clin. Drug Invest. 2015, 35, 307–317. 10.1007/s40261-015-0283-9. [DOI] [PubMed] [Google Scholar]
- Gordon E. M.; Duncton M. A. J.; Gallop M. A. Orally Absorbed Derivatives of the β-lactamase inhibitor avibactam. Design of novel prodrugs of sulfate-containing drugs. J. Med. Chem. 2018, 61, 10340–10344. 10.1021/acs.jmedchem.8b01389. [DOI] [PubMed] [Google Scholar]
- Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Järvinen T.; Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug Discovery 2008, 7, 255–270. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Wolfenden R.; Yuan Y. Monoalkyl sulfates as alkylating agents in water, alkylsulfatase rate enhancements, and the “energy-rich” nature of sulfate half-esters. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 83–86. 10.1073/pnas.0609644104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C. J.; Trout R.; Zulli A.; Mesaros E.; Jackson R.; Boyd S.; Liu B.; McLaughlin L.; Chatwin C.; Hamrick J.; Daigle D.; Pevear D.. Discovery of VNRX-7145: A Broad-Spectrum Orally Bioavailable β-Lactamase Inhibitor (BLI) for Highly Resistant Bacterial Infections (“Superbugs”). Presented at the National Meeting & Exposition of the American Chemical Society, Orlando, FL, 2019; Poster MEDI259.
- Hecker S. J.; Reddy K. R.; Lomovskaya O.; Griffith D. C.; Rubio-Aparicio D.; Nelson K.; Tsivkovski R.; Sun D.; Sabet M.; Tarazi Z.; Parkinson J.; Totrov M.; Boyer S. H.; Glinka T. W.; Pemberton O. A.; Chen Y.; Dudley M. N. Discovery of cyclic boronic acid QPX7728, an ultra broad-spectrum inhibitor of serine and metallo-β-lactamases. J. Med. Chem. 2020, 10.1021/acs.jmedchem.9b01976. [DOI] [PubMed] [Google Scholar]
- Tondi D.; Venturelli A.; Bonnet R.; Pozzi C.; Shoichet B. K.; Costi M. P. Targeting class A and C serine β-lactamases with a broad-spectrum boronic acid derivative. J. Med. Chem. 2014, 57, 5449–5458. 10.1021/jm5006572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachyra T.; Pechereau M. C.; Bruneau J. M.; Claudon M.; Frere J. M.; Miossec C.; Coleman K.; Black M. T. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non- β-lactam β-lactamase inhibitor. Antimicrob. Agents Chemother. 2010, 54, 5132–5138. 10.1128/AAC.00568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. G.; May-Dracka T. L.; Gagnon M. M.; Tommasi R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. 10.1021/jm501552x. [DOI] [PubMed] [Google Scholar]
- Laizure S. C.; Herring V.; Hu Z.; Witbrodt K.; Parker R. B. The role of human carboxylesterases in drug metabolism: have we overlooked their importance?. Pharmacotherapy 2013, 33, 210–222. 10.1002/phar.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab. Pharmacokinet. 2006, 21, 173–185. 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- Chanteux H.; Van Bambeke F.; Mingeot-Leclercq M.-P.; Tulkens P. M. Accumulation and oriented transport of ampicillin in Caco-2 cells from its pivaloyloxymethylester prodrug, Pivampicillin. Antimicrob. Agents Chemother. 2005, 49, 1279–1288. 10.1128/AAC.49.4.1279-1288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton O. A.; Noor R. E.; Kumar M. V. V.; Sanishvili R.; Kemp M. T.; Kearns F. L.; Woodcock H. L.; Gelis I.; Chen Y. Mechanism of proton transfer in class A β-lactamase catalysis and inhibition by avibactam. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 5818–5825. 10.1073/pnas.1922203117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D.; Mangani S.; Durand-Reville T.; Benvenuti M.; De Luca F.; Sanyal G.; Docquier J. D. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A. B.; Moussa S. H.; Carter N. M.; Gao N.; Miller A. A.. Cefpodoxime-ETX1317 Susceptibility Is Unaffected by Ceftazidime-Avibactam Resistance Mutations V240G, D179Y and D179Y/T243M in KPC-3 β-Lactamase. Presented at ASM Microbe, San Francisco, CA, 2019; Poster AAR-715.
- Nichols D. A.; Hargis J. C.; Sanishvili R.; Jaishankar P.; Defrees K.; Smith E. W.; Wang K. K.; Prati F.; Renslo A. R.; Woodcock H. L.; Chen Y. Ligand-induced proton transfer and low-barrier hydrogen bond revealed by X-ray crystallography. J. Am. Chem. Soc. 2015, 137, 8086–8095. 10.1021/jacs.5b00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J.; Tanudra A.; Chen A.; Hines D.; Tommasi R.; Mueller J. Pharmacokinetic/pharmacodynamic determination and preclinical pharmacokinetics of the β-lactamase inhibitor ETX1317 and its orally available prodrug ETX0282. ACS Infect. Dis. 2020, 6, 1378–1388. 10.1021/acsinfecdis.0c00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. A.; Shapiro A. B.; McLeod S. M.; Carter N. M.; Moussa S. H.; Tommasi R.; Mueller J. P. In vitro characterization of ETX1317, a broad-spectrum β-Lactamase inhibitor that restores and enhances β-lactam activity against multidrug-resistant Enterobacterales, including carbapenem-resistant strains. ACS Infect. Dis. 2020, 6, 1389–1397. 10.1021/acsinfecdis.0c00020. [DOI] [PubMed] [Google Scholar]
- Gerber A. U.; Craig W. A.; Brugger H.-P.; Feller C.; Vastola A. P.; Brandel J. Impact of dosing intervals on activity of gentamicin and ticarcillin against Pseudomonas aeruginosa in granulocytopenic mice. J. Infect. Dis. 1983, 147, 910–917. 10.1093/infdis/147.5.910. [DOI] [PubMed] [Google Scholar]
- Bulitta J. B.; Hope W. W.; Eakin A. E.; Guina T.; Tam V. H.; Louie A.; Drusano G. L.; Hoover J. L. Generating robust and informative nonclinical in vitro and in vivo bacterial infection model efficacy data to support translation to humans. Antimicrob. Agents Chemother. 2019, 63, e02307–18. 10.1128/AAC.02307-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose P. G.; Bhavnani S. M.; Rubino C. M.; Louie A.; Gumbo T.; Forrest A.; Drusano G. L. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin. Infect. Dis. 2007, 44, 79–86. 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- Weiss W. J.; Carter K.; Pulse M. E.; Nguyen P.; Valtierra D.; Peterson K. M.; Tommasi R.; Mueller J.; O’Donnell J. Efficacy of Cefpodoxime Proxetil and ETX0282 in a Murine UTI model with E. coli and K. pneumoniae. Presented at ECCMID, Amsterdam, The Netherlands, 2019; Poster P1991.
- McGuire H.; Bist S.; Bifulco N.; Zhao L.; Wu Y.; Huynh H.; Xiong H.; Comita-Prevoir J.; Dussault D.; Geng B.; Chen B.; Durand-Reville T.; Guler S.. β-Lactamase Inhibitor Compounds. Patent US9309245B2, 2016.
- Adam W.; Krebs O. The Nitroso Ene Reaction: A Regioselective and Stereoselective Allylic nitrogen functionalization of mechanistic delight and synthetic potential. Chem. Rev. 2003, 103, 4131–4146. 10.1021/cr030004x. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Comita-Prevoir J.; Sylvester M. 5-Carboxytetramethylrhodamine-ampicillin fluorescence anisotropy-based assay of Escherichia coli penicillin-binding protein 2 transpeptidase inhibition. ACS Infect. Dis. 2019, 5, 863–872. 10.1021/acsinfecdis.8b00351. [DOI] [PubMed] [Google Scholar]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; M07, 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- Chen Y.; Delmas J.; Sirot J.; Shoichet B.; Bonnet R. Atomic resolution structures of CTX-M β-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 2005, 348, 349–362. 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, D67, 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, D67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, D62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, D60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gudmundsson S.; Vogelman B.; Craig W. A. The in vivo postantibiotic effect of imipenem and other new antimicrobials. J. Antimicrob. Chemother. 1986, 18 (Suppl. E), 67–73. 10.1093/jac/18.Supplement_E.67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.