

SUMMARY
Cancer therapies kill tumors either directly or indirectly by evoking immune responses and have been combined with varying levels of success. Here, we describe a paradigm to control cancer growth that is based on both direct tumor killing and the triggering of protective immunity. Genetic ablation of serine protease inhibitor SerpinB9 (Sb9) results in the death of tumor cells in a granzyme B (GrB)-dependent manner. Sb9-deficient mice exhibited protective T cell-based host immunity to tumors in association with a decline in GrB-expressing immunosuppressive cells within the tumor microenvironment (TME). Maximal protection against tumor development was observed when the tumor and host were deficient in Sb9. The therapeutic utility of Sb9 inhibition was demonstrated by the control of tumor growth, resulting in increased survival times in mice. Our studies describe a molecular target that permits a combination of tumor ablation, interference within the TME, and immunotherapy in one potential modality.
Graphical Abstract
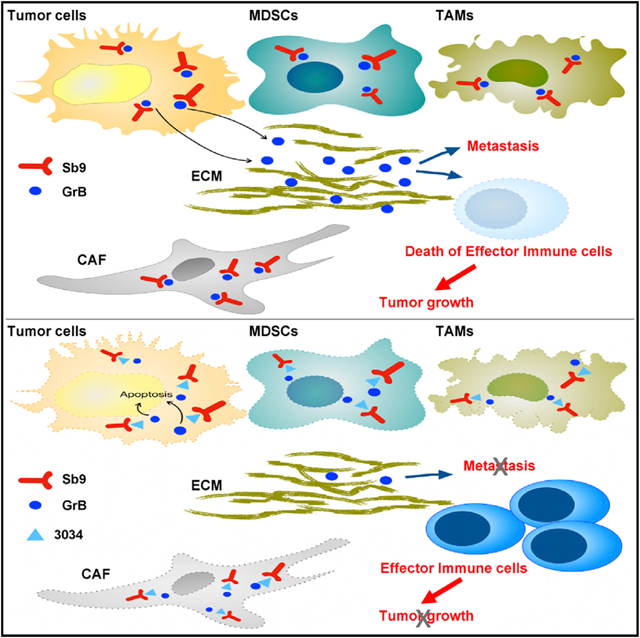
In Brief
SerpinB9 is important for tumor cell survival and for the presence of immunosuppressive cells in the tumor microenvironment, and a small-molecule inhibitor of SerpinB9 can reduce tumor growth and increase tumor immunogenicity in several mouse models of cancer.
INTRODUCTION
Serine proteases participate in a wide range of physiological processes, which are regulated by a large family of peptidase inhibitors referred to as serine protease inhibitors (serpins) (Silverman et al., 2001). Serpins act as a suicide substrate for a serine protease that results in a characteristic covalent inhibitory complex (Huntington et al., 2000; Mangan et al., 2008). In contrast to most serpins, which are extracellular, SerpinB9 (Sb9) (PI9 in human, Spi6 in mice) is a member of the ovalbumin family of serpins, which reside within the nuclei and cytoplasm of cells (Bird et al., 1998; Bots and Medema, 2008; Sun et al., 1996, 1997). Sb9 proteins are physiological inhibitors of granzyme B (GrB), which triggers apoptosis by activating caspases-3 and −8, following delivery into target cells by cytotoxic lymphocytes (CLs) (Pinkoski et al., 2001). Sb9 has been shown to protect pro-inflammatory CLs from self-inflicted damage by their own GrB (Hirst et al., 2003; Sun et al., 1996). Sb9 also protects other leukocytes, which are both pro-inflammatory (dendritic cells and neutrophils) (Medema et al., 2001b; Rizzitelli et al., 2012) or anti-inflammatory (regulatory T cells [Tregs] and myeloid-derived suppressor cells [MDSCs]) (Azzi et al., 2013; Kumar et al., 2016; Lindau et al., 2013), from GrB that either originates from CL or is produced endogenously.
Sb9 is also thought to protect tumor cells from GrB delivered by CLs, but this has not been tested directly in vivo (Bots and Medema, 2008; Mangan et al., 2008; Medema et al., 2001a). Previous studies have demonstrated the presence of GrB in some cancer cells, but additional functional and comprehensive in vivo studies need to be performed (D’Eliseo et al., 2010, 2016; Hu et al., 2003; Kontani et al., 2001; Pearson et al., 2014). Immunosuppressive tumor-associated macrophages (TAMs), MDSCs, and Tregs in the tumor microenvironment (TME) abet tumor progression and metastasis (Kumar et al., 2016; Lindau et al., 2013). The potential effect of GrB inhibition by Sb9 on both the anti-tu-mor cellular effectors (such as CL) and immunosuppressive components of the TME is not known (Quail and Joyce, 2013). Aside from the potential for immunomodulation, whether inhibition of Sb9 results in beneficial elimination of tumors either by direct killing or by increased host immunity remains to be determined. In addition to its implications on immune cells and the intrinsic survival of tumor cells, the Sb9-GrB axis can also have a major impact on the tumor stroma as well. Stromal cells, including cancer-associated fibroblasts (CAFs), constitute a major cellular component of the diverse TME and play a critical role in tumor development (Kalluri, 2016; Quail and Joyce, 2013). Stromal cells can create an optimal milieu by producing numerous growth factors, cytokines, and chemokines that promote tumor growth (Nilendu et al., 2018; Wei et al., 2018).
Here, we show that genetic ablation of Sb9 sensitized tumors to killing by not only CL-derived GrB, but also from endogenously produced GrB, which together resulted in the control of cancer in mice. The role of Sb9 in the anti-tumor host response was examined in Sb9 KO mice, which exhibited increased resistance to tumors. This was a consequence of impaired survival of immunosuppressive TAMs, MDSCs, Tregs, and CAFs in the TME that resulted in increased activity of anti-tumor CL. We developed a specific small molecule inhibitor of Sb9 and showed that treatment of mice could control tumor growth through direct sensitization to GrB and the activation of protective immunity.
RESULTS
Sb9 Is Required to Protect Melanoma Tumors from GrB-Induced Apoptosis
We examined the expression of Sb9 and GrB in several tumors of human and mouse. Sb9 (Figure 1A) and GrB (Figure 1B) were expressed in primary human and mouse malignant melanoma, breast adenocarcinoma, and lung adenocarcinoma. We also analyzed single-cell RNA sequencing data from previous studies (Guo et al., 2018; Jerby-Arnon et al., 2018; Slyper et al., 2020) to map the detailed expression of Sb9 and GrB in melanoma, breast cancer, and lung cancer of human origin. Sb9 and GrB were detected in these tumor cells, and their expression levels were measured in comparison to tumor-infiltrating CD4+ and CD8+ T cells as a positive control (Figure S1A). Additionally, mouse cancer cell lines (B16, mouse melanoma; 4T1, mouse breast cancer; and LLC1, mouse lung cancer) and human cancer cell lines (A375, human melanoma; A549, human lung cancer; and SK-BR3, human breast cancer) also expressed both Sb9 (Figure S1C) and GrB (Figure S1D). Then, B16 cells and LLC1 cells were implanted into UBC-GFP transgenic mice (expressing GFP under the direction of the human ubiquitin C promoter), as previously described (Andersson et al., 2009; Barrett et al., 2012; Kar et al., 2011), and the tumors were collected at day 17 post-implantation to compare GrB expression between the tumor and the host. Most GrB+ cells were GFP negative, indicating the presence of tumor-expressed GrB (Figure S1B). Inhibition of GrB by Sb9 via a classic serpin mechanism resulted in an SDS-resistant 62 kDa Sb9-GrB complex in B16 cells (Figures 1C and 1D; Mangan et al., 2016; Zhang et al., 2006). Western blot demonstrated that Sb9 was expressed mainly as a 47 kDa unbound monomer, while a smaller portion of Sb9 formed a 62 kDa Sb9-GrB complex in B16 cells (Figure 1C) (Mangan et al., 2016). We observed that almost all of the GrB exists as a 62 kDa complex with Sb9, in contrast to the 26 kDa monomeric GrB found in B16 cells (Figure 1D). The GrB RNAscope staining demonstrates the gene expression of GrB (green) in both B16 and 4T1 cells (Figure 1E). Furthermore, a GranToxiLux assay, which measures GrB activity in live cells, demonstrated that the GrB in B16 is functionally active (Figure 1F).
Figure 1. Sb9 Is Required to Protect Melanoma Tumors from GrB-Induced Apoptosis.
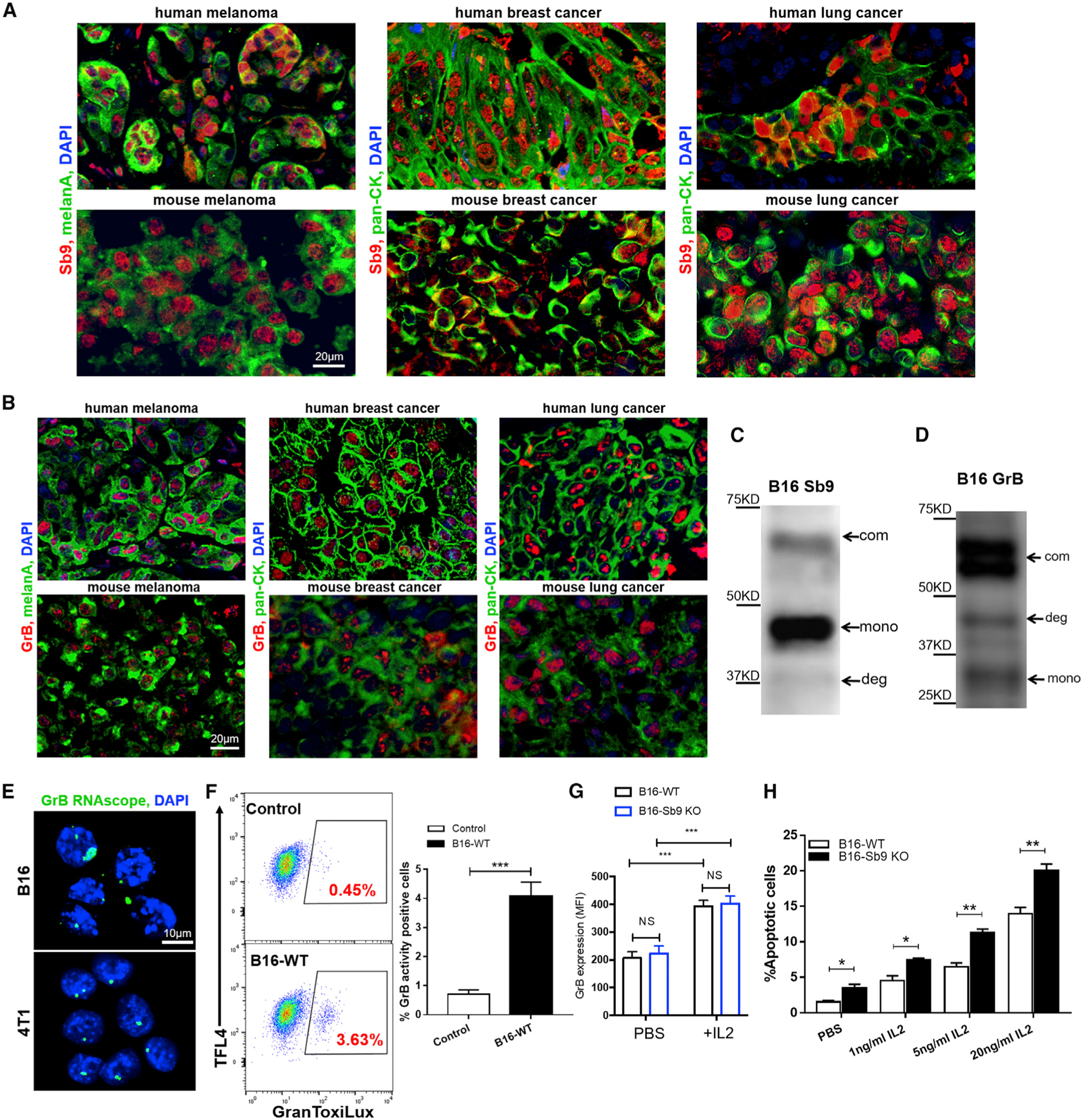
(A and B) Fluorescence micrographs show the presence of (A) Sb9 (red) and (B) GrB (red) in primary human and mouse melanoma (melanA, green), breast cancer (pan-CK, green), and lung cancer (pan-CK, green). DAPI (blue) was the nuclei. Scale bar, 20 μm.
(C and D) Western blotting shows three forms of Sb9 and GrB expression in B16 cells, indicated by arrows: Sb9-GrB complex (com), unbound Sb9/GrB (mono), and complex degradation (deg).
(E) GrB RNAscope staining shows the gene expression of GrB (green) by both B16 and 4T1 cells. DAPI (blue) was the nuclei. Scale bar, 10 μm.
(F) GranToxiLux assay displays the presence of GrB activity in B16 cells. ***p < 0.001.
(G) Flow cytometric analysis demonstrates that IL-2 (70 ng/mL) treatment for 48 h results in significantly higher GrB expression by B16-WT and B16-Sb9 KO cells. ***p < 0.001.
(H) Flow cytometry quantitative analysis reveals that treatment of B16-Sb9 KO cells with different concentrations of IL-2 (1, 5, 20 ng/mL) for 48 h results in higher percentage of apoptotic cells, as indicated by the annexin-V+ and viability− population. *p < 0.05, **p < 0.01. See also Figure S1.
To address the protection of Sb9 from GrB in melanoma cells, we disrupted the SERPINB9 gene using the CRISPR/Cas9 system. We targeted the 20-nucleotide sequence upstream of the protospacer-adjacent motif (PAM) sequence in exon 6 of the SERPINB9 gene (Figure S1E). Deletion of Sb9 in the B16 cells was confirmed by western blot (Figure S1F). The complex formation was hampered, suggesting that all GrB in the cell is no longer captured by Sb9. Residual Sb9 protein expression by B16-Sb9 knockout (KO) cells is not an infrequent observation (Figure S1F), due to common tetraploidy in these cells (Kendal et al., 1987; Smits et al., 2019). Sb9 RNAscope and immunofluorescence staining for Sb9 confirmed its lack of expression in B16-Sb9 KO cells (Figures S1G and S1H).
However, Sb9 KO did not affect the proliferation of B16 cells, as evidenced by a colony formation assay and the expression of Ki67 gene (Figures S1I and S1J; Li et al., 2015). Sb9 KO resulted in 2-fold increase (p = 0.016) in GrB activity (Figure S1K) and a corresponding 2.3-fold (p = 0.017) increase in GrB-specific apoptosis in B16-Sb9 KO cells compared to B16-wild-type (WT) cells (Figure 1H). Previous studies have reported that interleukin (IL)-2 induces GrB expression in cytotoxic T cells (CTL) and natural killer (NK) CLs (Tamang et al., 2006). We found that the mRNA level of GrB was significantly higher in IL-2-treated B16-WT cells and followed a dose-dependent response (Figure S1L). Additionally, the expression of GrB was also elevated significantly in IL-2-treated B16-WT and B16-Sb9 KO cells (Figures 1G and S1O). The cleaved caspase-3 (C-CAS3) expression was higher in B16-Sb9 KO cells compared to B16-WT and increased with IL-2 treatment (Figure S1P). The apoptosis rates (as indicated by the annexin-V+ and viability− population) of B16-WT and B16-Sb9 KO cells were significantly higher following treatment with multiple concentrations of IL-2 (Figure 1H), while B16-Sb9 and GrB KO cells showed no IL-2-induced apoptosis (Figure S1M). The GrB inhibitor 368050 also suppressed apoptosis following treatment with IL-2 (Figure S1N). Collectively, these results demonstrate that Sb9 is required to protect tumors from GrB-induced apoptosis.
Sb9 Acts Cell-Intrinsically to Control Tumor Growth In Vivo
To investigate the role of Sb9 in tumor progression and metastasis in vivo, B16-WT or B16-Sb9 KO cells were injected subcutaneously into C57BL/6 mice, then tumor growth was monitored to the size end points. The Sb9-disrupted tumors were ~4-fold smaller (p = 0.0001) than the B16-WT group at day 27 post-implantation (Figure 2A). The mice reached the size end points at 27 days and 39 days in the B16-WT and B16-Sb9 KO, respectively (Figure 2A). We also compared the sizes of B16-Sb9&GrB KO tumors with the B16-WT and B16-Sb9 KO tumors to assess the differential effects of tumor-derived and host-derived GrB in tumor growth. The B16-Sb9&GrB KO tumors grew faster than the B16-Sb9 KO tumors and slower than the B16-WT tumors (Figure S2A). The intermediate protection seen in B16-Sb9&GrB KO mice could be due to exposure to host-derived GrB. No difference was noted in the size of B16-Sb9 KO tumors derived from a single clone or those from a bulk cell population (Figure S2B). Staining of the melanoma sections with the melanoma marker MelanA at day 17 following implantation revealed a much smaller MelanA+ area in the B16-Sb9 KO group than the B16-WT group (Figures 2B and 2C). The median survival time (MST) of mice bearing B16-Sb9 KO tumors was significantly longer (p < 0.0001) than that of mice bearing B16-WT tumors (Figure S2C). B16-Sb9 KO tumors exhibited more apoptosis and higher expression of cleaved caspase-3 than the B16-WT tumors in vivo, while the proliferation rate was similar in both groups (Figures 2D–2F, S2D, and S2E).
Figure 2. Sb9 Acts Cell Intrinsically to Control Tumor Growth In Vivo.
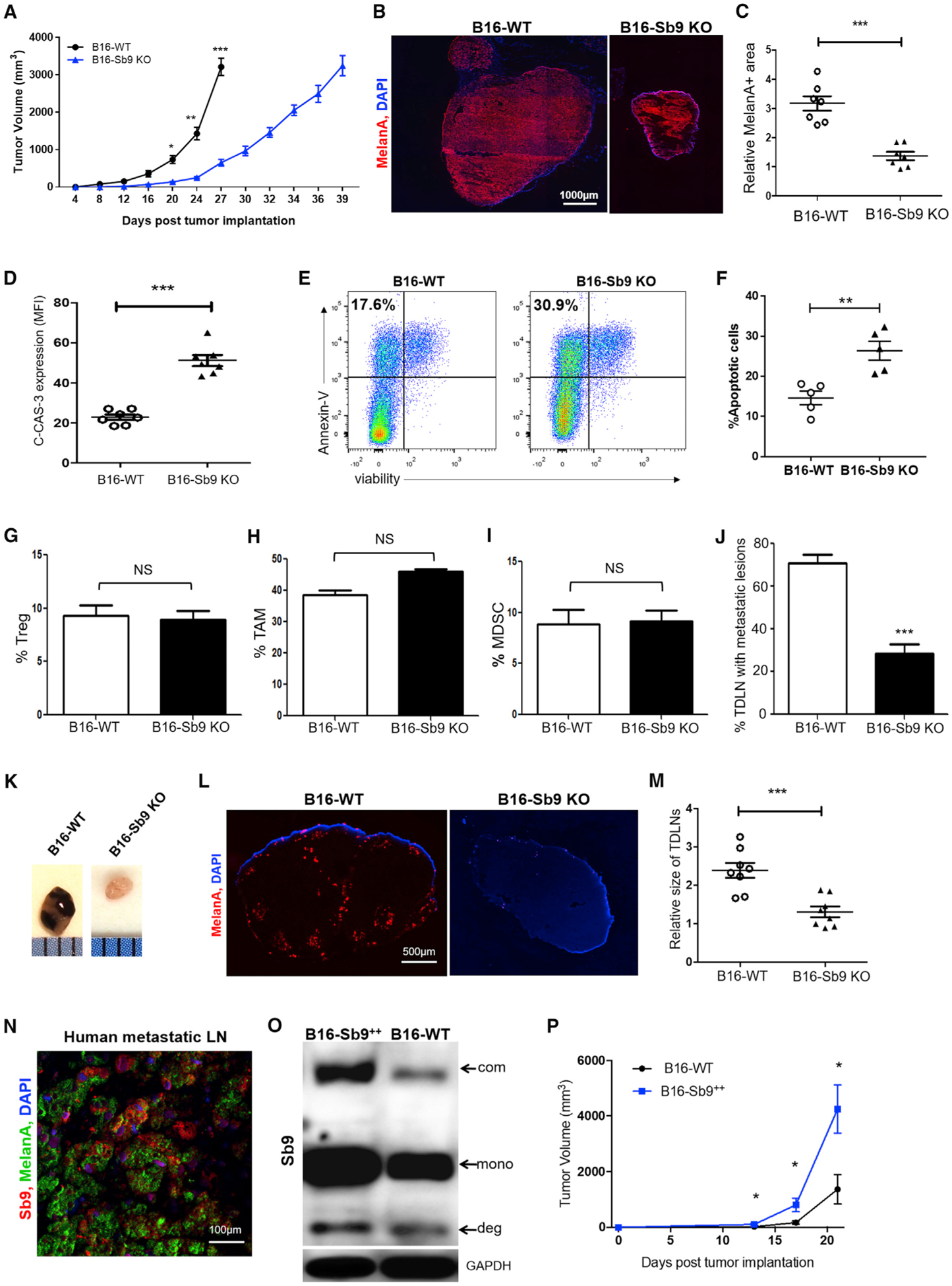
(A) Tumor growth curve through size end points (B16-WT, day 27; B16-Sb9 KO, day 39) indicates significantly slower growth rate of B16-Sb9 KO (n = 10) than B16-WT (n = 10) tumors. *p < 0.05, **p < 0.01, ***p < 0.001.
(B and C) Representative fluorescence micrographs of MelanA+ (red) melanoma cells demonstrates significantly smaller MelanA+ area in the B16-Sb9 KO than the B16-WT tumors at day 17 post-implantation. ***p < 0.001. Scale bar, 1000 μm.
(D) Semiquantitative analysis of fluorescence micrographs indicates a significantly higher density of cleaved caspase-3 (C-CAS-3)+ apoptotic cells in the B16-Sb9 KO than the B16-WT tumors at day 17 post-implantation. ***p < 0.001.
(E and F) Representative flow cytometry plots (E) and quantitative analysis (F) reveal higher percentage of apoptotic cells in the B16-Sb9 KO tumors, as indicated by annexin-V+ and viability− population, in comparison to the B16-WT tumors at day 17 post-implantation. **p < 0.01.
(G–I) Flow cytometric analysis demonstrates that the percentages of tumor-infiltrating CD4+CD25+Foxp3+ Treg populations (G), CD45+CD11b+F4/ 80+Ly6C−Ly6G− TAM populations (H), and CD45+CD11b+Gr1+CD3− MDSC populations (I) are similar between the B16-WT and B16-Sb9 KO melanoma tumors at day 17 post-implantation. NS (no significant difference).
(J and K) Comparison between tumor-draining lymph nodes (TDLNs) demonstrates significantly lower percentage of TDLN with metastatic lesions in mice implanted with B16-Sb9 KO (n = 8) tumors than B16-WT (n = 8) tumors. Representative photographs of TDLN from the B16-WT group and the B16-Sb9 KO group are provided (K). ***p < 0.001.
(L) Representative fluorescence micrographs demonstrate presence of MelanA (red) in TDLN of the B16-WT group and absence in TDLN of the B16-Sb9 KO group. Scale bar, 500 μm.
(M) Comparison between TDLNs demonstrates significantly smaller size of TDLNs in the B16-Sb9 KO group than that in the B16-WT group. ***p < 0.001.
(N) Fluorescence micrographs demonstrate the presence of Sb9 (red) and MelanA (green) in metastatic human melanoma lesions in LNs. DAPI (blue) was the nuclei. Scale bar, 100 μm.
(O) Western blotting indicates that the expression of Sb9 is significantly higher in B16-Sb92+ cells than B16-WT cells. Sb9-GrB complex (com), unbound Sb9 (mono), and complex degradation products or cleaved Sb9 (deg) are indicated by arrows.
(P) Tumor growth curve indicates more rapid growth of B16-Sb92+ (n = 14) than B16-WT (n = 14) mouse melanoma tumors in WT mice. *p < 0.05. See also Figure S2.
However, no difference was observed in the levels of Tregs, TAMs (M1 and M2), and MDSCs between the B16-WT and B16-Sb9 KO (Figures 2G–2I and S2F), indicating that Sb9 tumor expression had minimal impact on the proportion of immunosuppressive cell populations in the TME. Metastasis of B16-Sb9 KO tumors was also decreased, as evidenced by the decreased levels of melanoma cells in tumor-draining lymph nodes (TDLNs) (Figures 2J–2L). The size of TDLNs in the B16-Sb9 KO group was significantly (p = 0.0006) smaller than that in the B16-WT group (Figures 2K–2M). Additionally, we observed co-expression of Sb9 and MelanA in metastatic lesions of human melanoma in the LN (Figure 2N).
B16 cells overexpressing Sb9 (B16-Sb92+ cells) were generated, and Sb9 was confirmed by western blotting (Figure 2O). Following subcutaneous injection into C57BL/6 mice, we observed that the B16-Sb92+ tumors grew significantly more rapidly (3.2-fold, p = 0.017) at 21 days post-implantation in comparison to the B16-WT group (Figure 2P). We also utilized CRISPR/Cas9 to ablate Sb9 in other cell lines (4T1, LLC1, and A375) and observed significant growth inhibition of Sb9-disrupted mouse breast tumors, lung tumors, and human melanoma tumors after implantation (Figures S2G–S2I). The population of Nestin+ cells was also lower in B16-Sb9 KO sections (Figure S2J). Taken together, our findings strongly suggest that expression of Sb9 promotes tumor growth and metastases in vivo in a cell-intrinsic manner.
Sb9 Deletions Restore Host Immunity to Tumors and Disrupt Stroma in the TME
We investigated the role of Sb9 in the host immune response to tumors in Sb9 KO mice (Zhang et al., 2006). Melanoma tumors grew more slowly (2.7-fold, p = 0.0009, day 27) in Sb9 KO mice, as compared with WT mice (Figure 3A), resulting in a significantly longer (p = 0.0008) MST in Sb9 KO melanoma-bearing mice than WT mice (Figure 3B). The Sb9 KO mice implanted with the B16-WT tumors reached their size end point at day 33, as compared to day 27 for the WT mice (Figure 3A). However, maximal protection against melanoma development was observed when both tumor and host were deficient in Sb9 (Figure 3C). We monitored B16-Sb9 KO tumors in Sb9 KO mice until day 70, and they remained ~600 mm3 in size with a flattened growth trajectory (Figure 3C). The MST of Sb9 KO mice implanted with Sb9 KO tumors (Sb9 KO/Sb9 KO group) was much longer (p < 0.0001) than that of WT mice implanted with WT tumors (WT/WT group) (MST >110 days versus MST = 21 days) (Figure 3D). Additionally, we compared the proliferation rate (% of Ki67+ cells) of the WT/WT group and Sb9 KO/Sb9 KO group at day 24 and day 40 in vivo. No significant difference was found between the proliferation rate of the WT/WT group with that of the Sb9 KO/Sb9 KO group, either at day 24 or day 40 (Figure S3A). The apoptosis rate (% of c-cas3+ cells) of the Sb9 KO/Sb9 KO group was significantly higher than the WT/WT group at day 24, whereas that of the Sb9 KO/Sb9 KO group at day 40 was the highest (~40% apoptosis rate) (Figure S3B). Sb9 deficiency resulted in increased caspase-3 and GrB expression by tumors from the Sb9 KO/Sb9 KO group, as compared with those from the WT/WT group (Figures S3C and S3D).
Figure 3. Sb9 Deletions Restore Host Immunity to Tumors and Disrupt Stroma in the TME.
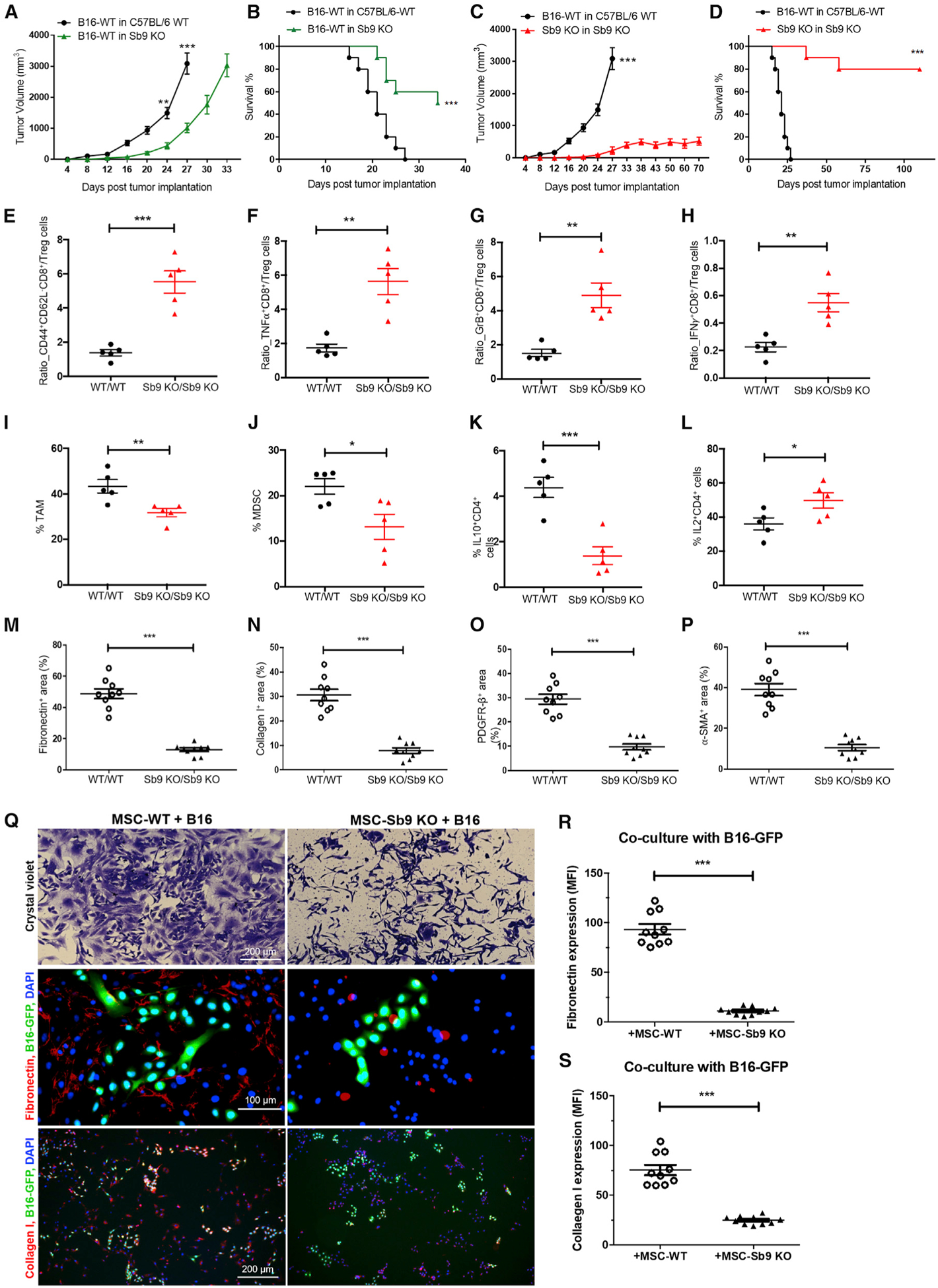
(A) Tumor growth curve demonstrates significantly slower growth of B16-WT tumors in Sb9 KO mice (n = 10) than C57BL/6 WT mice (n = 10) (size end points: B16-WT tumors in WT mice, day 27; B16-WT tumors in Sb9 KO mice, day 33). **p < 0.01, ***p < 0.001.
(B) Survival curve shows longer survival of Sb9 KO mice (n = 10) than C57BL/6 WT mice (n = 10) bearing B16-WT tumors. ***p < 0.001.
(C) Tumor growth curve displays significantly slower growth of B16-Sb9 KO tumors in Sb9 KO mice (n = 10, Sb9 KO/Sb9 KO) than B16-WT tumors in C57BL/6 WT mice (n = 10, WT/WT). ***p < 0.001.
(D) Survival curve shows longer survival of Sb9 KO mice bearing the B16-Sb9 KO tumors (n = 10, Sb9 KO/Sb9 KO) than C57BL/6 WT mice bearing the B16-WT tumors (n = 10, WT/WT). ***p < 0.001.
(E–H) Flow cytometric analyses demonstrate significantly higher ratios of CD44highCD62Llow CD8+effector memory T cells/Treg (E), TNFα+CD8+T cells/Treg (F), GrB+CD8+T cells/Treg (G), and IFNγ +CD8+T cells/Treg (H) in tumors from the Sb9 KO/Sb9 KO group than the WT/WT at day 17 post-implantation. **p < 0.01, ***p < 0.001.
(I and J) Flow cytometric analyses show significantly lower percentages of tumor-infiltrating CD45+CD11b+F4/80+Ly6C−Ly6G− TAM populations (I) and CD45+CD11b+Gr1+CD3− MDSC populations (J) in tumors from the Sb9 KO/Sb9 KO group than the WT/WT at 17 days post-implantation. *p < 0.05, **p < 0.01.
(K) Flow cytometric analysis shows significantly lower percentages of tumor-infiltrating IL-10+CD4+ population in tumors from the Sb9 KO/Sb9 KO group than the WT/WT at 17 days post-implantation. ***p < 0.001.
(L) Flow cytometric analysis shows significantly higher percentages of tumor-infiltrating IL-2+CD4+ population in tumors from the Sb9 KO/Sb9 KO group than the WT/WT at 17 days post-implantation. ***p < 0.001.
(M–P) Semiquantitative analysis of fluorescence micrographs demonstrates significantly smaller surface area occupied by fibronectin (M), collagen I (N), PDGFRβ+ fibroblasts (O), and α-SMA+ fibroblasts (P) in tumor sections from the Sb9 KO/Sb9 KO group than in the WT/WT at 17 days post-implantation. ***p < 0.001.
(Q–S) Crystal violet staining (Q, top) of MSC-WT cells co-cultured with B16-GFP cells for 48 h reveals multiple dense clusters comprised of both MSC-WT and B16 tumor cells, while co-cultured MSC-Sb9 KO and B16 cells are scattered and not situated in proximity to each other. Scale bar, 200 μm. Semiquantitative analysis of representative fluorescence micrographs shows significantly larger amounts of fibronectin (Q, middle; R) and collagen I (Q, bottom; S) produced by MSC-WT cells in co-culture with B16-GFP cells than the MSC-Sb9 KO and B16-GFP co-culture group after 48 h. DAPI (blue) was the nuclei. Scale bar, 100 μm and 200 μm. ***p < 0.001. See also Figure S3.
Next, we examined the effect of host deficiency of Sb9 on the TME of implanted melanomas. Tregs are potent suppressors of anti-tumor effector T cells and protected from cell-intrinsic GrB by endogenous Sb9 (Azzi et al., 2013). The ratios of effector CD8+ to Treg cells (p = 0.0156), CD44+CD62L− (effector memory) CD8+ to Treg cells (p = 0.0003), effector TNFα+CD8+ to Treg cells (p = 0.0012), effector GrB+CD8+ to Treg cells (p = 0.002), and effector interferon (IFN)γ+CD8+ to Treg cells (p = 0.0023) were all significantly higher in the sections from the Sb9 KO/Sb9 KO group, as compared with those from the WT/WT group (Figures 3E–3H and S3E).
In addition to Tregs, the recruitment of immunosuppressive TAMs and MDSCs also play a critical role in tumor progression (Kitamura et al., 2015; Ugel et al., 2015). Fluorescence micrographs demonstrate the expression of Sb9 in the TAMs (CD11b+, Ly-6C−) and MDSCs (CD11b+, Gr-1+) within melanoma and breast tumors (Figures S3G–S3J). The levels of TAMs (p = 0.0093) and MDSCs (p = 0.025) were also significantly lower in melanoma sections from the Sb9 KO/Sb9 KO group, as compared to the WT/WT group (Figure 3I and 3J). We conclude that loss of Sb9 in Tregs, TAMs, and MDSCs expose them to GrB-mediated killing within the TME, ultimately leading to restoration of the antitumor immune response and inhibition of tumor progression. This effect on immunosuppressive cells overall is apparently more dominant for anti-tumor immunity than any decrease in the survival of CLs. Furthermore, an increased level of effector IL-2+CD4+ cells and decreased levels of immunosuppressive IL-10+CD4+ cells and IL-10+CD8+ cells were found in the TME sections from the Sb9 KO/Sb9 KO group (Figures 3K, 3L, and S3F).
CAFs inhibit host resolution of solid cancers by preventing access of effector T cells to tumor cells (Kalluri, 2016). The number of CAFs in melanomas from the Sb9 KO/Sb9 KO group, as shown by the expression of fibronectin (4.1-fold, p < 0.0001), collagen-I (3.7-fold, p = 0.0002), PDGFR-β (3.3-fold, p = 0.0002), and α-SMA (3.9-fold, p < 0.0001), was dramatically reduced compared to the WT/WT group (Figures 3M–3P). To further assess the effect of Sb9 expression in the tumor stroma on the growth of tumors, we co-cultured B16-GFP cells with the mesenchymal stem cells (MSCs) isolated from the bone marrow of either C57BL/6-WT mice or Sb9 KO mice (Zhu et al., 2010). We investigated whether MSC-WT or MSC-Sb9 KO cells differ in their capacity to support B16 melanoma cells in vitro by assessing their growth together in culture plates. Micrographs of the co-cultured MSC-WT and B16-GFP cells revealed multiple dense clusters comprised of both MSC-WT and B16 cells, while the MSC-Sb9 KO and B16 cells were scattered and not situated in proximity to each other (Figure 3Q, top). Then, we assessed the accumulation of fibronectin and collagen I as surrogate markers of a fibrotic tumor microenvironment, which is a major barrier against the penetration of chemotherapeutic agents and constitutes a significant obstacle to their efficacy. We observed that MSC-WT cells produced fibronectin and collagen I more readily in co-culture with B16-GFP cells than the MSC-Sb9 KO cells group (Figures 3Q, middle and bottom, 3R, and 3S). These data indicate that the presence of Sb9 in stromal cells promotes their growth, synthesis of ECM fibers, and proliferative effect on B16 cells.
Small Molecule Inhibitors of Sb9 Evoke Protective Immunity to Tumors
A fragment-based drug discovery approach was used to identify a small organic molecule that binds to recombinant human Sb9 produced in E. coli (Figure S4A). To express soluble Sb9, an N-terminal fusion with maltose-binding protein (MBP) (Figure S4B) was constructed, purified, and confirmed by SDS-PAGE and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (MBP-Sb9 protein: 90.17 kDa) (Figures S4C and S4D). Functional activity of MBP-Sb9 was measured by its dose-dependent inhibitory effects on isolated GrB activity (Figure S4E). A thermal stability assay (TSA) was developed, showing an Sb9-specific transition at 51.5°C (Tm1) (Figure S4F), and used in a screen, which identified two compounds (ZT0587 and ZT0805) on the basis of a dose-dependent Tm1 shift (Figures 4A and S4G), with binding of fragment ZT0587 to MBP-Sb9 with a KD of 88 μM (Figure 4B). An increasingly complicated series of dihydroxybenzoic-like compounds was selected by an analog-by-catalog; one of these, compound 3034 (1,3-benzoxazole-6-carboxylic acid), exhibited direct binding to Sb9 with a KD of 273 μM as measured by surface plasmon resonance (SPR) (Figure 4B). We next evaluated in B16 cells the ability of top-ranking analogs to inhibit intracellular Sb9 as well as cell penetration, by measuring the induction of caspase-3 activity, a surrogate marker for increased GrB activity. Treatment with 3034 exhibited an ~4-fold increase in caspase-3 activity (p = 0.0003), compared to ZT0587 (Figure 4C). Additionally, the apoptosis rate (as indicated by the annexin-V+ and viability− population by fluorescence-activated cell sorting [FACS]) of B16 cells treated with 3034 was significantly higher than ZT0587 at both the 12-h and 24-h time points (Figure S4H).
Figure 4. Small Molecule Inhibitors of Sb9 Evoke Protective Immunity to Tumors.
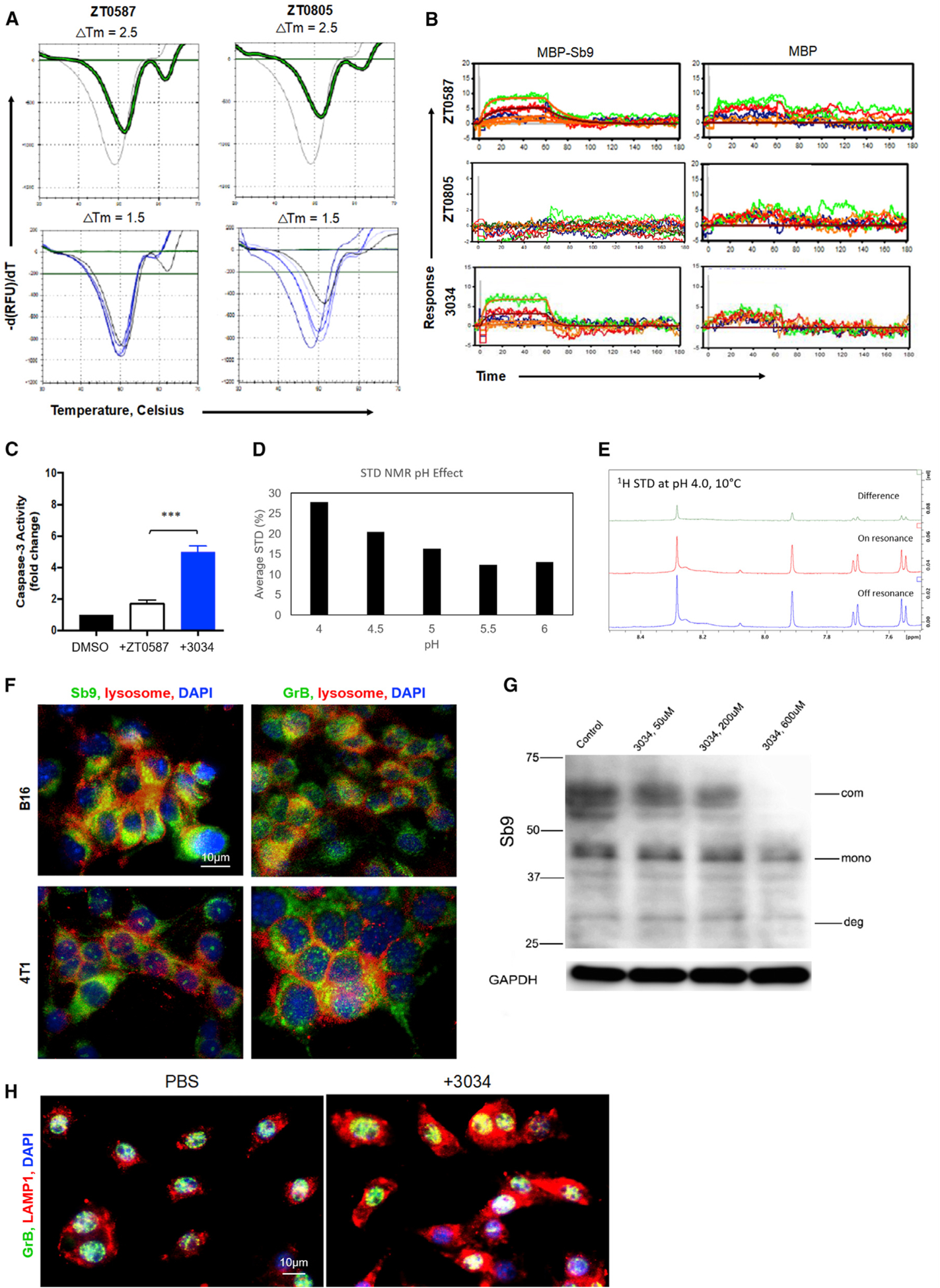
(A) The upper panels show the TSA results of the MBP-Sb9 protein incubated with 5 mM ZT0587 and ZT0805, or with 2.5% DMSO. The thermal shifts of the compounds ZT0587 and ZT0805 are shown in green against a baseline control shown in gray. The bottom panels showed the TSA results of the MBP-Sb9 incubated with ZT0587 or ZT0805 at different concentrations, including 5000, 1667, 556, 185, 62, and 21 μM. The concentration-response curves are plotted in a blue color scale (higher intensity is higher concentration) and the green plots represent the compound background signal.
(B) Sensorgrams of compounds ZT0587, ZT0805, and 3034 show the binding of compounds ZT0587 and 3034 to Sb9 (left), but not to MBP (right) by surface plasmon resonance (SPR).
(C) Comparison of caspase-3 activity among B16 melanoma cells treated with compounds ZT0587 and 3034 (200 μM) for 24 h demonstrated that compound 3034 induced highest activity, as determined by the EnzChek Caspase-3 Assay Kit. ***p < 0.001.
(D and E) Saturation-transfer difference (STD) NMR assays of compound 3034 (450 μM) and Sb9 protein (45 μM) at various pH values are provided. (D) Aromatic region of STD NMR spectrum showing compound 3034 in the presence of Sb9 protein at pH 4.0 and 10°C, exhibiting large saturation difference (green trace) comparing on-resonance (red) and off-resonance (blue) 1d 1H spectra. (E) Plots of averaged STD scores (%) of aromatic peaks showing binding of compound 3034 to MBP-Sb9 protein is enhanced significantly at lower pH in a pH-dependent manner.
(F) Fluorescence micrographs demonstrate the presence of Sb9 (green, left) and GrB (green, right) in the lysosomes (red) of B16 and 4T1 cells. DAPI (blue) was the nuclei. Scale bar, 10 μm.
(G) Western blot shows that compound 3034 reduces Sb9-GrB complex in a concentration-dependent manner in B16 cells. Sb9-GrB complex (com), unbound Sb9 (mono), and complex degradation products or cleaved Sb9 (deg) are indicated by arrows.
(H) Fluorescence micrographs indicate that compound 3034 significantly increases the expression of LAMP1 (red) and expands the lysosomes. DAPI (blue) was the nuclei. Scale bar, 10 μm. See also Figure S4.
Saturation-transfer difference (STD) nuclear magnetic resonance (NMR) assays demonstrated that compound 3034 bound to MBP-Sb9 protein and revealed that this interaction was significantly enhanced at lower pH in a pH-dependent manner (Figures 4D and 4E). With a pH as low as 4, lysosomes are the most acidic compartments within cells. Therefore, we hypothesized that the environment of lysosomes is conducive for binding between compound 3034 and Sb9. We showed that both Sb9 and GrB were co-localized in the lysosomes of B16 melanoma cells (Figure 4F), and we showed that compound 3034 reduced the expression of the Sb9-GrB complex by B16 cells in a concentration-dependent manner (Figure 4G). Interestingly, compound 3034 significantly increased LAMP1 expression and expanded lysosomes, suggesting that Sb9 activity is associated with lysosomal biogenesis (Figure 4H).
Treatment with a Small Molecule Inhibitor of Sb9 Suppresses Melanoma Progression In Vivo
We assessed the anti-tumor activity of compound 3034 in vivo. Injections of compound 3034 significantly reduced (3.7-fold, p = 0.0014) the size of B16 tumors implanted in the flanks of mice, compared to the control injected group (Figures 5A and S5A). Next, we examined the effect of compound 3034 on tumor immunity. The populations of CD44+CD62L− (effector memory) CD8+, effector GrB+CD8+, and effector IFNγ+CD8+ cells were all significantly higher in the melanoma sections from the 3034-treatment group, as compared with those from the control group (Figures 5B–5D). The levels of immunosuppressive Tregs, TAMs, and MDSCs (p = 0.025) were significantly lower in sections from the 3034-treatment group, as compared to the control group (Figures 5E–5G). Furthermore, an increased level of effector IFNγ +CD4+ cells and a decreased level of immunosuppressive IL-10+CD4+ cells and IL-10+CD8+ cells were found in the TME sections from the 3034-treatment group (Figures 5H, S5B, and S5C).
Figure 5. Treatment with a Small Molecule Inhibitor of Sb9 Suppresses Melanoma Progression In Vivo.
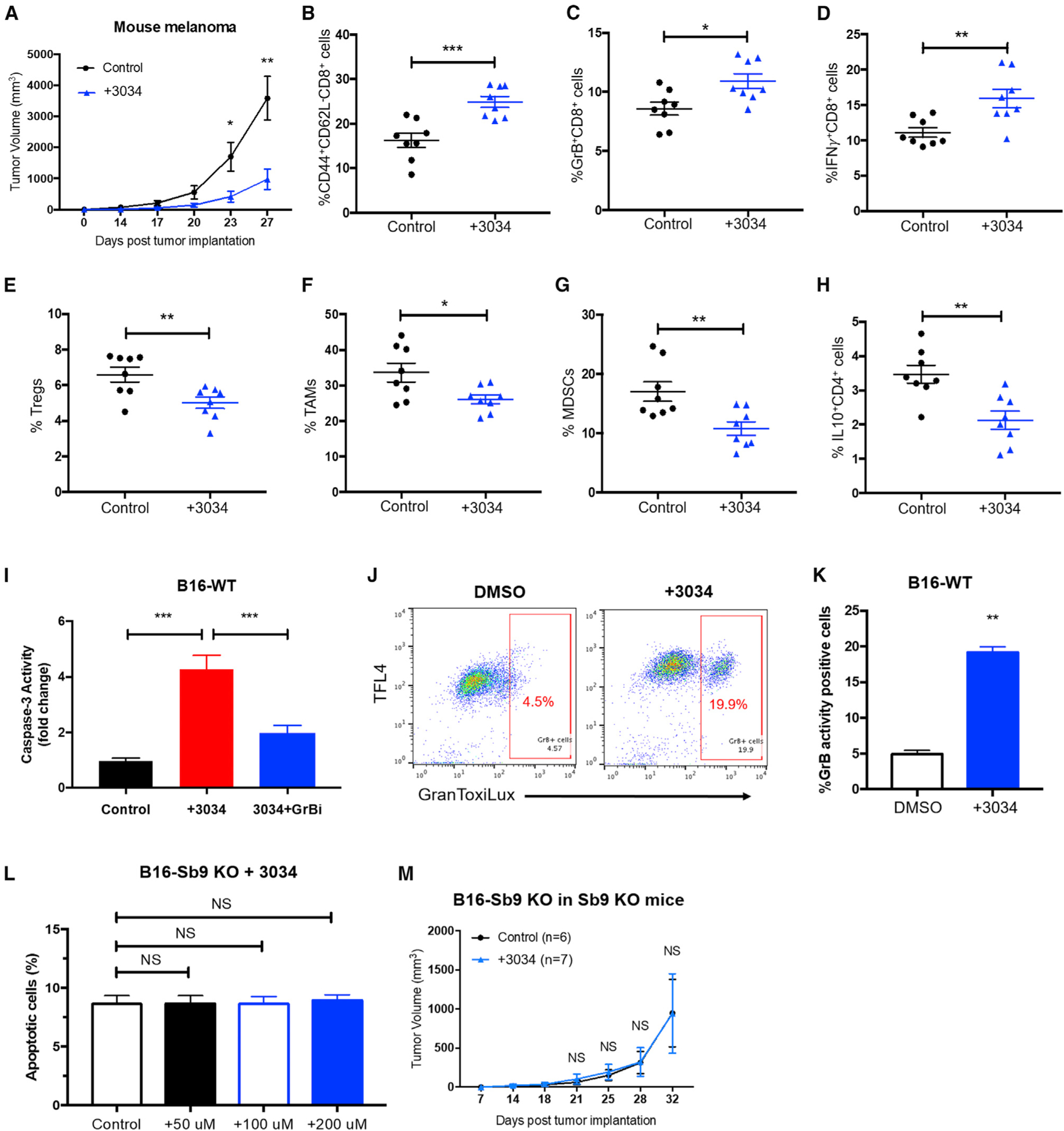
(A) Tumor growth curve demonstrates significantly slower growth of B16-WT tumors in the WT mice treated with Compound 3034 (300 μg intraperitoneally [i.p.] bid for 14 days following implantation) (n = 17) than the control group treated with 10% DMSO (n = 17). *p < 0.05, **p < 0.01.
(B–D) Flow cytometric analyses demonstrate significantly higher percentages of tumor-infiltrating CD44highCD62LlowCD8+ effector memory T cells (B), GrB+CD8+T cells (C), and IFNγ +CD8+T cells (D) in tumors from the 3034-treatment group than the control at day 17 post-implantation. *p < 0.05, **p < 0.01, ***p < 0.001.
(E–G) Flow cytometric analyses show significantly lower percentages of tumor-infiltrating CD4+CD25+Foxp3+ Treg populations (E), CD45+CD11b+F4/80+Ly6C−Ly6G− TAM populations (F), and CD45+CD11b+Gr1+CD3− MDSC populations (G) in tumors from the 3034-treatment group than the control at day 17 post-implantation. *p < 0.05, **p < 0.01.
(H) Flow cytometric analysis shows significantly lower percentages of tumor-infiltrating IL-10+CD4+population in tumors from the 3034-treatment group than the control at day 17 post-implantation. **p < 0.01.
(I) Caspase-3 activity assay demonstrates that compound 3034 (200 μM) induces GrB-mediated apoptosis in B16 cells, as determined by the EnzChek Caspase-3 Assay Kit. The GrB inhibitor (368050) was used at 10 μM. ***p < 0.001.
(J and K) GranToxiLux assay demonstrates increased activity of GrB in B16 cells treated with compound 3034 (200 μM) for 24 h. **p < 0.01.
(L) Quantitative flow cytometric analysis shows that compound 3034 does not induce additional apoptosis at various concentrations (50 μM, 100 μM, and 200 μM) in the B16-Sb9 KO cells, as indicated by annexin-V+ and viability dye− population. NS, no significant difference.
(M) Tumor growth curve indicates no difference in antitumor effect of compound 3034 on B16-Sb9 KO tumors in Sb9 KO mice (n = 7), as compared to the control (n = 6). NS, no significant difference. See also Figure S5.
To assess if the anti-tumor effects of compound 3034 were generalizable to human diseases, we also tested efficacy in an NSG humanized mouse model of A375. Compound 3034 reduced the size of human melanoma tumors significantly (2.1-fold, p = 0.0158), compared to controls (Figure S5D). Compound 3034 induced GrB-mediated apoptosis in B16 melanoma cells (Figure 5I). GranToxiLux assay demonstrated increased activity of GrB in B16 cells treated with compound 3034 (Figures 5J and 5K). We conclude that compound 3034 can inhibit Sb9 in melanoma tumor cells and render them susceptible to GrB-mediated apoptosis.
To evaluate the off-target effects of compound 3034 to Sb9, we treated the B16-Sb9 KO cells with various concentrations of compound 3034. No additional apoptotic effect was observed (Figure 5L). Furthermore, we did not observe antitumor effects of compound 3034 on the Sb9 KO mice bearing B16-Sb9 KO tumors, as compared to the control group (Figure 5M).
To further assess the in vivo toxicity of compound 3034, 300 μg of compound 3034 was administered twice a day intraperitoneally for 14 days. The CBC results showed no significant difference in WBC, HGB, and PLT between control and treatment groups (Figure S5E). The serum cells suggested that AST, ALT, CRE, and ALB levels were similar between the two groups (Figure S5F). H&E staining demonstrated that compound 3034 showed no toxicity to liver, kidney, lung, and heart tissues in mice (Figure S5G).
Treatment with an Sb9 Inhibitor Restrains Breast Tumor Growth In Vivo
To investigate the effect of compound 3034 on the tumor stroma, MSC-WT cells were treated with compound 3034 for 48 h. Compound 3034 treatment reduced the production of fibronectin and collagen I by MSC-WT cells significantly (Figures 6A and S6A). As expected, compound 3034 decreased the expression of fibronectin and collagen I by MSC-WT cells that were co-cultured with B16 cells (Figures 6B, 6C, and S6B).
Figure 6. Treatment with an Sb9 Inhibitor Restrains Breast Tumor Growth In Vivo.
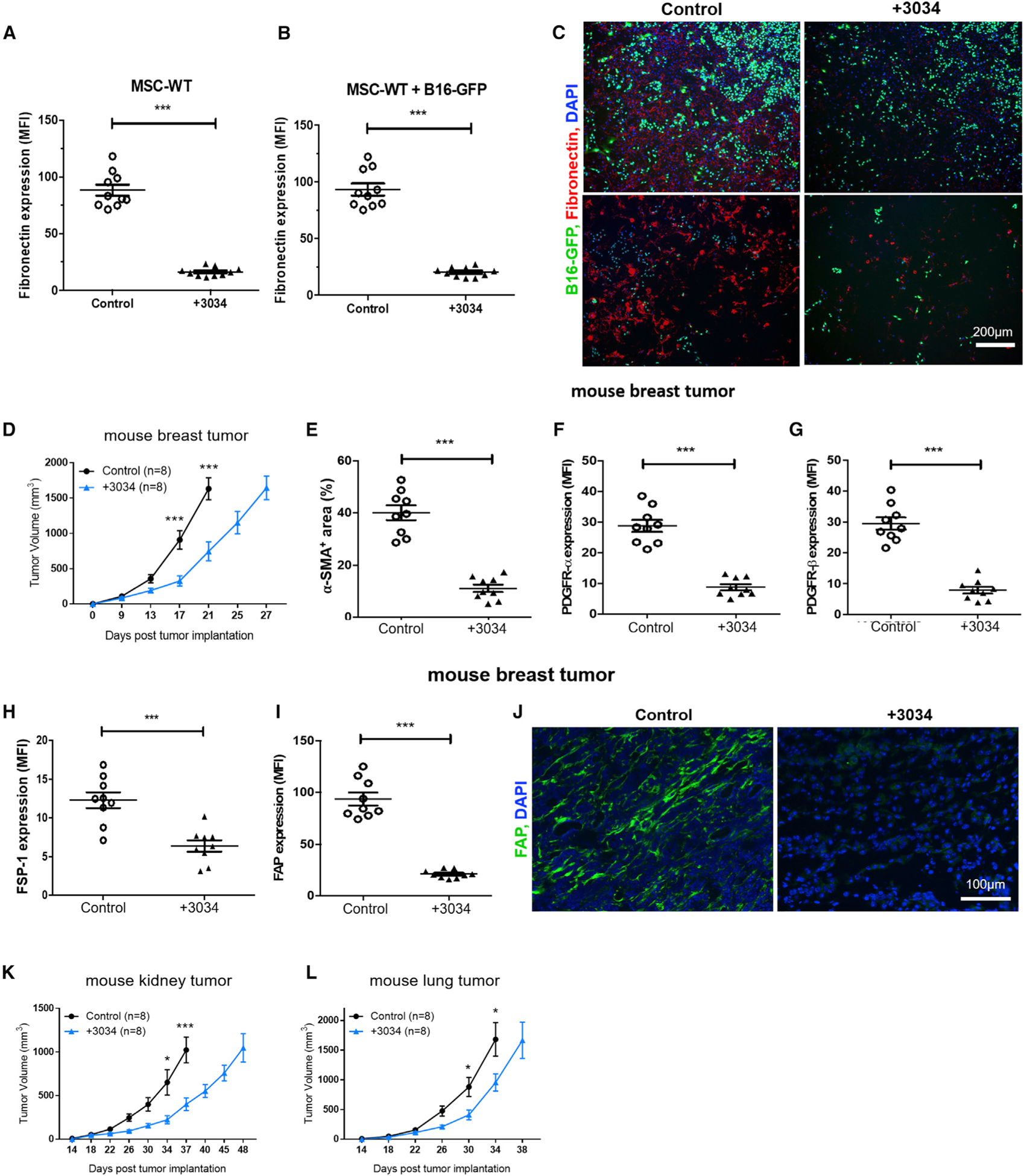
(A) Semiquantitative analysis of fluorescence micrographs show that compound 3034 reduces the production of ECM fibronectin by MSC-WT cells significantly.***p < 0.001.
(B and C) Representative fluorescence micrographs (C) and semiquantitative analysis (B) show that compound 3034 decreases significantly the expression of ECM fiber fibronectin by MSC-WT cells following co-culture with B16-GFP cells. ***p < 0.001. Scale bar, 200 μm.
(D) Tumor growth curve shows significantly slower growth of 4T1 tumors in BALB/c mice treated with compound 3034 (300 μg i.p. bid for 14 days following implantation) (n = 8) than the control treated with 10% DMSO (n = 8). ***p < 0.001.
(E–I) Semiquantitative analysis of fluorescence micrographs shows significantly lower expression (MFI) of cancer-associated fibroblast (CAF) markers α-SMA (E), PDGFRα (F), PDGFRβ (G), FSP-1 (H), and FAP (I) in breast tumor sections from the 3034-treatment group in comparison to the control group (10% DMSO) at day 17 post-implantation. ***p < 0.001.
(J) Representative fluorescence micrographs show significantly lower expression of cancer-associated fibroblast (CAF) marker FAP (green) in breast tumor sections from the compound 3034 treatment group in comparison to the control (10% DMSO) at day 17 post-implantation. DAPI (blue) was the nuclei. Scale bar, 100 μm.
(K) Tumor growth curve demonstrates significantly slower growth of Renca mouse kidney tumors in male BALB/c mice treated with compound 3034 (300 μg i.p. bid for 21 days following implantation) (n = 8) than the control treated with 10% DMSO (n = 8). *p < 0.05, ***p < 0.001.
(L) Tumor growth curve demonstrates significantly slower growth of LLC1 mouse lung tumors in C57BL/6 WT mice treated with compound 3034 (300 μg i.p. bid for 21 days following implantation) (n = 8) than the control group treated with 10% DMSO (n = 8). *p < 0.05. See also Figure S6.
Breast cancers exhibit a high degree of immunosuppression by their surrounding stroma (Kalluri, 2016; Lauricella et al., 2016). Sb9 is highly expressed in human primary breast tumor cells (Figures 1 and S6C) and in the stromal cells within the TME (Figures S6C and S6D). Therefore, we investigated whether compound 3034 could be used to treat mice implanted with 4T1 breast tumor cells. Treatment with compound 3034 significantly reduced (2.5-fold, p = 0.0006) the size of 4T1-derived tumors (Figure 6D) in comparison to the control group (the size end points were days 27 and 21, respectively). The MST of 4T1 tumors treated with 3034 was significantly longer (p < 0.0001) than the control group (29 days versus 22.5 days) (Figure S6E). Treatment with compound 3034 also resulted in reduced levels of stromal cells and CAFs (Figures 6E–6J).
Furthermore, we observed similar antitumor effects of compound 3034 in two other in vivo tumor models: Renca kidney tumor and LLC1 lung tumor (tumor size in Figures 6K and 6L; survival in Figures S6F and S6G). We conclude that inhibition of Sb9 by compound 3034 has potent anti-tumor effects in a broad spectrum of malignancies, including melanomas, as well as breast, lung, and kidney cancer.
DISCUSSION
The majority of malignant tumors are immunologically silent. In order to grow in an immune-competent host, tumor cells acquire genetic mutations and undergo epigenetic changes that result in immune-resistant phenotypes (Kather et al., 2018; Trujillo et al., 2018). In an effort to make tumors immunologically active, genotoxic therapies (such as radio- and chemotherapy) have been used as adjuvants for immunotherapy by evoking innate immune responses, such as type I-IFN signaling. However, these adjuvant strategies are hampered by the induction of suppressors of type I-IFN pathway by genotoxic agents (Trujillo et al., 2018). To circumvent this problem, combinations of up to 3 treatments may have to be used, complicating clinical trials. Progression of cancer is dependent upon both the biological characteristics of the cancer cells and the immunological status of the host (Gonzalez et al., 2018). The expression of GrB in immune cells is appreciated widely (Russell and Ley, 2002), but the findings of other studies (D’Eliseo et al., 2010, 2016; Hu et al., 2003; Kontani et al., 2001; Pearson et al., 2014) and ours highlight the potential importance of tumor-derived GrB as well. Our discovery of how these dual sources of GrB (both host- and cancer-derived) cause cancer cell death directly through both the inhibition of cancer cell-produced Sb9 and liberation of host anti-tumor immunity lays the groundwork for the singular modality to treat immuno-logically silent tumors. Future studies are required to determine the regional effect of tumor-derived GrB.
Remarkable heterogeneity is manifested by the distinct morphological and phenotypic profiles of different cancers, including various proliferative and metastatic capacities (Meacham and Morrison, 2013). This variety is likely to result in a range of adaptive behaviors by tumors to alterations in microenvironmental conditions and/or different capacities for malignant transformation (Tellez-Gabriel et al., 2016). Our data suggest that cancer cells with higher levels of Sb9 and/or GrB may adopt more aggressive phenotypes.
In this report, we used genetic ablation to demonstrate a direct requirement of Sb9 for tumor cell growth through inhibition of cell death caused by GrB. This is consistent with previous correlative studies in mice and humans (Bots et al., 2006; Medema et al., 2001a, 2001b). Along with others, we described the expression of Sb9 in both effector and immune-suppressive cells (Azzi et al., 2013; Mangan et al., 2008; Phillips et al., 2004). The ablation of Sb9 results in the impaired survival of CTL specific to intracellular pathogens (Zhang et al., 2006). However, we show that the aggregate effect of Sb9 ablation is increased CTL-based immunity to tumors in vivo. We conclude that the increased immunity to tumors in Sb9 KO mice is due more to the impaired survival of immuno-suppressive cells in the TME than any decrease in cell-intrinsic viability of GrB+ CTL. Sb9 is also known to inhibit caspase-1 (interleukin-1β-converting enzyme), which is involved in the inflammatory response by cleaving the precursors of inflammatory cytokines, including IL-1β, IL-18, and IL-33 (Annand et al., 1999; Young et al., 2000). The inflammatory microenvironment can directly modulate the number, function, migration, and maintenance of effector immune cells (Kim and Harty, 2014). Therefore, alteration of this milieu may constitute an alternative pathway by which Sb9 suppresses the inflammatory response and escapes from immune surveillance against tumors.
Notably, metastasis of melanoma to the TDLNs was significantly less frequent in the B16-Sb9 KO group. Given that the metastatic melanoma within the LN also expressed Sb9, one could speculate that Sb9 could protect metastatic niches against the CTL of LN, providing a mechanism for the spreading of neoplasms through lymphatics. Furthermore, various cancers have been associated with a heterogeneous and adaptive TME, and their growth can be driven by the local TME in which they thrive. Besides lymphocytes and other immune cells, stromal cells are a major cellular component of the diverse TME that plays a critical role in tumor development (Nilendu et al., 2018). Our finding that Sb9 regulates the function of the stroma is supported by our previous report for Sb9 in protecting mesenchymal stem cells (MSCs) from exogenous GrB (El Haddad et al., 2011a, 2011b). CAFs have received increasing attention as the key cellular player within the tumor stroma that supports the initiation, progression, and metastasis of cancers (Kalluri, 2016; Wei et al., 2018). Importantly in our melanoma mouse model, we found suppression of CAF activity with less accumulation and generation of matrix proteins as well as lower neovascularization in the Sb9 KO/Sb9 KO group. These findings suggest that lack of Sb9 in the stromal cells, including CAFs and MSCs, of the Sb9-deficient host could increase their susceptibility to the lethal effect of GrB released within the stroma. Notably, GrB has also been reported to be an extracellular substrate with ability to also cleave ECM materials, promoting tumor cell migration and metastasis (Prakash et al., 2014). This observation further highlights the importance of Sb9 inhibition in suppressing tumor metastasis. Altogether, pharmacologic inhibitors of Sb9 target tumors on multiple levels, inducing death of the tumor cells by their own GrB, downregulation of the immunosuppressive cells in the TME, and elimination of angiogenesis and the stroma, thereby rendering the TME less conducive for tumor growth and metastasis.
We regard this report as a starting point for the introduction of a small-molecule inhibitor of Sb9. Like any other field in its infancy, the field of Sb9 inhibition therapeutics requires more work for characterization and optimization of binding affinity as well as efficacy. For instance, one key challenge that remains is to generate drugs with improved affinity through high-throughput screening of fragments. Although the affinity of our compound remains in the expected range of small molecules, it may have much better affinity in vivo for many reasons. For example, the low pH of the lysosome confers a much higher affinity of compound 3034 to Sb9. Sb9 produced by the tumor likely undergoes significant post-translational modifications (not recapitulated in our screening assays) that may increase its affinity to compound 3034. Although the generation of selective ligands with high binding affinity defines the current paradigm, many low-affinity drugs could also be effective (Csermely et al., 2005; Lipton, 2006; Mestres and Gregori-Puigjané, 2009). In the case of Sb9, the development of a similar drug with a larger size and better affinity than 3034 may not result in an improvement of clinical efficacy, as its larger size may limit its cellular entry. Nonetheless, the fact that genetic or pharmacological inhibition of Sb9 in mice did not result in significant anomalies or side effects highlights the selectivity of the compound and its potential applicability for clinical translation. These findings also demonstrate the potential importance of disseminating our data to create the momentum needed to advance discoveries of Sb9 inhibitors. Immune checkpoint inhibitors are considered the most recent cancer therapeutic breakthrough (Peoples, 2019). However, Sb9 inhibition could be superior, due to its multifaceted actions on key players responsible for the growth and metastasis of tumors. Furthermore, Sb9 inhibitors may prove to be safer than immune checkpoint inhibitors (Peoples, 2019; Postow et al., 2018).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Reza Abdi (rabdi@rics.bwh.harvard.edu).
Materials Availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact author. This includes plasmids and proteins. All reagents will be made available on request after completion of a Material Transfer Agreement.
Data and Code Availability
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Chemicals and reagents
Dulbecco’s modified Eagle’s medium (DMEM), RPMI-1640 medium, fetal bovine serum (FBS), penicillin/streptomycin, and trypsin 0.25% were products of Hyclone, Thermo Scientific (Logan, UT). Dimethylsulfoxide (DMSO), Tris-HCl, sodium chloride, glycerol, and β-mercaptoethanol were purchased from Sigma-Aldrich (MO, USA). Zenobia Library 1 (352 compounds), Zenobia Library 2.2 (288 compounds), Life Chemical Fragments (436 compounds), and compound 3034 (1,3-benzoxazole-6-carboxylic acid) were obtained from Enamine LLC (NJ, USA). Recombinant murine IL-2 was purchased from Millipore Sigma (MA, USA). Granzyme B inhibitor I – Calbiochem (368050) was purchased from Millipore Sigma (MA, USA). The antibiotics puromycin, blasticidin, and G418 were purchased from InvivoGen (CA, USA).
Cell lines and cell culture
B16-F10 (mouse melanoma), Renca (mouse renal cancer), 4T1 (mouse breast cancer), LLC1 (mouse lung cancer), A375 (human melanoma), A549 (human lung cancer) and SK-BR3 (human breast cancer) cell lines were purchased from American Type Culture Collection (VA, USA). B16-F10, Renca, 4T1, A549 and SK-BR-3 cell lines were cultured in RPMI-1640 medium with 10% FBS and 1% penicillin/streptomycin. LLC1 and A375 cell lines were grown in DMEM medium with 10% FBS and 1% penicillin/streptomycin. Cells were maintained in a 37°C incubator at 5% CO2.
Human tumor samples
Human melanoma (ID: PA13AB6700), breast adenocarcinoma (ID: PA15478751), and non-small cell lung carcinoma (ID: PA00006545) slides were purchased from OriGene Technologies (MD, USA)
Mice
All animal experiments and methods were performed in accordance with the relevant guidelines and regulations approved by the Institutional Animal Care and Use Committee of Brigham and Women’s Hospital, Harvard Medical School, Boston, MA (protocol number: 2016N000167/04977). C57BL/6J (WT) (JAX#000664), BALB/c (WT) (JAX#000651), C57BL/6-Serpinb9tm1Arp/J (Spi6−/−) (JAX#008158) mice and C57BL/6-Tg(UBC-GFP)30Scha/J (JAX#004353) mice were purchased from Jackson Laboratory (Bar Har-bor, ME, USA) and used at 8–10 weeks of age. NOD.Cg-Prkdcscid/l2rgtm1Wjl/SzJ (NSG) (JAX#005557) mice were provided by Dr. Leonard D. Shultz from the Jackson Laboratory. All mice were used at 8–10 weeks of age (female mice) and were housed in sterilized and ventilated cages in a specific pathogen-free animal facility under a standard 12 h light/12 h dark cycle.
METHOD DETAILS
Immunofluorescence staining
5μm sections of tissue were cut by cryosectioning and stained with conjugated or purified antibodies. Purified antibodies were detected using secondary antibodies. The antibodies used include anti-Sb9 (PA5-51038, Invitrogen), anti-mouse GrB (16G6, #14-8822-82, Invitrogen), anti-human GrB (2C5, sc-8022, SCBT), anti-human GrB (B18.1, NBP1-97525, Novus), anti-MelanA (ab51061, Ab-cam), anti-α-SMA (D4K9N, #19245S, CST), anti-cleaved caspase-3 (Asp175, #9661, CST), anti-Collagen I (NB600-408, Novus), anti-Fibronectin (NBP1-91258, Novus), anti-PDGFR-α (APA5, ab90967, Abcam), anti-PDGFRβ (APB5, #14-1402-82, Invitrogen), anti-pan-cytokeratin (AE1/AE3, sc-81714, SCBT), anti-FAP (ab53066, Abcam), anti-FSP-1 (S100A4, ABF32, Millipore), FITC anti-CD11b (M1/70, #101206, Biolegend), FITC anti-Gr-1 (RB6-8C5, #108417, Biolegend), and anti-Ly-6C (HK1.4, #128012, Biolegend). DAPI (VECTASHIELD, Vector Laboratories) was used to counterstain the cell nuclei. The stained tissue sections were visualized using an EVOSTM FL Auto 2 Imaging System (Thermo Fisher Scientific) for whole images and a fluorescence confocal microscope (Nikon) for high-resolution images. Quantification was performed on 2–3 sections from at least 3 separate mice using image analysis softwares Celleste (Invitrogen) and ImageJ (NCBI, 1.8.0_112).
RNA in situ hybridization (RNAscope)
Granzyme B and Sb9 mRNA in situ hybridization (ISH) was measured with RNAscope assay (Advanced Cell Diagnostics, ACD, Hayward, CA), according to the manufacturer’s protocols. Briefly, 4T1 cells, B16 cells or B16-Sb9 KO cells were hybridized with Granzyme B probe (ACD, Cat. No. 490191), Sb9 probe (ACD, Cat. No. 578121), positive control probe (ACD, Cat. No.320881), and negative control probe (ACD, Cat. No. 320871) at 40°C for 2 h. Hybridization signals were amplified and visualized with RNA-scope® Multiplex Fluorescent Reagent Kit v2 (ACD, Cat. No. 323100). Images were captured with a fluorescence microscope (EVOSTM FL AUTO 2).
Western blotting
Lysates of cells and tissues were measured using the Bradford assay. Equal amounts of protein were separated by SDS-PAGE and transferred to a PVDF membrane. The membranes were immunoblotted with the following specific antibodies: anti-Sb9 (PA5-51038, Invitrogen), anti-mouse GrB (16G6, #14-8822-82, Invitrogen), anti-cleaved caspase-3 (Asp175, #9661, CST), anti–GAPDH (Abcam), anti-rabbit IgG-HRP (Abcam), anti-rat IgG-HRP (Abcam), using standard protocols. The blots were developed with West Dura chemiluminescent substrates using a Bio-Rad ChemiDoc imaging system.
Transfection and lentivirus transduction
Cells were transfected with Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. Briefly, cells were plated at 20–30% density in 12-well plates 24hrs prior to transfection. For plasmid transfection, the equivalent of 0.4 μg of plasmid per well of a 12-well plate was utilized. Plasmids utilized included Control Double Nickase Plasmid (control plasmid, sc-437281, SCBT), PI-9 Double Nickase Plasmid (h) (sc-404486-NIC, SCBT), granzyme B Double Nickase Plasmid (m) (sc-420745-NIC, SCBT) and serpinB9 expression plasmid in pcDNA3.1+/C-(K)DYK (OMu03805D, GenScript). After 24~72 hr incubation, selective medium was used to complete screening for successfully transfected cells. PI-9 Double Nickase Plasmid (h) is designed to disrupt gene expression by causing highly specific Cas9-mediated double nicking of the SERPINB9 (human) gene, which mimics a double-strand break. It consists of a pair of plasmids, each encoding a D10A mutated Cas9 nuclease and a unique, target-specific 20 nt guide RNA (gRNA). This pair of gRNAs target exon 3 of the SERPINB9 (human) gene, and their sequences are ACCTGCTGAGAACGGCCAAC and TCACTTC AGTGAGAAGCGAC. Control Double Nickase Plasmid was used as the negative control. To disrupt gene expression of the SER-PINB9 (mouse) gene, we targeted the 20-nucleotide sequence upstream of the protospacer-adjacent motif (PAM) sequence in exon 6 of the SERPINB9 (mouse) gene. The gRNA (sequence GCACCTCCTTCACATAGGCG) was cloned at the BsmBI site into the LentiCRISPRv2-blast (#98293, Addgene) vector, and pLentiCRIPSRv2 gRNA was subjected to sequence analysis to confirm the inserted oligonucleotide. Empty LentiCRISPRv2-blast vector was used as negative control. Lentiviral particles were generated by co-transfecting LentiCRISPRv2-blast plasmid into HEK293T cells with packaging plasmids pVSVg (#8485, Addgene) and psPAX2 (#12260, Addgene). For lentivirus transduction, cells were infected at either 20 or 40 MOI for 24 h with a Cas9-expressing lentivirus. Following blasticidin selection, we used the limiting dilution method in 96-well plates to select single-cell clones. The expression of Sb9 in the puromycin/G418/blasticidine-resistant clones was then analyzed by western blotting. Parental B16 cells were transduced with pLenti PGK GFP Puro encoding the enhanced green fluorescent protein (GFP) cDNA and the puromycin resistance gene (Puro) under the phosphoglycerate kinase (PGK) promoter. High GFP-expressing cells were selected using puromycin. The lentiviral vectors are self-inactivating vectors in which the viral enhancer and promoter have been deleted, and this transcription inactivation increases biosafety.
Reverse transcription (RT) and quantitative PCR
Total RNA was prepared from cells using the Quick-RNA MiniPrep kit (Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. cDNA was prepared from total RNA using High-Capacity cDNA Reverse Transcription Kit (Thermo). Following reverse transcription, quantitative PCR was performed using a cycling profile consisting of 95°C for 2min (Stage I), 40 cycles of 95°C for 20 s, 60°C for 30 s, and 70°C for 30 s (Stage II), and 65°C for 5 s (Stage III). Fold changes were determined by subtracting Cq values of the loading control from the Cq values of the gene of interest. The results were normalized to untreated controls. Primers used for real time quantitative PCR are listed as follows: mouse GAPDH-F: AGCCACATCGCTCAGACAC, mouse GAPDH-R: GCCCAATACGACCAAATCC; mouse GrB-F: ATGAAGATCCTCCTGCTACTGC, mouse-GrB-R: CCCACATATCGCCTCAGGCT; mouse Ki67-F: AGAGCCTTAGCAATAGCAACG, mouse-Ki67-R: GTCTCCCGCGATTCCTCTG.
Tumor implantation
Mice were anesthetized with 2% isoflurane with oxygen. B16-F10, Renca, A375, LLC1 and 4T1 cell lines were gently injected subcutaneously in the right flanks or 4th mammary glands of mice. 1×105 cells were injected per mouse for most of the experiments. 25×103 cells were injected per mouse for the experiments using Compound 3034. Upon 4hrs post-implantation, 300 μg of Compound 3034 was administered twice a day intraperitoneally for 14 days. Tumor growth was monitored three times per week by digital caliper (FisherbrandTMTraceableTMDigital Calipers).
Preparation of tumor tissues for flow cytometry
Tumor tissues from mice were minced into small pieces using a razor blade, then transferred to 70 μm cell strainers (BD) and separated mechanically using the plunger of a 5ml syringe. Cells that passed through the cell strainer were collected in a 50ml conical tube and resuspended in the RPMI-1640 complete growth medium. Then, the single-cell suspensions from tumors were ready for staining.
Flow cytometry
Cells were plated in 96-well round-bottom plates (Corning, NY) for intracellular cytokine staining and 96-well flat-bottom plates (Corning, NY) for cell-surface and intracellular transcription factor staining. The cell samples that underwent intracellular cytokine staining were first incubated with 100ng/ml PMA and 1μg/ml ionomycin (Sigma-Aldrich), and GolgiStopTM protein transport inhibitor (BD Bioscience) at 37°C for 4hr. All samples were washed with DPBS prior to incubation with Fixable Viability Dye eFluorTM 780 (Thermo Fisher Scientific) diluted 1:1000 in DPBS for 30min at 4°C. Then, the cells were washed with FACS buffer (DPBS + 2% fetal bovine serum +1 mM EDTA + 0.1% sodium azide) and incubated for 30min at 4°C with the following cell-surface antibodies: PB anti-CD4 (GK1.5, #100428, Biolegend), BV510 anti-CD8 (53–6.7, #100752, Biolegend), APC anti-CD44 (IM7, #103012, Biolegend), PE/Cy7 anti-CD62L (MEL-14, #104418, Biolegend), PE anti-CD25 (7D4, #558642, BD PharMingen), BV510 anti-CD45 (30-F11, #103138, Biolegend), PE/Cy7 anti-CD11b (M1/70, #101216, Biolegend), PB anti-F4/80 (BM8, #123124, Biolegend), PE anti-Ly-6G (1A8, #127608, Biolegend), FITC anti-Gr-1 (RB6-8C5, #108417, Biolegend), PerCP/Cy5.5 anti-Ly-6C (HK1.4, #128012, Biolegend), BV510 anti-CD4 (RM4-5, #100559, Biolegend), FITC anti-CD8 (53–6.7, #553031, BD PharMingen), APC anti-CD3 (17A2, #100236, Biolegend), PE anti-CD44 (BJ18, #338808, Biolegend), FITC anti-CD80 (16-10A1, #104706, Biolegend), PerCP/Cy5.5 anti-CD86 (GL-1, #105026, Biolegend), APC anti-CD163 (S15049I, #155305, Biolegend), PE/Cy7 anti-CD206 (C068C2, #141720, Biolegend), and PE/Cy7 anti-PD-L1 (10F.9G2, #124314, Biolegend). All of the cell-surface antibodies were diluted 1:300 in FACS buffer. The cells were permeabilized using the eBioscience Intracellular Fixation and Permeabilization Buffer Set (Thermo Fisher Scientific) for 30min at 4°C. Then they were incubated with the following intracellular antibodies: PerCP/Cy5.5 anti-FOXP3 (FJK-16 s, #45-5773-82, Invitrogen), PB anti-GrB (GB11, #515408, Biolegend), APC anti-IFNγ(XMG1.2, #505810, Biolegend), FITC anti-TNFα (MP6-XT22, #506304, Biolegend), PE anti-IL-2 (JES6-5H4, #554428, BD PharMingen) and PE/Cy7 anti-IL-10 (JES5-16E3, #505026, Biolegend). All of the intracellular antibodies were diluted 1:300 in the eBioscience Permeabilization Buffer (1x) (Thermo Fisher). Cells were washed once with Permeabilization Buffer and fixed in FACS buffer containing 1% formalin. Flow cytometry was performed using a BD FACS-CantoTM II flow cytometer (BD Biosciences). Analysis of flow cytometry results was performed via FlowJo software (FlowJo LLC, Ashland, OR).
Isolation and culture of mouse bone marrow-derived mesenchymal stem cells (MSCs)
Isolation and harvest of murine MSCs from either C57BL/6-WT mice or Sb9 KO mice were performed according to the protocol as described previously (Zhu et al., 2010).
Protein expression and purification
In an attempt to produce a soluble variant of Sb9, a construct was designed in which a hexa His-tagged maltose-binding protein (His-MBP) was added at the N terminus. Sb9 was soluble in multiple growth media conditions using this strategy. The condition (1mM IPTG, 37°C, 3hr) for best expression was selected for biomass production with 5L culture. The cells were lysed and clarified by centrifugation. The fusion protein was then captured by Ni-NTA affinity purification, such that it was eluted using high concentration imidazole. Then, the resulting protein was further purified by anionic exchange chromatography using a MonoQ column (GE Healthcare) and a NaCl gradient for elution. An amount of 31.4mg protein was obtained. The purified Sb9 protein was characterized by SDS-PAGE and MALDI-TOF mass spectrometry (MS). A construct of human serpinB9 covering residues 2–376 with N-terminal His-tag in the pFastBac1 vector (Invitrogen) was overexpressed in insect Trichoplusia ni cells adapted for suspension culture in ESF921 medium (Expression systems). Cells were grown at 27°C to a density of ~2 million cells per mL, infected with baculovirus containing serpinB9 construct, collected by centrifugation around 60hrs post-infection, and stored at −80°C. Cell pellets were microfluidized at 15000 psi in buffer A (25mM HEPES, pH 7.5, 200mM NaCl, 10% glycerol, 20mM Imidazole, 7mM BME) and the resulting lysate was centrifuged at 13,000 rpm for 30 min. Ni-NTA beads (QIAGEN) were mixed with lysate supernatant for 30 min and washed with buffer A and eluted with buffer B (25mM HEPES, pH 7.5, 200mM NaCl, 10% glycerol, 400mM Imidazole, 7mM BME). The sample was further purified by size exclusion chromatography using an Superdex-200 16/60 column (GE healthcare) in buffer C (20mM HEPES-7.5, 200mM NaCl, 10% glycerol, 0.5mM TCEP, 3mM DTT). Protein containing fractions were pooled, concentrated, and stored at −80°C.
MALDI-TOF mass spectrometry
Analyses were performed on a Finnigan Laser MAT 2000 time-of-flight mass spectrometer (Finnigan MAT, Hemel Hempstead, Herts, UK). The system used a nitrogen laser (337nm, 2ns pulse) to desorb ions from the sample specimen. The desorbed ions were accelerated to 20kV into a free long tube. The time recorded for a molecule to travel the length of the tube to a detector was proportional to the mass of the ion, which was its molecular weight. All spectra were obtained using the positive-ion mode. Standard stainless-steel targets (with a sample application area of about 3.14mm2) obtained from the manufacturer were employed for all analyses. The laser-mat software allowed the user to irradiate one of the four possible target regions or quadrants of about 0.02mm2. The spectra in this study were calibrated using instrumental calibration, based on the parameters determined from analysis of a number of standard proteins and peptides.
Thermal stability assay (TSA) and initial fragment (small organic molecules) screen
TSA is an analytic tool to estimate the overall stability of a protein by monitoring the shift in its melting temperature upon changing the buffer conditions or titrating various ligands. As the protein unfolds with increasing temperature in a thermal cycler (CFX384 Touch Real-Time PCR Detection System), SYPRO orange dye binds to the exposed hydrophobic core and fluoresces, leading to the signal that is measured in comparison to temperature to produce a melt curve. The rate of dye uptake peaks at the melting transition and signals the melting temperature (Tm). As an initial assay, MBP-Sb9 was tested at 1 and 0.25mg/ml. Four tests were performed at each concentration in Buffer A1 (20mM Tris-HCl pH8.0, 20mM sodium chloride, 5mM 2-mercaptoethanol and 10% glycerol): no additive, plus 10mM maltose, plus 5% glycerol, and plus 10% glycerol. Dye was added at 2x strength, and the readings were performed from 25 to 95°C at 0.5°C intervals. To set up the fragment screen assay, a plate was loaded with protein by manually pipetting 19 μL per well well-by-well. Then, 0.5 μL of fragment solutions were transferred from the 384-well library stock plate in a replicate manner, using an Integra ViaFlo II electronic pipette and mixing by swirling the pipette tips. This resulted in testing the Zenobia compounds at 5mM and the Life Chemicals compounds at 1.25mM, and all assay wells contained 2.5% DMSO. On all plates, at least 8 wells tested buffer alone and at least 24 wells tested 2.5% DMSO, with the latter used for the baseline measurement. Once a plate was set, it was sealed with foil and incubated with shaking for 1 hr at 25°C. Sypro Orange dye was added at 2x strength (Thermo S6650) using the ViaFlo pipette. The plate was sealed with clear adhesive, briefly shaken to disperse the dye, and measured using the BioRad C1000 thermal cycler with a CFX384 Touch Real-Time detector. The measurement cycle was performed from 25 to 95°C with a measurement interval of 0.5°C. One plate of fragments was tested on a given day. A commercially available library of over 1,000 compounds was tested on four plates. Structure-activity relationship (SAR) study was performed on the compound ZT0587 (3,4-dihydroxybenzoic acid) decorating the ring with various hydroxyl, methyl, amine, nitro, nitrile, carboxylic acid alternates and ring closure alternates. Compound 3034 (1,3-benzoxazole-6-carboxylic acid) was chosen based on its activity in the SPR, Caspase-3 activity assay, Granzyme B activity assay, STD-NMR assay.
Surface plasmon resonance (SPR)
Surface plasmon resonance (SPR) measures change in refractive index on a biosensor surface, as analytic mass accumulates via binding to an attached ligand. This allows derivation of affinity (KD) from measured kinetic rates of association (ka) and dissociation (kd). All assays were run at 25°C on a BiOptix 404pi. An NTA biosensor chip was used in this study to pre-concentrate 3μM MBP-Sb9 via an N-terminal His-tag. MBP-Sb9 was covalently immobilized with EDC/NHS in 20mM HEPES pH8.0, 150mM NaCl, 0.05% Tween-20, 5mM βME to 10,000 to 15,000 RUs. MBP was similarly immobilized on a separate channel as a negative control. Compounds were injected as 3-fold serial dilutions in duplicate starting at 200μM. Injections were done at 50μl/min for 60 s, followed by a 120 s dissociation in 20mM HEPES pH8.0, 150mM NaCl, 0.05% Tween-20, 5mM βME, 1% DMSO. A series of five DMSO standards between 0.5% and 1.5% were included with each run to correct for bulk refractive index. Sensorgrams were analyzed using Scrubber 2 software with a double reference and fit to a 1:1 Langmuir model to determine the interaction parameters KD, ka, and kd.
Caspase-3 activity assay
The EnzChek Caspase-3 Assay Kit was used to detect apoptosis by assaying for increases in caspase-3 activities. The basis for the assay is rhodamine 110 bis-(N-CBZ-L-aspartyl-L-glutamyl-L-valyl-L-aspartic acid amide) (Z-DEVD–R110). This substrate is a bisamide derivative of rhodamine 110 (R110) containing DEVD peptides covalently linked to each of R110’s amino groups, thereby suppressing the dye’s visible absorption and its fluorescence. Upon enzymatic cleavage, the nonfluorescent bisamide substrate is converted in a two-step process first to a fluorescent monoamide and then to an even more fluorescent R110. We used the lysate of 1 × 106 cells for each reaction. B16 cells were treated with compounds to induce apoptosis. A negative control was prepared by incubating cells in the absence of compounds. The cells were incubated with compounds for 24hr and washed in PBS. Each cell sample or control was resuspended in 50 μL of the 1x cell lysis buffer. Cells were lysed by incubating on ice for 30min, and the lysed cells were centrifuged to pellet the cellular debris. 50 μL of the supernatant was transferred from each sample to individual microplate wells. 50 μL of the 1x cell lysis buffer was used as a no-enzyme control to determine the background fluorescence of the substrate. 50 μL of the 2x substrate working solution was added to each sample and control. The microplate was covered, and the samples were incubated at room temperature for approximately 30min. The fluorescence was measured by a fluorescence microplate reader (excitation/emission ~496/520 nm) using excitation and emission filters.
GranToxiLux assay
The materials for this single cell-based fluorogenic cytotoxicity assay for the measurement of granzyme B activity in live cells are available commercially as the GranToxiLux kit from OncoImmunin. In brief, B16-WT cells were labeled with TFL4 dye (APC) and incubated with Granzyme B substrate from Vial GS (FITC). Cells untreated with the Granzyme B substrate were used as the negative control. Flow cytometry was performed using a BD FACSCantoTM II flow cytometer (BD Biosciences). Flow cytometric analysis was performed via FlowJo software (FlowJo LLC, Ashland, OR).
Granzyme B activity assay
The goal of these experiments was to find a compound that deters the ability of Sb9 to inhibit granzyme B proteolysis. To that end, a granzyme B activity assay was developed to test a number of compounds from the fragment screen hits. The assay is a commercially available kit, the Granzyme B Colorimetric Drug Discovery Kit by Enzo Life Sciences (BML-AK711). In this assay, granzyme B cleaves the peptide substrate Ac-IEPD-pNA to produce the chromophore p-nitroaniline, which is measured by absorbance at 405 nm. The measurements were performed with a LabSystems Multiskan RC plate reader. The appropriate volume of assay buffer was added to the wells of the microplate. 10 μL of tested compound was added to the designated wells. Then, 10 μL of Sb9 protein was added into the designated wells and pre-incubated with the compounds for 1 or 2 hr. 15 μL of GrB was added to the designated wells. Then this mixture was incubated with GrB for 30 min. The reaction was started by adding 50 μL of the 2x Ac-IEPD-pNA substrate solution. 100 ul of pNA at 50 μM was added as a calibration standard, which produced an A405 of ~0.3. The microplate was read continuously at A405 in a microplate-reading spectrophotometer. The reactions were run for 2hrs, and the reading interval was 10min. Lastly, we obtained the raw data and performed data analysis.
Saturation-Transfer Difference (STD) NMR assay
The Saturation-Transfer Difference (STD) NMR assay was used to evaluate the binding of Compound 3034 to Sb9. The STD NMR experiment was performed using 420 μM 3034 and 45 μM Sb9 protein in PBS buffer, pH7.0 with 10% D2O at 25°C, on a 600MHz Bruker Avance II spectrometer equipped with a Prodigy Cryoprobe. The saturation period was 3.0 s with irradiation at 0 ppm (on-resonance excitation) and −20 ppm (off-resonance excitation). The number of scans was 160 for the on and off-resonance spectra respectively. Spectra were processed using Topspin software and apodized with exponential multiplication with 1.0 Hz line-broadening.
Single cell RNA sequencing analysis
The single cell gene expression matrices and cell type annotation files of human melanoma, human breast cancer, and human non-small cell lung cancer were downloaded from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/), which are provided by previous literatures including (Jerby-Arnon et al., 2018) (GSE72056), (Slyper et al., 2020) (GSE140819) and (Guo et al., 2018) (GSE99254), respectively. Cell clustering was performed by a graph-based clustering approach provided by R package Seurat, clusters were visualized in a t-SNE plot. Gene expression levels of SERPINB9 (Sb9) and GZMB (GrB) in putative cancer cells and tumor-infiltrating T cells (CD4+ and CD8+) were extracted for comparation.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the data were presented as mean ± standard error from at least three times, each done in triplicate. The statistical significance between two groups was determined by Student’s t test, whereas the comparisons of multiple groups were carried out by one-way ANOVA, followed by Bonferroni’s post-test using GraphPad Prism7 software (GraphPad Software, Inc., CA). A probability value of *p < 0.05 was considered to be significant.
Supplementary Material
THE KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SerpinB9 | Invitrogen | PA5-51038 (RRID: AB_2636486) |
| mouse Granzyme B | Invitrogen | 14-8822-82 (RRID: AB_468530) |
| human Granzyme B | Santa Cruz | sc-8022 (RRID: AB_2232723) |
| human Granzyme B | Novus | NBP1-97525 (RRID: AB_11189027) |
| MelanA | Abcam | ab51061 (RRID: AB_880693) |
| α-SMA | Cell Signaling | 19245S (RRID: AB_2734735) |
| cleaved caspase-3 | Cell Signaling | 9661 (RRID: AB_2341188) |
| Collagen I | Novus | NB600-408 (RRID: AB_10000511) |
| Fibronectin | Novus | NBP1-91258 (RRID: AB_11039761) |
| PDGFRα | Abcam | ab90967 (RRID: AB_2049372) |
| PDGFRβ | Thermo Fisher | 14-1402-82 (RRID: AB_467493) |
| pan-cytokeratin | Santa Cruz | sc-81714 (RRID: AB_2191222) |
| FAP | Abcam | ab53066 (RRID: AB_880077) |
| FSP-1 | Millipore | ABF32 (RRID: AB_11203822) |
| LAMP1 | Abcam | ab25630 (RRID: AB_470708) |
| Ki67 | Abcam | ab15580 (RRID: AB_443209) |
| FITC CD11b | Biolegend | 101206 (RRID: AB_312789) |
| FITC Gr-1 | Biolegend | 108417 (RRID: AB_389309) |
| Nestin | Abcam | ab105389 (RRID: AB_10859398) |
| GAPDH | Abcam | ab181602 (RRID: AB_2630358) |
| rabbit IgG-HRP | Abcam | ab6721 (RRID: AB_955447) |
| rat IgG-HRP | Abcam | ab6734 (RRID: AB_955450) |
| PB CD4 | Biolegend | 100428 (RRID: AB_493647) |
| PE/Cy7 PD-L1 | Biolegend | 124314 (RRID: AB_10643573) |
| PE/Cy7 CD206 | Biolegend | 141720 (RRID: AB_2562248) |
| APC CD163 | Biolegend | 155305 (RRID: AB_2814059) |
| PerCP/Cy5.5 CD86 | Biolegend | 105026 (RRID: AB_893417) |
| FITC CD80 | Biolegend | 104706 (RRID: AB_313127) |
| APC CD3 | Biolegend | 100236 (RRID: AB_2561456) |
| FITC CD8 | BD Biosciences | 553031 (RRID: AB_394569) |
| PerCP/Cy5.5 Ly-6C | Biolegend | 128012 (RRID: AB_1659241) |
| BV510 CD4 | Biolegend | 100559 (RRID: AB_2562608) |
| FITC anti-Gr-1 | Biolegend | 108417 (RRID: AB_389309) |
| PE Ly-6G | Biolegend | 127608 (RRID: AB_1186099) |
| PB F4/80 | Biolegend | 123124 (RRID: AB_893475) |
| PE/Cy7 CD11b | Biolegend | 101216 (RRID: AB_312799) |
| PE CD25 | BD Biosciences | 558642 (RRID: AB_1645250) |
| BV510 CD45 | Biolegend | 103138 (RRID: AB_2563061) |
| BV510 CD8 | Biolegend | 100752 (RRID: AB_2563057) |
| PE/Cy7 CD62L | Biolegend | 104418 (RRID: AB_313103) |
| PerCP/Cy5.5 FOXP3 | Thermo Fisher | 45-5773-82 (RRID: AB_914351) |
| APC IFNγ | Biolegend | 505810 (RRID: AB_315404) |
| FITC TNFα | Biolegend | 506304 (RRID: AB_315425) |
| PE IL-2 | BD Biosciences | 554428 (RRID: AB_395386) |
| PE/Cy7 IL-10 | Biolegend | 505026 (RRID: AB_11150582) |
| APC CD44 | Biolegend | 103012 (RRID: AB_312963) |
| PE CD44 | Biolegend | 338808 (RRID: AB_2076578) |
| PB GrB | Biolegend | 515408 (RRID: AB_2562196) |
| Biological Samples | ||
| human melanoma | OriGene | PA13AB6700 |
| human breast adenocarcinoma | OriGene | PA15478751 |
| human non-small cell lung carcinoma | OriGene | PA00006545 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s modified Eagle’s medium (DMEM) | Hyclone | SH30022.01 |
| RPMI-1640 medium | Hyclone | SH30027.01 |
| ESF921 medium | Expression systems | 96-001-10 |
| fetal bovine serum | Hyclone | SH30071.03IH25-40 |
| Ni-NTA | Thermo Fisher | R90101 |
| penicillin/streptomycin | Hyclone | SV30010 |
| Dimethylsulfoxide (DMSO) | sigma | 472301-500ML |
| Zenobia Library 1 (352 compounds) | Enamine | N/A |
| Zenobia Library 2.2 (288 compounds), | Enamine | N/A |
| Life Chemical Fragments (436 compounds), | Enamine | N/A |
| compound 3034 (1,3-benzoxazole-6-carboxylic acid) | Enamine | N/A |
| compound ZT0587 (3,4-dihydroxybenzoic acid) | Enamine | N/A |
| Recombinant murine IL-2 | Sigma | 11271164001 |
| Granzyme B inhibitor I | Calbiochem | 368050 |
| Puromycin | InvivoGen | ant-pr-1 |
| G418 | InvivoGen | ant-gn-1 |
| Blasticidin | InvivoGen | ant-bl-05 |
| DAPI | Vector Laboratories | H-1800-10 |
| APC Annexin V | Biolegend | 640920 |
| Fixable Viability Dye eFluor™ 780 | Thermo Fisher | 65-0865-14 |
| West Dura chemiluminescent substrates | Thermo Fisher | 34075 |
| Lipofectamine 2000 | Invitrogen | 11668027 |
| PMA | Sigma | P1585-1MG |
| Ionomycin | Sigma | I0634-1MG |
| GolgiStop™ protein transport inhibitor | BD Bioscience | 554724 |
| IPTG | Sigma | I6758 |
| Imidazole | Sigma | 1336500 |
| Critical Commercial Assays | ||
| RNAscope® Multiplex Fluorescent Reagent Kit v2 | ACD | 323100 |
| Quick-RNA MiniPrep kit | Zymo | R1054 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | 4368814 |
| Intracellular Fixation and Permeabilization Buffer Set | Thermo Fisher | 88-8824-00 |
| The EnzChek® Caspase-3 Assay Kit | Thermo Fisher | E13184 |
| GranToxiLux kit | OncoImmunin | N/A |
| Granzyme B Colorimetric Drug Discovery Kit | Enzo Life | BML-AK711 |
| Experimental Models: Cell Lines | ||
| B16-F10 | ATCC | CRL-6475 |
| Renca | ATCC | CRL-2947 |
| 4T1 | ATCC | CRL-2539 |
| LLC1 | ATCC | CRL-1642 |
| A375 | ATCC | CRL-1619 |
| A549 | ATCC | CCL-185 |
| SK-BR-3 | ATCC | HTB-30 |
| HEK293T | ATCC | CRL-11268 |
| Trichoplusia ni | Kerafast | ENH127-FP |
| Probes and oligonucleotides | ||
| Granzyme B probe | ACD | 490191 |
| Sb9 probe | ACD | 578121 |
| mouse GAPDH-F: AGCCACATCGCTCAGACAC | This paper | N/A |
| mouse GAPDH-R: GCCCAATACGACCAAATCC | This paper | N/A |
| mouse GrB-F: ATGAAGATCCTCCTGCTACTGC, | This paper | N/A |
| mouse-GrB-R: CCCACATATCGCCTCAGGCT | This paper | N/A |
| mouse Ki67-F: AGAGCCTTAGCAATAGCAACG | This paper | N/A |
| mouse-Ki67-R: GTCTCCCGCGATTCCTCTG | This paper | N/A |
| Recombinant DNA | ||
| PI-9 Double Nickase Plasmid (human) | Santa Cruz | sc-404486-NIC |
| granzyme B Double Nickase Plasmid (mouse) | Santa Cruz | sc-420745-NIC |
| serpinB9 expression plasmid in pcDNA3.1+/C-(K)DYK | GenScript | OMu03805D |
| LentiCRISPRv2-blast | Addgene | 98293 |
| pVSVg | Addgene | 8485 |
| psPAX2 | Addgene | 12260 |
| pLenti PGK GFP Puro | Addgene | 19070 |
| pFastBac1 | Thermo Fisher | 10712024 |
| Software and Algorithms | ||
| FlowJo 10 | FlowJo | https://www.flowjo.com/ |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/ |
| Celleste | Invitrogen | N/A |
| ImageJ | NIH | NCBI, 1.8.0_112 |
| Scrubber 2 | Biologic | http://www.biologic.com.au/ |
Highlights.
SerpinB9 protects cancer cells from their own granzyme B
SerpinB9 expression in CAFs, MDSCs, and TAMs can promote tumor growth
Deletion of serpinB9 in both the tumor and host suppresses tumor growth markedly
SerpinB9 inhibition can target cancer cells, CAFs, MDSCs, and TAMs simultaneously
ACKNOWLEDGMENTS
This work was supported in part by the National Institute of Allergy and Infectious Diseases and Office of the Director of the NIH (R01-AI132963 to L.D.S. and R24-OD018259 to L.D.S. and D.L.G.), as well as by a National Cancer Institute core grant (CA034196 to L.D.S.).
Footnotes
DECLARATION OF INTERESTS
The authors declare they have no competing interests.
REFERENCES
- Andersson A, Yang SC, Huang M, Zhu L, Kar UK, Batra RK, Elashoff D, Strieter RM, Dubinett SM, and Sharma S (2009). IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J. Immunol 182, 6951–6958. [DOI] [PubMed] [Google Scholar]
- Annand RR, Dahlen JR, Sprecher CA, De Dreu P, Foster DC, Mankovich JA, Talanian RV, Kisiel W, and Giegel DA (1999). Caspase-1 (interleukin-1beta-converting enzyme) is inhibited by the human serpin analogue proteinase inhibitor 9. Biochem. J 342, 655–665. [PMC free article] [PubMed] [Google Scholar]
- Azzi J, Skartsis N, Mounayar M, Magee CN, Batal I, Ting C, Moore R, Riella LV, Ohori S, Abdoli R, et al. (2013). Serine protease inhibitor 6 plays a critical role in protecting murine granzyme B-producing regulatory T cells. J. Immunol 191, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LE, Granot Z, Coker C, Iavarone A, Hambardzumyan D, Holland EC, Nam HS, and Benezra R (2012). Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell 21, 11–24. [DOI] [PubMed] [Google Scholar]
- Bird CH, Sutton VR, Sun J, Hirst CE, Novak A, Kumar S, Trapani JA, and Bird PI (1998). Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol 18, 6387–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bots M, and Medema JP (2008). Serpins in T cell immunity. J. Leukoc. Biol 84, 1238–1247. [DOI] [PubMed] [Google Scholar]
- Bots M, Offringa R, and Medema JP (2006). Does the serpin PI-9 protect tumor cells? Blood 107, 4974–4975, author reply 4975. [DOI] [PubMed] [Google Scholar]
- Csermely P, Agoston V, and Pongor S (2005). The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol. Sci 26, 178–182. [DOI] [PubMed] [Google Scholar]
- D’Eliseo D, Pisu P, Romano C, Tubaro A, De Nunzio C, Morrone S, Santoni A, Stoppacciaro A, and Velotti F (2010). Granzyme B is expressed in urothelial carcinoma and promotes cancer cell invasion. Int. J. Cancer 127, 1283–1294. [DOI] [PubMed] [Google Scholar]
- D’Eliseo D, Di Rocco G, Loria R, Soddu S, Santoni A, and Velotti F (2016). Epitelial-to-mesenchimal transition and invasion are upmodulated by tumor-expressed granzyme B and inhibited by docosahexaenoic acid in human colorectal cancer cells. J. Exp. Clin. Cancer Res 35, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Haddad N, Heathcote D, Moore R, Yang S, Azzi J, Mfarrej B, Atkinson M, Sayegh MH, Lee JS, Ashton-Rickardt PG, and Abdi R (2011a). Mesenchymal stem cells express serine protease inhibitor to evade the host immune response. Blood 117, 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Haddad N, Moore R, Heathcote D, Mounayar M, Azzi J, Mfarrej B, Batal I, Ting C, Atkinson M, Sayegh MH, et al. (2011b). The novel role of SERPINB9 in cytotoxic protection of human mesenchymal stem cells. J. Immunol 187, 2252–2260. [DOI] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, and Werb Z (2018). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Hirst CE, Buzza MS, Bird CH, Warren HS, Cameron PU, Zhang M, Ashton-Rickardt PG, and Bird PI (2003). The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol 170, 805–815. [DOI] [PubMed] [Google Scholar]
- Hu SX, Wang S, Wang JP, Mills GB, Zhou Y, and Xu HJ (2003). Expression of endogenous granzyme B in a subset of human primary breast carcinomas. Br. J. Cancer 89, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington JA, Read RJ, and Carrell RW (2000). Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926. [DOI] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kar UK, Srivastava MK, Andersson A, Baratelli F, Huang M, Kickhoefer VA, Dubinett SM, Rome LH, and Sharma S (2011). Novel CCL21-vault nanocapsule intratumoral delivery inhibits lung cancer growth. PLoS ONE 6, e18758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kather JN, Suarez-Carmona M, Charoentong P, Weis CA, Hirsch D, Bankhead P, Horning M, Ferber D, Kel I, Herpel E, et al. (2018). Topography of cancer-associated immune cells in human solid tumors. eLife 7, e36967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendal WS, Wang RY, Hsu TC, and Frost P (1987). Rate of generation of major karyotypic abnormalities in relationship to the metastatic potential of B16 murine melanoma. Cancer Res. 47, 3835–3841. [PubMed] [Google Scholar]
- Kim MT, and Harty JT (2014). Impact of Inflammatory Cytokines on Effector and Memory CD8+ T Cells. Front. Immunol 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Qian BZ, and Pollard JW (2015). Immune cell promotion of metastasis. Nat. Rev. Immunol 15, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontani K, Sawai S, Hanaoka J, Tezuka N, Inoue S, and Fujino S (2001). Involvement of granzyme B and perforin in suppressing nodal metastasis of cancer cells in breast and lung cancers. Eur. J. Surg. Oncol 27, 180–186. [DOI] [PubMed] [Google Scholar]
- Kumar V, Patel S, Tcyganov E, and Gabrilovich DI (2016). The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 37, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauricella M, Carlisi D, Giuliano M, Calvaruso G, Cernigliaro C, Vento R, and D’Anneo A (2016). The analysis of estrogen receptor-α positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor PI-9: Possible regulatory mechanisms. Int. J. Oncol 49, 352–360. [DOI] [PubMed] [Google Scholar]
- Li LT, Jiang G, Chen Q, and Zheng JN (2015). Ki67 is a promising molecular target in the diagnosis of cancer (review). Mol. Med. Rep 11, 1566–1572. [DOI] [PubMed] [Google Scholar]
- Lindau D, Gielen P, Kroesen M, Wesseling P, and Adema GJ (2013). The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA (2006). Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat. Rev. Drug Discov 5, 160–170. [DOI] [PubMed] [Google Scholar]
- Mangan MS, Kaiserman D, and Bird PI (2008). The role of serpins in vertebrate immunity. Tissue Antigens 72, 1–10. [DOI] [PubMed] [Google Scholar]
- Mangan MS, Bird CH, Kaiserman D, Matthews AY, Hitchen C, Steer DL, Thompson PE, and Bird PI (2016). A Novel Serpin Regulatory Mechanism: SerpinB9 IS REVERSIBLY INHIBITED BY VICINAL DISULFIDE BOND FORMATION IN THE REACTIVE CENTER LOOP. J. Biol. Chem 291, 3626–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, and Morrison SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP, de Jong J, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, Franken KL, Hahne M, Albar JP, Melief CJ, and Offringa R (2001a). Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 98, 11515–11520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP, Schuurhuis DH, Rea D, van Tongeren J, de Jong J, Bres SA, Laban S, Toes RE, Toebes M, Schumacher TN, et al. (2001b). Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: differential modulation by T helper type 1 and type 2 cells. J. Exp. Med 194, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestres J, and Gregori-Puigjané E (2009). Conciliating binding efficiency and polypharmacology. Trends Pharmacol. Sci 30, 470–474. [DOI] [PubMed] [Google Scholar]
- Nilendu P, Sarode SC, Jahagirdar D, Tandon I, Patil S, Sarode GS, Pal JK, and Sharma NK (2018). Mutual concessions and compromises between stromal cells and cancer cells: driving tumor development and drug resistance. Cell Oncol. (Dordr.) 41, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JD, Zhang J, Wu Z, Thew KD, Rowe KJ, Bacani JT, and Ingham RJ (2014). Expression of granzyme B sensitizes ALK+ ALCL tumour cells to apoptosis-inducing drugs. Mol. Cancer 13, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoples GE (2019). Improving the outcomes of checkpoint inhibitors in breast cancer. Lancet Oncol. 20, 316–318. [DOI] [PubMed] [Google Scholar]
- Phillips T, Opferman JT, Shah R, Liu N, Froelich CJ, and Ashton-Rick-ardt PG (2004). A role for the granzyme B inhibitor serine protease inhibitor 6 in CD8+ memory cell homeostasis. J. Immunol 173, 3801–3809. [DOI] [PubMed] [Google Scholar]
- Pinkoski MJ, Waterhouse NJ, Heibein JA, Wolf BB, Kuwana T, Goldstein JC, Newmeyer DD, Bleackley RC, and Green DR (2001). Gran-zyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem 276, 12060–12067. [DOI] [PubMed] [Google Scholar]
- Postow MA, Sidlow R, and Hellmann MD (2018). Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med 378, 158–168. [DOI] [PubMed] [Google Scholar]
- Prakash MD, Munoz MA, Jain R, Tong PL, Koskinen A, Regner M, Kleifeld O, Ho B, Olson M, Turner SJ, et al. (2014). Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity 41, 960–972. [DOI] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzitelli A, Meuter S, Vega Ramos J, Bird CH, Mintern JD, Mangan MS, Villadangos J, and Bird PI (2012). Serpinb9 (Spi6)-deficient mice are impaired in dendritic cell-mediated antigen cross-presentation. Immunol. Cell Biol 90, 841–851. [DOI] [PubMed] [Google Scholar]
- Russell JH, and Ley TJ (2002). Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol 20, 323–370. [DOI] [PubMed] [Google Scholar]
- Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, Irving JA, Lomas DA, Luke CJ, Moyer RW, et al. (2001). The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem 276, 33293–33296. [DOI] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, et al. (2020). A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits AH, Ziebell F, Joberty G, Zinn N, Mueller WF, Clauder-Münster S, Eberhard D, Fälth Savitski M, Grandi P, Jakob P, et al. (2019). Biological plasticity rescues target activity in CRISPR knock outs. Nat. Methods 16, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Sun J, Bird CH, Sutton V, McDonald L, Coughlin PB, De Jong TA, Trapani JA, and Bird PI (1996). A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem 271, 27802–27809. [DOI] [PubMed] [Google Scholar]
- Sun J, Ooms L, Bird CH, Sutton VR, Trapani JA, and Bird PI (1997). A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9). J. Biol. Chem 272, 15434–15441. [DOI] [PubMed] [Google Scholar]
- Tamang DL, Redelman D, Alves BN, Vollger L, Bethley C, and Hudig D (2006). Induction of granzyme B and T cell cytotoxic capacity by IL-2 or IL-15 without antigens: multiclonal responses that are extremely lytic if triggered and short-lived after cytokine withdrawal. Cytokine 36, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez-Gabriel M, Ory B, Lamoureux F, Heymann MF, and Heymann D (2016). Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. Int. J. Mol. Sci 17, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo JA, Sweis RF, Bao R, and Luke JJ (2018). T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immuno-therapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res 6, 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, De Sanctis F, Mandruzzato S, and Bronte V (2015). Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Invest 125, 3365–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, Lin Q, Liu Y, Li Z, and Chen R (2018). Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 9, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JL, Sukhova GK, Foster D, Kisiel W, Libby P, and Schönbeck U (2000). The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1beta-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med 191, 1535–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Park SM, Wang Y, Shah R, Liu N, Murmann AE, Wang CR, Peter ME, and Ashton-Rickardt PG (2006). Serine protease inhibitor 6 protects cytotoxic T cells from self-inflicted injury by ensuring the integrity of cytotoxic granules. Immunity 24, 451–461. [DOI] [PubMed] [Google Scholar]
- Zhu H, Guo ZK, Jiang XX, Li H, Wang XY, Yao HY, Zhang Y, and Mao N (2010). A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat. Protoc 5, 550–560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and are available from the corresponding author upon request.