

Abstract
Cocaine-associated memories are critical drivers of relapse in cocaine-dependent individuals that can be evoked by exposure to cocaine or stress. Whether these environmental stimuli recruit similar molecular and circuit-level mechanisms to promote relapse remains largely unknown. Here, using cocaine- and stress-primed reinstatement of cocaine conditioned place preference to model drug-associated memories, we find that cocaine drives reinstatement by increasing the duration that mice spend in the previously cocaine-paired context whereas stress increases the number of entries into this context. Importantly, both forms of reinstatement require Cav1.2 L-type Ca2+ channels (LTCCs) in cells of the prelimbic cortex that project to the nucleus accumbens core (PrL→NAcC). Utilizing fiber photometry to measure circuit activity in vivo in conjunction with the LTCC blocker, isradipine, we find that LTCCs drive differential recruitment of the PrL→NAcC pathway during cocaine- and stress-primed reinstatement. While cocaine selectively activates PrL→NAcC cells prior to entry into the cocaine-paired chamber, a measure that is predictive of duration in that chamber, stress increases persistent activity of this projection, which correlates with entries into the cocaine-paired chamber. Using projection-specific chemogenetic manipulations, we show that PrL→NAcC activity is required for both cocaine- and stress-primed reinstatement, and that activation of this projection in Cav1.2-deficient mice restores reinstatement. These data indicate that LTCCs are a common mediator of cocaine- and stress-primed reinstatement. However, they engage different patterns of behavior and PrL→NAcC projection activity depending on the environmental stimuli. These findings establish a framework to further study how different environmental experiences can drive relapse, and supports further exploration of isradipine, an FDA-approved LTCC blocker, as a potential therapeutic for the prevention of relapse in cocaine-dependent individuals.
Introduction
Cocaine-associated memories are critical drivers of craving and relapse. In both humans and rodent models, relapse can be triggered by several environmental factors, including re-exposure to the drug itself or exposure to stressful stimuli, which act to renew the salience of drug-associated contexts or cues as well as cocaine-associated memories1. Understanding how different environmental stimuli trigger these cocaine-associated memories to induce relapse at a molecular and neural circuit level, and whether they recruit similar or distinct mechanisms, is crucial for the development of more effective pharmacotherapies to reduce relapse risk.
The Cav1.2 L-type calcium channel (LTCC) serves as a potential target for mediating relapse behavior. Cav1.2 channels are key mediators of long-term memory and synaptic plasticity2–4. Mutations in the gene CACNA1C, which codes for Cav1.2, have been associated with several neuropsychiatric disorders, including bipolar disorder and schizophrenia5, 6, which show high comorbidity with substance use disorders7, 8,9, and carriers of risk alleles demonstrate altered reward-related behaviors and associated neural activity10, 11. As several LTCC blockers, which target both Cav1.2 and the other brain-specific LTCC Cav1.3, are used clinically in the treatment of hypertension, studies have examined the potential efficacy of targeting LTCCs in addicted individuals6. Findings from these studies have been inconclusive, potentially due to small sample sizes or lack of placebo controls, but recent rodent studies demonstrate efficacy of pharmacological blockade of LTCCs by these antihypertensive medications in attenuating cocaine seeking behavior12, 13 and cocaine-associated memories14. In rodent models, Cav1.2 has proven critical for cocaine-related behaviors15–18 and mediates the effects of stress on brain and behavior19–21, suggesting that Cav1.2 channels could be a common mediator of the effects of cocaine and stress on relapse. However, the specific role of Cav1.2 channels in models of relapse has yet to be explored.
The prefrontal cortex (PFC), particularly via projections to the nucleus accumbens (NAc), has been implicated in reward-related behaviors, including relapse following exposure to cocaine and stress. Pharmacological manipulations and classical disconnection studies have implicated the projection from the prelimbic PFC (PrL) to the core of the NAc (NAcC) in relapse following exposure to cocaine challenge22–24 and acute stress25. However, the endogenous activity of this projection associated with relapse to either of these stimuli has not been characterized, and the role of LTCCs in the recruitment of this projection has not been explored. Intriguingly, human carriers of CACNA1C mutations demonstrate changes in PFC structure, function, and connectivity26, 27 suggesting that these mutations could lead to circuit-level dysfunction implicated in addiction and relapse.
Here we utilize cocaine- and stress-primed reinstatement of extinguished conditioned place preference (CPP), a preclinical model to study cocaine-associated memories, along with genetic knockout mice, pharmacology, and viral-mediated knockdown strategies to explore the role of Cav1.2 channels in reinstatement behavior. We demonstrate that cocaine and stress reinstate cocaine place preference via different CPP behavioral measures, but that both require Cav1.2 channels within the PrL→NAcC projection. Using in vivo fiber photometry calcium recordings to monitor PrL→NAcC activity, we find that both cocaine- and stress-primed reinstatement recruit the PrL→NAcC projection cells in an LTCC-dependent manner, however cocaine and stress priming induce different patterns of PrL→NAcC activity. Using projection-specific chemogenetics, we demonstrate the necessity of the PrL→NAcC projection for cocaine- and stress-primed reinstatement and the sufficiency of this projection to induce reinstatement in Cav1.2 deficient mice that typically do not demonstrate reinstatement. These data indicate that PrL→NAcC Cav1.2 channels are required for reinstatement of cocaine-associated memories following exposure to both cocaine and stress via recruitment of divergent behavioral and circuit-level mechanisms.
Materials and Methods
Animals
Male adult wild-type (WT) mice (C57/BL/6J; the Jackson Laboratory) and transgenic mutant mice on the C57BL/6J background were used for all experiments. Cav1.2 heterozygous (Cav1.2+/) mice and cacna1c-floxed mice were generated as previously described3, 28 and bred in the Weill Cornell Medicine facilities. Animals were group housed, maintained on a 12 hr light/dark cycle, and were given food and water ad libitum. All testing was conducted during the 12 hr light phase. Animals were assigned to groups based on their genotypes (non-randomized) or were randomly assigned to experimental groups (randomized). All animal procedures were conducted in accordance with the rules of Weill Cornell Medicine Institutional Animal Care and Use Committee following the National Institutes of Health guide for the care and use of Laboratory animals.
Drugs
For cocaine conditioned place preference (CPP) experiments, cocaine hydrochloride (Sigma-Aldrich) was dissolved in 0.9% saline at a concentration of 1.0 mg/ml to be injected intraperitoneally (i.p.) at 0.01 ml/g body weight for a final dosage of 10 mg/kg. Isradipine (Alomone Labs) was dissolved in saline (16% ethanol) at a concentration of 0.12 mg/ml to be injected i.p. at 0.01 ml/g body weight for a final dosage of 1.2 mg/kg. Isradipine or vehicle (16% ethanol in saline) was injected 25 minutes prior to behavioral testing. For chemogenetic experiments, clozapine-N-oxide (CNO, Enzo Life Sciences) was dissolved in saline at a concentration of 0.3 mg/ml to be injected I.P. at 0.01 ml/g body weight for a final dosage of 3.0 mg/kg. CNO or saline was injected 45 minutes prior to behavioral testing.
Cocaine Conditioned Place Preference
A modified cocaine CPP protocol (outlined in Fig 1A) was performed in a three-chamber apparatus (Med Associates Inc.). During the baseline test, mice were placed into the grey central chamber of a three-chamber CPP apparatus for a one minute habituation period. Mice were then given free access to all three chambers for 20 minutes and the time spent in each chamber was recorded. For the following three days, in the morning, mice were given a systemic injection of saline (0.01ml/g body weight, i.p.) and were confined to the outer chamber that they preferred during the baseline test for 20 minutes. In the afternoon, mice were given a systemic injection of cocaine (10mg/kg, i.p) and were confined to the opposite outer chamber for 20 minutes. On the following day (acquisition test), mice were given free access to all three chambers and the amount of time spent in each chamber was recorded. Following acquisition, mice went through extinction training, as described previously15, for 3-9 days, during which they were given free access to all three chambers without any drug exposure. Mice were then tested for cocaine-primed reinstatement or stress-primed reinstatement. During cocaine-primed reinstatement, mice were injected with an acute cocaine priming injection (10mg/kg, i.p.) and placed immediately into the CPP apparatus. During stress-primed reinstatement, mice were exposed to a set of fifteen unpredictable footshocks (0.7mA, 1sec) in a fear conditioning chamber (Coulbourn, Whitehall, PA, USA) over the course of 5 minutes, and were then placed immediately into the CPP apparatus. For all CPP experiments, data are presented as preference score (time spent in the cocaine-paired chamber minus time spent in the saline-paired chamber).
Figure 1.
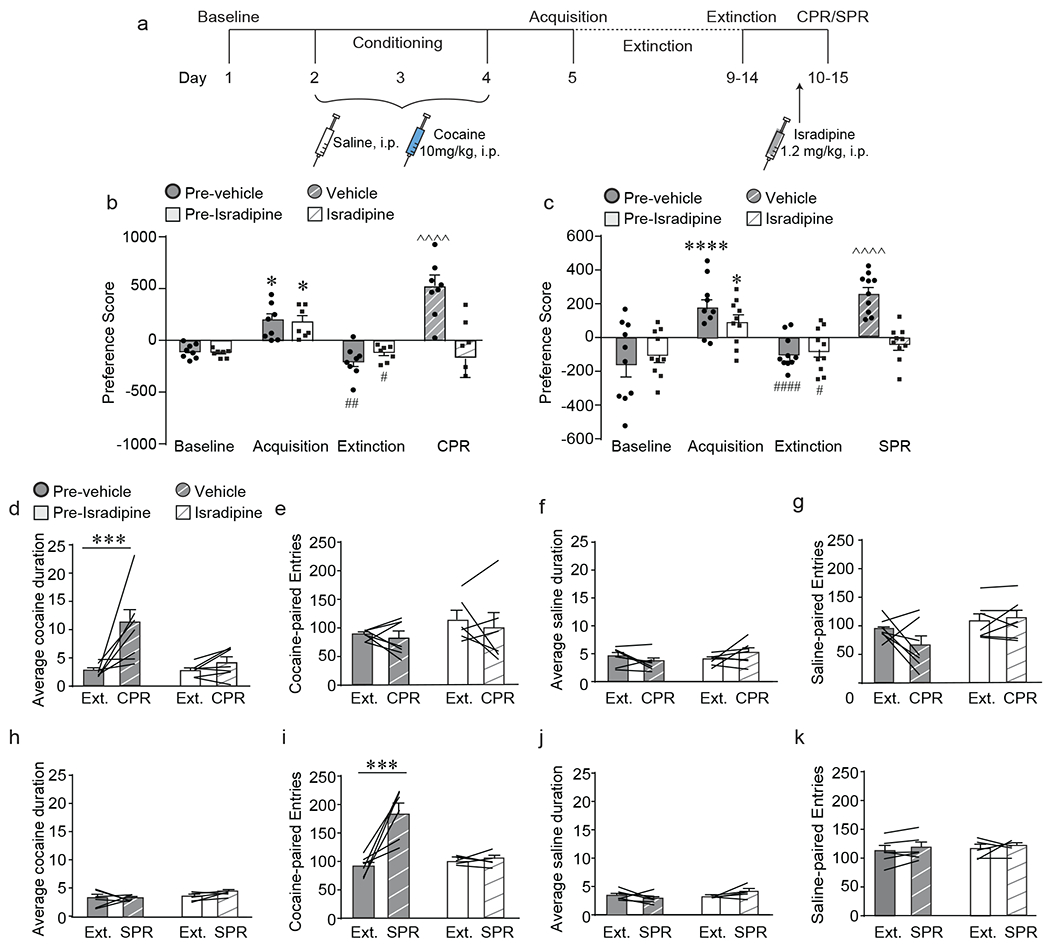
LTCCs are required for cocaine- and stress-primed reinstatement of cocaine conditioned place preference, a) Experimental timeline of cocaine conditioned place preference (CPP) acquisition, extinction and cocaine-primed (CPR) or stress-primed (SPR) reinstatement protocol and isradipine treatment, b) Prior to treatment with either vehicle (pre-vehicle) or isradipine (pre-isradipine), all mice acquired (*p < 0.05, Bonferroni post-hoc baseline vs. acquisition) and extinguished (##p < 0.01, #p< 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. Cocaine-primed reinstatement (CPR) significantly increased preference score in vehicle-treated mice (^^^^p < 0.0001, Bonferroni post-hoc extinction vs. CPR; n = 8) but not in isradipine-treated mice (n = 7). c) Prior to treatment with either vehicle (pre-vehicle) or isradipine (pre-isradipine), all mice acquired (****p < 0.0001, *p < 0.05, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, #p< 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. Stress-primed reinstatement (SPR) significantly increased preference score in vehicle-treated mice (^^^^p < 0.0001, Bonferroni post-hoc extinction vs. SPR; n = 10) but not in isradipine-treated mice (n = 10). d) CPR significantly increased average cocaine-paired duration in vehicle-treated mice (***p < 0.001, Bonferroni post-hoc extinction vs. CPR; n = 7) but not in isradipine-treated mice (n = 7). e) CPR has no effect on number of cocaine-paired entries. f) CPR had no effect on average saline-paired duration or g) number of saline-paired entries. h) SPR had no effect on average cocaine-paired duration. i) SPR significantly increased the number of cocaine-paired entries in vehicle-treated mice (***p < 0.001, Bonferroni post-hoc extinction vs. SPR; n = 6) but not in isradipine-treated mice (n = 5). j) SPR had no effect on average saline-paired duration, or k) number of saline-paired entries. Data are presented as mean + SEM.
Elevated plus maze
The elevated plus maze was performed as previously described20. Briefly, mice were placed onto a plus-shaped maze with two open arms (length 30.5cm) and two arms enclosed by walls (15.2 cm height). Mice were allowed to freely explore the apparatus for 5 minutes and time spent in the open arms was recorded using AnyMaze software (Stoelting).
Spontaneous Alternation
The spontaneous alternation task was used to measure working memory as previously described20. Mice were placed into a Y-shaped apparatus with three arms (33cm x 7.6 cm x 38.1 cm) and were allowed to freely explore for 5 minutes. A spontaneous alternation was recorded when mice entered all three arms of the maze without repeating entry into the same arm. Percentage of spontaneous alternations was calculated by dividing number of spontaneous alternations by the total number of alternations and multiplying by 100.
Corticosterone ELISA
An ELISA was utilized to measure circulating corticosterone levels in serum. Trunk blood was collected and incubated at room temperature for 60 minutes. Samples were spun at 1200 x g for 15 min and supernatant was collected and stored at −80 degrees. Corticosterone levels were analyzed using the corticosterone high sensitivity EIA (Immunodiagnostic Systems, AC-15F1).
Region-specific gene knockdown
Focal knockdown of cacna1c within bilateral prelimbic or infralimbic cortices was achieved using stereotaxic microinjection as previously described15, 29. Mice were anesthetized with 2% isoflurane and placed into a stereotaxic apparatus. Adeno-associated viral vectors (AAV2/2, Vector BioLabs) expressing Cre recombinase (AAV-Cre-GFP) or GFP (AAV-GFP) was microinjected into bilateral PrL or IL of cacna1c-floxed mice at a rate of 0.05 μl/min for a total volume of 0.20μl/hemisphere. The stereotaxic coordinates used for PrL were anteroposterior (AP): +2.0 mm, mediolateral (ML): ±0.25 mm, and dorsoventral (DV): −2.25 mm and for IL were AP: +1.5 mm, ML: ±0.35 mm, and DV: −3.2 mm. Behavioral experiments began a minimum of 4 weeks following surgery to allow for maximum viral expression. Fluorescence immunohistochemistry for GFP (as described below) was used to confirm injection placement. A small proportion of mice (<15%) with GFP expression outside of the PrL or IL was excluded from behavioral analysis.
Quantitative Real-Time PCR (QPCR)
Mice were decapitated and brains were dissected and sectioned on a 1mm brain block. GFP labeled tissue was visualized using GFP goggles (BLS-Ltd.com). RNA was isolated from brain tissue and QPCR was performed as previously described16. Cacna1c (QuantiTect Primer assay QT00150752; Qiagen) and Gapdh (QuantiTect Primer assay QT01658692; Qiagen) mRNA levels were measured using mRNA-specific primers. Cycle threshold (Ct) values for target genes were normalized to the Gapdh. Each experiment was performed in triplicate, and the values were averaged.
Circuit-specific gene knockdown
Knockdown of cacan1c within bilateral NAcC-projecting PrL cells was achieved using stereotaxic surgery as described above. A flp-dependent AAV expressing Cre recombinase (AAV-fDIO-Cre-GFP) was injected into bilateral PrL (AP: 2.0 mm, ML: ±0.25 mm, DV: −2.25 mm) and a retrograde AAV expressing flp (pAAV-EF1a-mCherry-IRES-Flpo) was injected into bilateral NAcC (AP: +1.0mm, ML: ±1.20 mm, DV: −4.75 mm) of cacna1c-floxed mice, leading to selective expression of Cre recombinase in PrL cells projecting to the NAcC. pAAV-EF1a-mCherry-IRES-flpo (Addgene viral prep # 55634-AAVrg; http://n2t.net/addgene:55634 ; RRID:Addgene_55634) was a gift to Addgene from Karl Deisseroth. Behavioral experiments began a minimum of 4 weeks following surgery to allow for maximum viral expression. All animals underwent both cocaine-primed and stress-primed reinstatement (CPR and SPR, respectively) with a minimum of 1 day of extinction in between. The order of reinstatement test was counterbalanced between animals.
Histological Preparation
Following behavior, mice were transcardially perfused with 40ml of 0.1M phosphate buffer (PB) followed by 40ml of 4% paraformaldehyde at pH 7.4. Brains were dissected and post-fixed in 4% PFA overnight and stored in 0.1M PB with 30% sucrose and 0.1% sodium azide for a minimum of 72 hours prior to sectioning.
GFP Immunohistochemistry
Fixed brains were sectioned at 50μm using a microtome and placed in cryoprotectant solution (30% sucrose and 30% ethylene glycol in PB)30 at −20°C. Sections were washed twice in 0.1M PB with 0.3% tritonX-100. Next, sections were incubated in blocking solution (0.1M PB with 1% normal goat serum and 1% BSA) for 1hr at room temperature followed by primary antibody incubation with chicken anti-GFP (1:2000, Aves Laboratories) in blocking solution overnight at room temperature. Sections were then incubated with Alexa Fluor 488 goat anti-chicken IgG secondary antibody in 0.1M PB with 0.3% tritonX-100 for 1hr at room temperature. Sections were imaged using an epifluorescence microscope (Leica DM550B with Leica Application Suite Advanced Fluorescence 3.0.0 build 8134 software).
RNAscope in situ hybridization
Fixed brains were sectioned at 30μM using a cryostat and placed in RNAse-free cryoprotectant (Milner et al., 2011). Sections were mounted in PBS onto charged Superfrost slides (Fisher Scientific) and were allowed to air dry for ~18 hours. All incubations took place at 40°C in the HybEZ oven using the EZ-Batch system (ACDbio). Sections were incubated in target retrieval buffer (Cat No. 322001, ACDbio) for 12min and washed two times with deionized water (dH20). Sections were then incubated in Protease IV (Cat No. 322336, ACDbio) for 30min, washed five times with dH20, and incubated in either positive probe control for Polr2a (Cat No. 320881, ACDbio) or cacna1c probe (Cat No. 445451, ACDbio) for two hrs. Two washes occurred in-between the following incubations with wash buffer (Cat No. 320058). Sections were incubated for 30 min with AMP1 (Cat No. 323101), AMP2 (Cat No. 323102) and AMP3 (Cat No. 323103). Next, sections were incubated in HRP C1 (Cat No. 323104) for 15 min and CY5 (1:1000) in TSA buffer (Cat No. 322809). Sections were next incubated in an HRP blocker (Cat No. 323107) for 15 min. In order to examine co-localization of GFP-tagged virus with cacan1c mRNA, immunohistochemistry for GFP was performed (as described above) on the same slides as RNAscope. Slides were cover slipped with Prolong Gold containing DAPI (Thermo Scientific). Images were taken using an Olympus 1X81 confocal microscope with Fluoview FV1000 software.
Quantitative analysis to measure Cav1.2 mRNA levels was performed as previously described for in situ hybridization31 with the following modifications. Quantitation was performed using Image J software (NIH, Bethesda, MD). GFP-labeled neurons were visualized under a 60x oil immersion lens and confocal images collected at low fluorescent intensity to identify discrete Cav1.2-labeled mRNA dots, for AAV-GFP control and Flp-FRT Cav1.2 knockout mice. Using Image J, the soma of each GFP-labeled neuron was outlined, and the overlying dots (representing Cav1.2 transcripts) were counted. Both the area of the neuronal soma, μm2, and the number of dots present were recorded. The intensity of labeling of each neuron was computed as dots/1000μm2. Two-three animals were used for each condition, and 26-30 neurons were counted per animal.
In vivo fiber photometry calcium recording
Fiber photometry32, 33 was performed to measure in vivo calcium dependent activity dynamics during cocaine CPP. A Cre-dependent AAV expressing GCaMP6s (AAV1.Syn.DIO.GCaMP6s, Penn Vector Core) was injected unilaterally in PrL (AP: +2.00 mm, ML: −0.25 mm, DV: −2.25 mm) and a retrograde AAV expressing Cre recombinase (retro-AAV2-CAG-Cre, UNC Vector Core, Ed Boy den) was injected into the ipsilateral NAcC (AP: +1.0 mm, ML: −1.20 mm, DV: −4.75 mm), thereby expressing GCaMP6s only in those PrL neurons that project to the NAcC. During the same surgery, a 400μm diameter optical fiber (Doric) was implanted above the PrL (AP: +2.00 mm, ML: −0.25 mm, DV: −2.15 mm) and was secured with Metabond. Prior to behavioral testing, mice were habituated to a patch cord attached to the implanted optical fiber for 1 minute in their home cage. During behavioral testing, fiber photometry recordings were taken during the first 10 minutes of the baseline test, acquisition test, extinction test, and reinstatement tests. Behavior was recorded by a video camera above the CPP apparatus and behavior was hand-scored by a blinded investigator. Following behavioral testing, fluorescence microscopy was utilized to confirm GCAMP6s expression and optical fiber placement. A small proportion of mice (<10%) with fiber placement or GCaMP6s expression outside of the PrL was excluded from fiber photometry analysis.
To excite GCaMP6s, light from a 470nm LED (Thorlabs, M470F3) modulated at a frequency of 521 Hz was passed through a filter (Semrock, FF02-472/30), reflected by a dichroic (Semrock, FF495-Di03) and coupled to a 0.48 NA, 400um core optical fiber patch cord (Doric). Emitted fluorescence traveled back through the patch cord, passed through the dichroic, a filter (Semrock, FF01-535/50), and was focused onto a photodetector (Newport, Model 2151). The modulated signal passed from the photodetector to a RP2.1 real-time processor (Tucker Davis Technologies) where it was demodulated and low-pass filtered using a corner frequency of 15 Hz. TTL pulses denoting the start of behavioral trials were passed to the processor in real time for alignment of calcium recordings to behavioral measures.
Pre-Entry Analysis:
Data were analyzed using a custom MATLAB (Mathworks) script. Raw fluorescence signals were first detrended to account for any photobleaching by fitting a third degree polynomial and subtracting this polynomial from the raw signal trace. Next, fluorescence signal was normalized (transformed to ΔF/F) by taking the median value of the overall trace, subtracting this median value from all data points and then dividing by the median. These ΔF/F values were then translated such that 0% ΔF/F was equivalent to the 10th percentile of the data points. To quantify the fluorescence level prior to zone entries during CPP, we calculated the mean ΔF/F over the five seconds prior to the zone entry time (as determined from behavioral scoring) for every zone entry during behavioral testing.
Event Frequency Analysis:
Data were analyzed using custom MATLAB (Mathworks) scripts. Fluorescence signals were normalized (transformed to ΔF/F) by taking the median value of the signal during a 20 second (+/− 10 seconds) window around each data point, subtracting this median from the data point and then dividing by the median. Significant calcium events were identified as periods of time in which ΔF/F rose above 2.91 median absolute deviations (MADs) from baseline32 and remained at least 2 MADs above baseline. The total number of these identified events in each behavioral session was divided by the duration of time of that session to determine the overall event frequency. To determine the event frequency specific to each CPP chamber during a session, the total number of calcium events that initiated while the animal was present in a chamber was divided by the duration of time the animal spent in that chamber during the behavioral session.
Chemogenetic manipulation of neural circuitry
Chemogenetics was utilized to manipulate neural projections during behavioral testing. In WT mice, a Cre-dependent AAV expressing the hM4Di inhibitory DREADD (AAV2-hSyn-DIO-hM4DGi-mCherry34) or the hM3Dq excitatory DREADD (AAV2-hSyn-DIO-hM3Dq-mCherry34) was injected into bilateral PrL (AP: +2.00 mm, ML: ±0.25 mm, and DV: −2.25 mm) and a retrograde AAV expressing Cre recombinase (retro-AAV-CAG-Cre) was injected into bilateral NAcC (AP: +1.0 mm, ML: ±1.20 mm, and DV: −4.75 mm). In Cav1.2+/− mice, a Cre-dependent AAV expressing the hM3Dq excitatory DREADD (AAV2-hSyn-DIO-hM3DGq-mCherry) was injected into bilateral PrL (AP: +2.00 mm, ML: ±0.25 mm, and DV: −2.25 mm) and a retrograde AAV expressing Cre recombinase (rAAV-CAG-Cre) was injected into bilateral NAcC (AP: +1.0 mm, ML: ±1.20 mm, and DV: −4.75 mm). pAAV-hSyn-DIO-hM4D(Gi)-mCherry (Addgene viral prep # 44362-AAV2; http://n2t.net/addgene:44362; RRID:Addgene_44362) and pAAV-hSyn-DIO-hM3D(Gq)-mCherry (Addgene viral prep # 44361-AAV2; http://n2t.net/addgene:44361 ; RRID:Addgene_44361), gifts to Addgene from Bryan Roth, were purchased from Addgene, Watertown, MA. Clozapine-N-oxide (CNO, 3.0 mg/kg, I.P.) was injected 45 minutes prior to the start of behavioral testing to activate the DREADD. Two control groups were utilized; one group was injected with the Cre-dependent DREADD only into the PrL (no retro-AAV injected into the NAcC) and treated with CNO 45 minutes prior to behavioral testing (Sham + CNO). The second group was injected with both the Cre-dependent DREADD (into the PrL) and the retrograde Cre recombinase (into the NAcC) and later treated with saline 45 prior to behavioral testing (DREADD + vehicle). A small proportion of mice (<10%) with DREADD expression outside of the PrL was excluded from behavioral analysis.
Statistical Analysis
Statistical analyses were conducted using Graphpad Prism 7.0 or MATLAB (Mathworks) custom scripts. For analysis of behavioral data, a two-way repeated measures (RM) ANOVA was utilized. For behavioral data with significant interactions, Bonferroni corrected post-hoc tests were conducted. For pre-entry fiber photometry experiments, a mixed linear effects model was utilized to compare the average amplitude five seconds prior to CPP chamber entry, with the animal modeled as a random effect and CPP chamber (cocaine vs. saline) was modeled as a fixed effect. For event frequency fiber photometry analysis, two-way RM-ANOVA was used. Pearson’s correlation coefficients were used to quantify associations between behavioral and fiber photometry data. When comparing two sets of normally distributed data, unpaired two-tailed t-tests were utilized. Equality of variances were tested with an F-test (unpaired t-tests) and the Brown-Forsythe and Bartlett tests (ANOVAs). Sample sizes were based on previous work in the lab and are similar to other published work in the field. Exact sample sizes for each experiment are indicated in figure legends. Statistical significance was determined by a p value <0.05. All data are displayed in figures as mean + standard error of the mean (SEM).
Results
Pharmacological blockade of L-type calcium channels attenuates cocaine- and stress-primed reinstatement of cocaine place preference via different behavioral mechanisms
We utilized cocaine conditioned place preference (CPP), in which mice are conditioned to associate a particular context with the rewarding effects of cocaine to model cocaine-associated memories. Once conditioned, mice demonstrate a preference for the cocaine-associated context that can be extinguished following several drug-free exposures. Cocaine-associated memories can be reinstated by exposure to various stimuli, including cocaine and stress, in order to model the effects of these environmental insults on relapse behavior.
We first tested the effect of pharmacological blockade of L-type calcium channels (LTCCs) on reinstatement of cocaine place preference following a priming injection of cocaine or exposure to intermittent footshock stress. C57BL/6J mice were trained and tested in cocaine CPP and extinction, and 25 minutes prior to either the cocaine- or stress-primed reinstatement test (CPR and SPR, respectively), mice were treated with the LTCC inhibitor isradipine (1.2mg/kg, i.p.) or vehicle (Fig. 1a). This dose of isradipine was chosen based on previously published work demonstrating behavioral and electrophysiological efficacy in blocking LTCCs12, 14. In a cohort tested for cocaine-primed reinstatement, all mice acquired and extinguished cocaine CPP. In vehicle-treated mice, a priming injection of cocaine reinstated preference for the previously cocaine-paired chamber, an effect that was attenuated by pre-treatment with isradipine (Fig. 1b; interaction (drug x day) F (3,39) = 12.15, p < 0.0001, two-way RM-ANVOA). In a cohort tested for stress-primed reinstatement, all mice acquired and extinguished cocaine CPP. During the reinstatement test, priming with acute stress reinstated preference for the previously cocaine-paired chamber in vehicle-treated mice but not in isradipine-treated mice (Fig. 1c; interaction (drug x day) F(3, 54) = 7.98, p = 0.0002, two-way RM-ANOVA). These data indicate that acute LTCC signaling is necessary for reinstatement of cocaine-associated memories induced by cocaine and stress priming.
An increase in preference for the cocaine-paired chamber can be driven by either an increase in the time spent in the cocaine-paired chamber during each entry (duration) or by an increase in number of entries into the cocaine-paired chamber. We thus evaluated the effect of cocaine or stress priming on these specific CPP behavioral measures. Interestingly, cocaine and stress reinstated preference for the cocaine-paired chamber by altering different behaviors: cocaine-primed reinstatement increased the average duration that mice spent in the cocaine-paired chamber (Fig. 1d; interaction drug x day F(1,12) = 5.91, p = 0.03, two-way RM-ANOVA) with no change in the number of cocaine-paired entries (Fig. 1e), whereas stress-primed reinstatement increased the number of entries into the cocaine-paired chamber (Fig. 1i; interaction drug x day F(1,9) = 15.11, p = 0.004, two-way RM-ANOVA) without altering the average duration (Fig. 1h). Pretreatment with isradipine blocked these increases in duration and entries (Fig. 1d, i), indicating a role of LTCCs in mediating the behavioral changes driving reinstatement of cocaine-associated memories. No change in average duration (Fig. 1f, j) or number of entries (Fig. 1g, k) was observed in the saline-paired chamber in vehicle or isradipine-treated mice during either cocaine or stress-primed reinstatement. Additionally, no difference in locomotor activity was observed between vehicle and isradipine-treated mice for either stimulus (Supp. Fig 1a, CPR: main effect of day F(1,12) = 40.18, p < 0.0001; Supp. Fig. 1b, SPR: main effect of day F(1,9) = 94.36, p < 0.0001, two-way RM-ANOVA ). Interestingly, neither average duration (Supp. Fig. 1c) nor number of entries (Supp. Fig. 1d) appear to be the driving factor during acquisition of cocaine place preference, suggesting that different mechanisms are driving the increase in preference score observed during acquisition versus reinstatement. Taken together, the above data demonstrate that cocaine and stress reinstate cocaine-associated memories by altering different CPP behavioral measures and suggests this could occur via divergent mechanisms, although both requiring LTCCs.
Focal deletion of Cav1.2 in prelimbic cortex inhibits cocaine- and stress-primed reinstatement of cocaine-associated memories
Given that Cav1.2 channels represent the primary LTCC isoform expressed in the brain35, 36, and as CACNA1C has been linked to reward processing6, 10, 11, we next tested the role of Cav1.2 channels in reinstatement behavior. Cav1.2 heterozygous (Cav1.2+/−) mice were trained and tested in cocaine CPP (Fig. 2a; Cav1.2 heterozygous animals were utilized as homozygous knockout mice are embryonic lethal37). In a cohort tested for cocaine-primed reinstatement, both Cav1.2+/− mice and their wild-type littermates (WT) acquired and extinguished cocaine CPP. However, cocaine priming reinstated preference for the cocaine-paired chamber in WT but not in Cav1.2+/− mice (Fig. 2b; interaction (group x day), F(3,33) = 3.14, p = 0.04, two-way RM-ANOVA). Similarly, in a cohort tested for stress-primed reinstatement, all mice acquired and extinguished cocaine CPP, but acute stress reinstated preference for the cocaine-paired chamber in WT but not Cav1.2+/− mice (Fig. 2c; interaction (group x day), F(3,42) = 8,54, p = 0.0002, two-way RM-ANOVA). This lack of stress-primed reinstatement in Cav1.2+/− mice was not due to a lack of stress-responsivity, as Cav1.2+/− mice demonstrated the expected increase in anxiety-like behavior in the elevated plus maze (Supp. Fig 2a; main effect of stress F(1,30) = 13.88, p < 0.008, two-way ANOVA), behavioral deficits in the spontaneous alternation task (Supp. Fig. 2b; main effect of stress F(1,30) = 13.4, p = 0.001, two-way ANOVA), and increased serum corticosterone (Supp. Fig. 2c; main effect of stress, F(1,18) = 46.27, p < 0.0001, two-way ANOVA) following exposure to acute stress. These data indicate that Cav1.2 LTCCs are required for both cocaine- and stress-primed reinstatement of cocaine-associated memories.
Figure 2.
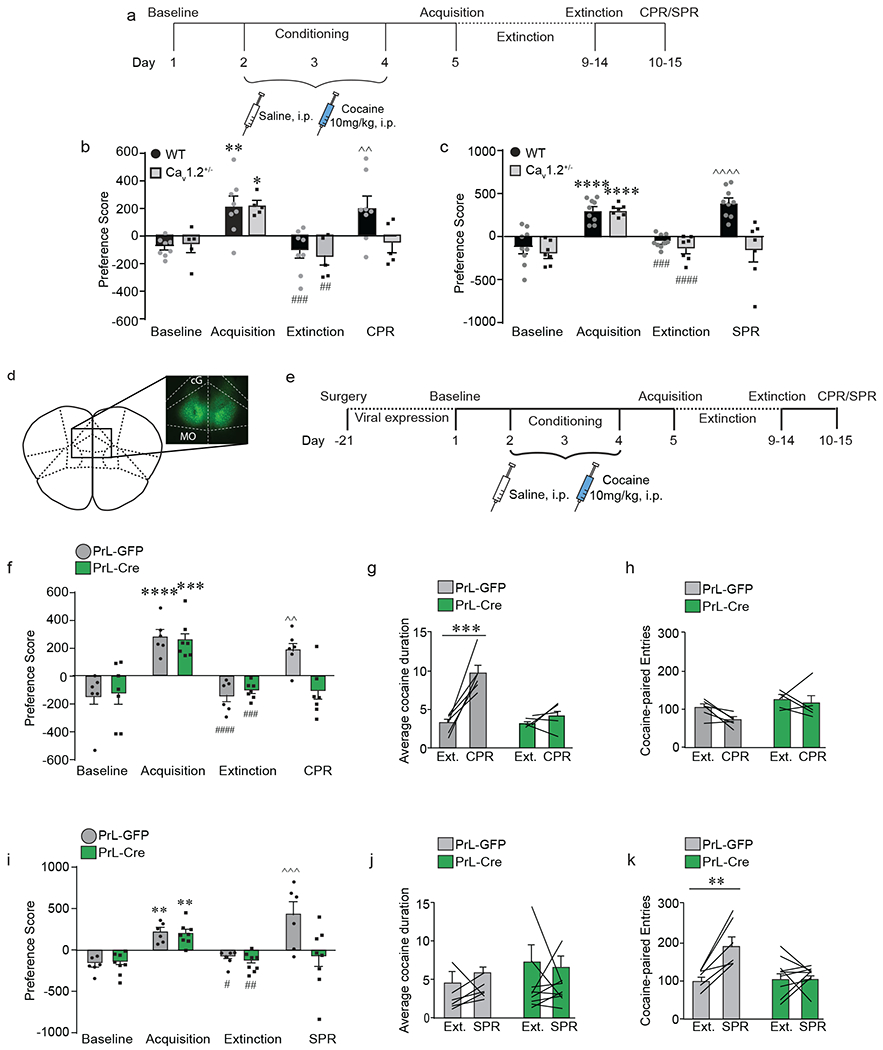
Prelimbic (PrL) cortex Cav1.2 channels are required for stress- and cocaine-primed reinstatement of cocaine conditioned place preference. a) Experimental timeline for surgery and behavioral experiments. b) Both wild-type (WT) and Cav1.2 heterozygous (Cav1.2+/−) mice acquired (**p < 0.01, *p < 0.05, Bonferroni post-hoc baseline vs. acquisition) and extinguished (###p < 0.001, ##p < 0.01, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. CPR significantly increased preference score in WT ^^p < 0.01, bBonferroni post-hoc extinction vs. SPR; n = 8) but not in Cav1.2+/− mice (n = 5). c) Both WT and Cav1.2+/− mice acquired (****p < 0.0001, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, ###p < 0.001, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. SPR significantly increased preference score in WT (^^^^p < 0.0001, Bonferroni post-hoc extinction vs. SPR; n = 9) but not in Cav1.2+/− mice (n = 7). d) Representative image of bilateral GFP expression in cacan1c-floxed mice who were injected with either AAV2-GFP or AAV2-Cre to knockdown Cav1.2 expression in bilateral PrL. Cg, cingulate cortex, MO, medial orbitofrontal cortex. e) Experimental timeline of viral injection and cocaine CPP. Following 21 days for viral expression, mice were tested using the cocaine CPP protocol. f) Mice injected with AAV2-GFP (PrL-GFP) or AAV2-Cre (PrL-Cre) into bilateral PrL acquired (****p < 0.0001, ***p < 0.001, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, ###p < 0.001, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. CPR significantly increased preference score in PrL-GFP mice (^^p < 0.001, Bonferroni post-hoc extinction vs. CPR; n = 6) but not in PrL-Cre mice (n = 7). g) CPR significantly increased the average cocaine-paired duration in PrL-GFP mice (***p < 0.001, Bonferroni post-hoc extinction vs. CPR; n = 6) but not in PrL-Cre mice (n = 5). h) CPR had no effect on the number of cocaine-paired entries in either PrL-GFP or PrL-Cre mice. i) PrL-GFP and PrL-Cre mice acquired (**p < 0.01, Bonferroni post-hoc baseline vs. acquisition) and extinguished (##p < 0.01, #p < 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. SPR significantly increased preference score in PrL-GFP mice (^^^p < 0.001, Bonferroni post-hoc extinction vs. CPR; n = 6) but not in PrL-Cre mice (n = 8). j) SPR had no effect on average cocaine duration in either group. k) SPR significantly increased the number of cocaine-paired entries in PrL-GFP (**p < 0.01, Bonferroni post-hoc extinction vs. SPR; n = 6) but not in PrL-Cre mice (n = 9). Data are presented as mean + SEM.
We next tested the role of Cav1.2 channels in the PFC, as this brain region has been implicated in several models of reinstatement behavior, with activation of the prelimbic (PrL) subregion shown to promote reinstatement38, 39 and activation of the infralimbic (IL) region found to reduce reinstatement40–42. Additionally, neuropsychiatric-linked SNPs in CACNA1C have been associated with changes in structure, function, and connectivity within the PFC in humans26, 43, 44, underscoring the significance of Cav1.2 function within this region.
To this end, we generated focal knockdown of Cav1.2 within either the PrL (Fig. 2d; Supp. Fig. 3a) or IL (Supp. Fig. 4a) by bilateral stereotaxic injection of AAV-Cre in cacan1c-floxed mice and tested them in cocaine CPP (Fig. 2e). Quantitative real time PCR indicated an ~50% knockdown (AAV-GFP, 1.00 ± 0.06 vs. AAV-Cre 0.52 ± 0.04; p < 0.0001). Focal deletion of Cav1.2 within the PrL had no effect on acquisition or extinction, but significantly attenuated reinstatement of cocaine place preference following both cocaine priming (Fig. 2f; interaction (Cav1.2 x day) F(3,33) = 4.05, p = 0.015, two-way RM-ANOVA) and stress priming (Fig. 2i; interaction (Cav1.2 x day) F(3,36) = 6.04, p = 0.002, two-way RM-ANOVA). In contrast, while focal deletion of Cav1.2 within the IL (Supp. Fig 4a) did not impact acquisition, it blocked extinction behavior (Supp. Fig 4b; interaction (Cav1.2 x day) F(2,36) = 3.05, p = 0.06, two-way RM-ANOVA), consistent with a role for this subregion in extinction of cocaine-associated behaviors40–42.
When examining the duration and number of entries, we again found that cocaine priming in control mice reinstated place preference by increasing the average duration mice spent in the cocaine-paired chamber, and focal knockdown of PrL Cav1.2 blocked this increase (Fig 2g; interaction (Cav1.2 x day) F(1,9) = 13.18, p = 0.006, two-way RM-ANOVA). As before (Fig 1e–g) no change was observed in the number of cocaine-paired entries (Fig. 2h), saline-paired duration (Supp. Fig. 4c), or in the number of saline-paired entries (Supp. Fig. 4d). Alternatively, stress reinstated place preference by increasing the number of entries into the cocaine-paired chamber, an effect that was abolished in mice with focal knockdown of PrL Cav1.2 (Fig. 2k interaction (Cav1.2 x day) F(1,13) = 10.13, p = 0.007, two-way RM-ANOVA). No change was observed in the average cocaine-paired duration (Fig. 2j), the number of saline-paired entries (Supp. Fig. 4f), or average saline-paired duration (Supp. Fig. 4e). These data indicate that Cav1.2 channels in the PrL are required for both cocaine- and stress-primed reinstatement of cocaine place preference via increasing duration and number of entries, respectively.
Given our finding that Cav1.2 channels within the PrL are critical for cocaine- and stress-primed reinstatement, we next aimed to identify downstream targets of the PrL that mediate reinstatement. Previous literature suggests projections from the PrL to the nucleus accumbens core (NAcC), the primary target of the PrL in the nucleus accumbens45, are necessary for reinstatement22–25, but the role of LTCCs within this projection has yet to be explored. To test the necessity of Cav1.2 channels within the PrL→NAcC projection, Cav1.2 was selectively knocked-down within PrL cells that project to the NAcC by injecting a Flp-dependent virus co-expressing Cre recombinase and GFP (AAV-fDIO-Cre-GFP) into bilateral PrL and a retrograde virus expressing Flp (retro-AAV-Flp) into bilateral NAcC of cacna1c-floxed mice (Fig. 3a, Supp. Fig. 5a). Control mice were injected with AAV-fDIO-Cre-GFP in bilateral PrL, but did not receive retro-AAV-Flp into the NAcC. RNAscope in situ hybridization in combination with GFP immunohistochemistry confirmed knockout of Cav1.2 mRNA in GFP-positive cells using this viral strategy (Fig. 3c, Supp. Fig 5b, c); PrL-NAcC Cav1.2 KO mice had significantly lower expression of cacna1c mRNA in GFP-tagged cells compared to control mice expressing AAV-GFP (Fig. 3d, t(3) = 11.85, p = 0.001, unpaired t-test). Mice were tested in cocaine CPP (Fig. 3b). Selective deletion of Cav1.2 within the PrL→NAcC projection did not alter acquisition or extinction of cocaine CPP, however it did block both CPR and SPR (Fig. 3e; trending interaction (Cav1.2 x day), F(4,84) = 1.70, p = 0.16; main effect of day, F(4,84) = 16.39, p < 0.0001, two-way RM-ANOVA). These data indicate that Cav1.2 channels within the PrL→NAcC projection are required for both cocaine- and stress-primed reinstatement of cocaine-associated memories.
Figure 3.
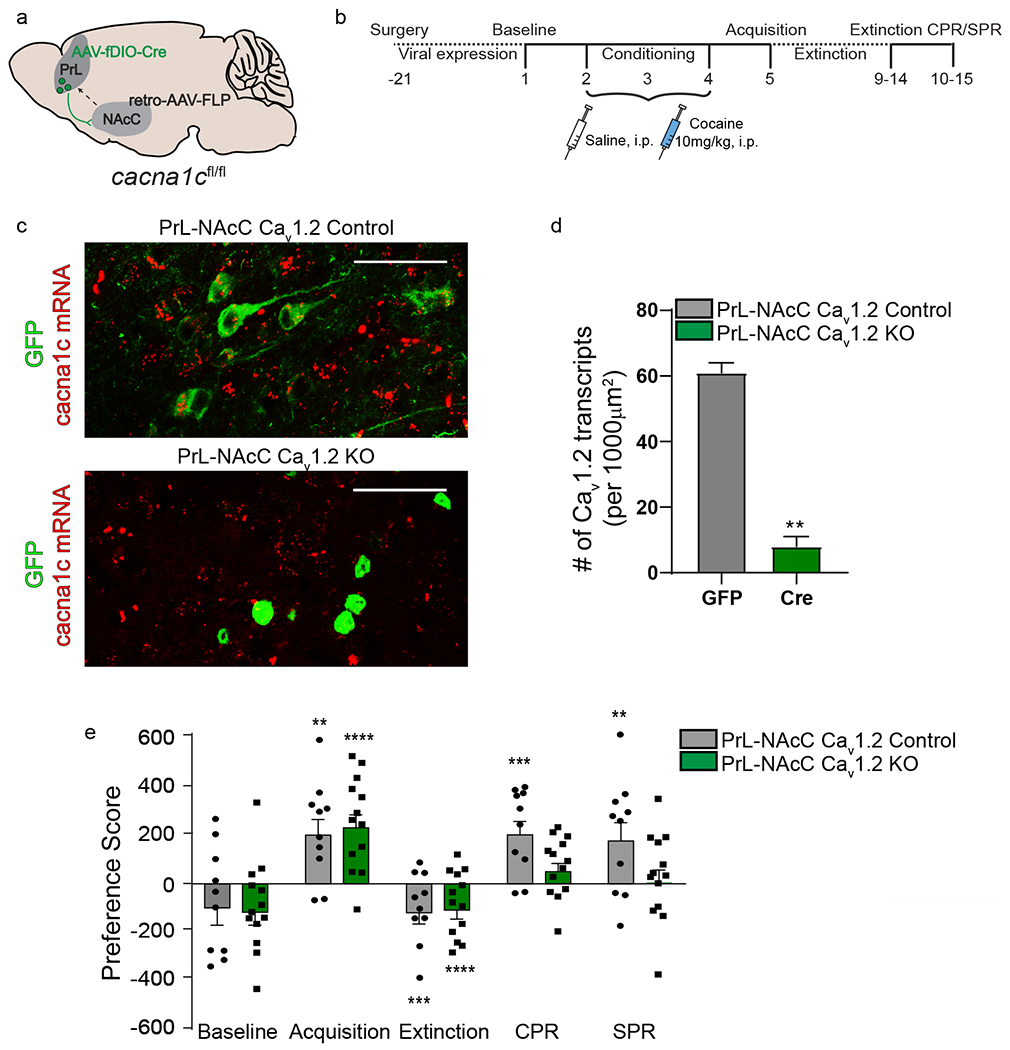
Cav1.2 within the PrL→NAcC projection is required for reinstatement, a) cacan1c-floxed mice were injected with a Flp-dependent AAV expressing Cre recombinase (AAV-fDIO-Cre-GFP) into bilateral PrL and a retrograde AAV expressing Flp (retro-AAV-Flp) into bilateral NAcC to knockdown cacna1c expression selectively in PrL→NAcC cells, b) Experimental timeline for surgery and behavioral experiments, c) Representative images of RNAscope in situ hybridization showing cacna1c mRNA (red) and GFP-tagged cells (green) in the PrL of control mice injected with AAV-GFP (Top- PrL-NAcC Cav1.2 control) and experimental cacna1c floxed mice injected with AAV-fDIO-Cre-GFP into the PrL and retro-AAV-Flp into the NAcC (bottom-PrL-NAcC Cav1.2 KO). Scale bar = 50μm d) Quantitative analysis of cacna1c mRNA transcripts. Mice with Cav1.2 knocked out in PrL cells projecting to the NAcC have significantly less cacna1c mRNA in GFP+ cells in the PrL compared to controls (**p< 0.01, unpaired t-test; GFP, n = 2 mice, 26-30 cells/mouse, Cre, n = 3 mice, 30 cells/mouse), e) Control mice (PrL-NAcC Cav1.2 Control) and mice with knockdown of Cav1.2 within the PrL→NAcC projection (PrL-NAcC Cav1.2 KO) acquired (****p < 0.0001, **p < 0.01) and extinguished (****p < 0.0001, ***p < 0.001) cocaine CPP. CPR significantly increased preference score in PrL-NAcC Cav1.2 control mice (***p < 0.001, n = 10) but not in PrL-NAcC Cav1.2 KO mice (n = 13). Similarly, SPR significantly increased preference score in PrL-NAcC Cav1.2 control mice (**p < 0.01) but not in PrL-NAcC Cav1.2 KO mice. Data are presented as mean + SEM.
Cocaine and stress priming recruit differing patterns of PrL→NAcC projection activity via LTCCs
The PrL → NAcC projection is known to be important for reinstatement behavior in a variety of models, but whether this projection is endogenously recruited during cocaine- or stress-primed reinstatement, and the role of LTCCs therein, has yet to be explored. Therefore, we utilized in vivo calcium recordings via fiber photometry32 to acquire real-time activity of the PrL→NAcC projection during behavior in the CPP task. GCAMP6s was selectively expressed in PrL neurons projecting to the NAcC by injecting a Cre-dependent virus expressing GCAMP6s into the PrL and a retrograde virus expressing Cre recombinase into the NAcC (Fig. 4a, b). An optical fiber was implanted above the PrL in order to record changes in Ca2+-dependent fluorescence (Supp. Fig. 3b), a proxy for neuronal activity in PrL→NAcC cells. Fiber photometry recordings of PrL→NAcC were obtained during the baseline, acquisition, extinction, and reinstatement tests (Fig. 4c).
Figure 4.
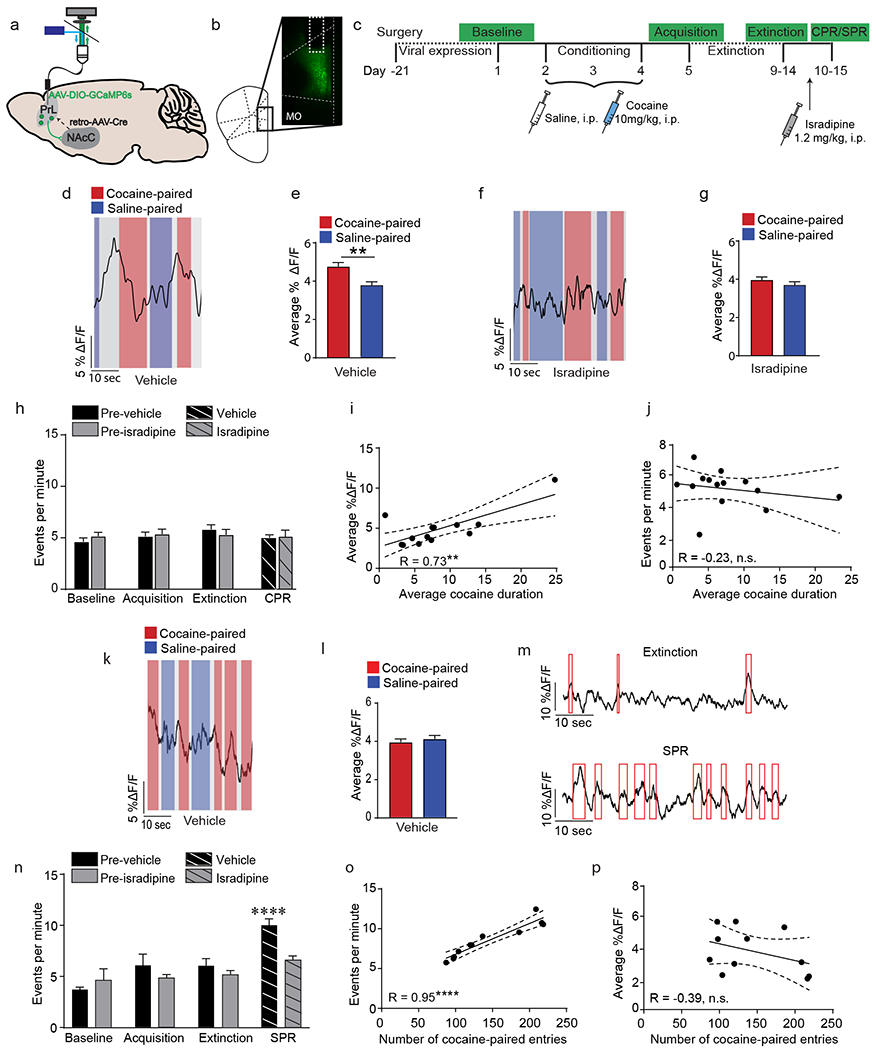
Cocaine- and stress-primed reinstatement recruit the PrL→NAcC projection, a) C57BL/6J mice were injected with a Cre-dependent AAV expressing GCaMP6s into unilateral PrL and a retrograde AAV expressing Cre recombinase was injected into ipsilateral NAcC. An optic fiber was implanted above the PrL, which could be attached to a patch cord to record Ca2+ signals, b) Representative image of GCaMP6s expressing in the PrL. Optic fiber location is outlined in white. MO, medial orbitofrontal cortex; Cg, cingulate cortex, c) Experimental timeline for fiber photometry recording during cocaine CPP. Ca2+ imaging was conducted during the baseline test, acquisition test, extinction test, and CPR/SPR. Mice were treated with vehicle or isradipine to block LTCCs prior to CPR/SPR recording, d) Representative fiber photometry trace of a vehicle-treated mouse during CPR showing Ca2+ transients prior to cocaine-paired entries (red) but not saline-paired entries (blue). Grey, middle CPP chamber, e) The average amplitude (% ΔF/F) of PrL→NAcC fiber photometry signal 5 seconds prior to entry into the cocaine-paired chamber was significantly higher as compared to entries into the saline-paired chamber in vehicle-treated mice (**p < 0.01, linear mixed effects model, number of entries = 454, number of mice = 7). f) Representative fiber photometry trace of an isradipine-treated mouse during CPR. g) The average amplitude of PrL→NAcC fiber photometry signal 5 seconds prior to entry did not differ between cocaine-paired entries and saline-paired entries in isradipine-treated mice (linear mixed effects model; number of entries = 468, number of mice = 6). h) There was no change in the number of events per minute observed during any day of CPP testing (vehicle, n = 7; isradipine, n = 7). i) The average amplitude of PrL→NAcC fiber photometry signal was significantly positively correlated with the average cocaine-paired duration during CPR (**p < 0.01, n = 13). j) The number of events per minute was not correlated with the average cocaine-paired duration during CPR (n = 13). k) Representative fiber photometry trace of a vehicle-treated mouse during SPR. 1) The average amplitude of PrL→NAcC fiber photometry signal 5 seconds prior to entry did not differ between cocaine-paired entries and saline-paired entries during SPR (linear mixed effects model; number of entries = 840, number of animals = 6). m) Representative fiber photometry traces of a vehicle-treated mouse during extinction and SPR. Red boxes indicate significant calcium events, n) SPR significantly increased the events per minute in vehicle-treated mice (****p < 0.0001, Bonferroni post-hoc extinction vs. SPR; n = 6) but not in isradipine-treated mice (n = 5). o) The number of events per minute was significantly positively correlated with the number of cocaine-paired entries during SPR (****p < 0.0001, n = 11). p) The average fiber photometry signal 5 seconds prior to entry into the cocaine-paired chamber was not correlated with the number of entries into the cocaine-paired chamber (n = 11). Data are presented as mean + SEM.
We found that during the cocaine-primed reinstatement test, increases in Ca2+ activity were observed preceding entry into the cocaine-paired chamber (Fig. 4d; Supp. Fig. 6a). When quantified, the calcium signal preceding cocaine-paired entries was significantly higher than signal preceding saline-paired entries (Fig. 4e; t(2,17) = 0.22, p = 0.009, linear mixed-effects model). This difference in entry-related signal was not observed during the baseline, acquisition, or extinction tests (Supp. Fig. 6 b, c, d). Additionally, this increase in PrL→NAcC activity preceding cocaine-paired entries was not accompanied by an increase in overall frequency of Ca2+ transients (Fig. 4h). To test if LTCCs are required for this increase in PrL→NAcC activity, mice were pretreated with isradipine (1.2mg/kg, i.p.) prior to the cocaine priming injection. Pre-treatment with isradipine attenuated the increase in pre-entry activity (Fig. 4f, g, Supp. Fig. 6e), without altering overall frequency of Ca2+ transients (Fig. 4h), indicating that the recruitment of the PrL→NAcC projection during cocaine-primed reinstatement is LTCC-dependent.
It was surprising to us that cocaine-primed reinstatement increased the average duration of time that mice spent in the cocaine-paired chamber while the observed increase in PrL→NAcC activity occurred immediately preceding entry and not while the animal was in the chamber. Therefore, we next examined the correlation between PrL→NAcC activity and the average duration in the cocaine-paired chamber. Surprisingly, the PrL→NAcC signal preceding entry into the cocaine-paired chamber demonstrated a significant positive correlation with the average cocaine-paired duration for each mouse (Fig. 4i; r = 0.73, p = 0.004), with no correlation found between PrL→NAcC activity and average saline-paired duration (Supp. Fig. 6f). Additionally, no correlation was observed between the overall frequency of PrL→NAcC calcium events during cocaine-primed reinstatement and the average cocaine-paired duration (Fig. 4j), demonstrating that increased PrL→NAcC projection activity preceding entry into the cocaine-paired chamber is predictive of increased time spent in that chamber during cocaine-primed reinstatement.
In contrast to the effect of cocaine priming on PrL→NAcC projection activity, in a cohort tested for stress-primed reinstatement, no change in PrL→NAcC activity was observed preceding entry into the cocaine-paired chamber (Fig. 4k, l, Supp. Fig. 6g). However, stress-primed reinstatement significantly increased the overall frequency of calcium events as compared to that seen during the extinction test (Fig. 4m, n; significant interaction (day x drug) F(3,27) = 3.05, p = 0.04), with a similar increase observed when the mouse was in both the saline-paired and cocaine-paired chamber (Supp. Fig. 6h; main effect of day F(3,20) = 15.11, p < 0.0001), indicating that stress-primed reinstatement persistently increases PrL→NAcC activity throughout the reinstatement test. This increase in event frequency was specific to the cocaine place preference context, as footshock stress had no effect on PrL→NAcC activity in a home cage setting (Supp. Fig. 6i).
To examine the role of LTCCs on this stress-primed increase in event frequency, mice were pretreated with isradipine (1.2mg/kg, i.p.) prior to footshock stress. Isradipine significantly attenuated the increase in event frequency to the level seen during extinction (Fig. 4n; interaction (Day x Drug), F(3,27) = 3.05, p = 0.046, two-way RM-ANOVA), demonstrating that the persistent increase in PrL→NAcC projection activity requires LTCCs. Next, when examining the relationship between event frequency and number of entries into the cocaine-paired chamber, a behavioral parameter we show to be specific to the effects of stress-primed reinstatement, we found a positive correlation between event frequency and the number of entries into the cocaine-paired chamber (Fig. 4o; r = 0.95, p < 0.0001) but not the saline-paired chamber (Supp. Fig. 6j). Furthermore, no correlation was observed between PrL→NAcC signal prior to cocaine-paired entries and the number of cocaine-paired entries (Fig. 4p), highlighting the difference in PrL→NAcC recruitment by cocaine and stress priming. Importantly, during both cocaine- and stress-primed reinstatement, treatment with isradipine specifically blunted the behaviorally-evoked PrL→NAcC signal changes and had no effect on baseline PrL→NAcC activity recorded in the animal’s home cage (Supp. Fig. 6k, l, m), indicating that isradipine does not reduce detectable calcium, but instead functions by blocking PrL→NAcC signal changes specific to cocaine- and stress-primed reinstatement.
Chemogenetic inhibition of the PrL→NAcC projection attenuates CPP behavioral patterns that drive cocaine- and stress-primed reinstatement of cocaine place preference
Given our observation that cocaine- and stress-primed reinstatement increase PrL→NAcC projection activity, we next utilized chemogenetics to inhibit this projection during reinstatement. The hM4Di inhibitory DREADD was selectively expressed in PrL neurons projecting to the NAcC in C57BL/6J mice (Fig. 5a, b; Supp. Fig. 3c). Mice were trained and tested in cocaine CPP, and 45 minutes prior to the reinstatement test, animals were injected with CNO (3mg/kg, i.p.) to activate the hM4Di DREADD and selectively inhibit the PrL→NAcC pathway (Fig. 5c). Control groups included mice expressing hM4Di in the PrL→NAcC pathway that were injected with saline to control for the effects of viral expression (hM4Di control) and mice that received sham surgery that were injected with CNO to control for off-target behavioral effects of CNO administration (sham control). These control groups showed no statistical differences in behavior (Supp. Fig. 7a, CPR: main effect of day F(3,12) = 27.08, p < 0.0001, two-way RM-ANOVA; Supp. Fig. 7b, SPR: main effect of day F(3,27) = 20.24, p < 0.0001, two-way RM-ANOVA), and were therefore combined for analysis described below.
Figure 5.
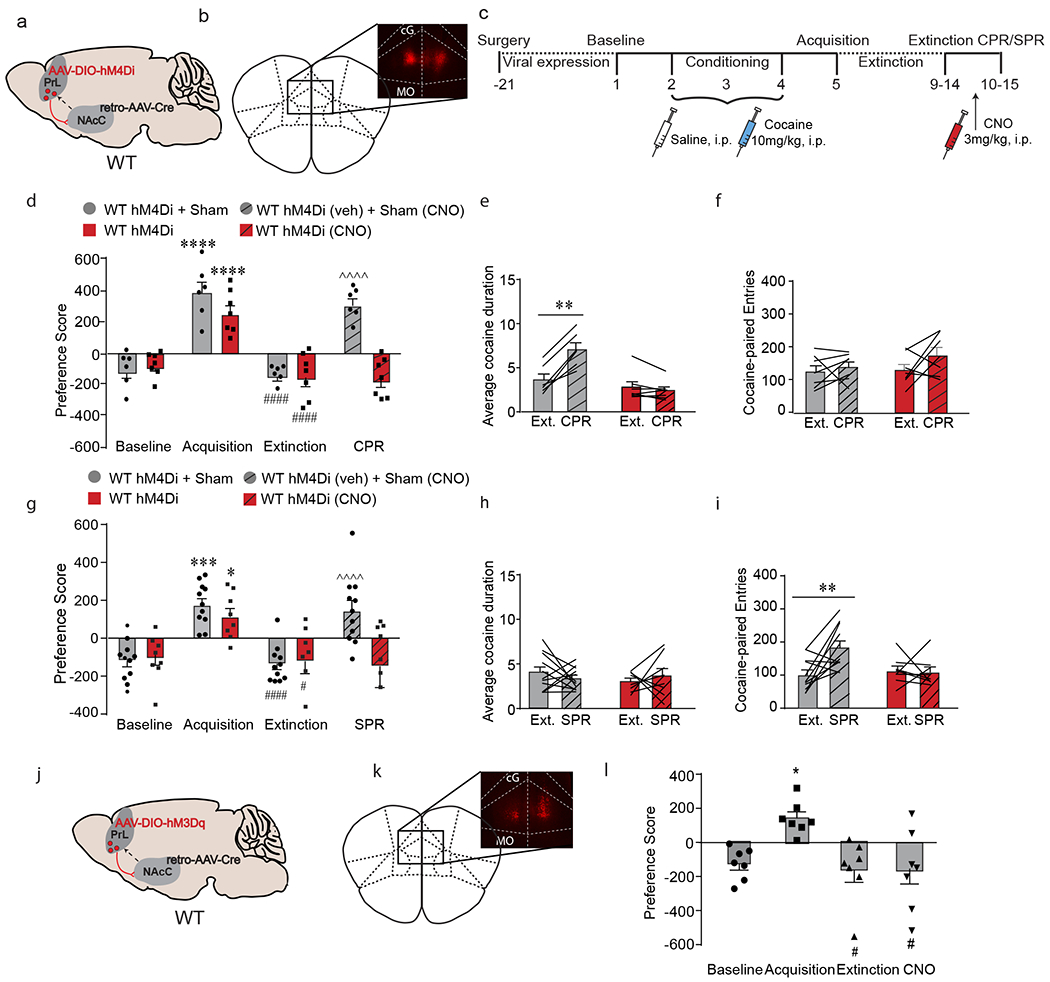
PrL→NAcC projections are necessary but not sufficient for cocaine- and stress-induced reinstatement, a) C57BL/6J mice were injected with a mCherry-tagged Cre-dependent AAV expressing the inhibitory DREADD hM4Di into bilateral PrL and a retrograde AAV expressing Cre recombinase into bilateral NAcC. b) Representative image of mCherry staining in bilateral PrL. c) Experimental timeline of DREADD injection and cocaine CPP. Following 21 days for viral expression, mice were run through the cocaine CPP protocol and prior to reinstatement, mice were injected with CNO to inhibit the PrL→NAcC projection, d) Both sham and hM4Di-expressing mice acquired (****p < 0.0001, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. CPR significantly increased preference score in control mice (^^^^p < 0.0001, Bonferroni post-hoc extinction vs. CPR; n = 6) but not in hM4Di-expressing mice injected with CNO (n = 7). e) CPR significantly increased the average cocaine-paired duration in control mice (**p < 0.01, Bonferroni post-hoc extinction vs. CPR; n = 7) but not in hM4Di-expressing mice treated with CNO (n = 7). f) CPR had no effect on number of cocaine-paired entries, g) Both sham and hM4Di-expressing mice acquired (***p < 0.001, *p < 0.05, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, #p < 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. SPR significantly increased preference score in control mice (^^^^p < 0.0001, Bonferroni post-hoc extinction vs. SPR; n = 11) but not in hM4Di-expressing mice injected with CNO (n = 8). h) SPR had no effect on average cocaine-paired duration, i) SPR significantly increased the number of cocaine-paired entries in control mice (**p < 0.01, Bonferroni post-hoc extinction vs. SPR; n = 11) but not in hM4Di-expressing mice treated with CNO (n = 8). j) C57BL/6J mice were injected with an mCherry-tagged Cre-dependent AAV expressing the excitatory DREADD hM3Dq into bilateral PrL and a retrograde AAV expressing Cre recombinase into bilateral NAcC. k) Representative image of mCherry staining in bilateral PrL. 1) All mice acquired (*p < 0.05, Bonferroni post-hoc baseline vs. acquisition) and extinguished (#p < 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. Treatment with CNO in hM3Dq-expressing mice had no effect on preference score (n = 7). Data are presented as mean + SEM.
In a cohort tested for cocaine-primed reinstatement, all mice acquired and extinguished cocaine CPP. During the cocaine-primed reinstatement test, chemogenetic inhibition of PrL→NAcC attenuated reinstatement behavior (Fig. 5d, interaction (DREADD x day) F(3, 33) = 11.48, p < 0.0001, two-way RM-ANOVA), confirming that activity of the PrL→NAcC projection is necessary for cocaine-primed reinstatement of cocaine-associated memories. As observed previously, cocaine priming increased the average duration in the cocaine-paired chamber, and chemogenetic inhibition of the PrL→NAcC projection attenuated reinstatement by decreasing this duration (Fig. 5e; interaction (DREADD x day) F(1,11) = 54.16, p < 0.0001, two-way RM-ANOVA), without altering cocaine-paired entries (Fig. 5f) or saline-paired duration (Supp. Fig. 7c).
In a cohort tested for stress-primed reinstatement, all mice acquired and extinguished cocaine CPP. During the stress-primed reinstatement test, chemogenetic inhibition of the PrL→NAcC projection attenuated reinstatement behavior (Fig. 5g; interaction (DREADD x day) F(3,51) = 4.20, p = 0.01, two-way RM-ANOVA), demonstrating that the PrL→NAcC projection is necessary for stress-primed reinstatement of cocaine-associated memories. As observed previously, stress priming increased the number of entries into the cocaine-paired chamber, and chemogenetic inhibition of PrL→NAcC attenuated reinstatement by decreasing the number of entries (Fig. 5i; interaction (DREADD x day), F(1,17) = 5.62, p = 0.03), without altering cocaine-paired duration (Fig. 5h) or saline-paired entries (Supp. Fig. 7d). Taken together, these data demonstrate the necessity of PrL→NAcC for both cocaine- and stress-primed reinstatement and the associated changes in CPP behavioral measures.
Increasing PrL→NAcC activity in the absence of cocaine or stress priming is not sufficient to induce reinstatement of cocaine place preference
Next, in order to test if activating the PrL→NAcC projection in the absence of the cocaine and stress priming is sufficient to reinstate cocaine place preference behavior, we utilized chemogenetics to excite the PrL→NAcC projection during the reinstatement test. The hM3Dq excitatory DREADD was selectively expressed in PrL neurons projecting to the NAcC in C57BL/6J (Fig. 5j, k). Mice were trained and tested in the cocaine CPP protocol, and 45 minutes prior to the reinstatement test, animals were injected with CNO (3mg/kg, i.p.) to activate the PrL→NAcC circuit. Chemogenetic activation of PrL→NAcC did not reinstate preference for the cocaine-paired chamber (fFig. 5l; F (2.427, 14.56) = 6.123, p = 0.009, one-way RM-ANOVA), indicating that activation of PrL→NAcC is not sufficient to induce reinstatement behavior in the absence of cocaine or stress.
Chemogenetic activation of the PrL→NAcC projection in Cav1.2-deficient mice reinstates cocaine place preference following cocaine and stress priming
Given our findings that systemic pharmacological inhibition of LTCCs attenuated cocaine- and stress-primed reinstatement (Fig. 1b, c) and reduced the required PrL→NAcC activity during reinstatement (Fig. 4g, n), we next tested whether chemogenetic activation of PrL→NAcC in Cav1.2+/− mice is sufficient to reinstate preference for the previously cocaine-paired chamber following either cocaine or stress priming. The hM3Dq excitatory DREADD was selectively expressed in PrL neurons projecting to the NAcC in Cav1.2+/− mice (Fig. 6a, b). Mice were trained and tested in the cocaine CPP protocol, and 45 minutes prior to the reinstatement test (Fig. 6c), mice were injected with CNO (3 mg/kg, i.p.). Control mice expressing the hM3Dq DREADD in PrL→NAcC (hM3Dq control) were injected with vehicle and sham control mice were injected with CNO (Sham control) 45 minutes prior to the reinstatement test. No statistical difference was observed between control groups during behavior (Supp. Fig. 8a, CPR: main effect of time, F(3,18) = 9.87, p = 0.0005, two-way RM-ANOVA; Supp. Fig. 8b, SPR: main effect of time, F(3,18) = 8.02, p = 0.01, two-way RM-ANOVA), therefore groups were combined for the analysis described below.
Figure 6.
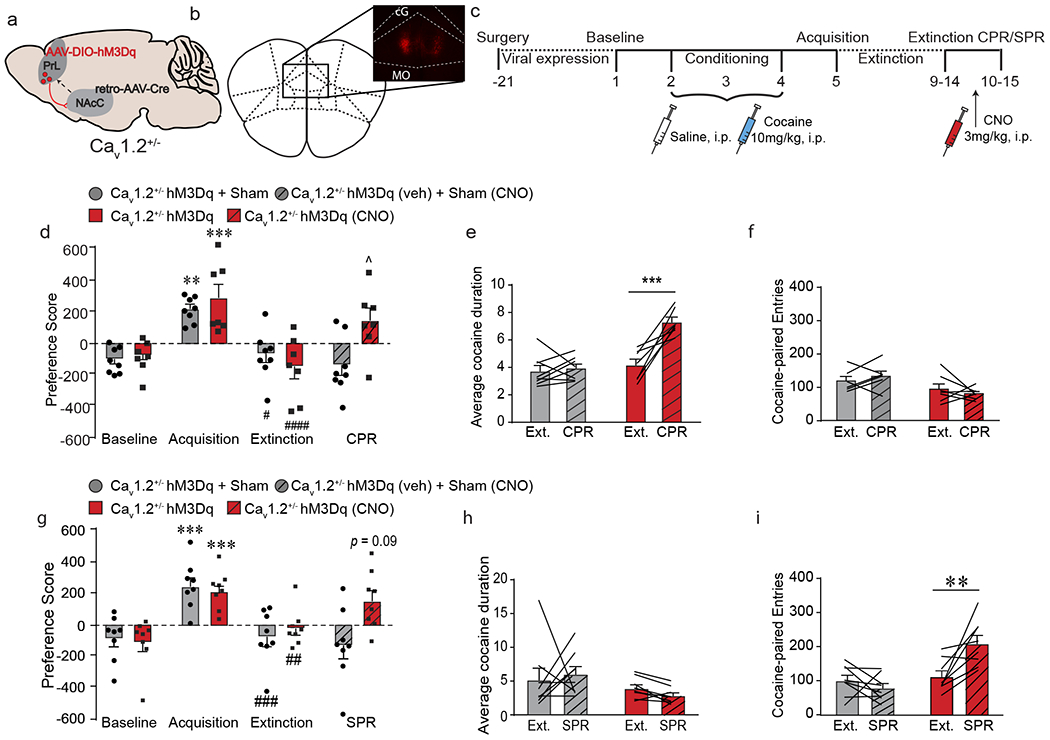
Activation of PrL→NAcC projections is sufficient to induce cocaine- and stress-primed reinstatement in Cav1.2+/− mice, a) Cav1.2+/− mice were injected with mCherry-tagged Cre-dependent AAV expressing the excitatory DREADD hM3Dq into bilateral PrL and a retrograde AAV expressing Cre recombinase into bilateral NAcC. b) Representative image of mCherry staining in bilateral PrL. c) Experimental timeline of viral injection and cocaine CPP. Following 21 days for viral expression, mice were run through the cocaine CPP protocol and prior to reinstatement, mice were injected with CNO to activate the PrL→NAcC projection, d) Control (hM3Dq + Sham) and hM3Dq-expressing mice acquired (***p < 0.001, **p < 0.01, Bonferroni post-hoc baseline vs. acquisition) and extinguished (####p < 0.0001, #p < 0.05, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. CPR had no effect on preference score in control Cav1.2+/− mice (n = 8), but significantly increased preference score in Cav1.2+/− mice expressing hM3Dq treated with CNO (^p < 0.05, Bonferroni post-hoc extinction vs. CPR; n = 7). e) CPR significantly increased average cocaine-paired duration in Cav1.2+/− mice expressing hM3Dq treated with CNO (***p < 0.001, Bonferroni post-hoc extinction vs. CPR; n = 7) but not in Cav1.2+/− control mice (n = 8). f) CPR had no effect on number of cocaine-paired entries in either group. g) Control (hM3Dq + Sham) and hM3Dq-expressing mice acquired (***p < 0.001, Bonferroni post-hoc baseline vs. acquisition) and extinguished (###p < 0.001, ##p < 0.01, Bonferroni post-hoc acquisition vs. extinction) cocaine CPP. SPR had no effect on preference score in control Cav1.2+/− mice (n = 8), but induced a trending increase in preference score in Cav1.2+/− mice expressing hM3Dq treated with CNO (p = 0.09, Bonferroni post-hoc extinction vs. CPR; n = 8). h) SPR had no effect on average cocaine-paired duration in either group. i) SPR significantly increased the number of cocaine-paired entries in Cav1.2+/− mice expressing hM3Dq treated with CNO (**p < 0.01, Bonferroni post-hoc extinction vs. SPR; n = 8) but not in Cav1.2+/− control mice (n = 8). Data are presented as mean + SEM.
In a cohort tested for cocaine-primed reinstatement, all mice acquired and extinguished cocaine CPP. As expected, cocaine failed to reinstate place preference in Cav1.2+/− control mice, whereas chemogenetic activation of PrL→NAcC restored reinstatement behavior by significantly increasing the preference score (Fig. 6d; interaction (DREADD x day) F(3,39) = 3,60, p = 0.02, two-way RM-ANOVA), and the average duration spent in the cocaine-paired chamber (Fig. 6e; interaction dreadd x day F(1,12) = 13.75, p = 0.003, two-way RM-ANOVA), without altering the number of cocaine-paired entries (Fig. 6f) or average saline-paired duration (Supp. Fig. 8c), demonstrating that activation of PrL→NAcC in Cav1.2+/− mice is sufficient to induce cocaine-primed reinstatement.
In a cohort tested for stress-primed reinstatement, all mice acquired and extinguished cocaine CPP. As expected, stress failed to reinstate place preference in Cav1.2+/− control mice, but chemogenetic activation of the PrL→NAcC projection induced a trending increase in preference score following stress priming (Fig. 6g; interaction (DREADD x day) F(3,42) = 4.60, p = 0.007, two-way RM-ANOVA) and a significant increase in the number of entries into the cocaine-paired chamber (Fig. 6i; interaction (DREADD x day), F(1,14) = 10.72, p = 0.006, two-way RM-ANVOA), with no change observed in average cocaine-paired duration (Fig. 6h) or saline-paired entries (Supp. Fig. 8d), indicating that activation of the PrL→NAcC projection in Cav1.2+/− mice is sufficient to induce stress-primed reinstatement.
Discussion
Cocaine-associated memories are critical drivers of craving and relapse in addicted individuals. Understanding how different environmental stimuli, such as re-exposure to a drug of abuse or stressful experiences can reinstate these cocaine-associated memories is crucial for the development of better treatment options to prevent relapse. Here we demonstrate that reinstatement of cocaine associated-memories following cocaine or stress priming is driven by different patterns of behavior and PrL→NAcC activity and requires Cav1.2 channels within the PrL→NAcC projection.
The prefrontal cortex is a critical hub for reward-related processing, with different prefrontal subregions being implicated in extinction and reinstatement behaviors following various environmental stimuli38–41. Using focal knockdown of the Cav1.2 LTCC within the PrL and IL subregions of the prefrontal cortex, we demonstrate that Cav1.2 channels within the PrL are required for reinstatement, while Cav1.2 within the IL is required for extinction of cocaine-associated memories. Interestingly, Cav1.2 in these subregions was not required for acquisition, highlighting the selective recruitment of these channels during extinction and reinstatement. These findings are consistent with the known role of the PrL and IL in reinstatement and extinction, respectively, and identify Cav1.2 channels as a common molecular mediator of cocaine-associated memories within these prefrontal subregions.
In this study, we demonstrate that cocaine-primed reinstatement is driven by an increase in the duration of time spent in the cocaine-paired chamber while stress-primed reinstatement is driven by an increased number of entries into the cocaine-paired chamber, both requiring PrL Cav1.2 channels. These different behavioral effects suggest that different mechanisms may be recruited by cocaine- and stress-primed reinstatement. Previous studies have implicated the neural circuit from PrL to NAcC in reinstatement behavior following both cocaine and stress priming. Thus, we combined behavioral analysis with fiber photometry in vivo calcium imaging to demonstrate that cocaine-primed reinstatement recruits the PrL→NAcC projection prior to entry into the cocaine-paired chamber, and that this activity positively correlates with the increase in the duration mice spend in this chamber. In contrast, stress-primed reinstatement persistently recruits the PrL→NAcC projection, and this persistent activity positively correlates with the increase in the number of entries into the cocaine-paired chamber. It was surprising to us that during cocaine-primed reinstatement the transient increase in PrL→NAcC activity observed prior to entry into the cocaine-paired chamber correlated with the duration that the mouse spent in this chamber even after this PrL→NAcC activity subsided. One possible explanation for this finding is that transient increases in the PrL→NAcC projection could lead to more persistent activity of the NAcC postsynaptically, which might drive increased duration in the chamber. Future studies using optogenetic inhibition timed precisely to the seconds preceding chamber entry will address whether the increased activity preceding cocaine-paired entries can alter the duration of stay in that chamber. Using pharmacology, we demonstrate that the recruitment of the PrL→NAcC projection and the particular CPP behavioral measures that are altered during cocaine- and stress-primed reinstatement are dependent upon LTCC signaling.
One possible explanation for the different patterns of PrL→NAcC projection activity observed is that different presynaptic inputs to the PrL are recruited by cocaine versus stress and thus differentially modulate the activity of PrL projection neurons. Previous studies have shown that cocaine-primed reinstatement requires dopamine release within the PrL24 while norepinephrine signaling is required for stress-primed but not cocaine-primed reinstatement of cocaine CPP46. As reward-predictive cues elicit phasic activation of prefrontal dopamine release47, and reinstatement is associated with a renewal of the salience of cocaine-predictive cues1, an increase in phasic dopamine release prior to entry into the cocaine-paired chamber might reflect an enhanced response to cocaine-associated contextual cues, which could account for the entry-related phasic activity of the PrL→NAcC projection we observe during cocaine-primed reinstatement, a question that will be addressed in future studies.
Alternatively, acute stress is known to enhance norepinephrine release within the prefrontal cortex48, which can drive persistent activity within this region49. Interestingly, in humans, blockade of beta adrenergic receptors (β2AR) is able to block a stress-induced shift from goal-directed to habitual behavior50. We show that stress-primed reinstatement is associated with an increase in cocaine-paired entries, potentially reflecting habitual entry behavior, and suggesting that norepinephrine release during stress-primed reinstatement might modulate both the persistent PrL→NAcC projection activity and the correlated increase in habitual cocaine-paired entries observed in the current studies. In support of these hypotheses, studies have shown regulation of Cav1.2 channels by both dopamine D117 and β2ARs51, suggesting that LTCCs may mediate stimulus-specific reinstatement behavioral patterns and neural activity by differential recruitment of these two signaling pathways. Alternatively, this persistent increase in PrL activity could be caused by a decrease in inhibition. This hypothesis is supported by findings that stress-potentiated cocaine-primed reinstatement of self-administration is driven by a CB1 receptor-mediated disinhibition of the PrL52.
Interestingly, we did not observe any changes in fiber photometry recordings of the PrL→NAcC projection during acquisition or extinction of cocaine CPP. This is consistent with previous findings that the PrL is selectively required for reinstatement but not for acquisition or extinction of cocaine CPP39, and suggests that the PrL→NAcC projection is endogenously recruited during reinstatement but not during these other phases of cocaine CPP. This finding, along with our finding that changes in cocaine-paired duration and cocaine-paired entries are observed only during reinstatement and not during acquisition, suggests that distinct mechanisms are likely mediating the increase in cocaine place preference during acquisition and reinstatement.
In addition to characterizing the recruitment of the PrL→NAcC projection activity during cocaine- and stress-primed reinstatement, we additionally find, using direct chemogenetic inhibition, that the PrL→NAcC projection is required for both cocaine- and stress-primed reinstatement, as well as for the behavioral changes observed during these different types of reinstatement. Interestingly, using chemogenetic activation, we demonstrate that the PrL→NAcC projection is not sufficient to induce reinstatement in the absence of cocaine or stress, but does induce reinstatement in Cav1.2+/−mice in response to both cocaine and stress priming. This indicates that the other neural circuits required for reinstatement are likely intact in Cav1.2-deficient mice and suggests that in this global, genetic model of Cav1.2 deficiency, activation of the PrL→NAcC projection is sufficient to restore reinstatement behavior.
Conclusions
Our findings demonstrate that LTCCs are required for reinstatement of cocaine-associated memories following priming with cocaine or stress via modulation of the projection from PrL to NAcC. These findings support clinical evidence that pharmacological blockade of LTCCs may help attenuate craving, a critical factor leading to risk for relapse6. Additionally, we demonstrate for the first time that the Cav1.2 LTCC isoform is required for reinstatement, highlighting the potential efficacy of and need for isoform-specific pharmacotherapies.
Supplementary Material
Acknowledgements
We would like to thank Nii Addy, Kristen Pleil, and Francis Lee for their input and edits on the manuscript. This work was supported by grants to A.M.R. from the National Institute of Drug Abuse (5R01DA029122), The New York Weill Cornell Alumni Council Award, and the Paul Fund along with grants to C.L. from the National Institute of Mental Health, the One Mind Institute, The Hartwell Foundation, the Rita Allen foundation, the Klingenstein-Simons Foundation Fund, and the Brain and Behavior Research Foundation, grants to T.L.K from NIAAA (U24AA025475 and U01AA020911), and grants to T.A.M from NIH (DA08259 and HL098351). C.C.B. was supported by a T32 grant from NIDA (T32DA039080-01), a TL1 grant from the National Center for Advancing Translation Sciences (TL1TR002386), and the Frank & Blanche Mowrer Memorial Fellowship. R. N. F. was supported by a T32 grant from NIGMS (T32GM007739) and an NRSA from NIMH (F30MH115622). C.E.B. was supported by a T32 grant from NIDA (T32DA039080-01) and the Frank & Blanche Mowrer Memorial Fellowship. B.S.H. was supported by an NSF GRFP fellowship (ID# 2015174265, Award# 1257284) and the Jacques Cohenca Predoctoral Fellowship. R.B. was supported by a T32 grant from NIGMS (T32GM007739) and an NRSA from NIMH (F30MH117939).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Stewart J Review. Psychological and neural mechanisms of relapse. Philos Trans R Soc Lond B Biol Sci 2008; 363(1507): 3147–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White JA, McKinney BC, John MC, Powers PA, Kamp TJ, Murphy GG. Conditional forebrain deletion of the L-type calcium channel Ca V 1.2 disrupts remote spatial memories in mice. Learn Mem 2008; 15(1): 1–5. [DOI] [PubMed] [Google Scholar]
- 3.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 2005; 25(43): 9883–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev 2015; 67(4): 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, Owen MJ et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog Neurobiol 2015; 134: 36–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabir ZD, Martinez-Rivera A, Rajadhyaksha AM. From Gene to Behavior: L-Type Calcium Channel Mechanisms Underlying Neuropsychiatric Symptoms. Neurotherapeutics 2017; 14(3): 588–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cassidy F, Ahearn EP, Carroll BJ. Substance abuse in bipolar disorder. Bipolar Disord 2001; 3(4): 181–188. [PubMed] [Google Scholar]
- 8.Green AI, Brown ES. Comorbid schizophrenia and substance abuse. J Clin Psychiatry 2006; 67(9): e08. [PubMed] [Google Scholar]
- 9.Post RM, Kalivas P. Bipolar disorder and substance misuse: pathological and therapeutic implications of their comorbidity and cross-sensitisation. Br J Psychiatry 2013; 202(3): 172–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lancaster TM, Heerey EA, Mantripragada K, Linden DE. CACNA1C risk variant affects reward responsiveness in healthy individuals. Transl Psychiatry 2014; 4: e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wessa M, Linke J, Witt SH, Nieratschker V, Esslinger C, Kirsch P et al. The CACNA1C risk variant for bipolar disorder influences limbic activity. Mol Psychiatry 2010; 15(12): 1126–1127. [DOI] [PubMed] [Google Scholar]
- 12.Addy NA, Nunes EJ, Hughley SM, Small KM, Baracz SJ, Haight JL et al. The L-type calcium channel blocker, isradipine, attenuates cue-induced cocaine-seeking by enhancing dopaminergic activity in the ventral tegmental area to nucleus accumbens pathway. Neuropsychopharmacology 2018; 43(12): 2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du C, Volkow ND, You J, Park K, Allen CP, Koob GF et al. Cocaine-induced ischemia in prefrontal cortex is associated with escalation of cocaine intake in rodents. Mol Psychiatry 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Degoulet M, Stelly CE, Ahn KC, Morikawa H. L-type Ca(2)(+) channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol Psychiatry 2016; 21(3): 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgdorf CE, Schierberl KC, Lee AS, Fischer DK, Van Kempen TA, Mudragel V et al. Extinction of Contextual Cocaine Memories Requires Cav1.2 within D1R-Expressing Cells and Recruits Hippocampal Cav1.2-Dependent Signaling Mechanisms. J Neurosci 2017; 37(49): 11894–11911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q et al. Cav1.2 L-type Ca(2)(+) channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Ca(v)1.3 channels. J Neurosci 2011; 31(38): 13562–13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE et al. Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 2010; 30(50): 17051–17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terrillion CE, Dao DT, Cachope R, Lobo MK, Puche AC, Cheer JF et al. Reduced levels of Cacna1c attenuate mesolimbic dopamine system function. Genes Brain Behav 2017; 16(5): 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dedic N, Pohlmann ML, Richter JS, Mehta D, Czamara D, Metzger MW et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry 2018; 23(3): 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bavley CC, Fischer DK, Rizzo BK, Rajadhyaksha AM. Cav1.2 channels mediate persistent chronic stress-induced behavioral deficits that are associated with prefrontal cortex activation of the p25/Cdk5-glucocorticoid receptor pathway. Neurobiol Stress 2017; 7: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terrillion CE, Francis TC, Puche AC, Lobo MK, Gould TD. Decreased Nucleus Accumbens Expression of Psychiatric Disorder Risk Gene Cacna1c Promotes Susceptibility to Social Stress. Int J Neuropsychopharmacol 2017; 20(5): 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 2003; 23(8): 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK et al. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci 2002; 22(7): 2916–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 2001; 21(21): 8655–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci 2004; 24(7): 1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soeiro-de-Souza MG, Lafer B, Moreno RA, Nery FG, Chile T, Chaim K et al. The CACNA1C risk allele rs1006737 is associated with age-related prefrontal cortical thinning in bipolar I disorder. Transl Psychiatry 2017; 7(4): e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabir ZD, Lee AS, Rajadhyaksha AM. L-type Ca(2+) channels in mood, cognition and addiction: integrating human and rodent studies with a focus on behavioural endophenotypes. J Physiol 2016; 594(20): 5823–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee AS, Ra S, Rajadhyaksha AM, Britt JK, De Jesus-Cortes H, Gonzales KL et al. Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol Psychiatry 2012; 17(11): 1054–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bavley CC, Rice RC, Fischer DK, Fakira AK, Byrne M, Kosovsky M et al. Rescue of Learning and Memory Deficits in the Human Nonsyndromic Intellectual Disability Cereblon Knock-Out Mouse Model by Targeting the AMP-Activated Protein Kinase-mTORC1 Translational Pathway. J Neurosci 2018; 38(11): 2780–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milner TA, Waters EM, Robinson DC, Pierce JP. Degenerating processes identified by electron microscopic immunocytochemical methods. Methods Mol Biol 2011; 793: 23–59. [DOI] [PubMed] [Google Scholar]
- 31.Rajadhyaksha A, Husson I, Satpute SS, Kuppenbender KD, Ren JQ, Guerriero RM et al. L-type Ca2+ channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci 2004; 24(34): 7464–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A et al. Natural neural projection dynamics underlying social behavior. Cell 2014; 157(7): 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lerner TN, Shilyansky C, Davidson TJ, Evans KE, Beier KT, Zalocusky KA et al. Intact-Brain Analyses Reveal Distinct Information Carried by SNc Dopamine Subcircuits. Cell 2015; 162(3): 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 2011; 121(4): 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 1993; 123(4): 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U et al. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 2009; 75(2): 407–414. [DOI] [PubMed] [Google Scholar]
- 37.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem 2000; 275(50): 39193–39199. [DOI] [PubMed] [Google Scholar]
- 38.Capriles N, Rodaros D, Sorge RE, Stewart J. A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2003; 168(1-2): 66–74. [DOI] [PubMed] [Google Scholar]
- 39.Zavala AR, Weber SM, Rice HJ, Alleweireldt AT, Neisewander JL. Role of the prelimbic subregion of the medial prefrontal cortex in acquisition, extinction, and reinstatement of cocaine-conditioned place preference. Brain Res 2003; 990(1-2): 157–164. [DOI] [PubMed] [Google Scholar]
- 40.Augur IF, Wyckoff AR, Aston-Jones G, Kalivas PW, Peters J. Chemogenetic Activation of an Extinction Neural Circuit Reduces Cue-Induced Reinstatement of Cocaine Seeking. J Neurosci 2016; 36(39): 10174–10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters J, LaLumiere RT, Kalivas PW. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci 2008; 28(23): 6046–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutman AL, Nett KE, Cosme CV, Worth WR, Gupta SC, Wemmie JA et al. Extinction of Cocaine Seeking Requires a Window of Infralimbic Pyramidal Neuron Activity after Unreinforced Lever Presses. J Neurosci 2017; 37(25): 6075–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, McIntosh AM, He Y, Gelernter J, Blumberg HP. The association of genetic variation in CACNA1C with structure and function of a frontotemporal system. Bipolar Disord 2011; 13(7-8): 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paulus FM, Bedenbender J, Krach S, Pyka M, Krug A, Sommer J et al. Association of rs1006737 in CACNA1C with alterations in prefrontal activation and fronto-hippocampal connectivity. Hum Brain Mapp 2014; 35(4): 1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berendse HW, Galis-de Graaf Y, Groenewegen HJ. Topographical organization and relationship with ventral striatal compartments of prefrontal corticostriatal projections in the rat. J Comp Neurol 1992; 316(3): 314–347. [DOI] [PubMed] [Google Scholar]
- 46.Vranjkovic O, Hang S, Baker DA, Mantsch JR. beta-adrenergic receptor mediation of stress-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: roles for beta1 and beta2 adrenergic receptors. J Pharmacol Exp Ther 2012; 342(2): 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellwood IT, Patel T, Wadia V, Lee AT, Liptak AT, Bender KJ et al. Tonic or Phasic Stimulation of Dopaminergic Projections to Prefrontal Cortex Causes Mice to Maintain or Deviate from Previously Learned Behavioral Strategies. J Neurosci 2017; 37(35): 8315–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finlay JM, Zigmond MJ, Abercrombie ED. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience 1995; 64(3): 619–628. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, Seguela P. Norepinephrine drives persistent activity in prefrontal cortex via synergistic alpha1 and alpha2 adrenoceptors. PLoS One 2013; 8(6): e66122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwabe L, Hoffken O, Tegenthoff M, Wolf OT. Preventing the stress-induced shift from goal-directed to habit action with a beta-adrenergic antagonist. J Neurosci 2011; 31(47): 17317–17325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patriarchi T, Buonarati OR, Hell JW. Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by beta2 adrenergic receptor/PKA and Ca(2+)/CaMKII signaling. EMBO J 2018; 37(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McReynolds JR, Doncheck EM, Li Y, Vranjkovic O, Graf EN, Ogasawara D et al. Stress Promotes Drug Seeking Through Glucocorticoid-Dependent Endocannabinoid Mobilization in the Prelimbic Cortex. Biol Psychiatry 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.