

Abstract
The centriole duplication cycle normally ensures that centriole number is maintained at two centrioles per G1 cell. However, some circumstances can result in an aberrant increase in centriole number—a phenotype that is particularly prevalent in several types of cancer. Following an artificial increase in centriole number without tetraploidization due to transient overexpression of the kinase PLK4, human cells return to a normal centriole number during the proliferation of the population. We examine the mechanisms responsible for this return to normal centriole number at the population level in human retinal pigment epithelial cells. We find that the return to normal centriole number in the population of induced cells cannot be explained by limited duplication of centrioles, instability of extra centrioles, or by grossly asymmetric segregation of extra centrioles in mitosis. However, cells with extra centrioles display heterogenous phenotypes including extended cell cycle arrest, longer interphase durations, and death, which overall results in a proliferative disadvantage relative to normal cells in the population. Although about half of cells with extra centrioles in a population were able to divide, the extent of the disadvantages conferred by other fates is sufficient to account for the observed rate of return to normal centriole number. These results suggest that only under conditions of positive selection for cells with extra centrioles, continuous generation of such centrioles, or alleviation of the disadvantageous growth phenotypes would they be maintained in a population.
INTRODUCTION
The centrosome is the main microtubule organizing center in animal cells, participates in the formation of the mitotic spindle, and provides the platform for primary cilium formation (Nigg and Raff, 2009; Conduit et al., 2015). Cells normally have one centrosome (two centrioles) during G1, which duplicates during S-phase to result in two centrosomes (four centrioles) in G2/mitosis (Nigg and Stearns, 2011; Nigg and Holland, 2018). In cycling cells, centriole number is strictly regulated by the centriole duplication cycle, which allows the formation of a single new centriole (pro-centriole) from an already existing one (Nigg and Stearns, 2011; Nigg and Holland, 2018). Extra centrioles are commonly found in human cancers and have been linked to advanced tumor grade, poor prognosis, and metastasis (Chan, 2011). In tissue culture cells, transient overduplication of centrioles in a cell population after initial centriole depletion by drug treatment is followed by a return to the normal number of centrioles per cell within a few generations at the population level (Wong et al., 2015), which occurs through an uncharacterized mechanism.
Overexpression of PLK4, the kinase necessary for canonical centriole duplication (Bettencourt-Dias et al., 2005; Habedanck et al., 2005), is sufficient to generate extra centrioles in diploid human cells (Kleylein-Sohn et al., 2007), a condition known as centriole amplification (CA), without inducing tetraploidy as CA generated through cytokinesis failure does. Both overexpression of a PLK4 mutant unable to be autophosphorylated—a process which leads to its ubiquitination and destruction—and CA due to induced cytokinesis failure lead to p53-dependent cell cycle arrest (Holland et al., 2012; Fava et al., 2017). Furthermore, p53 negatively regulates PLK4 activity and thus acts to prevent CA (Nakamura et al., 2013).
While previous work supports p53 activation in populations of cells with extra centrioles (CA-cells), it remained unanswered if there is heterogeneity in response to CA in individual CA-cells, that is, whether every CA-cell undergoes p53-dependent arrest in the next cell cycle. In addition to immediate and irreversible cell cycle arrest, we consider four different, not mutually exclusive, mechanisms that could facilitate the return to a normal number of centrioles following amplification. First, that not all extra centrioles are able to duplicate in a single cell cycle. This would limit duplication to a subset of centrioles and reduce the number by segregation over time. Second, that extra centrioles are eliminated. Such elimination could be specific, as in oocytes of most animals (Delattre and Gonczy, 2004), or due to instability of centrioles caused by a defect in their structure (Wang et al., 2011, 2017). Third, that centrioles are segregated asymmetrically at mitosis. Extra centrioles are typically clustered at the poles of a bipolar spindle (Ring et al., 1982; Kwon et al., 2008), and they could be clustered asymmetrically at spindle poles, generating daughter cells with different numbers of centrioles. This might occur with some specificity, creating one cell with the correct centriole number in a single division, or might occur stochastically, yielding such a normal cell by chance. Fourth, a small number of cells in the population with normal centriole number could have a proliferation advantage over CA-cells and overtake the population.
By examining the fate of individual cells and of extra centrioles produced by transient overexpression of PLK4, we found that all extra centrioles are able to duplicate, that they are retained during division cycles when cells do not arrest, and that the majority of mitoses results in relatively symmetric segregation of the centrioles to the two daughter cells. Our results show that the main drivers to the return to a normal number of centrioles are heterogeneous phenotypes that disadvantage cells with extra centrioles, resulting in reduced proliferation and allowing the cells with a normal number in the population to predominate over time.
RESULTS AND DISCUSSION
To generate diploid cells with extra centrioles, we induced overexpression of the centriole duplication regulator PLK4 in a clonal human hTERT-RPE-1 GFP-Centrin-2 tetON-PLK4 cell line (Hatch et al., 2010). These cells constitutively express a centriole marker, GFP-Centrin-2, to allow visualization of centrioles in living cells and express hPLK4 under control of doxycycline, in addition to the endogenous PLK4. After 48 h of treatment with 1 μg/ml doxycycline, PLK4 transcript levels were elevated 238 ± 9.8% in these cells in comparison to control cells of the same genotype treated with DMSO as measured by RT-qPCR. We first determined a suitable time course of induction of extra centrioles and characterized the return to normal centriole number, shown in Figure 1A. Cells were treated with either doxycycline or DMSO as a control for 48 h, followed by drug washout, and centriole number was analyzed on subsequent days via immunofluorescence staining of CP110 and endogenous Centrin-2-GFP fluorescence (Figure 1B). Cells were classified as having either normal centriole numbers (2–4) or extra centrioles (5+). Induction of PLK4 for 48 h resulted in the generation of extra centrioles in more than 90% of cells, as opposed to less than 10% of control cells with extra centrioles (Figure 1C). Following doxycycline washout, centriole number returned to normal over the course of about 10 d (Figure 1C), in accordance with previous results documenting centriole reduction after amplification (Wong et al., 2015; Baudoin et al., 2020; Galofré et al., 2020). Centriole number remained constant following DMSO washout (Supplemental Figure S1), with the starting population showing 5% CA that never increased throughout the time course. The cells were seeded at a low density after washout and were reseeded as they became confluent, such that control cells could go through approximately 12 generations over the course of the experiment. Furthermore, PLK4 protein level returned to normal between 48 and 72 h after washout of doxycycline (Figure 1, D and D’).
FIGURE 1:

Centriole number returns to normal after initial amplification. (A) Schematic representation of experimental treatment of hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells. Cells were treated with 1 μg/ml doxycycline or 1:1000 (vol/vol) DMSO for 48 h. Drugs were subsequently washed out, and cells were analyzed at time points afterward. (B) Representative images of hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 DMSO-treated cells (Control, left) and doxycycline-treated cells (DOX, right). Cells were stained for CP110, and GFP-centrin-2 was visualized by endogenous fluorescence. Nuclear DNA was visualized with DAPI in the merged low-magnification image. In the merged insets, CP110 is shown in red and GFP-centrin-2 in green. Scale bar = 20 μm. Insets 1–4 show enlargements of the selected areas. Scale bar is 2 μm. (C) Centriole number over time after doxycycline washout. The graph shows calculated means from seven independent experiments with SEM (n ≥ 150 for each experiment). (D) Western blot of PLK4 and actin protein levels after washout of doxycycline or DMSO in hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells. (D’) Quantification of D. PLK4 is normalized to total protein. Error bars represent the SEM from two independent experiments.
A simple mechanism to explain the ability of cells to return to a normal number of centrioles would be limited or no duplication of centrioles, such that the number would decline after each cell division. The formation of nascent daughter centrioles, termed procentrioles, is marked by the presence of SASS6, a component of the centriole cartwheel that is present only in procentrioles (Strnad et al., 2007). Cells with extra centrioles were analyzed 5 d after doxycycline washout for the presence of paired centrioles with two centriole markers, GFP-Centrin-2 and CP110, which label the distal ends of centrioles (Chen et al., 2002). We found that cells either had all centrioles as unpaired singlets (as in G1 cells) or had all centrioles as paired doublets, and that SASS6 was associated with one of the two centrioles in each doublet (94% ± 2 of cells, N = 3 experiments, n = 791 cells in total), consistent with these representing a centriole and a procentriole (Figure 2A). Furthermore, centriole rosettes were not observed, which would have indicated ongoing PLK4 overexpression (Lopes et al., 2015). These results demonstrate that all centrioles in a cell can be competent for duplication under the conditions tested, which suggests that cycling cells have a greater capacity for centriole duplication than is normally exercised in a single cycle and thus that there is no limiting component within the range of centriole numbers we have observed.
FIGURE 2:

Extra centrioles are competent for duplication and are not subject to elimination. (A) Representative image of a cell with extra centrioles 5 d after washout of doxycycline showing doublets with associated SASS6 and other centriole markers, as indicated. GFP-centrin-2 is shown as endogenous fluorescence and SASS6 and CP110 by antibody staining. DAPI staining of nuclear DNA is also shown in the merged low-magnification image. In the merged inset, CP110 is shown in cyan and SASS6 in red. SASS6 was associated with one centriole in each doublet in 94% ± 2 of cells. N = 3 experiments for a total of n = 791 cells. Scale bar = 10 μm; 5 μm for inset. (B) Maximum intensity projection of selected images from Supplemental Figure S2 showing a dividing hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cell in which segregation of centrioles (GFP-centrin-2) is observed after doxycycline washout. Live imaging was performed on a spinning-disk confocal microscope. Z-stack = 0.5 μm. Scale bar = 20 μm. Insets scale bar = 2 μm. (C) Graph of the change (Δ) in centriole number in daughter cells compared with the mother cell from which they were derived. Cells were imaged live over 3 d, and centrioles were counted at the beginning and the end of the imaging session. To calculate the Δ value, the centriole number in the mother cell was subtracted from the summed centriole number in daughter cells divided by 2 to account for duplication. The graph represents the mean from four independent experiments with SEM, and n numbers are reported on top of each bar for each category as a total from all five independent experiments.
Another mechanism that could account for the restoration of the normal number of centrioles is elimination of extra centrioles. We used live imaging of cells after doxycycline washout to observe the fate of centrioles through mitotic divisions. If the extra centrioles induced by PLK4 overexpression are stable, then their number should be equal in a mother cell and the two resulting daughter cells. In contrast, if centriole elimination occurs, then the number of centrioles in the daughter cells will be less than in the mother cell. Figure 2B shows a sequence derived from a maximum projection of a confocal stack of images (more frames are shown in Supplemental Figure S2) in which the mother cell has eight GFP-Centrin-2 foci; since this cell divides soon afterward and thus is presumably in G2, these likely represent a bright centriole and an engaged (Tsou and Stearns, 2006) dimmer procentriole not resolvable at this point in the cell cycle. The two daughter cells that result from division also have a total of eight foci, one having six and the other two. The insets in Figure 2B show that after the disengagement that occurs in mitosis (Tsou and Stearns, 2006), each of the foci in the daughter cell can be seen to consist of two centrioles, one bright and one dim; thus, one daughter inherited 12 centrioles and the other 4—both states of CA for G1 cells.
We then analyzed 32 such dividing cells over the course of 3 d using an automated widefield fluorescence microscope and found no evidence for selective elimination or loss of centrioles (Figure 2C). The majority of the analyzed cells (83%) underwent one mitotic division during the course of the imaging session; the remaining 17% comprises cells that underwent two subsequent divisions, as well as cells that completed a first division giving rise to two cells, one of which divided once again and the other which never divided while imaged. The adopted resolution does not allow the discrimination between cells that entered S phase by the end of the imaging session. Nonetheless, we quantified the number of centrioles in the resulting daughter cells and compared it to the original cell at the beginning of the session (Figure 2C). These data show that in most dividing cells, all centrioles could be accounted for pre- and post-division. In those cells in which there was a difference, there were most often more centrioles in the daughter cells; we consider it likely that this difference is attributable to the inherent variation of cell cycle time and initiation of centriole duplication following division. Our conclusion regarding the lack of elimination is further supported by our experiment looking at microtubule nucleation from the extra centrioles (Figure 3A). Loss of microtubule nucleation can in fact be a cause of centriole loss during division (Godinho and Pellman, 2014). Our results concur with those of Baudoin et al. (2020), who also did not observe centriole elimination during mitosis in tetraploid cells (Baudoin et al., 2020).
FIGURE 3:

Extra centrioles are competent for nucleating microtubules and are mostly segregated symmetrically in mitosis. (A) Representative image of microtubule regrowth at 2.5 min postnocodazole washout in an hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cell with extra centrioles. Cells were treated with doxycycline for 48 h, and analysis was carried out from 1 d following washout. Cells were incubated with 2 μg/ml nocodazole for 1 h, then incubated without nocodazole for 2.5 min. Cells were stained for α-tubulin (red) and CP110 (cyan), and GFP-centrin-2 (green) was visualized by endogenous fluorescence. Nuclear DNA was visualized with DAPI in the merged image. Of the 2922 analyzed cells, 95% ± 3 of analyzed cells showed microtubule nucleation from all centrosomes. Scale bar = 10 μm. (B) Centriole segregation in recently divided pairs of daughter cells, as identified by presence of midbody (arrowheads). The left panel shows an example of asymmetric division (11 + 5 centrioles). The right panel shows an example of symmetric division (5 + 6 centrioles). Cells were stained for α- and ϕ-tubulin, and GFP-centrin-2 was visualized by endogenous fluorescence. DAPI staining of nuclear DNA and α-tubulin are only shown in the merged low-magnification image. Scale bar = 20 μm; 2 μm for insets. (C) Quantification of symmetry of centriole segregation in hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells fixed 1 h after mitotic shake-off. The Δ centriole # refers to the absolute difference in centriole number between the two daughters identified by the presence of the midbody. The results are pooled from eight independent experiments for a total of more than 1000 analyzed cells. n numbers for each category are included on top of each bar. (D) Quantification of symmetric and asymmetric segregation of centrioles. Calculations were obtained from the same experiments as in C, and centrioles numbers were rounded to the next even number when odd centrioles were counted. Centrioles # refers to the total number of centrioles in the two daughter cells identified by the presence of the midbody. Symmetric refers to an equal number of centrosomes in each daughter cell ± 2. Asymmetric refers to situations in which a daughter cell inherited >2 centrosomes compared with the sister cell. Overall frequency refers to the frequency of symmetric or asymmetric segregation over the total number of cells that showed symmetric or asymmetric segregation, respectively; n numbers are reported at the top of each bar.
A third possible mechanism that could account for restoration of centriole number is asymmetric segregation of centrioles in mitosis. Centrioles segregate as part of centrosomes on the mitotic spindle in most animal cells, and this segregation requires that centrioles acquire pericentriolar material in a centriole-centrosome conversion step (Wang et al., 2011). In Drosophila epithelial cells, extra centrioles can become inactivated, losing microtubule nucleation capacity (Sabino et al., 2015), which might result in failure to segregate accurately. Based on the timeline of our experiments (Figure 1A), we would expect that most of the extra centrioles generated would have had the opportunity to undergo the centriole-centrosome conversion. We tested this by assessing their ability to nucleate microtubules in a microtubule regrowth assay. We found that in most cases, all centrioles nucleated microtubules, showing that they are active as centrosomes (95% ± 3 of analyzed cells; n = 2922 cells). Figure 3A shows a typical cell with extra centrioles, in which all centrioles were active for nucleation. This suggests that selective inactivation is unlikely to be a factor in restoration of centriole number. Our result is in accordance with the findings by Rhys et al. (2018) which showed that amplified centrosomes in MCF10A cells do not lose γ-tubulin and pericentrin (Rhys et al., 2018), as occurs with centrosome inactivation in Drosophila (Basto et al., 2008; Sabino et al., 2015).
We next examined centriole segregation directly, quantifying segregation into daughter cells following synchronization of cells in mitosis by mitotic shake-off. Daughter cells from a recent division were identified by the presence of a midbody, and centrioles were counted in each cell (Figure 3B). The data were quantified as the absolute difference in centriole number between two daughter cells. Centrioles were segregated symmetrically in approximately 70% of the analyzed cells (Figure 3C). The remaining 30% show asymmetric segregation, with decreasing frequency with more uneven segregation that is best described as a Poisson distribution centered on λ = 1.3 (R2 = 0.93). Furthermore, the frequency of asymmetric segregation increased with centriole number (Figure 3D).
Given that none of the direct mechanisms of centriole number homeostasis were consistent with our observations, we considered the possibility that cells with extra centrioles might have a selective growth disadvantage relative to normal cells. Consistent with previous work (Holland et al., 2012), we found that hTERT-RPE-1 GFP-Centrin-2 tetON-PLK4 cells induced to have extra centrioles proliferated more slowly as a population than control-treated cells (Figure 4A, p < 0.001). To further understand the nature of this reduced-growth phenotype, we employed several single-cell-level assays of proliferation. EdU incorporation into DNA was used as a measure for cell cycle progression into S phase. The top two panels in Figure 4B show the time course of labeling and analysis. In cells that experienced doxycycline treatment but had the normal number of centrioles, the percent of cells entering S phase reached approximately 65% after 24 h of EdU incubation at all three assay timepoints. In contrast, the percent of cells with extra centrioles entering S phase was similar to normal cells on day 1, but decreased when assayed on subsequent days, falling to 36% on day 3 (Figure 4B). This increased with longer incubation in EdU to account for a potentially longer cell cycle but did not reach the same level as in normal cells. Thus, the fraction of cells with extra centrioles that enter the cell cycle declines over time.
FIGURE 4:

CA results in increased cell cycle length. (A) Growth curves of hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells either treated with 1 μg/ml doxycycline (DOX washout) for 48 h followed by washout or treated with DMSO as a control followed by washout (DMSO washout). Each point represents the mean + SEM of three independent experiments. Significance was calculated by one-way ANOVA; p < 0.0001. (B) DNA replication at times after CA. DNA replication was assayed by EdU incorporation. The top panel shows a schematic of the experiment; EdU incubation was initiated at the indicated time and continued until fixation at 24 or 48 h after addition. The bottom panel shows EdU+ vs. EdU- cells along with centriole number (2–4 (normal) vs. 5+ (amplified)) as mean + SEM of three independent experiments. (C) Observed events in live imaging of hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells. Cells were observed over 3 d, and cells that are indicated as “no divisions” did not divide in the observed period; 5+ refers to cells with amplified centrioles, and 2–4 refers to cells with the normal number of centrioles. Total number of cells in each category is indicated above each bar. (D, E) Duration of mitosis and interphase in hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells with normal number (2–4) centrioles vs. those with amplified centrioles (5+). Time of cell cycle phase was calculated from live-imaging sequences. The graph shows mean values; significance was assessed by unpaired two-tailed t test; for mitosis, p < 0.0001; for interphase, p < 0.0005. (F) Immunoblot of p53 and actin (loading control) in TP53–/– cells, hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells without doxycycline, and hTERT-RPE-1 GFP-centrin-2 tetON-Plk4 cells with 48 h 1 μg/ml doxycycline pretreatment. Cells were treated with Nutlin3a as a positive control for p53 activation. See Supplemental Figure S3A for the uncropped blot. Error bars represent SEM from three independent experiments. Significance was determined via a Welch’s t test; ***p < 0.0001. (G) Quantification of cells with visible nuclear enrichment of p53 after washout of doxycycline following 48 h treatment. Shown are the percentages of p53+ cells within the population of cells with normal (2–4, teal line) centriole numbers or amplified centrioles (5+, purple line). Data were pooled from three independent experiments.
We next used time-lapse imaging to observe cell division histories in cells with extra centrioles. hTERT-RPE-1 GFP-Centrin-2 tetON-PLK4 cells were imaged over 3 d, recording both phase and GFP fluorescence at 5 min intervals in cells that experienced doxycycline treatment but had the normal number of centrioles (n = 51) and those with extra centrioles (n = 214). These sequences revealed several features of cell division of cells with extra centrioles (Figure 4C). First, only 51% of the cells with extra centrioles divided over 3 d of observation, whereas 100% of normal cells divided. Second, of those cells with extra centrioles that divided, only 3% divided into more than two cells, consistent with previous reports of centrosome clustering in CA-cells (Ring et al., 1982; Kwon et al., 2008), whereas none of the normal cells underwent a multipolar division. Lastly, 8% of the cells with extra centrioles died at some point during imaging, assessed by dramatically increased blebbing and detachment from the substrate, whereas none of the normal cells suffered this fate.
We also used the live-cell imaging sequences to determine the length of mitosis (Figure 4D) and interphase (Figure 4E) in cells with normal or extra centrioles within the same treated population. The duration of mitosis was measured as the time from the beginning of rounding up to the appearance of two distinctly separate cell bodies. Mitotic duration was significantly longer in cells with extra centrioles, 45.5 ± 1.5 min (n = 104), compared with cells with normal centrioles, 29.9 ± 1.2 min (n = 51); p < 0.0001. This is in accordance with previous results in both Drosophila and mammalian cells where increased mitosis time was observed (Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008). Interphase duration was also significantly longer in cells with extra centrioles, 1405 ± 52.3 min (n = 27), compared with cells with normal centrioles, 1151 ± 26.5 min (n = 21); p < 0.0005. Although we have not characterized the mechanism of the cell cycle arrest and/or delay that many CA-cells undergo, we note that previous findings by other groups showed that CA-cells divide aberrantly to generate aneuploidy at higher frequency than cells without extra centrioles (Ganem et al., 2009; Nicholson et al., 2015) which can result in cell cycle arrest or cell death (Nicholson et al., 2015),
Because of previous reports of p53 stabilization in centrosome-amplified cells (Holland et al., 2012; Fava et al., 2017), we quantified p53 protein levels hTERT-RPE-1 GFP-Centrin-2 tetON-PLK4 with or without doxycycline treatment. TetON-PLK4 cells treated with doxycycline for 48 h followed by 3 d washout showed 33% increased level of p53 protein compared with DMSO-treated cells. These cells are able to stabilize p53 to much higher levels; treatment with the MDM1 inhibitor Nutlin3a increased the p53 level more than 8-fold. As a negative control, no p53 was detected in RPE-1-TP53–/– cells. (Figure 4F). Due to the only modest stabilization of p53, especially in comparison to published results from cells overexpressing an autoregulation deficient PLK4, we quantified presence of nuclear p53 in individual TetON-PLK4 cells treated with doxycycline for 48 h followed by washout. A higher percentage of cells with amplified (5+) centrioles had visible nuclear p53 than cells with normal (2–4) centrioles; however, not all cells with extra (5+) centrioles showed nuclear p53 (Figure 4G), which is in agreement with the only modest increase in p53 level we observed via Western blot. It’s important to note that WT RPE-1 cells without the TetON-PLK4 insert showed increases in nuclear p53 in the initial days after doxycycline washout, suggesting that doxycycline treatment itself leads to some nuclear p53 accumulation (Supplemental Figure S3B), which has been previously reported (Fujioka et al., 2004). Some cells with CA do show nuclear p53, but the nuclear accumulation does not occur in all cells simultaneously after overexpression of WT PLK4.
An important question is how the results we describe in cultured cells may relate to cancer cells in vivo. Despite the disfunction of the p53 pathway in many human cancers (Mantovani et al., 2019), some cancers have an intact p53 pathway, like the RPE-1 model system that we employ in this work. To predict cancers to which our observations might apply, we assessed alterations (including mutation, fusion, amplification, deep deletion, or multiple alterations) of the p53 pathway genes TP53, CDKN1A, CDKN2A, MDM2, MDM4, and RB1 across 32 cancer types from the Pan-Cancer project from The Cancer Genome Atlas (TCGA; Supplemental Figure S4A). Next, we examined PLK4 mRNA levels across the same 32 cancer types from the same Pan-Cancer project (Figure 5A). The three cancer types with the highest median PLK4 mRNA expression were cervical cancer, testicular germ cell cancer, and AML, which all had less than 20% of cases with genomic alterations in TP53, CDKN1A, CDKN2A, MDM2, MDM4, and RB1 (Figure 5A; Supplemental Figure S4A). Furthermore, these cancer types also showed above-average SASS6 and STIL mRNA levels (Supplemental Figure S4, B and C), each of which can cause centriole overduplication when overexpressed in tissue culture cells (Leidel et al., 2005; Tang et al., 2011) and together are transcript signatures of CA in cancer cells (De Almeida et al., 2019).
FIGURE 5:

PLK4 and p53 relationship in cancers and summary of findings. (A) PLK4 expression in human cancers. Data are represented as beeswarm plots showing mRNA expression levels and organized from left to right in order of increasing median PLK4 expression; medians are denoted by orange lines. Transcriptomic and genomic data, obtained from the TCGA Research Network PanCanAtlas and visualized with cBioPortal, are RNA-seq data from Illumina HiSeq_RNASeqV2. Percentages of cancers with altered p53 pathway transcripts (TP53, CDKN1A, CDKN2A, MDM2, MDM4, and RB1) are also from the PanCanAtlas and includes mutation, fusion, amplification, deep deletion, and multiple alterations (see also Supplemental Figure S4). (B) log2(PLK4 mRNA) in testicular germ cell tumors with or without genomic and transcriptomic alterations to TP53. Transcriptomic and genomic data, obtained from the TCGA Research Network PanCanAtlas, are RNA-seq data from Illumina HiSeq_RNASeqV2. (C) Comparison between calculated population growth (theoretical) and experimental (empirical) data. The model considers 10% of normal cells (2–4 centrioles) in the starting population. In the equations, N(t) represents the population size of normal (n) cells or ca (centriole amplified) cells; α represents the proportion of cells that divide; δ represents the rate of mitoses per day; ϕ represents the death rate in the population. See Experimental Procedures for details. (D) Schematic to summarize fates of cells with CA.
In the case of cervical cancer, many such cancers have a p53 pathway that is inactivated by HPV viral proteins E6 and E7 (Zur Hausen, 2002); therefore, although mutations in the p53 pathway in the cancer cells themselves are not common, we discounted these cancers as likely to have nonfunctional p53 pathways. The negative regulators of p53, MDM2 and MDM4 are frequently overexpressed in AML (Supplemental Figure S4D; Bueso-Ramos et al., 1993). However, there was no significant correlation between transcript levels of PLK4 with either MDM2 or MDM4 in AML (Supplemental Figure S4, E and F). Testicular germ cell cancers typically retain wild-type p53, and centrosome amplification has been reported in these tumors (Mayer et al., 2003). Furthermore, TP53 mRNA levels did not correlate with PLK4 mRNA levels in testicular germ cell cancer (Supplemental Figure S4G), and the small subset of p53-deficient tumors did not show higher PLK4 than the majority with predicted functional p53 (Figure 5B). In addition to its relevance to population-level centriole number homeostasis, our work may also be relevant to testicular germ cell cancer and other cancers in which PLK4 overexpression and CA occur in cells with an intact p53 pathway.
Our results suggest that a population containing cells with extra centrioles returns to normal numbers via proliferation of cells with normal centriole numbers preexisting within the population or generated by relatively rare divisions that asymmetrically segregate centrioles. A simple mathematical description matching the parameters of division and proliferation that we characterized in Figure 4 would be sufficient to explain the change in the population shown in Figure 1C. These equations take into consideration the difference in cell cycle length, the fraction of cells that die each generation, and the fraction of cells that enter the cell cycle. Using the parameters determined in Figure 4, we find that this description accurately recapitulates the population dynamics of the experimental cultures (Figure 5C). Through examination of single cells rather than populations, we found that individual cells with amplified centrioles do not all arrest immediately in the next cell cycle. Instead, cells showed heterogeneity in response to CA (Figure 5D). Recent reports by Baudoin et al. (2020) and Galofré et al. (2020) focused on the fates of extra centrosomes in tetraploid or near-tetraploid cells generated through cytokinesis failure (Baudoin et al., 2020) or naturally occurring in cancer cells (Galofré et al., 2020). Baudoin et al. (2020) observed a loss of extra centrosomes from a population of cells and concluded that asymmetric divisions are the main factor leading to the return to normal number of centrosomes. In our experiments, we did not observe frequent asymmetric divisions, although we did observe these events rarely. However, we need to take into account the two different experimental systems employed by our study and those of Baudoin et al. (2020) and Galofré et al. (2020). Cytokinesis failure as a method to generate CA also causes tetraploidy, which itself has been reported to cause whole-chromosome missegregation and chromosomal rearrangements, p53-dependent cell cycle arrest, and/or cell death (Fujiwara et al., 2005; Ganem et al., 2014). The generation of extra centrioles by PLK4 overexpression as we used here leads to variable centriole numbers, as opposed to the exact number found in cells that failed cytokinesis, but does not directly alter ploidy. Additionally, we conducted our experiments in p53-proficient cells, as opposed to the RPE-1 p53–/– or cancer cells employed in the other experiments. Nonetheless, both works share similar overall observations regarding the return to a normal number of a population with extra centrosomes.
In summary, we have found that CA in populations of human RPE-1 cells is resolved by selection against cells with amplified centrioles based on their growth disadvantage and not by centriole elimination, centriole duplication failure, or grossly asymmetric centriole segregation. These results suggest that only under conditions of positive selection for cells with extra centrioles, continuous generation of such centrioles, or alleviation of the disadvantageous growth phenotypes would they be maintained in a population.
EXPERIMENTAL PROCEDURES
Cell lines and cell culture
hTERT-RPE-1 GFP::CETN2 TetON::PLK4 (Hatch et al., 2010) and hTERT-RPE-1 TP53–/– were a gift from Meng-Fu Bryan Tsou (Memorial Sloan Kettering Cancer Center). All RPE-1 cells were cultured in DMEM/F-12 (with 15 mM HEPES, phenol red, and L-glutamine; Corning) supplemented with 10% Cosmic calf serum (CCS; HyClone). Cells were maintained at 37°C under 5% CO2. For the PLK4TetO cells, single clones were isolated via fluorescence-activated cell sorting and characterized for centrioles amplification capabilities. One single clone was selected for further analysis.
Cells were routinely tested for mycoplasma contamination using pooled forward primers: 5′-CGCCTGAGTAGTACGTTCGC, 5′-CGCCTGAGTAGTACGTACGC, 5′-TGCCTGAGTAGTACATTCGC, 5′-TGCCTGGGTAGTACATTCGC, 5′-CGCCTGGGTAGTACATTCGC, 5′-CGCCTGAGTAGTATGCTCGC; and pooled reverse primers: 5′-GCGGTGTGTACAAGACCCGA, 5′-GCGGTGTGTACAAAACCCGA 5′-GCGGTGTGTACAAACCCCGA.
Immunofluorescence
Cells were grown on poly-l-lysine-coated #1.5 glass coverslips (Electron Microscopy Sciences). Cells were washed with phosphate-buffered saline (PBS), then fixed with 20°C methanol for 15 min. Coverslips were then washed with PBS and blocked with PBS-BT (3% bovine serum albumin, 0.1% Triton X-100, 0.02% sodium azide in PBS) for 30 min. Coverslips were incubated with primary antibodies diluted in PBS-BT for 45 min, washed with PBS-BT, and incubated with secondary antibodies, and DAPI was diluted in PBS-BT for 45 min and then washed again. Samples were mounted using Mowiol (Polysciences) in glycerol containing 1,4,-diazobicycli-[2.2.2] octane (Sigma-Aldrich) antifade. For EdU staining, the Click-iT Cell Reaction Kit (Thermo Fisher; Cat. No. C10269) was used according to the manufacturer’s instructions. Images were acquired on a Zeiss Axiovert 200 M microscope (Carl Zeiss, Jena, Germany) with Plan Apo Chromat 63×/1.4 NA objective and CCD camera (Orca ER; Hamamatsu Photonics, Hamamatsu, Japan).
Antibodies for immunofluorescence
Primary antibodies used for immunofluorescence: rabbit anti-CP110 (1:200, Proteintech); mouse IgG2b anti-SASS6 (1:200, Santa Cruz Biotech); rabbit IgG anti-gamma-tubulin, clone AK-15 (1:200, Sigma-Aldrich); mouse IgG1 anti-alpha-tubulin, clone DM1A (1:1000, Sigma-Aldrich); mouse IgG2a anti-p53, clone DO-1 (1:1000, Santa Cruz); mouse IgG1 anti-polyglutamylated-tubulin, clone GT335 (1:1000, Sigma-Aldrich); mouse IgG2b anti-centrin3, clone 3E6 (1:1000, Novus Biological). For immunofluorescence, Alexa Fluor–conjugated secondary antibodies (Thermo-Fisher) were diluted 1:2000.
SDS–PAGE and Western blot
Samples were lysed directly into 1× Laemmlli buffer, boiled, and reduced with BME (p53) or not reduced (PLK4), then separated by 12% (p53) or 7.5% (PLK4,) SDS–PAGE resolving (5% stacking) gels and transferred to nitrocellulose membrane at 100 V for 1.5 h at 4°C in 20% ethanol-containing transfer buffer. When used, Revert total Protein Stain (Li-Cor) was used as instructed by manufacturer before blocking. Blots were blocked in 5% nonfat dried milk (Safeway) in TBST (Tris-buffered saline + 1% Tween-20) for 1 h, shaking at RT. Primary and secondary antibody staining was performed in blocking solution for 2 h each, shaking at RT, or overnight, shaking at 4 °C (PLK4). Blots were imaged using a Li-Cor Odyssey.
Primary antibodies used for Western blot: anti-β1-actin, clone AC-74 (1:10,000, Sigma-Aldrich); anti-p53, clone DO-1 (1:5000, Santa Cruz); anti-PLK4, clone 6H5 (1:2500, EMD Millipore). Secondary antibodies used for Western blot: donkey anti-mouse IgG (H+L), IRDye 800CW conjugated (1:15,000, Li-Cor Biosciences); donkey anti-rabbit IgG, IRDye 800CW conjugated (1:15,000, Li-Cor Biosciences). Ladders shown in uncropped blots are Precision Plus Dual Color Standards (Bio-Rad).
Cell treatments and assays
To induce centrosome amplification, hTERT-RPE-1 GFP-Centrin-2 TetON-Plk4 were treated with 1 μg/ml doxycycline hydrochloride (DOX; Thermo Fisher Scientific; Cat. No. BP2653) in DMSO for 48 h. For washout experiments, the medium was removed, and cells were washed 3× with PBS before adding fresh medium.
For microtubule regrowth, 1 d prior to the experiment cells were seeded on poly-l-lysine-coated #1.5 glass coverslips. Cells were treated with 2 μg/ml nocodazole for 1 h, washed with cold PBS, incubated with warm complete medium, and fixed in methanol at indicated time points.
For mitotic shake-off, cells were grown in 10-cm plates. Medium was removed, and plates were subjected to gentle shaking. Medium was added to collect detached cells and discarded. After 30 min, the procedure was repeated to plate cells on poly-l-lysine-coated #1.5 glass coverslips. Cells were fixed with methanol and stained.
To assay proliferation, 104 cells from each condition were seeded in 24-well plates after doxycycline washout. Three wells were counted with a hemocytometer each day until the end of the experiment.
For stabilization of p53 treatment, 10 μM Nutlin3a (Selleckchem) was added to cells 48 h before lysis in 1× Laemmlli buffer.
Time-lapse imaging
hTERT-RPE-1 GFP-Centrin-2 tetON-PLK4 cells were diluted 1:10 with WT hTERT-RPE-1 cells to facilitate long-term tracking of the fluorescent cells as they migrated and divided. The cells were seeded onto glass-bottom dishes (World Precision Instruments) 1 d prior to imaging; 30 min prior to imaging, the medium was changed to phenol-free DMEM-F12 (Life Technologies) supplemented with 10% CCS. Images were acquired every 5 min on a Keyence digital optical microscope (Keyence) with a Nikon S Plan Fluor ELWD 20×/0.45 NA objective. Still images were taken using a Nikon Plan Apo 60×/1.40 NA objective. Cells were maintained in a humidified chamber at 37°C under 5% CO2 during image acquisition. Alternatively, cells were imaged every 15 min as 0.5-μm Z-stacks using a Zeiss Axio Observer microscope with a confocal spinning-disk head (Yokogawa), Plan Apo Chromat 63×/1.4 NA objective, and a Cascade II:512 EM-CCD camera (Photometrics) run with MicroManager software (Edelstein et al., 2014). Statistical analyses were performed using GraphPad Prism.
Determination of single cell p53 status
After 48 h treatment with DMSO or doxycycline, cells were washed 3× in PBS and then fixed on the indicated day after washout. Fixed cells were stained for p53 and centriole markers (centrin, polyglutamylated-tubulin) as well as DAPI. Cells with visible p53-labeled nuclei were considered p53+ and cells without a distinct nuclear outline in the p53 channel were considered p53–. Centriole number was determined by counting the centrin/polyglytamylated-tubulin copositive foci.
RT-qPCR
After 48 h treatment with DMSO or doxycycline, RNA was extracted using Trizol (Invitrogen) following the manufacturer’s instructions. cDNA was synthesized using Maxima First Strand Synthesis (Thermo) following the manufacturer’s instructions. RT-qPCR was performed using SYBR Green (Bio-Rad) following the manufacturer’s instructions. Primers used were:
GAPDH (forward) 5′-ACATCGCTCAGACACCATG
GAPDH (reverse) 5′-TGTAGTTGAGGTCAATGAAGGG
PLK4 (forward) 5′-AGACCACCCTTCGACACTGA
PLK4 (reverse) 5′-GTCCTTGGCCTCTATTGACAAA
Reactions were performed in triplicate on N = 2 samples per condition.
Theoretical calculations
Theoretical population dynamics were considered under a discrete model, since we experimentally measured cell number per day. Calculations were performed where cell populations are represented by the generalized equation:
in which N is the population size, 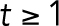 is the time in days, α is the proportion of cells that divide, δ is the division rate per day, and ϕ is the rate of death in the population. Values for α, δ, and ϕ were derived form the live imaging data:
is the time in days, α is the proportion of cells that divide, δ is the division rate per day, and ϕ is the rate of death in the population. Values for α, δ, and ϕ were derived form the live imaging data:
| α | δ | ϕ | |
|---|---|---|---|
| Normal centrioles (n) | 1 | 1.219 | 0 |
| Amplified centrioles (ca) | 0.51 | 0.993 | 0.08 |
δ was calculated for each population using the averages of interphase, 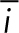 , and mitosis,
, and mitosis, 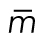 lengths:
lengths:
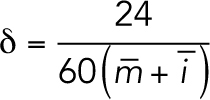 |
Conversion from CA-cells to normal cells was considered negligible since centrioles in CA-cells duplicated. Consideration of the empirical values results in the generalized equation for the population of cells with normal centriole numbers,  :
:
and, for the population of CA-cells,  :
:
Supplementary Material
Acknowledgments
The results published here in Figure 5 and Supplemental Figure S4 are in whole or part based upon data generated by TCGA Research Network: https://www.cancer.gov/tcga. Cell sorting/flow cytometry analysis for this project was done on instruments in the Stanford Shared FACS Facility. This work was supported by National Institutes of Health (NIH) grant R35GM130286 to T.S. K.F. was supported by the National Institute of General Medical Sciences of the NIH under award number T32GM007276. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations used:
- CA
centriole amplification
- CCS
Cosmic calf serum
- DOX
doxycycline hydrochloride
- PBS
phosphate-buffered saline
- TBS
Tris-buffered saline.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-04-0195) on September 23, 2020.
REFERENCES
- Basto R, Brunk K, Vinadogrova T (2008). Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032–1042. 10.1016/j.cell.2008.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoin NC, Nicholson JM, Soto K, Martin O, Chen J, Cimini D (2020). Asymmetric clustering of centrosomes defines the early evolution of tetraploid cells. Elife. 10.7554/eLife.54565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, Carmo N, Balloux F, Callaini G, Glover DM (2005). SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 15, 2199–2207. 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- Bueso-Ramos CE, Yang Y, DeLeon E, McCown P, Stass SA, Albitar M (1993). The human MDM-2 oncogene is overexpressed in leukemias. Blood 82, 2617–2623. 10.1182/blood.V82.9.2617.2617 [DOI] [PubMed] [Google Scholar]
- Chan JY (2011). A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7, 1122–1144. 10.7150/ijbs.7.1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD (2002). CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3, 339–350. 10.1016/S1534-5807(02)00258-7 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW (2015). Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 16, 611–624. 10.1038/nrm4062 [DOI] [PubMed] [Google Scholar]
- De Almeida BP, Vieira AF, Paredes J, Bettencourt-Dias M, Barbosa-Morais NL (2019). Pan-cancer association of a centrosome amplification gene expression signature with genomic alterations and clinical outcome. PLoS Comput Biol 15, e1006832. 10.1371/journal.pcbi.1006832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre M, Gönczy P (2004). The arithmetic of centrosome biogenesis. J Cell Sci 117, 1619–1630. 10.1242/jcs.01128. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N (2014). Advanced methods of microscope control using μManager software. J Biol Methods 1, e10. 10.10.14440/jbm.2014.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, Villunger A (2017). The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31, 34–45 10.1101/gad.289728.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka S, Schmidt C, Sclabas GM, Li Z, Pelicano H, Peng B, Yao A, Niu J, Zhang W, Evans DB, et al. (2004). Stabilization of p53 is a novel mechanism for proapoptotic function of NF-κB. J Biol Chem 279, 27549–27559. 10.1074/jbc.M313435200 [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D (2005). Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047. 10.1038/nature04217 [DOI] [PubMed] [Google Scholar]
- Galofré C, Asensio E, Ubach M, Torres IM, Quintanilla I, Castells A, Camps J (2020). Centrosome reduction in newly-generated tetraploid cancer cells obtained by separase depletion. Sci Rep 10, 1–12. 10.1038/s41598-020-65975-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu S-Y, O’rourke KP, Arnaud J, Yimlamai D, Thé M, Camargo FD, Pellman D (2014). Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation Cell 158, 833–848 10.1016/j.cell.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Pellman D (2014). Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc B Biol Sci 369, 20130467. 10.1098/rstb.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA (2005). The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 7, 1140–1146. 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T (2010). Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol 191, 721–729. 10.1083/jcb.201006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW (2012). The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev 26, 2684–2689. 10.1101/gad.207027.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA (2007). Plk4-induced centriole biogenesis in human cells. Dev Cell 13, 190–202. 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D (2008). Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 22, 2189–2203. 10.1101/gad.1700908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Delattre M, Cerutti L, Baumer K, Gönczy P (2005). SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat Cell Biol 7, 115–125. 10.1038/ncb1220 [DOI] [PubMed] [Google Scholar]
- Lopes CAM, Jana SC, Cunha-Ferreira I, Zitouni S, Bento I, Duarte P, Gilberto S, Freixo F, Guerrero A, Francia M, et al. (2015). PLK4 trans-autoactivation controls centriole biogenesis in space. Dev Cell 35, 222–235. 10.1016/j.devcel.2015.09.020 [DOI] [PubMed] [Google Scholar]
- Mantovani F, Collavin L, Del Sal G (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ 26, 199–212. 10.1038/s41418-018-0246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer F, Stoop H, Sen S, Bokemeyer C, Oosterhuis JW, Looijenga LHJ (2003). Aneuploidy of human testicular germ cell tumors is associated with amplification of centrosomes. Oncogene 22, 3859–3866. 10.1038/sj.onc.1206469 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Saito H, Takekawa M (2013). SAPK pathways and p53 cooperatively regulate PLK4 activity and centrosome integrity under stress. Nat Commun 4. 10.1038/ncomms2752 [DOI] [PubMed] [Google Scholar]
- Nicholson JM, Macedo JC, Mattingly AJ, Wangsa D, Camps J, Lima V, Gomes AM, Dória S, Ried T, Logarinho E, Cimini D (2015). Chromosome mis-segregation and cytokinesis failure in trisomic human cells. Elife 4. 10.7554/eLife.05068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ (2018). Once and only once: Mechanisms of centriole duplication and their deregulation in diseases. Nat Rev Mol Cell Biol 19, 297–312. 10.1038/nrm.2017.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009). Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678. 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Stearns T (2011). The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13, 1154–1160. 10.1038/ncb2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhys AD, Monteiro P, Smith C, Vaghela M, Arnandis T, Kato T, Leitinger B, Sahai E, McAinsh A, Charras G, Godinho SA (2018). Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J Cell Biol 217, 195–209. 10.1083/jcb.201704102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring D, Hubble R, Kirschner M (1982). Mitosis in a cell with multiple centrioles. J Cell Biol 94, 549–556. 10.1083/jcb.94.3.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino D, Gogendeau D, Gambarotto D, Nano M, Pennetier C, Dingli F, Arras G, Loew D, Basto R (2015). Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol 25, 879–889. 10.1016/j.cub.2015.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gönczy P (2007). Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell 13, 203–213. 10.1016/j.devcel.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CJC, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, Chang CW, Wu KS, Tang TK (2011). The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J 30, 4790–4804. 10.1038/emboj.2011.378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou MFB, Stearns T (2006). Mechanism limiting centrosome duplication to once per cell cycle. Nature 442, 947–951. 10.1038/nature04985 [DOI] [PubMed] [Google Scholar]
- Wang JT, Kong D, Hoerner CR, Loncarek J, Stearns T (2017). Centriole triplet microtubules are required for stable centriole formation and inheritance in human cells. Elife 6. 10.7554/eLife.29061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W-J, Soni RK, Uryu K, Bryan Tsou M-F (2011). The conversion of centrioles to centrosomes: essential coupling of duplication with segregation. J Cell Biol 193, 727–739. 10.1083/jcb.201101109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. (2015). Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160. 10.1126/science.aaa5111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Loncˇarek J, Khodjakov A, Rieder CL (2008). Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat Cell Biol 10, 748–751. 10.1038/ncb1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur Hausen H (2002). Papillomaviruses and cancer: From basic studies to clinical application. Nat Rev Cancer 2, 342–350. 10.1038/nrc798 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.