

SUMMARY
Generating mammalian cells with desired mitochondrial DNA (mtDNA) sequences is enabling for studies of mitochondria, disease modeling, and potential regenerative therapies. MitoPunch, a high-throughput mitochondrial transfer device, produces cells with specific mtDNA-nuclear DNA (nDNA) combinations by transferring isolated mitochondria from mouse or human cells into primary or immortal mtDNA-deficient (ρ0) cells. Stable isolated mitochondrial recipient (SIMR) cells isolated in restrictive media permanently retain donor mtDNA and reacquire respiration. However, SIMR fibroblasts maintain a ρ0-like cell metabolome and transcriptome despite growth in restrictive media. We reprogrammed non-immortal SIMR fibroblasts into induced pluripotent stem cells (iPSCs) with subsequent differentiation into diverse functional cell types, including mesenchymal stem cells (MSCs), adipocytes, osteoblasts, and chondrocytes. Remarkably, after reprogramming and differentiation, SIMR fibroblasts molecularly and phenotypically resemble unmanipulated control fibroblasts carried through the same protocol. Thus, our MitoPunch “pipeline” enables the production of SIMR cells with unique mtDNA-nDNA combinations for additional studies and applications in multiple cell types.
Graphical Abstract

In Brief
Patananan and colleagues demonstrate a pipeline for transferring isolated mitochondria into mtDNA-deficient recipient cells. mtDNA-depleted fibroblasts permanently retain acquired non-native mtDNA through cell fate transitions. Initially, mitochondrial recipients show mtDNA-deficient cell transcriptome and metabolome profiles, with improvement to control profiles by reprogramming to pluripotency and subsequent differentiation.
INTRODUCTION
Mammalian mitochondria are cellular power plants with additional roles in apoptosis, reactive oxygen species (ROS) and Fe-S cluster generation, Ca2+ regulation, and metabolite production (Patananan et al., 2018). Each mitochondrion contains >1,100 nucleus-encoded and imported proteins (Calvo et al., 2016) with numerous copies of a circular ~16.5-kilobase pair (kbp) mitochondrial genome (mtDNA) encoding 13 proteins required for electron transport chain (ETC) activity and respiration. As many as 1:5,000 people have mtDNA mutations that impair high-energy-demand tissues and contribute to debilitating diseases, including cancer, diabetes, and metabolic syndromes (Schaefer et al., 2004). In addition, cells may contain a mixture of different mtDNA sequences, a situation termed heteroplasmy, with up to 1 in 8 asymptomatic individuals carrying an unsuspected pathogenic mtDNA mutation (Elliott et al., 2008; Rebolledo-Jaramillo et al., 2014). Thus, an ability to controllably manipulate mtDNA sequences could enable studies of mitochondria and potentially develop disease models or therapies for mtDNA disorders.
In human reproduction, several types of mitochondrial replacement strategies were developed to exchange pathogenic mtDNA in a zygote with non-detrimental mtDNA from a healthy donor oocyte. These approaches have potential for preventing transmission of mtDNA disorders from carrier mothers to their children (Wolf et al., 2015; Wolf et al., 2019). However, in vitro methods to change mtDNA sequences within somatic cells and tissues remain limited (Patananan et al., 2016). Cell fusions that produce “cybrids” permanently retain donor mitochondria (Wong et al., 2017), although fusion partners are typically transformed cells that cannot be reprogrammed. Also, endonucleases imported into mitochondria can shift heteroplasmy ratios to alter mitochondria and cell functions by targeting specific sequences for destruction. However, these endonucleases are laborious to produce, are limited to certain mtDNA sequences, are inefficient, and do not yield homoplasmy (Campbell et al., 2018; Yahata et al., 2017; Yang et al., 2018). Of note, a recent and exciting development using a bacterial cytidine deaminase, DddA, to edit mtDNA single-base sequences is tempered by low efficiency and an undesirable off-target rate (Mok et al., 2020).
Several methods transfer isolated mitochondria into mtDNA-deficient cells, known as ρ0 (rho null) cells, to restore respiration (Kim et al., 2018; Nzigou Mombo et al., 2017). In addition, some studies reported endoyctosis of mitochondria by mammalian cells (Clark and Shay, 1982; Kesner et al., 2016). However, these studies were not concerned with rescuing ρ0 cells and generating stable isolated mitochondrial recipient (SIMR) clones that permanently retain donor mtDNA (Kesner et al., 2016; Kitani et al., 2014; Sun et al., 2019). A recent study did produce a limited number of SIMR clones by coincubating high concentrations of isolated HEK293T donor mitochondria with ρ0 osteosarcoma cells (Patel et al., 2017). We (Dawson et al., 2020) and others (Ali Pour et al., 2020) have recently reported similar findings in which cells are capable of endocytosing exogenous mitochondria and even altering metabolic functions for a limited period of time (~1 week), but these exogenous mtDNAs are lost over time. To address this problem, we previously developed a photothermal nanoblade to stably transfer small quantities of isolated mitochondria into ρ0 osteosarcoma cells (Wu et al., 2016). Unfortunately, the nanoblade is laborious and low throughput, and two of three SIMR clones reported did not reset the ρ0 cell metabolome. A technique that generates many non-transformed stable clones is desirable to examine novel mtDNA-nuclear DNA (nDNA) combinations through reprogramming to pluripotency and differentiation into multiple cell types.
Here, we describe a simple-to-use mitochondrial transfer technique called “MitoPunch” to rapidly generate numerous non-transformed SIMR clones. We apply MitoPunch to implement a pipeline that demonstrates donor mtDNA functions in recipient host primary cells at different cell fates. Our study establishes this resource pipeline to generate primary SIMR cells using non-immortalized materials. We also measure the status of the metabolome, transcriptome, and biophysical properties of SIMR cells with defined mtDNA-nDNA combinations to guide future studies generating somatic cells with desired mtDNA-nDNA combinations.
RESULTS
MitoPunch Generates SIMR Cells with a Range of Cell Types and mtDNAs
MitoPunch is a massively parallel, pressure-driven, large cargo transfer platform based on prior photothermal nanoblade and biophotonic laser-assisted cell surgery tool (BLAST) technologies (Sercel et al., 2020; Wu et al., 2016; Wu et al., 2015). Mito-Punch uses a mechanical plunger to physically deform a pliable polydimethylsiloxane (PDMS) reservoir containing isolated mitochondria suspended in phosphate-buffered saline (1× PBS [pH 7.4]) (Figure 1A). Plunger activation propels the suspended cargo within the PDMS delivery chamber through a porous membrane containing numerous 3-μm-diameter holes on which a confluent layer of adherent cells is grown. This action directly forces isolated mitochondria into the cytosol of recipient cells.
Figure 1. MitoPunch Is a Versatile Mitochondrial Transfer Technology.

(A) Schematic representation of the MitoPunch mitochondrial transfer platform.
(B) Images of crystal-violet-stained SIMR colonies from coincubation or MitoPunch delivery of either 1× PBS (pH 7.4) (sham control) or isolated HEK293T cell mitochondria into 143BTK− ρ0 osteosarcoma cells after selection in uridine-depleted medium. Data are the means ± SD of three technical replicates. (C) OCR measurements for ~1.5 × 104 143BTK−, 143BTK− ρ0, WT cybrid, MELAS cybrid, 143BTK− ρ0+MELAS SIMR, and 143BTK− ρ0+WT SIMR cells by Seahorse XF96 Extracellular Flux Analyzer. Values were calculated by standard procedures (see STAR Methods). Data are the means ± SD of four technical replicates.
(D) OCR measurements for ~1.5 × 104 L929 ρ0 and L929 ρ0 SIMR cells generated with mitochondria from C57BL/6 mouse tissues. Data are the means ± SD of 12 technical replicates (L929 ρ0 cells had four technical replicates).
(E) OCR measurements for ~1.5 × 104 L929 ρ0 and L929 ρ0 SIMR cells generated by transferring isolated mitochondria alone or in combinations from mouse cybrids with non-overlapping mtDNA mutations (Mito 1, Mito 2, and Mito 3). Data are the means ± SD of eight technical replicates.
Statistical significance for (B)–(E) by unpaired, two-tailed Student’s t test. *p ≨ 0.05; **p ≨ 0.01; ***p ≨ 0.001; ****p ≨ 0.0001. See also Figure S1.
To demonstrate MitoPunch generation of SIMR cells, we transferred isolated mitochondria from ~1.5 × 107 HEK293T cells into ~2 × 105 143BTK− ρ0 osteosarcoma cells. Post-transfer, we select for and isolate SIMR colony clones with permanently retained donor mtDNA using uridine-deficient media. This selection is enabling because respiration-defective ρ0 cells have inactive dihydroorate dehydrogenase and depend on exogenous uridine or restored respiration for pyrimidine biosynthesis (Grégoire et al., 1984). Compared to the coincubation of the same amount of isolated mitochondria with cells (Clark and Shay, 1982; Kesner et al., 2016), only 143BTK− ρ0 cells with HEK293T mitochondria from MitoPunch transfer (143BTK− ρ0+HEK293T) permanently retained donor mtDNA and survived uridine-deficient media selection (Figure 1B). In a representative set of mitochondrial transfer experiments, MitoPunch generated ~75 independent crystal-violet-stained SIMR clones in comparison to no clones obtained by coincubation (Figure 1B).
We next examined whether MitoPunch could generate SIMR clones with defined mtDNA-nDNA pairs that transfer features of mitochondrial disease. We isolated mitochondria from cybrid cells containing either an A3243G mtDNA substitution commonly associated with mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) or wild-type (WT), non-mutant mtDNA from the same individual (Picard et al., 2014). The A3243G point mutation is in the tRNALEU gene and results in altered production and assembly of ETC complexes with impaired oxidative phosphorylation (Chomyn et al., 1992; Sasarman et al., 2008). Following MitoPunch into 143BTK− ρ0 recipients and 2 weeks of selection, two of several dozen independent SIMR clones that permanently retained MELAS (143BTK− ρ0+MELAS) or WT (143BTK− ρ0+WT) mtDNA were tested for oxygen consumption rate (OCR) using the Seahorse Extracellular Flux Analyzer. Results showed 143BTK−ρ0+MELAS clones had significantly impaired basal respiration, maximal respiration, and spare respiratory capacity compared to patient-matched 143BTK− ρ0+WT and native 143BTK− control cells (Figure 1C), indicating stable mtDNA transfer of the primary metabolic deficit of the MELAS phenotype.
We then expanded mitochondrial donor and recipient cell pairings beyond these initial studies to demonstrate the versatility of MitoPunch. As examples, mitochondria were isolated from harvested C57BL/6 mouse tissues and MitoPunch transferred into C3H/An-derived L929 ρ0 immortalized fibroblasts. Two weeks of selection yielded dozens of SIMR clones from each mitochondrial source. SIMR clones generated with high-energy-demand heart, lung, or muscle-derived mitochondria showed the most robust respiratory profiles, in contrast to SIMR clones that received low-energy-demand spleen- or kidney-derived mitochondria (Figure 1D). We also evaluated MitoPunch delivery of heteroplasmic mtDNA mixtures into cells. Mitochondria isolated from mouse cybrid lines containing mtDNA mutations in the cytochrome B (mt-Cytb), NADH dehydrogenase subunit 4 (mt-nd4), and NADH dehydrogenase subunit 6 (mt-nd6) genes were MitoPunch transferred individually or in 1:1 mixtures by protein content into L929 ρ0 fibroblasts. SIMR cells with a single source of mutant mtDNA continued to show severe respiratory impairments (Figure 1E). In contrast, SIMR cells with a mixture of non-overlapping mutant mtDNAs showed markedly improved respiratory profiles, strongly suggesting that both mtDNAs were stably maintained (Figure 1E). Thus, MitoPunch and selection is a versatile approach for generating human or mouse SIMR cells with desired mtDNA-nDNA pairs. Co-transfer of multiple mtDNA types into the same recipient cell also provides a simple method to examine complementation for mutant mtDNA mixtures.
MitoPunch Generates Non-Transformed, Non-Malignant SIMR Cells
To obtain SIMR cells with mtDNA-nDNA combinations using non-immortalized recipient cells, we established a human fibroblast mitochondrial recipient pipeline. Hayflick-limited BJ foreskin (BJ) fibroblasts, neonatal dermal fibroblasts (NDFs), and adult dermal fibroblasts (ADFs) were treated for 3 weeks with FDA-approved 2′,3′-dideoxycytidine (ddC) (Nelson et al., 1997) to deplete endogenous mtDNA. Primary ρ0 human fibroblasts had undetectable mtDNA (Figures S1A and S1B) and cellular respiration (Figures S1C and S1D) by qPCR and Seahorse assay, respectively. Because ddC could cause nDNA alterations, we examined BJ ρ0 fibroblasts by whole-genome sequencing and identified only a few non-synonymous mutations at 0.6 mutations per megabase, on average, with no chromosomal breaks and no changes in DNA copy number (Table S1).
Subsequently, mitochondria isolated from a human peripheral blood mononuclear cell lot (PBMC1) were transferred into fresh ρ0 fibroblasts, followed by an empirical and reproducible selection protocol with uridine-deficient galactose medium. From 5–10 BJ fibroblasts, NDFs, or ADFs, ρ0+PBMC1 SIMR clones were isolated that showed the correct mtDNA-nDNA sequence pairs and human leukocyte antigen (HLA) recipient cell haplotypes (Figures 2A–2C). Primary, non-immortal SIMR clones were also obtained from independent PBMC2 and HEK293T cell mitochondrial transfers. We observed variable efficiencies for HEK293T cell and the PBMC2 mitochondrial transfers, whereas ADFs ρ0+PBMC2 did not yield clones (Figure S1E). Analysis of the bulk culture representing 23 BJ ρ0+HEK293T SIMR clones confirmed the correct mtDNA-nDNA pairing and HLA haplotype (Figures S1F and S1G).
Figure 2. Generation of SIMR Fibroblasts.

(A) Selection workflow (in days) for generating SIMR cells from ρ0 primary human fibroblasts and SIMR clone generation efficiency data. Mitochondria from ~3 × 107 peripheral blood mononuclear cells (PBMC1) were MitoPunch transferred into BJ ρ0, NDF ρ0, or ADF ρ0 recipient fibroblasts. After selection, SIMR colonies were stained with crystal violet and quantified. Clone counts from a single representative mitochondrial transfer into ~1 × 105 recipient ρ0 fibroblasts are indicated.
(B) D-loop hypervariable region mtDNA sequences from native BJ, PBMC1, and BJ ρ0+PBMC1 SIMR fibroblasts. Arrows denote single-nucleotide polymorphisms.
(C) Major histocompatibility complex (MHC) class I HLA A, B, and C locus genotyping using OptiType v.1.3.1 for native BJ, BJ ρ0, and BJ ρ0+PBMC1 SIMR fibroblasts.
(D) OCR measurements for ~1.5 3 104 native BJ, BJ ρ0, and BJ ρ0+PBMC1 SIMR fibroblasts by the Seahorse XF96 Extracellular Flux Analyzer. Values were calculated by standard procedures (see STAR Methods). Data are the means ± SD of four technical replicates. Statistical significance by unpaired, two-tailed Student’s t test. *p ≨ 0.05; ***p ≨ 0.001
(E) Representative images of native BJ, BJ ρ0, and BJ ρ0+PBMC1 SIMR fibroblasts immunostained for double-stranded DNA (dsDNA) (green) and TOM20 (red) with colocalization indicated (yellow). Images (1003) were acquired on a Leica SP8 confocal microscope. Scale bars, 15 μm.
We examined the respiratory function of BJ ρ0+PBMC1 and BJ ρ0+HEK293T SIMR fibroblasts by Seahorse assay, which showed statistically improved basal and maximal respiration and spare respiratory capacity for both SIMR cell types compared to BJ ρ0 fibroblasts, albeit remaining lower than levels for control BJ fibroblasts (Figures 2D and S1H). Immunofluorescence (IF) microscopy showed BJ ρ0 fibroblasts with a fragmented mitochondrial network morphology lacking mtDNA-containing nucleoids, as observed previously for ρ0 cells (Kukat et al., 2008) (Figure 2E). In contrast, native BJ fibroblasts showed a reticular mitochondrial network with dozens of nucleoids per cell (Figure 2E). By IF nucleoid speckle numbers, both BJ ρ0+PBMC1 and BJ ρ0+HEK293T SIMR cells appeared to restore mtDNA content to levels equivalent to or exceeding that of native BJ fibroblasts (Figures 2E and S1I). SIMR cell mitochondria showed a reticular mitochondrial network morphology similar to that of native BJ fibroblasts, although with denser and more swollen mitochondria (Figures 2E and S1I). Despite SIMR fibroblasts permanently retaining donor mtDNA, OCR and IF suggest that assimilation of transferred mtDNA results in cells with features in between those of BJ ρ0 and native BJ fibroblasts.
SIMR Fibroblasts Are Reprogrammable
We reprogrammed BJ ρ0+PBMC1 and BJ ρ0+HEK293T SIMR fibroblasts along with native BJ fibroblasts using OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28 RNAs and quantified for TRA-1–60+ staining clones. In two independent experiments, native BJ fibroblasts yielded an average of 136 reprogrammed TRA-1–60+ clones (0.068% efficiency), compared to 21 (0.011%) and three (0.0015%) clones for BJ ρ0+PBMC1 and BJ ρ0+HEK293T cells, respectively (Figures 3A and S1J). Three unique reprogrammed clones of BJ ρ0+PBMC1-iPSCs (1, 2, and 11) and BJ ρ0+HEK293T-iPSCs (1, 2, and 4) were tested for pluripotency biomarkers and stained positive for OCT3/4 and SOX2 transcription factors by flow cytometry, as did BJ-induced pluripotency stem cell (iPSC) control, but not native BJ fibroblasts, as expected (Figures 3B and S1K). Conversely, the differentiated cell biomarker CD44 (Quintanilla et al., 2014) was negative in all reprogrammed BJ ρ0+PBMC1-iPSC, BJ ρ0+HEK293T-iPSC, and control BJ-iPSC clones and immunostained only the native BJ fibroblasts (Figures 3B and S1K). BJ-iPSC and all SIMR-iPSC reprogrammed clones were also SSEA-4+ (Abujarour et al., 2013) and OCT4+ by IF (Figures 3C and S1L). Seahorse assays of BJ ρ0+PBMC1-iPSC and BJ ρ0+HEK293T-iPSC clones showed minimal or no statistical differences in basal respiration, maximal respiration, and spare respiratory capacity compared to the native BJ-iPSC control (Figures 3D and S1M). Thus, SIMR fibroblast reprogramming generated iPSCs with donor mtDNA.
Figure 3. SIMR Fibroblasts Can Be Reprogrammed.

(A) Native BJ and SIMR fibroblasts reprogrammed to iPSCs with TRA-1–60+ clones counted by microscopy. Data are the means of biological duplicates. Data for BJ fibroblast control are the same data as in Figure S3A.
(B) Flow cytometry of pluripotency biomarkers SOX2 and OCT3/4, and fibroblast biomarker CD44. Immunostained samples are shown in color with isotype negative controls in gray. Representative data for native BJ fibroblasts and BJ-iPSCs, and for BJ ρ0+PBMC1-iPSC cells. Data for the native
BJ fibroblasts and BJ-iPSCs shown here are the same as in Figure S3B.
(C) Representative phase contrast and IF microscopy images of native BJ fibroblast (negative control), BJ-iPSC (positive control), and three BJ ρ0+PBMC1-iPSC clones immunostained for pluripotency biomarkers SSEA-4 and OCT4. Scale bars, 100 μm.
(D) OCR measurements for ~1.5 × 104 native BJ-iPSCs and BJ ρ0+PBMC1-iPSC clones 1, 2, and 11. Data for BJ-iPSC control are the same as in Figure S3D. Data are the means ± SD of four technical replicates. Statistical significance by unpaired, two-tailed Student’s t test. *p ≨ 0.05; **p ≨ 0.01
Other studies have shown iPSC reprogramming of fibroblasts from individuals with pathogenic mtDNA mutations (Cherry et al., 2013; Folmes et al., 2013; Hämäläinen et al., 2013; Kang et al., 2016; Kodaira et al., 2015; Ma et al., 2015; Matsubara et al., 2018; Pek et al., 2019; Perales-Clemente et al., 2016; Russell et al., 2018; Yang et al., 2018), but this has not been attempted for SIMR fibroblasts with donated, non-native mutant mtDNA. Therefore, mitochondria containing an A3243G MELAS mtDNA mutation or WT mtDNA were isolated, followed by MitoPunch transfer into BJ ρ0 fibroblasts and selection. BJ ρ0+MELAS SIMR fibroblasts showed impaired proliferation during reprogramming and did not yield iPSCs (data not shown). Therefore, we switched to NDF ρ0 recipient fibroblasts and generated SIMR fibroblasts using isolated MELAS (NDF ρ0+MELAS), WT (NDF ρ0+WT), or NDF (NDF ρ0+NDF) mitochondria. Seahorse assays showed that NDF ρ0+MELAS fibroblasts had a significant reduction in basal respiration, maximal respiration, and spare respiratory capacity compared to native NDF, NDF ρ0+NDF, and NDF ρ0+WT fibroblasts (Figure S1N). Restriction fragment length polymorphism (RFLP) PCR analyses confirmed the generation of homoplasmic NDF ρ0+MELAS SIMR fibroblasts (Figure S1O). We also generated ~25%–50% heteroplasmic NDF ρ0+MELAS/WT fibroblasts, which was verified by RFLP analyses (Figure S1O). Homoplasmic NDF ρ0+MELAS fibroblasts underwent RNA-based reprogramming as described earlier, but all developing iPSC clones spontaneously differentiated (Figure S1P). Reprogramming of NDF ρ0+MELAS/WT heteroplasmic fibroblasts (Figures S1O) yielded 20 iPSC clones, but all clones retained only WT mtDNA by RFLP analysis (Figures S1P and S1Q). To examine whether the reprogramming method influenced mutant mtDNA SIMR-iPSC generation, NDF ρ0+MELAS fibroblasts underwent integrating DNA, lentiviral, and Sendai virus reprogramming strategies. In all cases, NDF ρ0+MELAS cells spontaneously differentiated despite early signs of reprogramming (Table S2). In addition, no NDF ρ0+MELAS-iPSCs were obtained when reprogramming was performed with additional uridine supplementation, antioxidant N-acetylcysteine, a Rho-associated protein kinase (ROCK) inhibitor, or low oxygen tension (data not shown). Similar results were also obtained with all four reprogramming strategies for NDF ρ0 SIMR fibroblasts containing additional mtDNA mutations including a cytochrome B deletion, a Kearns-Sayre common deletion, and A8344G or T8993G mtDNA substitutions (Table S2). Thus, SIMR fibroblasts readily maintain a large variety of mtDNA sequences, in contrast to SIMR-iPSCs, which can be generated only with non-detrimental mtDNA sequences. Further biochemical investigations are needed to determine how mtDNA sequences dictate SIMR reprogramming, whereas native mutant mtDNA fibroblasts can be reprogrammed (Hämäläinen et al., 2013; Ma et al., 2015; Pek et al., 2019).
SIMR-iPSCs Produce Functional, Differentiated Cell Types
We next determined whether SIMR-iPSCs with isogenic nuclei and non-native donor mtDNAs could differentiate. We chose to examine defined medium differentiation of mesenchymal stem cells (MSCs) because of their relevance to potential therapies and current use in over 850 clinical trials (Hsu et al., 2016). A BJ-iPSC control, BJ ρ0+PBMC1-iPSCs, and BJ ρ0+HEK293T-iPSCs were differentiated into MSCs and validated with an antibody panel against surface biomarkers established by the International Society for Cellular Therapy (ISCT) (Dominici et al., 2006). Flow cytometry verified that the BJ-MSC control, BJ ρ0+PBMC1-MSC clones, and BJ ρ0+HEK293T-MSC clones were positive for MSC biomarkers CD73, CD90, and CD105, and negative for a cocktail of non-MSC biomarkers, including CD11b, CD19, CD34, CD45, and HLA-DR (Figures 4A and S1R). BJ-MSCs and all SIMR-MSC clones from both mtDNA donors adhered to plastic, consistent with ISCT criteria for MSCs (Figures 4B and S1S).
Figure 4. SIMR iPSCs Produce MSCs with Trilineage Differentiation Potential.
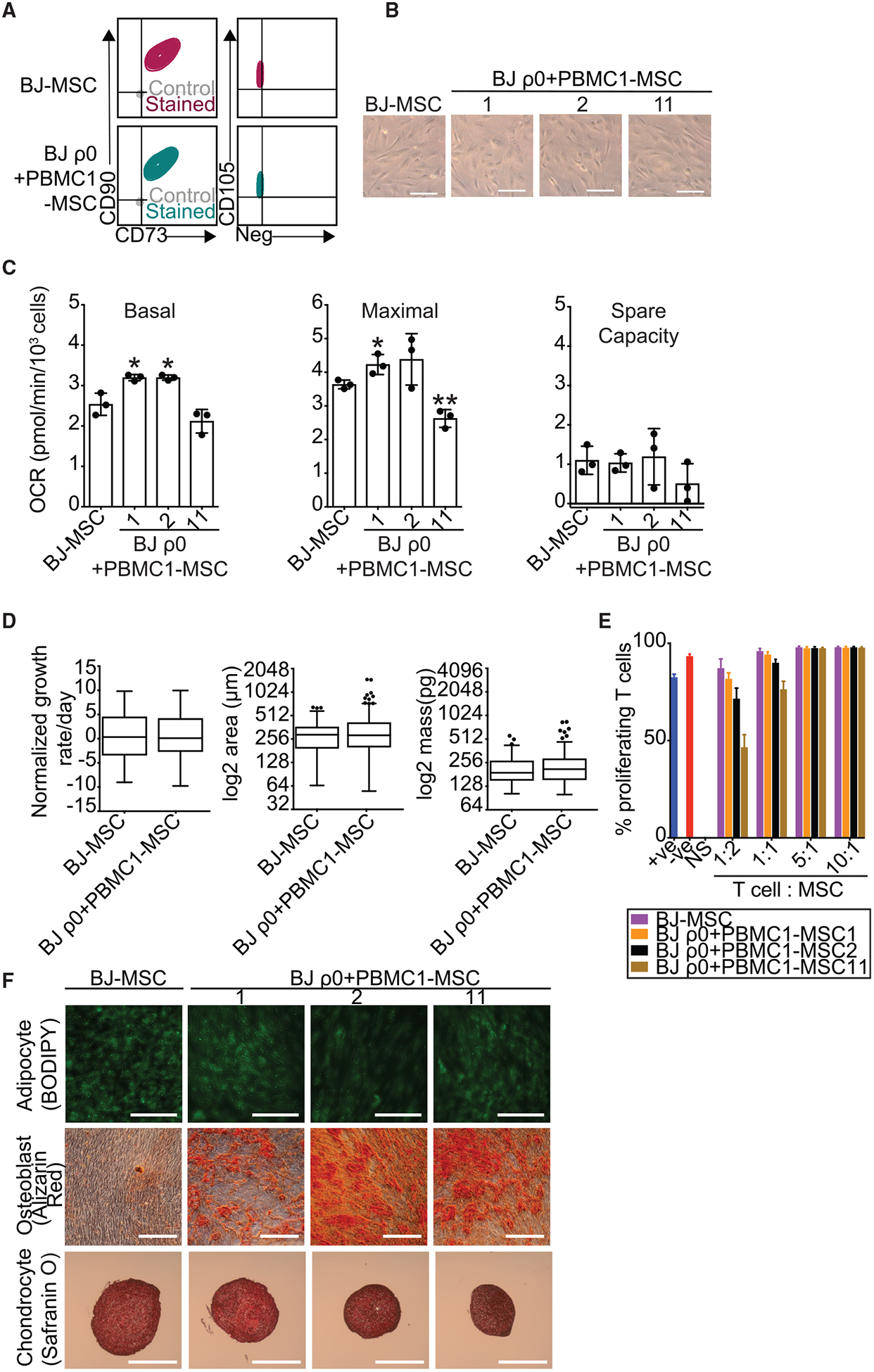
(A) Flow cytometry of MSC biomarkers CD73, CD90, and CD105, and a cocktail of negative MSC biomarkers. Immunostained samples are indicated in color, with isotype negative controls in gray. Data for BJ-MSC control are the same data as in Figure S5A. Representative clones for native BJ-MSCs and BJ ρ0+PBMC1-MSCs are indicated.
(B) Bright-field microscopy showing unmanipulated BJ-MSC and BJ ρ0+PBMC1-MSC clones 1, 2, and 11 adhering to plastic at 203 magnification (scale bars, 100 mm). Data for BJ-MSC control are the same data as in Figure S5B.
(C) OCR measurements for ~1.5 × 104 native BJ-MSCs and BJ ρ0+PBMC1-MSC clones 1, 2, and 11. Data for BJ-MSC control are the same as in Figure S5C. Data are the means ± SD of three technical replicates. Statistical significance by unpaired, two-tailed Student’s t test. *p ≨ 0.05; **p ≨ 0.01.
(D) Quantitative phase microscopy of native BJ-MSCs and a 1:1:1 mix of BJ ρ0+PBMC1-MSC clones 1, 2, and 11. Data for BJ-MSC control are the same as in Figure S5D. Shown are box-and-whisker Tukey plots with outliers identified. Data were averaged from 77 and 172 cells for native BJ-MSCs and BJ ρ0+PBMC1- MSCs, respectively. Statistical significance by Welch’s t test.
(E) T cells were added into native BJ-MSC or BJ ρ0+PBMC1-MSC clone 1, 2, or 11 cultures at 1:2, 1:1, 5:1, and 10:1 T cell:MSC ratios. After 5 days of co-culture, T cell proliferation was measured using a CFSE dye dilution assay by flow cytometry. The data labeled “NS” (no stimulus) denote T cells without CD3/CD28 bead activation. The data labeled “-ve” (negative) denote no addition of MSCs to stimulated T cells. The data labeled “+ve” (positive) denote a 1:1 addition of myeloid-derived suppressor cells to T cells. Data are the means ± SD of three technical replicates.
(F) Trilineage differentiation of native BJ-MSCs and BJ ρ0+PBMC1-MSC clones 1, 2, and 11. Representative sections were fixed and stained with 1 μM Bodipy 493/503, 1% alizarin red S, and 0.1% Safranin O, respectively. Shown are adipocytes (first row, 20×; scale bars, 100 μm), osteocytes (second row, 20×; scale bars, 200 μm), and chondrocytes (fourth row, 5×; scale bars, 500 μm).
See also Figure S1.
Seahorse assays of BJ-MSC control, BJ ρ0+PBMC1-MSC clones, and BJ ρ0+HEK293T-MSC clones revealed no or mild differences in basal respiration, maximal respiration, and spare respiratory capacity, indicating that respiratory changes are mutable for ρ0 fibroblasts after MitoPunch with reprogramming and differentiation (Figures 4C and S1T). Quantitative phase microscopy (QPM) was used to examine key cellular biophysical properties in SIMR MSCs and detected minimal to no differences in cell growth rate, area, and biomass among the BJ-MSC control, BJ ρ0+PBMC1-MSC clones, and BJ ρ0+HEK293T-MSC clones (Figures 4D and S1U). The function of SIMR-MSC clones and the BJ-MSC control was compared by co-culture with human PBMC-isolated T cells in a standard immunosuppression assay, which measures MSC clinical immunomodulatory performance (Djouad et al., 2003; Ghannam et al., 2010). All BJ ρ0+PBMC1-MSC and BJ ρ0+HEK293T-MSC clones repressed T cell proliferation (Figures 4E and S1V). BJ ρ0+PBMC1-MSC clone 11 showed the greatest immunosuppression and reduction in T cell proliferation, whereas no large differences were detected between the remaining SIMR-MSC clones and the BJ-MSC control. Finally, we performed directed trilineage differentiation of SIMR-MSCs into adipocytes, osteoblasts, and chondrocytes to demonstrate the clinical potential of MitoPunch-engineered lines. The BJ-MSC control, BJ ρ0+PBMC1-MSCs, and BJ ρ0+HEK293T-MSCs all formed these three MSC-differentiated lineages (Figures 4F and S1W). Adipocytes and chondrocytes were phenotypically similar between the BJ control and SIMR clones, whereas SIMR osteoblasts tended to qualitatively produce more calcium deposits. Thus, our mitochondrial transfer strategy enables the generation of iPSCs, MSCs, and further differentiated cell types from ρ0 fibroblasts by stable incorporation of specific, non-detrimental, and non-native donor mtDNAs.
SIMR Cell Metabolism and RNA Transcript Changes with Fate Transitions
We used ultra-high-performance liquid chromatography-mass spectrometry (UPHLC-MS) to quantify 154 steady-state metabolites in native BJ, BJ ρ0, BJ ρ0+PBMC1 clones, and BJ ρ0+HEK293T clones at fibroblast, iPSC, and MSC fates. Hierarchical clustering showed distinct, grouped profiles for fibroblasts, iPSCs, and MSCs independent of mitochondrial transfer status (Figures S2A and S2B; Table S3). Principal component analysis (PCA) of metabolite data also showed three main clusters representing fibroblast, iPSC, and MSC fates but no clear differences between SIMR and native control cells within each fate (Figures S2C and S2D). Metabolite set variation analysis (MSVA) and Euclidean distance analysis of the BJ ρ0+PBMC1-iPSC and BJ ρ0+PBMC1-MSC clones showed similar metabolite pathway profiles to themselves and to their respective BJ-iPSC and BJ-MSC controls (Figures S2A, S2E, and S2F). In contrast, BJ ρ0+HEK293T-iPSC clones 1 and 2 clustered separately from clone 4 and the BJ-iPSC control for several metabolic pathways, particularly purine, pyrimidine, glutathione, and ethanol metabolism (Figures S2B, S2E, and S2F). This separation in BJ ρ0+HEK293T-iPSC clones was no longer present upon further differentiation to BJ ρ0+HEK293T-MSC clones (Figures S2B, S2E, and S2F). In summary, steady-state metabolite analyses showed that SIMR cells are comparable to native control cells, with only a few differences that are resolved upon iPSC reprogramming and differentiation to MSCs.
We utilized RNA sequencing (RNA-seq) to evaluate whole-transcriptome profiles for SIMR cells at fibroblast, iPSC, and MSC fates. DESeq2 was used to identify significant differentially expressed genes (DEGs), defined as genes showing an absolute log 2-fold change > 0.5 and adjusted p < 0.05. For both BJ ρ0+PBMC1 and BJ ρ0+HEK293T SIMR cells, the greatest number of DEGs compared to native BJ control cells with an adjusted p < 0.05 occurred at the fibroblast fate (Figures 5A and S3A). RNA-seq identified 1741, 194, and 224 elevated and 1827, 68, and 115 repressed DEGs by comparing BJ ρ0+PBMC1 cells to native BJ parent cells at the fibroblast, iPSC, and MSC fates, respectively (Figure 5A; Table S4). Transcriptomic analysis of the independently generated BJ ρ0+HEK293T cells similarly identified 1,377, 537, and 239 elevated and 1,564, 648, and 210 repressed DEGs compared to native BJ parent cells at fibroblast, iPSC, and MSC fates, respectively (Figure S3A; Table S4).
Figure 5. Transcriptome Features of SIMR and Native mtDNA Cells.

(A) Number of DEGs for BJ ρ0+PBMC1 cells compared to native BJ cells at fibroblast, iPSC, and MSC fates.
(B) Hierarchical clustering of nuclear-encoded MitoCarta2.0 database genes from native BJ, BJ ρ0, and BJ ρ0+PBMC1 cells at fibroblast, iPSC, and MSC fates.
(C) Normalized, batch-adjusted read counts shown as box-and-whisker Tukey plots for 13 MitoCarta2.0-annotated mtDNA-encoded genes for native BJ, BJ ρ0, and BJ ρ0+PBMC1 cells at the fibroblast, iPSC, and MSC fates. Statistical significance was by Welch’s t test.
(D) MitoXplorer-categorized DEGs for BJ ρ0+PBMC1 compared to native BJ cells at the fibroblast, iPSC, and MSC fates divided into the 38 mitochondrial processes.
See also Figures S2, S3, S4, and S5 and Tables S3, S4, and S5.
Reactome pathway enrichment analysis of DEGs showed diverse pathways altered in SIMR fibroblast transcript profiles compared to those in native BJ fibroblasts, including those associated with extracellular matrix organization and the complement cascade (Figure S3B; Table S5). Differential expression and pathway enrichment analyses comparing all SIMR-iPSCs and SIMR-MSCs to native BJ-iPSC and BJ-MSC controls, respectively, identified a dramatically smaller number of DEGs, with overrepresented pathways driven primarily by a cluster of histone transcripts (Figures S3C and S3D; Table S5).
Further detailed transcriptome analyses uncovered metabolic pathway differences based on cell condition and fate. Somatic cell reprogramming to iPSCs requires a metabolic shift from predominantly oxidative phosphorylation to mainly glycolysis, which corresponds with all BJ ρ0+PBMC1-iPSC and BJ ρ0+HEK293T-iPSC clones showing elevated expression of glycolysis-associated transcripts by gene set variation analysis (GSVA) (Figures S4A and S4B). Additionally, GSVA showed increased expression of ETC transcripts in BJ ρ0, BJ ρ0+PBMC1, and BJ ρ0+HEK293T SIMR fibroblasts compared to that in native BJ fibroblasts (Figures S4A and S4B). However, immunoblots for succinate dehydrogenase (SDHB; complex II), ubiquinol-cytochrome c reductase core protein 2 (UUQCRC2; complex III), cytochrome C oxidase II (MT-COXII; complex IV), and ATP synthase F1 subunit alpha (ATP5A; complex V) demonstrated the opposite result, with ETC proteins in SIMR fibroblasts reduced compared to those in the native BJ fibroblast control (Figure S4C). Overall, whole-transcriptome data analysis showed that initial large differences between SIMR clones and native control cells at the fibroblast fate progressively dissipated during reprogramming and differentiation.
We examined the RNA transcript levels of 1,158 nuclear genes listed in the MitoCarta2.0 database that encode proteins that localize to the mitochondria. Hierarchical clustering analysis of transcripts from these genes identified a separation of BJ ρ0+PBMC1, BJ ρ0+HEK293T, and BJ ρ0 transcripts away from the native BJ transcripts at the fibroblast fate (Figures 5B and S3B). Pathway analysis of DEGs between these two groups showed an enrichment for genes encoding ETC proteins in the native BJ fibroblasts (Table S4). Of note, this differential clustering was not observed at the iPSC and MSC fates (Figures 5B and S5A).
Closer examination of mtDNA-encoded transcripts only demonstrated, as anticipated, that BJ ρ0 cells have dramatically lowered expression of all 13 coding gene transcripts compared to transcripts from SIMR and native fibroblast cells (Figures 5C and S5B). Additionally, both BJ ρ0+PBMC1 and BJ ρ0+HEK293T fibroblasts showed significantly reduced expression of the 13 mtDNA-encoded genes compared to native BJ fibroblasts (Figures 5C and S5B). By contrast, no significant difference was observed in mtDNA transcript levels after reprogramming SIMR and control fibroblasts to iPSCs, followed by differentiation to MSCs (Figures 5C and S5B).
We then used the MitoXplorer pipeline (Yim et al., 2020) to quantify the representation of 38 distinct mitochondrial processes within the identified DEGs. As anticipated from abolished respiration, BJ ρ0 fibroblasts showed DEGs for 29 mitochondrial processes compared to native BJ fibroblasts, especially within oxidative phosphorylation, mitochondrial genome translation, and amino acid metabolism processes (Figure 5D). Similarly, BJ ρ0+PBMC1 and BJ ρ0+HEK293T fibroblasts had altered gene expression profiles in 30 and 29 mitochondrial processes, respectively, in comparison to native BJ fibroblasts (Figures 5D and S5C). Further analysis at this fate using MitoXplorer highlighted differences between the two SIMR lines, as the BJ ρ0+PBMC1 had fewer DEGs for mtDNA-associated oxidative phosphorylation and mitochondrial genome translation compared to BJ ρ0+HEK293T fibroblasts. Furthermore, both SIMR fibroblast lines had more DEGs associated with nuclear-encoded mitochondrial translation and calcium signaling and transport than the BJ ρ0 line, when compared to native BJ fibroblasts. Of note, the number of affected mitochondrial processes was dramatically reduced by reprogramming, with only 7 and 16 processes exhibiting DEGs in BJ ρ0+PBMC1-iPSCs and BJ ρ0+HEK293T-iPSCs, respectively (Figures 5D and S5C). Finally, BJ ρ0+PBMC1-MSCs and BJ ρ0+HEK293T-MSCs exhibited only 3 and 8 mitochondrial processes with DEGs, respectively, with only ROS defense similarly altered between the two SIMR-MSC lines and MSC control (Figures 5D and S5C). These data uncover transcriptomic alterations to translation, among additional mitochondrial processes, at the fibroblast fate as a potential difference maker for exogenous mtDNA function in SIMR cells. Combined, the results show that, although SIMR cell mitochondrial function becomes more similar to that of the BJ control with reprogramming and differentiation, differences still exist that are based on the transferred mtDNA sequence.
DISCUSSION
Here, we use MitoPunch to report on a rapid, versatile pipeline to generate transformed or non-immortal cells with specific mtDNA-nDNA combinations, an advance with certain advantages over cybrid technology (Patananan et al., 2016; Wong et al., 2017), un-controlled selection in physiologic mitochondrial “bottlenecks” (Latorre-Pellicer et al., 2019), or time-consuming screens for cells with desired mtDNA mutations (Fayzulin et al., 2015; Lorenz et al., 2017). We show that the transcriptome and metabolome of SIMR fibroblasts resemble those of ρ0 matched recipient cells and that reprogramming to pluripotency followed by differentiation resets these profiles to closely resemble those of unmanipulated control cells. Our studies would be difficult or impossible using other mitochondrial transfer approaches, such as those that use immortal cell lines incapable of reprogramming. Although it is also possible to generate SIMR cells with the nanoblade and microinjection, these low-throughput methods suffer practical and experimental limitations. In contrast, MitoPunch is an accessible approach to generate numerous clones with desired, stable mtDNA-nDNA combinations within 2 weeks.
SIMR clone formation was achieved for all ρ0 recipient fibroblasts studied. Of note, some mtDNA-nDNA combinations produced SIMR clones at lower efficiencies than other combinations, which could only be detected using a high-throughput platform like MitoPunch. In contrast to 143BTK− ρ0+HEK293T SIMR cells that produced ~75 clones, MitoPunch transfers into primary fibroblasts yielded fewer SIMR clones, possibly related to the Hayflick limit or a sub-optimal response to the MitoPunch procedure. For example, ADF ρ0 recipients grew the slowest, reached senescence shortly after mtDNA depletion (data not shown), and resulted in the fewest SIMR clones. On average, twice as many BJ ρ0+HEK293T clones were obtained compared to BJ ρ0+PBMC1 clones, which may be from a more compatible mtDNA-nDNA pairing and more favorable metabolic profile (Latorre-Pellicer et al., 2016). SIMR fibroblasts also showed a 10-fold reduction in reprogramming efficiency compared to native fibroblast controls. A similar reduction was observed in mouse embryo fibroblast reprogramming with non-native mtDNA, perhaps from a lower mtDNA-nDNA compatibility (Latorre-Pellicer et al., 2019).
Evidence for incomplete mtDNA-nDNA assimilation in SIMR fibroblasts was observed in transcriptome data that showed hundreds of DEGs between SIMR and unmanipulated fibroblasts. Also, MitoCarta2.0- and mtDNA-encoded transcripts were most similar for SIMR and ρ0 fibroblasts, despite SIMR cell culture in restrictive medium requiring ETC activity for growth and survival. These data suggest that exogenous mtDNA in SIMR fibroblasts do not fully communicate and influence the nDNA, despite being able to support a selectable level of ETC activity and adequate synthesis of metabolites for cell proliferation. Supporting evidence for this suggestion includes that ddC exposure yielded minimal nDNA damage and that the metabolome and transcriptome profiles of SIMR fibroblasts are mostly reset to unmanipulated BJ-iPSC profiles in SIMR-iPSCs. Our results agree with a report showing that ρ0 cells have altered metabolism and an epigenome that can only be partially reset by cybrid formation (Smiraglia et al., 2008). Our study provides a platform for investigating the resetting of ρ0 cell transcription and metabolism after stable mtDNA transplantation and subsequent cell fate changes. Further work is needed to determine whether cells that receive exogenous mtDNA by other forms of mitochondrial transfer also have disrupted mtDNA transcription profiles.
In summary, we provide a proof-of-principle mitochondrial transfer pipeline to generate cells of different fates with specific mtDNA-nDNA combinations, including clonal lines with genome pairings not found in nature. Future studies will generate SIMR-derived cells representing high-energy-demand tissues, such as cardiomyocytes or neurons, and will investigate the current inability to generate SIMR-iPSCs containing mutant mtDNAs to enable patient-specific disease and drug screening models with isogenic nuclei for mtDNA diseases. Furthermore, our results show that the interpretation of mitochondrial transfer experiments must consider that cells initially generated may not show complete mtDNA-nDNA integration and subsequent restoration of cellular pathways. This is particularly salient for cells that lack subsequent reprogramming potential, such as transient or transformed mitochondrial transfer cell lines, since our results show that reprogramming and differentiation are required for resetting the nDNA expression profile. Finally, this mitochondrial transfer pipeline bypasses the evolutionary pairwise selection of mtDNAs and nDNAs in cells to expand upon the repertoire of genomic combinations present in the human population and generates a library of cells at various fates with defined mtDNA-nDNA combinations and unique functional properties for research and potential therapeutic applications.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Michael A. Teitell (mteitell@mednet.ucla.edu).
Materials Availability
All materials generated in this study are available upon reasonable request to the Lead Contact, Dr. Michael A. Teitell.
Data and Code Availability
All raw RNA-Seq reads, transcript abundance values, and processed gene count matrices are submitted to the NCBI Gene Expression Omnibus (GEO). The accession number for the RNA-seq reads reported in this paper is GEO: GSE115871. All other data are available upon request. All software used is available either commercially or as freeware. All custom code is available on GitHub at https://bitbucket.org/ahsanfasih/mitoDesigner/src/master/.
EXPERIMENT MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T cells expressing mitochondria-targeted DsRed protein (pMitoDsRed, Clontech Laboratories) were made as previously described (Miyata et al., 2014). Primary, non-transformed human fibroblast sources include BJ (ATCC, Cat. # CRL-2522), ADF (ATCC, Cat. # PCS-201–012), and NDF (ATCC, Cat. # PCS-201–010). Isogenic cybrid cell lines derived from the same patient containing either the homoplasmic A3243G MELAS substitution or homoplasmic WT sequence were obtained from Douglas Wallace (Children’s Hospital of Philadelphia Research Institute). An alternative A3243G MELAS cybrid cell line, in addition to A8344G MERRF and Dcytochrome B 3.0 cybrid cell lines, were from Carlos Moraes (University of Miami). Two primary A3243G MELAS fibroblast lines were from Anu Suomalainen Wartiovaara (University of Helsinki). Primary fibroblasts associated with Leigh Syndrome (T8993G, Cat. # GM13411) and Kearns Sayre Syndrome (common deletion, Cat. # GM06225) were obtained from the Coriell Repository.
BJ, NDF, ADF, and HEK293T-DsRed cells were grown at 37°C and 5% CO2 in complete media containing DMEM (Corning, Cat. # 10013CV) supplemented with 10% Fetal Bovine Serum (FBS, Hyclone, Cat. # SH30088.03HI0), penicillin-streptomycin (Corning, Cat. # 30–002-CI), GlutaMax (ThermoFisher, Cat. # 35050–061), and non-essential amino acids (MEM NEAA, ThermoFisher, Cat. # 11–140-050). BJ ρ0, NDF ρ0, ADF ρ0, MELAS, MERRF, Δcytochrome B 3.0, Leigh Syndrome, and Kearns Sayre Syndrome cells were grown in complete media supplemented with 50 μg/ml uridine (Sigma, Cat. # U3003). iPSCs were grown on matrigel (Corning, Cat. # 356234) coated plates in mTeSR1 media (StemCell Technologies, Cat. # 85850) according to the manufacturer’s protocol. MSCs were grown in defined, MesenCult-ACF media (StemCell Technologies, Cat. # 05449) following the manufacturer’s protocol. Cells tested negative repeatedly for mycoplasma using a universal mycoplasma detection kit (ATCC, Cat. # 30–1012K).
Human Tissues
The following human tissues were used: PBMC1 (PBMCs from leukopak donor 351, Caucasian female, 42 year old, Donor ID: D326351, HemaCare Corp) and PBMC2 (PBMCs from leukopak donor 298, Hispanic/Latino male, 25 year old, Donor ID: D316153, HemaCare Corp).
METHOD DETAILS
mtDNA Depletion and qPCR Verification
A 1000x stock of ddC (Sigma, Cat. # D5782) was prepared in water and added to BJ, ADF, and NDF cells grown in complete media with 50 μg/ml uridine to an appropriate final concentration. Cells were passaged every 3–4 d with fresh ddC added over 3 weeks. Following ddC treatment, total DNA was extracted (QIAGEN, Cat. # 69504) and mtDNA quantified using SYBR Select Master Mix for CFX (Life Technologies, Cat. # 4472942). mtDNA-encoded MT-ND1 was amplified with the following primers: forward: CCCTAAAACCCGCCACATCT; reverse: CGATGGTGAGAGCTAAGGTC. mtDNA levels were normalized to nucleus-encoded GAPDH using the following primers: forward: TGCACCACCAACTGCTTAGC; reverse: GGCATGGACTGTGGTCATGAG. RPLP0 served as an alternative nucleus-encoded gene for normalization using the following primers: forward: CGACCTGGAAGTCCAAC-TAC; reverse: ATCTGCTGCATCTGCTTG. qPCR was run on a BioRad CFX Thermal Cycler using the following protocol: 1) 50°C for 2 min, 2) 95°C for 2 min, and 3) 40 cycles at 95°C for 10 s and 60°C for 45 s. Samples were compared by calculating ΔΔCT and fold differences.
Mitochondrial Transfer into ρ0 Recipients
Mitochondria were harvested from HEK293T-DsRed cells, PBMCs (PBMC1 or PBMC2), or other cell types using a Qproteome Mitochondria Isolation Kit (QIAGEN, Cat. # 37612) following the manufacturer’s protocol. Mitochondrial pellets were re-suspended in 1x PBS, pH 7.4, at 1 mg total protein/ml. Mitochondrial suspensions were delivered into ρ0 cells using MitoPunch.
The MitoPunch platform is a force-based mitochondrial transfer device. Briefly, a 5V solenoid (Sparkfun, Cat. # ROB-11015) is mounted on a threaded plug (Thor Labs, Cat. # SM1PL) and inserted into a threaded cage plate (Thor Labs, Cat. # CP02T). Above the solenoid, assembly rods (Thor Labs, Cat. # ER3) support an upper plate (Thor Labs, Cat. # CP02). The upper plate holds a custom machined aluminum washer (outer diameter, 25 mm; inner diameter, 10 mm) that supports a deformable PDMS (10:1 ratio of Part A base: Part B curing agent) fluid reservoir above the solenoid. The PDMS reservoir is composed of a bottom circular layer (25 mm diameter, 0.67 mm height) chemically bonded to an upper circular ring layer (outer diameter, 25 mm; inner diameter, 10 mm; height, 1.30 mm) and can hold approximately 120 μl of isolated mitochondrial suspension. Cells are seeded onto a porous membrane with 3 μm pores (Corning, Cat. # 353181) 24 h prior to mitochondrial transfer.
To perform mitochondrial transfer, this membrane with adherent cells is secured on top of the PDMS reservoir using an upper plate (Thor Labs, Cat. # CP02). The solenoid is controlled by a 5V power supply mini board (Futurlec, Cat. # MINIPOWER) and powered by a 12V, 3 Amp DC power supply (MEAN WELL, Cat. # RS-35–12). The activated solenoid strikes the center of the PDMS chamber, deforming the bottom circular layer by approximately 1.3 mm. This deformation rapidly injects the mitochondrial suspension through the membrane and into the monolayer cell culture on the opposite side. A tunable MitoPunch prototype was developed by NanoCav LLC with variable plunger force which improves SIMR generation efficiency especially in replication-limited fibroblasts.
As a comparison to MitoPunch, we performed isolated mitochondria coincubation control experiments. An equal number of coincubation recipient cells were seeded alongside MitoPunch recipients in 12 well dishes instead of the porous membrane. After ~24 h, an equal volume of mitochondrial isolate as loaded into the PDMS reservoir for MitoPunch was pipetted into the cell medium of each coincubation recipient well and incubated at 37°C for 2 h before being released, collected, and plated on 10 cm dishes for selection as described below.
For human fibroblasts and mouse recipients, cells were grown in complete media with 50 μg/mL uridine for 4 d following mitochondria delivery and on day 5 post-delivery, cells were shifted to uridine-free complete media prepared with 10% dialyzed FBS (Life Technologies, Cat. # 26400–044). On day 8 post-delivery, cells were shifted to glucose-free, galactose-containing medium (DMEM without glucose, GIBCO, Cat. # 11966025) supplemented with 10% dialyzed FBS and 4.5 g/l galactose. Colonies emerged at approximately 10 d post-delivery and cells were shifted back to uridine-free medium before colonies were counted by microscopy or isolated using cloning rings. For human 143BTK– ρ0 recipients, cells were grown in complete media with 50 μg/mL uridine following mitochondria delivery and shifted to uridine-free complete media prepared with 10% dialyzed FBS on day 3 post-delivery, and clones emerged approximately 10 d post-delivery and were quantified.
Confocal microscopy
Cells, ~1×105, were plated on glass coverslips (Zeiss, Cat. # 474030–9000) in 6 well dishes in 2 mL of media and cultured for approximately 24 h. The media was aspirated and cells then fixed by incubation of 1 mL freshly diluted 4% paraformaldehyde (Thermo Fisher Scientific, Cat. # 28906) in 1x PBS, pH 7.4, for 15 min at RT. Paraformaldehyde was removed and cells were washed 3x with PBS, and then washed 3x with PBS with 5 min RT incubation during each wash. Cells were permeabilized by a 10 min RT incubation with 0.1% Triton X-100 (Sigma, Cat. # X100). Permeabilized cells were washed 3x with PBS and then blocked by incubation for 1 h at RT with 2% bovine serum albumin (BSA) dissolved in PBS. After blocking, cells were incubated with primary antibodies at 1:1000 dilution in 2% BSA blocking buffer against dsDNA (Abcam, Cat. # ab27156) and TOM20 (Abcam, Cat. # ab78547), and then washed 3x with 5 min RT incubation with PBS. After washing, cells were incubated for 1 h with secondary antibodies (Invitrogen, Cat. #’s A31573 and A21202) diluted 1:100 in 2% BSA blocking buffer protected from light at RT, and washed 3x with 5 min incubations with PBS. To mount coverslips on slides, samples were removed from the 6 well dish, dipped in deionized water, dried with a Kimwipe, and mounted using ProLong Gold Antifade Mountant with DAPI (Invitrogen, Cat. # P3691) on microscope slides (VWR, Cat. # 48311–601). Mounted samples were allowed to dry protected from light at RT for 48 h prior to imaging with a Leica SP8 confocal microscope.
Mitochondrial Oxygen Consumption Measurements
OCR was measured using a Seahorse XF96 Extracellular Flux Analyzer (Agilent). For fibroblasts or MSCs, 1 – 2 ×105 cells per well were seeded onto a V3 96-well plate (Agilent, Cat. # 101085–004) and grown overnight before analysis. iPSCs were treated similarly but plated on matrigel-coated V3 plates. A mitochondrial stress test quantified OCR at basal respiration and after the sequential addition of mitochondrial inhibitors oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and rotenone.
Mitochondrial Isolation from Mouse Tissues and Delivery
Spleen, liver, lung, bone marrow, heart, skeletal muscle, and kidney were harvested from an ~8 month-old female C57BL/6 mouse. Briefly, tissue was dissociated by passage through a cell strainer using the plunger of a syringe, and mitochondria were isolated from dissociated tissue using the Qproteome Mitochondria Isolation Kit (QIAGEN, Cat. # 37612) following the manufacturer’s protocol. Mitochondrial suspensions were delivered into L929 ρ0 fibroblasts using MitoPunch. L929 ρ0 recipient cells were grown in complete media supplemented with 50 μg/mL uridine for 4 d following mitochondria delivery. On day 5 post-delivery, cells were shifted to uridine-free complete media prepared with 10% dialyzed FBS (Life Technologies, Cat. # 26400–044). On day 8 post-delivery, cells were shifted to glucose-free, galactose-containing medium (DMEM without glucose, GIBCO, Cat. # 11966025) supplemented with 10% dialyzed FBS and 4.5 g/l galactose. Colonies emerged at approximately 10 d post-delivery and cells were shifted back to uridine-free medium before colonies were counted by microscopy or isolated using cloning rings.
iPSC Reprogramming
Reprogramming of fibroblast lines to iPSCs was done using the StemRNA-NM Reprogramming kit (Stemgent, Cat. # 00–0076) following the manufacturer’s protocol. Briefly, fibroblasts were plated on a matrigel (Corning, Cat. # 356234) coated 6-well plate at 2×105 cells/well on 0 d. Daily transfections of non-modified (NM)-RNA reprogramming cocktail were performed for 4 d using Lipofectamine RNAiMAX (ThermoFisher, Cat. # 13778100). On 10–12 d, iPSC colonies were identified by staining with TRA-1–60 antibody (Stemgent, Cat. # 09–0068). TRA-1–60+ iPSC colonies were picked and re-plated on matrigel coated 12-well plates and maintained in mTeSR 1 medium (StemCell Technologies, Cat. # 85850). Alternative reprogramming strategies for fibroblasts included using ReproRNA-OKSGM (Stem Cell Technologies, Cat. # 05930), STEMCCA Lentiviral (MilliporeSigma, Cat. #SCR510), and Cyto-Tune-iPS 2.0 Sendai (Fisher Scientific, Cat. # A16517) kits according to the manufacturers’ protocols.
MSC Differentiation
MSC lines were generated from iPSCs using the STEMdiff Mesenchymal Progenitor Kit (StemCell Technologies, Cat. # 05240) following the manufacturer’s protocol over the course of 21 d. Briefly, iPSCs were dispersed as single cells, plated at ~5 × 104 cells/cm2, and cultured for 2 d on Matrigel with mTeSR1 medium before the medium was changed to STEMdiff -ACF Mesenchymal Induction Medium. STEMdiff -ACF Mesenchymal Induction Medium was changed daily for 3 d, and on day 4, the medium was changed to MesenCult -ACF Plus Medium. Cells were fed again with MesenCult -ACF Plus Medium on day 5. On day 6, cells were collected with Gentle Cell Dissociation Reagent (StemCell Technologies, Cat. # 07174) and passaged onto plastic plates with MesenCult -ACF Plus Medium with 10 μM ROCK inhibitor (Y-27632; Stem Cell Technologies, Cat. # 72304). Daily half-medium changes were made for ~1 week when cells were ~80% confluent. Cells were further passaged by dissociation with ACF Enzymatic Dissociation Solution and resuspended in MesenCult -ACF Plus Medium before further analysis.
Human mtDNA D-Loop Sequencing
Total DNA was extracted from 1×106 cells using the QIAGEN DNasy Blood and Tissue kit. PCR was performed using Phusion high-fidelity PCR master mix with HF buffer (NEB, Cat. # M0531S) and the following primers: forward – TTCCAAGGACAAATCAGA-GAAAAAGT, reverse – AGCCCGTCTAAACATTTTCAGTGTA. PCR was run on an Eppendorf vapo.protect thermal cycler at 1) 98°C for 2 min, 2) 30 cycles at 98°C for 15 s, 58°C for 30 s, 72°C for 30 s, and 3) 72°C for 5 min. PCR products were run on a 0.8%–1% agarose TAE gel, extracted with the QIAGEN QIAQuick Gel Extraction kit (QIAGEN, Cat. # 28704), and Sanger sequenced using the same PCR primers.
ROS Quantification
CellROX Green Flow Cytometry Assay Kit (ThermoFisher, Cat. # C10492) was used according to the manufacturer’s protocol. Briefly, 7.5×104 cells were plated in a 6-well plate ~24 h prior to measurements. 250 μM tert butyl hydroperoxide (TBHP) and 750 μM CellROX reagent were added to the cells ~2 h and 1 h prior to quantification, respectively. Cells were released using Accutase, washed once with FACS buffer (5% FBS in 1x DPBS, pH 7.4), and quantified using a LSRFortessa flow cytometer (BD Bioscience).
iPSC Flow Cytometry
iPSCs were harvested by 15 min RT incubation with Gentle Cell Dissociation Reagent. Cells were centrifuged at 300 x g for 5 min, washed in 1ml DPBS + 10% FBS, and re-suspended in 100 μL BD Perm/Fix Buffer (BD Bioscience). Cells were incubated at 4°C for 15 min and washed twice in DPBS + 10% FBS. Following the second wash, cells were incubated in 50μl DPBS + 10% FBS containing conjugated antibodies. Antibodies used were OCT3/4 AlexaFluor488 (BD Bioscience 561628 1:10), SOX2 V450 (BD Bioscience 561610 1:10), Mouse IgG1 κ Isotype Control AlexaFluor488 (BD Biosciences 557782 1:10), Mouse IgG1, κ Isotype Control V450 (BD Bioscience 560373 1:10), and CD44 PE (BD Bioscience 562245 1:21). Cells were incubated with conjugated antibodies for 30 min and then washed twice in DPBS + 10% FBS. Data was acquired on a LSRFortessa flow cytometer (BD Bioscience) and analyzed using FlowJo software (FlowJo, LLC).
MSC Flow Cytometry
MSCs were harvested by 5 min, 37°C incubation with Accutase (BD Biosciences). Cells were centrifuged at 300 x g for 5 min, washed in 1ml DPBS + 10% FBS, and re-suspended in DPBS + 10% FBS at 5×106 cells/ml. Cells were incubated in 100μl DPBS + 10% FBS for 30 min at 4°C with the antibodies provided in the Human MSC Analysis Kit (BD Biosciences, Cat. # 562245) for 30 min and then washed twice in DPBS + 10% FBS. Data was acquired on a LSRFortessa flow cytometer (BD Bioscience) and analyzed using FlowJo software (FlowJo, LLC).
Fluorescence Microscopy
iPSCs were cultured on matrigel-coated 6-well plates and fixed with 4% paraformaldehyde for 10 min. Blocking was done for 1 h in 1x PBS, pH 7.4, with 5% FBS and 0.3% Triton X-100. Cells were stained with SSEA4 (eBioscience, Cat. # 12–8843-42) and OCT4 (eBioscience, Cat. # 53–5841-82) antibodies, and Hoechst 33342 dye (ThermoFisher, Cat. # R37605) overnight at 4°C in blocking buffer. Phase contrast and fluorescence images were obtained with a Zeiss Axio Observer Z1 microscope and Hamamatsu EM CCD camera (Cat. # C9100–02).
MSC Immunosuppression Assay
MSC inhibition of T cell proliferation was performed as described previously (Hsu et al., 2015). Briefly, MSCs were plated in a 12-well plate the day before assay. PBMCs were isolated by Ficoll gradient from a healthy de-identified leukopak donor. CD4+ T cells were isolated from PBMCs using the CD4+ T cell Isolation Kit (Miltenyi Biotec, Cat. # 130–096-533) and labeled with CFDA SE (ThermoFisher, Cat. # V12883). Labeled CD4+ T cells were stimulated with Dynabeads Human T-activator CD3/CD28 (ThermoFisher, Cat. # 11131D) at a ratio of one bead per T cell. T cells were added into MSC cultures at the following T cell:MSC ratios: 1:2, 1:1, 5:1, and 10:1. After 5 d of co-culture, T cell proliferation was measured using CFSE signature dye dilution by flow cytometry.
Tri-lineage Differentiation
Adipocytes, osteoblasts, and chondrocytes were generated from MSCs. For adipocyte differentiation, MSCs between passages 3 – 4 were plated on 6-well plates with MesenCult-ACF Basal Medium (StemCell Technologies, Cat. # 05449) at 4 – 5 3 105 cells per well. Differentiation was performed using the MesenCult Adipogenic Differentiation Kit (StemCell Technologies, Cat. # 05412) according to the manufacturer’s protocol. Media changes were done every 3 – 4 d until 13 d. For osteogenic lineage differentiation, MSCs between passages 3 – 4 were plated on a 6-well plate with MesenCult-ACF Basal Medium (StemCell Technologies) at 3 – 4 × 104 cells per well. Differentiation was performed using MesenCult Osteogenic Differentiation medium (StemCell Technologies, Cat. # 05465) according to the manufacturer’s protocol. Medium changes were done every 3 – 4 d until 13 d. For 3-D pellet chondrogenic differentiation, MSCs were first released from T25 flasks using ACF Enzymatic Dissociation/Inhibition Solution (StemCell Technologies, Cat. # 05426) and collected in polypropylene tubes at 2.5 – 3 × 106 cells per tube with MesenCult-ACF Chondrogenic Differentiation Medium (StemCell Technologies, Cat. # 05455) according to the manufacturer’s protocol. Medium changes were done every 3 – 4 days until 13 d.
Tri-lineage Differentiation Analyses
Osteogenic differentiation was assayed by staining cells with 1% Alizarin Red solution. Medium was removed from cells grown on 6-well plates and cells were washed 3 times with 1X DPBS. Cells were fixed in 4% PFA in 1X DPBS at 4°C for 15 min prior to 15 min incubation with 1% alizarin red at RT. Alizarin red solution was aspirated and the cells were imaged using a standard inverted microscope.
Adipogenic differentiation was assayed by staining cells with 0.1% Bodipy solution. Medium was removed from cells grown on 6-well plates and cells were washed 3x with 1X DPBS. Cells were fixed in 4% PFA in 1X DPBS at 4°C for 15 min and washed twice with 1X DPBS prior to a 10 min incubation with 0.1% Bodipy at RT. Bodipy solution was aspirated and the cells were washed with 1X DPBS prior to acquiring phase contrast and fluorescence images with a Zeiss Axio Observer Z1 microscope and Hamamatsu EM CCD camera (Cat. # C9100–02).
Chondrocyte differentiation was assayed by staining chondrogenic spheroids and spheroid sections with 0.1% Safranin O solution. For staining whole spheroids, the medium was removed from the spheroids and they were fixed in 4% PFA for 15 min at RT. The spheroids were washed twice with 1x PBS, pH 7.4, before 15 min incubation with 0.1% Safranin O solution at RT. The stained spheroids were washed twice with 1ml water and transferred by serological pipette to a 48-well dish for imaging. For spheroid section staining, spheroids were fixed in 10% formalin for 18 h, washed twice in water, and placed in 70% ethanol. Spheroids were microtome sectioned by the UCLA Translational Pathology Core Laboratory, tissue placed on microscope slides. Sections were deparaffinized and rehydrated by washes in xylenes, ethanol, and water. Unstained sections were stained with hematoxylin and eosin or 0.1% Safranin O for 10 min at RT prior to washing in ethanol. Sections were imaged under a standard inverted microscope.
UPHLC-MS Metabolomics Processing
Ultra-high-performance liquid chromatography mass spectrometry (UHLPC-MS) was performed as described previously (Xiao et al., 2018) to quantify metabolites from ~7×105 cells. Briefly, cells were rinsed with cold 150 mM ammonium acetate, pH 7.3, followed by addition of ice-cold 80% methanol. Cells were detached with scrapers, transferred into microcentrifuge tubes, and 1 nmol D/L-norvaline added. After vortexing, the suspension was centrifuged at 4°C at maximum speed. The supernatant was transferred into a glass vial, metabolites dried down under vacuum using an EZ-2Elite evaporator at 30°C, and re-suspended in 70% acetonitrile. To normalize samples, pellets were re-suspended in 58 mM Tris-HCl, pH 6.8, 5% glycerol, and 17 mg/ml sodium dodecyl sulfate and quantified by BCA protein assay (ThermoFisher, Cat. # 23225).
Metabolites were separated on a Luna NH2 (150 mm x 2 mm, Phenomenex) column using 5 mM NH4AcO, pH 9.9 (buffer A), acetonitrile (buffer B), and the following gradient: initially at 15% buffer B, 18 min gradient to 90% buffer B, 9 min isocratic at 90% buffer B, 7 min isocratic at 15% buffer B. Samples were analyzed with an UltiMate 3000RSLC (Thermo Scientific) coupled to a Q Exactive mass spectrometer (Thermo Scientific) run with polarity switching (+3.50 kV / 3.50 kV) in full scan mode and m/z range of 65–975. Metabolites were quantified with TraceFinder 3.3 using accurate mass measurements (≤3 ppm) and retention times of pure standards.
RNA Extraction
Fibroblasts, iPSCs, and MSCs were grown in biological triplicates and technical duplicates to 70 – 80% confluence and purified using the RNeasy Mini Kit (QIAGEN, Cat. # 74104) and RNase-free DNase (QIAGEN, Cat. # 79254) following the manufacturer’s protocols. All samples showed a A260/280 ratio > 1.99 (Nanodrop; Thermo Scientific). Prior to library preparation, quality control of the RNA was performed using the Advanced Analytical Technologies Fragment Analyzer (Advanced Analytical, Inc.) and analyzed using PROSize 2.0.0.51 software. RNA Quality Numbers (RQNs) were computed per sample between 8.1 and 10, indicating intact total RNA per sample prior to library preparation.
RNA-Seq Library Preparation
Strand-specific ribosomal RNA (rRNA) depleted RNA-Seq libraries were prepared from 1 μg of total RNA using the KAPA Stranded RNA-Seq Kit with Ribo-Erase (Kapa Biosystems, Roche). Briefly, rRNA was depleted from total RNA samples, the remaining RNA was heat fragmented, and strand-specific cDNA was synthesized using a first strand random priming and second strand dUTP incorporation approach. Fragments were then A-tailed, adapters were ligated, and libraries were amplified using high-fidelity PCR. All libraries were prepared in technical duplicates per sample (n = 60 samples, 120 libraries total), and resulting raw sequencing reads merged for downstream alignment and analysis. Libraries were paired-end sequenced at 2×150 bp on an Illumina NovaSeq 6000.
Restriction-Fragment Length Polymorphism Heteroplasmy Assay
To quantify relative levels of mtDNA containing the A3243G substitution, total DNA was isolated from cells using the DNeasy Blood and Tissue kit (QIAGEN, Cat. # 69504). PCR amplification of the MELAS region to generate a 634 bp product was performed using the following primers: forward – CCTCGGAGCAGAACCCAACCT and reverse – CGAAGGGTTGTAGTAGCCCGT. ApaI digestion (NEB Biolabs, Cat. # R0114S) of the PCR product was performed according to manufacturer’s protocol for 2 h at 25°C, and deactivated at 65°C for 20 min. Sample was separated on a 2.5% agarose gel at 100V for 1 h.
QUANTIFICATION AND STATISTICAL ANALYSIS
mtDNA Depletion and qPCR Verification
Statistical details are provided in each figure legend.
Mitochondrial Oxygen Consumption Rate Measurements
Statistical details are provided in each figure legend.
Metabolomics Data Analysis
Data analysis, including principal components analysis (PCA) and clustering, was performed using the statistical language R v3.6.0 and Bioconductor v3.9.0 packages (Huber et al., 2015; R Core Team, 2017). Metabolite abundance was normalized per mg of protein content per metabolite extraction, and metabolites not detected were set to zero. Metabolite normalized amounts were log transformed and then scaled and centered into Z-scores for relative comparison using R base function scale() with parameters “scale = TRUE, center = TRUE”. Heatmaps and Euclidean distance similarity plots were created using the Z-scores in R package pheatmap v1.0.12, and hierarchical clustering was performed using the Euclidean distance measure.
PCA was performed using R packages FactoMineR v2.2 and factoextra v1.0.6. PC scores computed from normalized metabolite counts with function PCA() using parameters “scale.unit = TRUE, ncp = 10, graph = FALSE”.
Pathway-level metabolite set enrichment analysis was performed using R Bioconductor package GSVA v1.32.0 (Hänzelmann et al., 2013). Metabolite normalized abundances were standardized using a log2(normalized amounts + 1) transformation, and metabolites per sample were converted to a pathways per sample matrix using function gsva() with parameters “method = gsva, rnaseq = FALSE, abs.ranking = FALSE, min.sz = 5, max.sz = 500”. GSVA pathway enrichment scores were then extracted and significance testing for multiple transfer conditions was calculated using R Bioconductor package limma v3.40.6, as described above. Pathway metabolite sets were constructed using the KEGG Compound Database and derived from the existing Metabolite Pathway Enrichment Analysis (MPEA) toolbox (Kanehisa et al., 2012; Kankainen et al., 2011).
RNA-Seq Pre-Processing
Fibroblasts, iPSCs, and MSCs were each sequenced in biological triplicates and technical duplicates (n = 60 total samples) to account for variation in extraction and culturing. Raw sequencing reads were converted into fastq files and filtered for low quality reads and Illumina sequencing adaptor contamination using bcl2fastq (Illumina). Reads were then quasi-mapped and quantified to the Homo sapiens GENCODE 28 (GRCh38.p12, Ensembl 92, April 2018) transcriptome using the alignment-free transcript level quantifier Salmon v0.9.1 (Harrow et al., 2012; Mudge and Harrow, 2015; Patro et al., 2017). A quasi-mapping index was prepared using parameters “salmon index -k 31 –type quasi”, and comprehensive transcript level estimates were calculated using parameters “salmon quant -l A –seqBias –gcBias–discardOrphansQuasi”. Transcript level counts were collapsed to gene level (HGNC) counts, transcripts per million abundances (TPM) and estimated lengths using R Bioconductor package tximport v1.12.3 (Soneson et al., 2015).
Differential Gene Expression Analysis
The resulting sample gene count matrix was size factor normalized and analyzed for pairwise differential gene expression using R Bioconductor package DESeq2 v1.18.1. Expression changes were estimated using an empirical Bayes procedure to generate moderated fold change values with design “~ Batch + Sample,” modeling batch effect variation due to day of RNA extraction (Huber et al., 2015; Love et al., 2014). Significance testing was performed using the Wald test, and resulting P values were adjusted for multiple testing using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). DEGs were filtered using an adjusted false discovery rate (FDR) q value < 0.05 and an absolute log2 transformed fold-change > 0.05.
MitoXplorer Analysis
Differential expression analysis was performed using DESeq2, specifying an absolute log2 transformed fold change threshold > 0.5. Result lists including all genes were uploaded to the MitoXplorer 1.0 pipeline (http://mitoxplorer.ibdm.univ-mrs.fr/index.php) comparing SIMR-fibroblasts, -iPSCs, or -MSCs to the corresponding cell fate of the BJ control. DEGs were filtered using a log2 transformed fold-change threshold of 0.05. Subsequently, the number of upregulated and downregulated DEGs for each mitochondrial process were counted.
Gene Expression PCA and hierarchal clustering
Variance stabilized transform (VST) values in the gene count matrix were calculated and plotted for PCA using R Bioconductor packages DESeq2, FactoMineR, and factoextra, as described in the metabolomics methods (Huber et al., 2015; Love et al., 2014). For PCA of nucleus-encoded mitochondrial protein and mtDNA transcripts, relevant transcripts were extracted using localization evidence derived from MitoMiner v4.0, subsetting VST matrices using genes listed in MitoCarta 2.0 (Calvo et al., 2016; Smith and Robinson, 2016). Clonal heatmaps were prepared using R Bioconductor packages pheatmap v1.0.8 and gplots v3.0.1 (Warnes et al., 2016; Kolde, 2015). Heirarchal clustering was performed using R based function hclust and plotted using the dendextend package.
Metabolic Transcript Gene Set Variation Analysis (GSVA)
GSVA on metabolic transcripts was performed similarly to metabolomics data as noted above. Pathway-level metabolic gene set enrichment analysis was performed using R Bioconductor package GSVA v1.32.0 function gsva() with parameters “method = gsva, rnaseq = FALSE, abs.ranking = FALSE, min.sz = 5, max.sz = 500” using a log2(TPM + 1) transformed gene expression matrix (Hänzelmann et al., 2013). GSVA pathway enrichment scores per sample were extracted and assessed for significance using R Bioconductor package limma v3.40.0, as described above except with a Benjamini-Hochberg adjusted P value threshold = 0.01. Pathway metabolite sets were constructed using the KEGG PATHWAY Database, utilizing gene sets annotated to the metabolic pathways overview map HSA01100 (Kanehisa et al., 2012). Significance testing across clones and conditions for each gene set were calculated using Kruskal-Wallis ANOVA.
Gene Set Overrepresentation Analysis (ORA)
DEGs were extracted and analyzed for pathway/gene ontology (GO) term overrepresentation using the R Bioconductor package clusterProfiler v3.12.0 and ReactomePA v1.28.0, using a background gene set of all genes expressed with at least one read count in the sample gene count matrix (Yu and He, 2016; Yu et al., 2012). Overrepresented Reactome/KEGG pathways and GO terms were identified across DEG lists and conditions using clusterProfiler function compareCluster() with significance testing cutoffs of p < 0.05, and an adjusted FDR < 0.25.
HLA Class I Genotyping
MHC Class I HLA genotypes were identified using OptiType v1.3.1(Szolek et al., 2014). All raw RNA-Seq sample FASTQs were aligned to the HLA Class I reference transcriptome packaged in OptiType using BWA MEM v0.7.17 with standard parameters (Li and Durbin, 2010). HLA subset reads were then analyzed for Class I genotype using OptiType in paired-end RNA mode with standard parameters.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| OCT3/4 | BD Bioscience | Cat#561628, RRID: AB_10895977 | |
| SOX2 | BD Bioscience | Cat#561610, RRID: AB_10712763 | |
| Mouse IgG1 κ Isotype Control | BD Bioscience | Cat#557782, RRID: AB_396870 | |
| Mouse IgG1, κ Isotype Control | BD Bioscience | Cat#560373, RRID: AB_1645606 | |
| SSEA4 | eBioscience | Cat#12-8843-42, RRID: AB_11151520 | |
| OCT4 | eBioscience | Cat#53-5841-82, RRID: AB_1210530 | |
| TRA-1-60 | Stemgent | Cat#09-0068, RRID: AB_2233143 | |
| TOMM20 | Abcam | Cat#ab78547 RRID: AB_2043078 | |
| dsDNA | Abcam | Cat#ab27156 RRID: AB_470907 | |
| Biological Samples | |||
| LP351 Human PBMCs (PBMC1) | HemaCare | Donor ID: D326351 | |
| LP298 Human PBMCs (PBMC2) | HemaCare | Donor ID: D316153 | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| 2′,3′-dideoxycytidine | Sigma | D5782, CAS 7481-89-2 | |
| Dialyzed FBS | Life Technologies | Cat#26400-044 | |
| DMEM without glucose | GIBCO | Cat#11966025 | |
| Matrigel | Corning | Cat#356234 | |
| Lipofectamine RNAiMAX | ThermoFisher | Cat#13778100 | |
| mTeSR 1 | StemCell Technologies | Cat#85850 | |
| Phusion high-fidelity PCR master mix with HF buffer | NEB | Cat#M0531S | |
| Gentle Cell Dissociation Reagent | StemCell Technologies | Cat#07174 | |
| Accutase BD Biosciences | Cat#561527 | ||
| Hoechst 33342 | ThermoFisher | Cat#R37605 | |
| Apa1 | NEB BioLabs | Cat#R0114S | |
| Critical Commercial Assays | |||
| DNeasy Blood & Tissue kit | QIAGEN | Cat#69504 | |
| SYBR Select Master Mix for CFX | Life Technologies | Cat#4472942 | |
| Qproteome Mitochondria Isolation kit | QIAGEN | Cat#37612 | |
| StemRNA-NM Reprogramming kit | |||
| ReproRNA-OKSGM | Stem Cell Technologies | Cat#05930 | |
| STEMCCA Constituitive Polycistronic (OKSM) Lentivirus Reprogramming kit | MilliporeSigma | Cat#SCR510 | |
| CytoTune-iPS 2.0 Sendai Reprogramming kit | Fisher Scientific | Cat#A16517 | |
| STEMdiff Mesenchymal Progenitor Kit | StemCell Technologies | Cat#05240 | |
| QIAGEN QIAQuick Gel Extraction kit | QIAGEN | Cat#28704 | |
| Fixation/Permeabilization Solution Kit with BD GolgiPlug | BD Bioscience | Cat#555028 | |
| Human MSC Analysis Kit | BD Bioscience | Cat#562245 | |
| V3 96-well plate | Agilent | Cat#101085-004 | |
| CD4+ T cell isolation kit | Miltenyi Biotec | Cat#130-096-533 | |
| Vybrant CFDA SE. Cell Tracer Kit | ThermoFisher | Cat#V12883 | |
| Dynabeads Human T-activator CD3/CD28 | ThermoFisher | Cat#11131D | |
| MesenCult-ACF Basal Medium | StemCell Technologies | Cat#05449 | |
| MesenCult Adipogenic Differentiation Kit | StemCell Technologies | Cat#05412 | |
| MesenCult Osteogenic Differentiation medium | StemCell Technologies | Cat#05465 | |
| ACF Enzymatic Dissociation/Inhibition Solutions | StemCell Technologies | Cat#05426 | |
| MesenCult-ACF Chondrogenic Differentiation Medium | StemCell Technologies | Cat#05455 | |
| Y-27632 | Stem Cell Technologies | Cat#72304 | |
| BCA protein assay | ThermoFisher | Cat#23225 | |
| CellROX Green Flow Cytometry Assay Kit | ThermoFisher | Cat#C10492 | |
| KAPA Stranded RNA-Seq Kit with Ribo- Erase | Kapa Biosystems, Roche | Cat#07962304001 | |
| Deposited Data | |||
| RNaseq count matrices and raw reads | This paper | GEO: GSE115871 | |
| Metabolite relative amounts | This paper | N/A | |
| Experimental Models: Cell Lines | |||
| HEK293T | ATCC | Cat#CRL-3216 | |
| BJ Foreskin Fibroblast | ATCC | Cat#CRL-2522 | |
| Primary Dermal Fibroblast; Normal, Human, Adult | ATCC | Cat#PCS-201-012 | |
| Primary Dermal Fibroblast Normal; Human, Neonatal | ATCC | Cat#PCS-201-010 | |
| Leigh Syndrome ATP Synthase 6 (T8993G) Fibroblast | Coriell Institute | Cat#GM13411 | |
| Kearns-Sayre Syndrome (common deletion) Fibroblast | Coriell Institute | Cat#GM06225 | |
| MELAS (A3243G) Cybrid (CL3) | Gift from Douglas Wallace (Children’s Hospital of Philadelphia Research Institute) | N/A | |
| Wildtype Cybrid (CL9) | Gift from Douglas Wallace (Children’s Hospital of Philadelphia Research Institute) | N/A | |
| MELAS (A3243G) Cybrid | Gift from Carlos Moraes (University of Miami) | N/A | |
| MERRF (A8344G) Cybrid | Gift from Carlos Moraes (University of Miami) | N/A | |
| D Cytochrome B 3.0 Cybrid | Gift from Carlos Moraes (University of Miami) | N/A | |
| L929 ρ0 Mouse Fibroblast | Gift from Jose Antonio Enriquez Dominguez (Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC)) | N/A | |
| Oligonucleotides | |||
| ND1 forward -CCCTA AAACCCGCCACATCT | IDT | N/A | |
| ND1 reverse - CGAT GGTGAGAGCTAAGGTC | IDT | N/A | |
| GAPDH Forward - TGCAC CACCAACTGCTTAGC | IDT | N/A | |
| GAPDH Reverse - GGCA TGGACTGTGGTCATGAG | IDT | N/A | |
| RPLP0 Forward -CGA CCTGGAAGTCCAACTAC | IDT | N/A | |
| RPLP0 Reverse -ATCT GCTGCATCTGCTTG | IDT | N/A | |
| D Loop Forward - TTCCAA GGACAAATCAGAGAAAAAGT | IDT | N/A | |
| D Loop Reverse - AGCC CGTCTAAACATTTTCAGTGTA | IDT | N/A | |
| RFLP Forward - CCTC GGAGCAGAACCCAACCT | IDT | N/A | |
| RFLP Reverse - CGAA GGGTTGTAGTAGCCCGT | IDT | N/A | |
| Software and Algorithms | |||
| FlowJo Software Version 10.4.2 | FlowJo, LLC | N/A | |
| Salmon v0.9.1 | (Harrow et al., 2012; Mudge and Harrow, 2015; Patro et al., 2017) | https://github.com/COMBINE-lab/salmon/releases | |
| Statistical Language R v3.6.0 | (R Core Team, 2017) | https://www.r-project.org/ | |
| Bioconductor v3.9.0 | (Huber et al., 2015) | https://bioconductor.org/news/bioc_3_3_release/ | |
| R Bioconductor package tximport v1.12.3 | (Soneson et al., 2015) | https://support.bioconductor.org/p/106345/ | |
| R Bioconductor package DESeq2 v1.24.0 | (Huber et al., 2015; Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| R package ggpubr v0.1.6 | (Kassambara, 2017) | https://www.rdocumentation.org/packages/ggpubr/versions/0.1.6 | |
| R package pheatmap v1.0.12 | (Warnes et al., 2016) | https://www.rdocumentation.org/packages/pheatmap/versions/1.0.8 | |
| R package gplots v3.0.1 | (Kolde, 2015) | https://cran.r-project.org/web/packages/gplots/index.html | |
| R package FactoMineR v2.2 | (Husson, 2020) | https://www.rdocumentation.org/packages/FactoMineR/versions/1.34 | |
| R package factoextra v1.0.6 | (Kassambara, 2019) | https://www.rdocumentation.org/packages/factoextra/versions/1.0.5 | |
| R Bioconductor package Mfuzz v2.38.0 | (Kumar and Futschik, 2007) | https://bioc.ism.ac.jp/packages/3.6/bioc/html/Mfuzz.html | |
| R package ggplot2 v3.2.0 | (Wickham, 2019) | https://cran.r-project.org/web/packages/ggplot2/index.html | |
| R Bioconductor package GSVA v1.32.0 | (Hanzelmann et al., 2013) | https://anaconda.org/bioconda/bioconductor-gsva | |
| R Bioconductor package limma v3.40.6 | (Benjamini and Hochberg, 1995; Ritchie etal., 2015) | https://www.bioconductor.org/install/ | |
| R Bioconductor package clusterProfiler v3.12.0 | (Yu and He, 2016) | https://bioconductor.org/packages/3.7/bioc/vignettes/clusterProfiler/inst/doc/clusterProfiler.html | |
| R Bioconductor package ReactomePA v1.28.0 | (Yu et al., 2012) | https://anaconda.org/bioconda/bioconductor-reactomepa | |
| TraceFinder v3.3 | ThermoFisher | Cat#OPTON-30493 | |
| MitoXplorer 1.0 | (Yim et al., 2020) | http://mitoxplorer.ibdm.univ-mrs.fr | |
Highlights.
We report a “proof-of-principle” mitochondrial transfer pipeline by MitoPunch
MitoPunch generates cells with unique mtDNA-nDNA pairs, regardless of cell source
Replacement mtDNA in non-immortal cells remains stable with cell fate conversions
Enables studies of mtDNA-nDNA interactions with reprogramming and differentiation
ACKNOWLEDGMENTS
A.N.P. is supported by the NIH (T32CA009120) and American Heart Association (18POST34080342). A.J.S. is supported by the NIH (T32GM007185 and T32CA009120). K.P. is supported by the Broad Center of Regenerative Medicine and Stem Cell Research at UCLA, the David Geffen School of Medicine, the NIH (P01GM099134), and a Faculty Scholar grant from the HHMI. M.A.T. is supported by the Air Force Office of Scientific Research (FA9550-15-1-0406), the NIH (R01GM114188, R01GM073981, R01CA185189, R21CA227480, R01GM127985, and P30CA016042), and CIRM (RT3-07678). We thank Rebeca Acin-Perez, Linsey Stiles, and Orian Shirihai of the UCLA Metabolomics Core for help with Seahorse XF Analyzer assays. We thank Felicia Codrea from the UCLA Flow Cytometry Core, in addition to Laurent Bentolila and Matthew Schibler from the California Nanosystems Institute Advanced Light Microscopy/Spectroscopy Laboratory for their insight and assistance. We thank Jonathan Wanagat for his critical insights during manuscript preparation. We also acknowledge Drs. Idoya Lahortiga and Luk Cox for their Library of Science and Medical Illustrations (https://www.somersault1824.com/).
Footnotes
DECLARATION OF INTERESTS
M.A.T. and P.-Y.C. are co-founders, board members, shareholders, and consultants for NanoCav, a private start-up company working on mitochondrial transfer and quantitative phase microscopy techniques and applications. T.-H.W. was an employee of NanoCav and is employed by ImmunityBio, and S.R. and K.R.N. are board members of NanoCav and employed by Immunity-Bio. The other authors report no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108562.
REFERENCES
- Abujarour R, Valamehr B, Robinson M, Rezner B, Vranceanu F, and Flynn P (2013). Optimized surface markers for the prospective isolation of high-quality hiPSCs using flow cytometry selection. Sci. Rep 3, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Pour P, Kenney MC, and Kheradvar A (2020). Bioenergetics Consequences of Mitochondrial Transplantation in Cardiomyocytes. J. Am. Heart Assoc 9, e014501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Methodol 57, 289–300. [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44 (D1), D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JM, Perales-Clemente E, Ata H, Vidal-Folch N, Liu W, Clark KJ, Xu X, Oglesbee D, Nelson TJ, and Ekker SC (2018). Engineering targeted deletions in the mitochondrial genome. bioRxiv 10.1101/287342. [DOI] [Google Scholar]
- Cherry AB, Gagne KE, McLoughlin EM, Baccei A, Gorman B, Hartung O, Miller JD, Zhang J, Zon RL, Ince TA, et al. (2013). Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells 31, 1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, Lai ST, Nonaka I, Angelini C, and Attardi G (1992). MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 89, 4221–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, and Shay JW (1982). Mitochondrial transformation of mammalian cells. Nature 295, 605–607. [DOI] [PubMed] [Google Scholar]
- Dawson ER, Patananan AN, Sercel AJ, and Teitell MA (2020). Stable retention of chloramphenicol-resistant mtDNA to rescue metabolically impaired cells. Sci. Rep 10, 14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, Noël D, and Jorgensen C (2003). Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 102, 3837–3844. [DOI] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D.j., and Horwitz E (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, and Chinnery PF (2008). Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet 83, 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayzulin RZ, Perez M, Kozhukhar N, Spadafora D, Wilson GL, and Alexeyev MF (2015). A method for mutagenesis of mouse mtDNA and a resource of mouse mtDNA mutations for modeling human pathological conditions. Nucleic Acids Res 43, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CDL, Martinez-Fernandez A, Perales-Clemente E, Li X, McDonald A, Oglesbee D, Hrstka SC, Perez-Terzic C, Terzic A, and Nelson TJ (2013). Disease-causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells 31, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannam S, Bouffi C, Djouad F, Jorgensen C, and Noël D (2010). Immunosuppression by mesenchymal stem cells: mechanisms and clinical applications. Stem Cell Res. Ther 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire M, Morais R, Quilliam MA, and Gravel D (1984). On auxotrophy for pyrimidines of respiration-deficient chick embryo cells. Eur. J. Biochem 142, 49–55. [DOI] [PubMed] [Google Scholar]
- Hämäläinen RH, Manninen T, Koivumäki H, Kislin M, Otonkoski T, and Suomalainen A (2013). Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. USA 110, E3622–E3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, Liu KJ, Chao YY, Sytwu HK, and Yen BL (2015). Assessment of the immunomodulatory properties of human mesenchymal stem cells (MSCs). J. Vis. Exp (106), e53265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Wu YT, Yu TH, and Wei YH (2016). Mitochondria in mesenchymal stem cell biology and cell therapy: From cellular differentiation to mitochondrial transfer. Semin. Cell Dev. Biol 52, 119–131. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson F (2020). FactoMineR v2.2 (RDocumentation)
- Kanehisa M, Goto S, Sato Y, Furumichi M, and Tanabe M (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y, et al. (2016). Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 18, 625–636. [DOI] [PubMed] [Google Scholar]
- Kankainen M, Gopalacharyulu P, Holm L, and Oresic M (2011). MPEA–metabolite pathway enrichment analysis. Bioinformatics 27, 1878–1879. [DOI] [PubMed] [Google Scholar]
- Kassambara A (2017). R package ggpubr v0.1.6 (RDocumentation)
- Kassambara A (2019). factoextra v1.0.6 (RDocumentation)
- Kesner EE, Saada-Reich A, and Lorberboum-Galski H (2016). Characteristics of Mitochondrial Transformation into Human Cells. Sci. Rep 6, 26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Hwang JW, Yun CK, Lee Y, and Choi YS (2018). Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep 8, 3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani T, Kami D, Matoba S, and Gojo S (2014). Internalization of isolated functional mitochondria: involvement of macropinocytosis. J. Cell. Mol. Med 18, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaira M, Hatakeyama H, Yuasa S, Seki T, Egashira T, Tohyama S, Kuroda Y, Tanaka A, Okata S, Hashimoto H, et al. (2015). Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio 5, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2015). Package ‘pheatmap’ - Pretty Heatmaps (CRAN: R-Project)
- Kukat A, Kukat C, Brocher J, Schäfer I, Krohne G, Trounce IA, Villani G, and Seibel P (2008). Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res 36, e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L, and Futschik ME (2007). Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, Calvo E, Aix E, González-Guerra A, Logan A, et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. [DOI] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Lechuga-Vieco AV, Johnston IG, Hämäläinen RH, Pellico J, Justo-Méndez R, Fernández-Toro JM, Clavería C, Guaras A, Sierra R, et al. (2019). Regulation of Mother-to-Offspring Transmission of mtDNA Heteroplasmy. Cell Metab 30, 1120–1130.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Mlody B, Singh M, Semtner M, Mah N, Auré K, et al. (2017). Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 20, 659–674.e9. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Folmes CD, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, et al. (2015). Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524, 234–238. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Kanda H, Imamura H, Inoue M, Noguchi M, Hosoda K, Kakizuka A, and Nakao K (2018). Analysis of mitochondrial function in human induced pluripotent stem cells from patients with mitochondrial diabetes due to the A3243G mutation. Sci. Rep 8, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Steffen J, Johnson ME, Fargue S, Danpure CJ, and Koehler CM (2014). Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Proc. Natl. Acad. Sci. USA 111, 14406–14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, et al. (2020). A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, and Harrow J (2015). Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm. Genome 26, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson I, Hanna MG, Wood NW, and Harding AE (1997). Depletion of mitochondrial DNA by ddC in untransformed human cell lines. Somat. Cell Mol. Genet 23, 287–290. [DOI] [PubMed] [Google Scholar]
- Nzigou Mombo B, Gerbal-Chaloin S, Bokus A, Daujat-Chavanieu M, Jorgensen C, Hugnot JP, and Vignais ML (2017). MitoCeption: Transferring isolated human MSC mitochondria to glioblastoma stem cells. J. Vis. Exp (120), 55245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patananan AN, Wu TH, Chiou PY, and Teitell MA (2016). Modifying the mitochondrial genome. Cell Metab 23, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patananan AN, Sercel AJ, and Teitell MA (2018). More than a powerplant: the influence of mitochondrial transfer on the epigenome. Curr. Opin. Physiol 3, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Rorbach J, Downes K, Szukszto MJ, Pekalski ML, and Minczuk M (2017). Macropinocytic entry of isolated mitochondria in epidermal growth factor-activated human osteosarcoma cells. Sci. Rep 7, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pek NMQ, Phua QH, Ho BX, Pang JKS, Hor JH, An O, Yang HH, Yu Y, Fan Y, Ng SY, and Soh BS (2019). Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis 10, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Clemente E, Cook AN, Evans JM, Roellinger S, Secreto F, Emmanuele V, Oglesbee D, Mootha VK, Hirano M, Schon EA, et al. (2016). Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J 35, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, et al. (2014). Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 111, E4033–E4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RH Jr., Asprer JST, Vaz C, Tanavde V, and Lakshmipathy U (2014). CD44 is a negative cell surface marker for pluripotent stem cell identification during human fibroblast reprogramming. PLoS ONE 9, e85419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing)
- Rebolledo-Jaramillo B, Su MS, Stoler N, McElhoe JA, Dickins B, Blankenberg D, Korneliussen TS, Chiaromonte F, Nielsen R, Holland MM, et al. (2014). Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 111, 15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell OM, Fruh I, Rai PK, Marcellin D, Doll T, Reeve A, Germain M, Bastien J, Rygiel KA, Cerino R, et al. (2018). Preferential amplification of a human mitochondrial DNA deletion in vitro and in vivo. Sci. Rep 8, 1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasarman F, Antonicka H, and Shoubridge EA (2008). The A3243G tRNA-Leu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet 17, 3697–3707. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Taylor RW, Turnbull DM, and Chinnery PF (2004). The epidemiology of mitochondrial disorders–past, present and future. Biochim. Biophys. Acta 1659, 115–120. [DOI] [PubMed] [Google Scholar]
- Sercel AJ, Patananan AN, Man T, Wu T-H, Yu AK, Guyot GW, Rabizadeh S, Niazi KR, Chiou P-Y, and Teitell MA (2020). Stable transplantation of human mitochondrial DNA by high-throughput, pressurized mitochondrial delivery. bioRxiv 10.1101/2020.09.15.298174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, and Singh KK (2008). A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther 7, 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AC, and Robinson AJ (2016). MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res 44 (D1), D1258–D1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Liu X, Wang B, Wang Z, Liu Y, Di C, Si J, Li H, Wu Q, Xu D, et al. (2019). Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 9, 3595–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, and Kohlbacher O (2014). OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, et al. (2016). gplots: Various R Programming Tools for Plotting Data (CRAN: R-Project; ). [Google Scholar]
- Wickham H (2019). ggplot2 v3.2.0 (RDocumentation)
- Wolf DP, Mitalipov N, and Mitalipov S (2015). Mitochondrial replacement therapy in reproductive medicine. Trends Mol. Med 21, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DP, Mitalipov PA, and Mitalipov SM (2019). Principles of and strategies for germline gene therapy. Nat. Med 25, 890–897. [DOI] [PubMed] [Google Scholar]
- Wong RCB, Lim SY, Hung SSC, Jackson S, Khan S, Van Bergen NJ, De Smit E, Liang HH, Kearns LS, Clarke L, et al. (2017). Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging (Albany NY) 9, 1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Wu TH, Clemens DL, Lee BY, Wen X, Horwitz MA, Teitell MA, and Chiou PY (2015). Massively parallel delivery of large cargo into mammalian cells with light pulses. Nat. Methods 12, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TH, Sagullo E, Case D, Zheng X, Li Y, Hong JS, TeSlaa T, Patananan AN, McCaffery JM, Niazi K, et al. (2016). Mitochondrial transfer by photothermal nanoblade restores metabolite profile in mammalian cells. Cell Metab 23, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, Zhang QC, Aghajanirefah A, Cosgun KN, Sadras T, et al. (2018). B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173, 470–484.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata N, Matsumoto Y, Omi M, Yamamoto N, and Hata R (2017). TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep 7, 15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wu H, Kang X, Liang Y, Lan T, Li T, Tan T, Peng J, Zhang Q, An G, et al. (2018). Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 9, 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim A, Koti P, Bonnard A, Marchiano F, Durrbaum M, Garcia-Perez C, Villaveces J, Gamal S, Cardone G, Perocchi F, et al. (2020). mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res 48, 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, and He QY (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw RNA-Seq reads, transcript abundance values, and processed gene count matrices are submitted to the NCBI Gene Expression Omnibus (GEO). The accession number for the RNA-seq reads reported in this paper is GEO: GSE115871. All other data are available upon request. All software used is available either commercially or as freeware. All custom code is available on GitHub at https://bitbucket.org/ahsanfasih/mitoDesigner/src/master/.