

Abstract
Background
KBG syndrome is a rare autosomal dominant genetic disease mainly caused by pathogenic variants of ankyrin repeat domain-containing protein 11 (ANKRD11) or deletions involving ANKRD11. Herein, we report a novel de novo heterozygous frameshift ANKRD11 variant via whole exome sequencing in a Chinese girl with KBG syndrome.
Case presentation
A 2-year-2-month-old girl presented with a short stature and developmental delay. Comprehensive physical examinations, endocrine laboratory tests and imaging examination were performed. Whole‐exome sequencing and Sanger sequencing were used to detect and confirm the variant associated with KBG in this patient, respectively. The pathogenicity of the variant was further predicted by several in silico prediction tools. The patient was diagnosed as KBG syndrome with a short stature and developmental delay, as well as characteristic craniofacial abnormalities, including a triangular face, long philtrum, wide eyebrows, a broad nasal bridge, prominent and protruding ears, macrodontia of the upper central incisors, dental crowding, and binocular refractive error. Her skeletal anomalies included brachydactyly, fifth finger clinodactyly, and left-skewed caudal vertebrae. Electroencephalographic results generally showed normal background activity with sporadic spikes and slow wave complexes, as well as multiple spikes and slow wave complexes in the bilateral parietal, occipital, and posterior temporal regions during non-rapid-eye-movement sleep. Brain MRI showed a distended change in the bilateral ventricles and third ventricle, as well as malformation of the sixth ventricle. Whole exome sequencing revealed a novel heterozygous frameshift variant in the patient, ANKRD11 c.1366_1367dup, which was predicted to be pathogenic through in silico analysis. The patient had received physical therapy since 4 months of age, and improvement of gross motor dysfunction was evident.
Conclusions
The results of this study expand the spectrum of ANKRD11 variants in KBG patients and provide clinical phenotypic data for KBG syndrome at an early age. Our study also demonstrates that whole exome sequencing is an effective method for the diagnosis of rare genetic disorders.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-00920-3.
Keywords: KBG syndrome, ANKRD11, Frameshift variant, Physical therapy, Whole-exome sequencing, Case report
Background
KBG syndrome (MIM # 148050) is a rare autosomal dominant genetic disease that was first described by Herrmann in 1975 [1]. The typical phenotypes include intellectual disability, developmental delay, macrodontia of the upper central incisors, characteristic craniofacial features, hearing loss, skeletal abnormalities, and short stature [2–6]. The prevalence of this disease remains unknown. To date, more than 200 patients with KBG syndrome have been reported worldwide [7]. However, it is likely that this syndrome is underdiagnosed, since many of the features are mild and do not specific to this syndrome [2]. The genetic causes of KBG syndrome include single nucleotide variants and small indels of ANKRD11 or microdeletions of 16q24.3 involving ANKRD11, accounting for approximately 83% and 17% of cases, respectively [8–12]. ANKRD11 encodes ankyrin repeat domain-containing protein 11, an ankyrin repeat domain-containing cofactor that plays essential roles in embryogenesis, skeletogenesis, and bone turnover [13, 14]. To date, more than 300 variants in ANKRD11 have been identified in cases of KBG syndrome according to ClinVar database, and the majority of these variants occurred de novo [8].
In this study, we report the analysis of a de novo heterozygous frameshift ANKRD11 variant in a Chinese girl with KBG syndrome and the corresponding phenotypes.
Case presentation
The patient was a Chinese girl who was born at 37+3 weeks gestation via spontaneous vaginal delivery after an uneventful pregnancy. Her birth length and weight were 45.0 cm (− 2.2 SD) and 2200 g (− 2.5 SD) respectively, and her head circumference was 31 cm (− 2.4 SD). The height of her mother and father were 168 cm and 155 cm, respectively. After birth, the patient showed feeding difficulty, short stature (Fig. 1), motor dysfunction, and developmental delay (Table 1). She began to walk and talk at 17 and 25 months, respectively. No hearing or behavioral issue was noted.
Fig. 1.

Evaluation of height and weight of the patient. a Height of patient at the age period between 0 and 24 months (measured in lying down state). b Height of patient at the age period between 24 and 34 months (measured in standing state). c Weight of patient. The triangles and squares indicate the height and weight measurements of the patient, respectively
Table 1.
Neuropsychological evaluation of the patient
| Age (M) | Gross motor | Fine motor | Adaptive strengths | Language | Social behaviors | General evaluation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DQ | MA | DQ | MA | DQ | MA | DQ | MA | DQ | MA | DQ | MA | |
| 15.3 | 65 | 10.0 | 65 | 10.0 | 72 | 11.0 | 59 | 9.0 | 72 | 11.0 | 67 | 10.2 |
| 21.0 | 93 | 19.5 | 71 | 15.0 | 71 | 15.0 | 71 | 15.0 | 57 | 12.0 | 73 | 15.3 |
| 25.6 | 94 | 24.0 | 70 | 18.0 | 64 | 16.5 | 74 | 19.0 | 59 | 15.0 | 72 | 18.5 |
| 33.4 | 90 | 30.0 | 76 | 25.5 | 72 | 24.0 | 67 | 22.5 | 76 | 25.5 | 76 | 25.5 |
| 37.6 | 96 | 36.0 | 76 | 28.5 | 76 | 28.5 | 60 | 22.5 | 76 | 28.5 | 77 | 28.8 |
DQ = MA/M × 100%. DQ ≥ 130: exellent; DQ = 115–129: good; DQ = 85–114: average; DQ = 70–84: intermediate; DQ ≤ 70: poor
M month, DQ developmental quotient, MA mental age
At the age of 2 years and 2 months, she received a comprehensive physical examination, which showed some suspect craniofacial features, including a triangular face, long philtrum, wide eyebrows, broad nasal bridge, prominent and protruding ears, macrodontia of the upper central incisors (dental crown: 7.0 mm × 7.7 mm), dental crowding, and binocular refractive error (Fig. 2). Other physical examination findings included brachydactyly, fifth finger clinodactyly, delayed anterior fontanel closure, and an abnormal caudal vertebrae shape (Fig. 2).
Fig. 2.
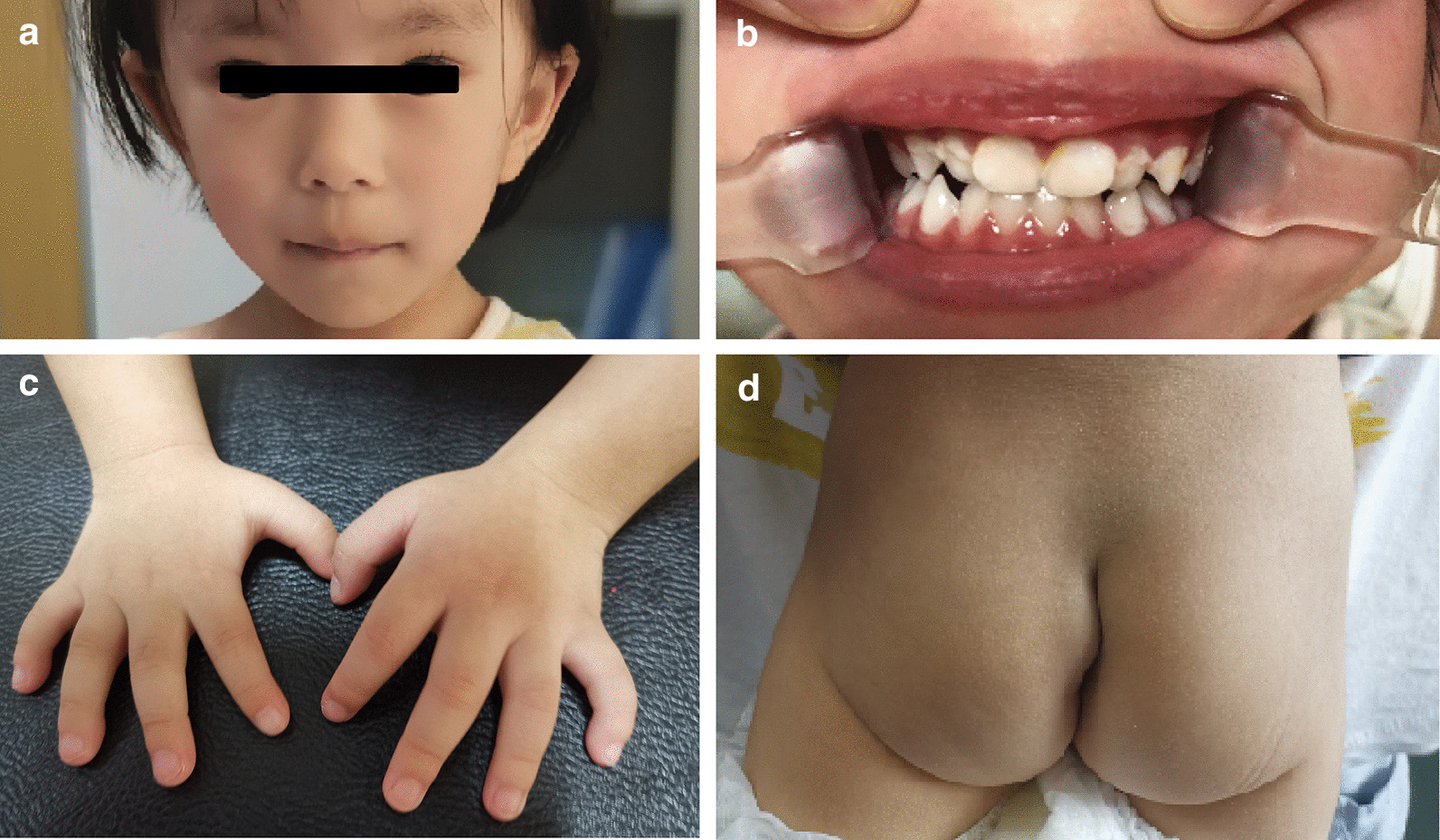
Physical examination of the patient. a Craniofacial appearances. b Macrodontia of upper central incisors (dental crown: 7.0 mm × 7.7 mm) and dental crowding. c Fifth fingers clinodactyly. d Abnormal caudal vertebra shape
Her karyotype was normal. Similarly, no abnormality was noted in tests for growth hormone, IGF‐1, IGFBP‐3, serum calcium, serum phosphorus, thyroid-stimulating hormone, parathyroid hormone, 25-hydroxy-vitamin D, prolactin, cortisol, adrenocorticotropic hormone, glycosylated hemoglobin, or glucose tolerance. In addition, amino acid, organic acid, and acylcarnitine spectra in blood and urine samples were also normal. Electroencephalographic results showed generally normal background activity with sporadic spikes and slow wave complexes, as well as multiple spikes and slow wave complexes in the bilateral parietal, occipital, and posterior temporal regions during non-rapid-eye-movement sleep. However, according to her parents’ observations, there was no indication of epilepsy.
Ultrasonic examinations showed no significant findings in the heart, liver, gallbladder, pancreas, spleen, adrenal glands, uterus, and ovaries. Radiographic examinations revealed delayed bone age (by approximately 8 months) and confirmed brachydactyly, fifth finger clinodactyly, and left-skewed sacral vertebrae (Fig. 3). Brain MRI showed a distended change in the bilateral ventricles and third ventricles, as well as malformation of the sixth ventricle (Fig. 3).
Fig. 3.
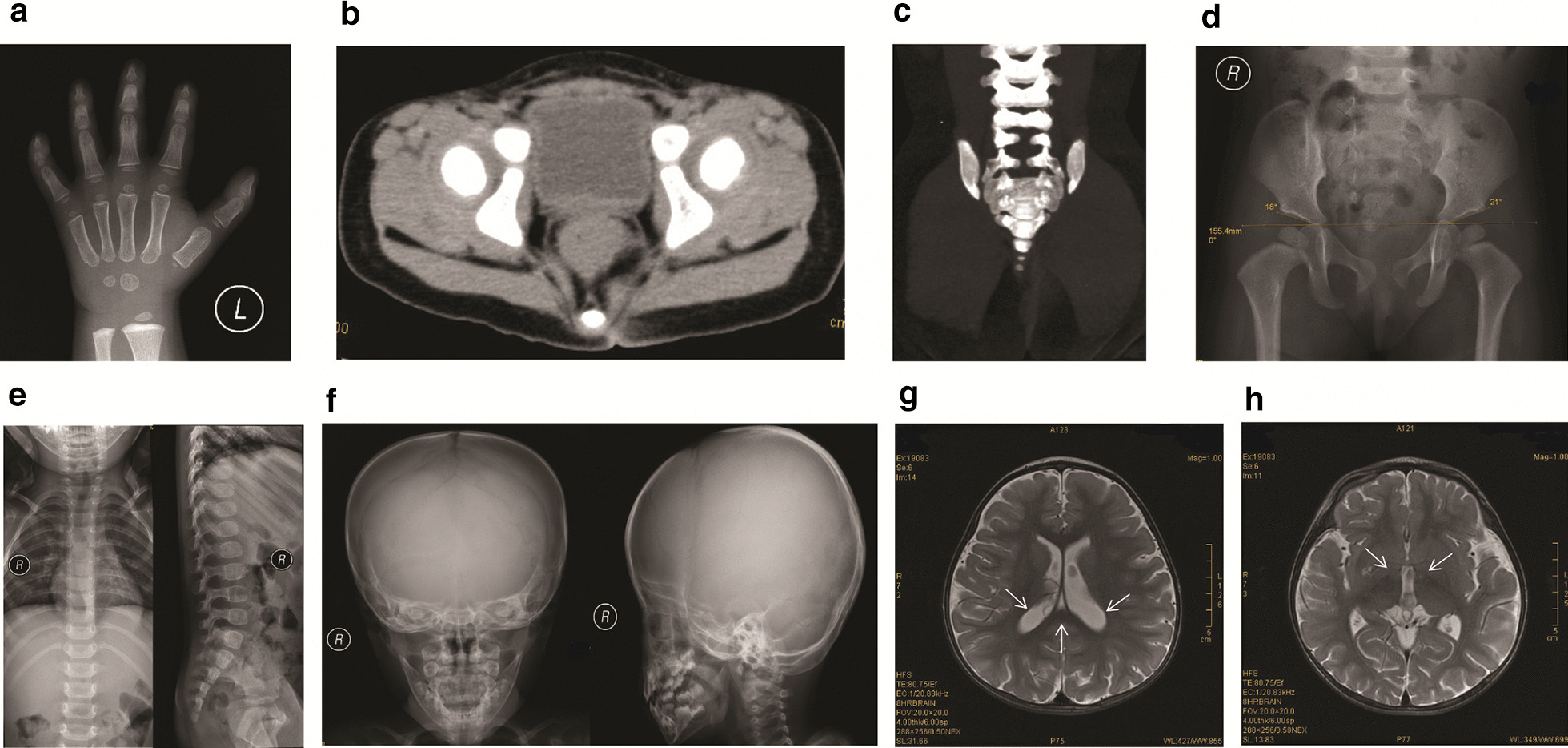
Imaging of the patient at the age of 2 years and 2 months. a Clinodactyly fifth finger. b Left-skewed caudal vertebra noted by the axial computed tomography (CT) scan. c Left-skewed caudal vertebra noted by the two-dimensional CT reconstruction. No abnormalities were noted in hip (d), main body of spine (e), and the skull (f). g Distended changes in bi-lateral ventricles (g) and the third ventricle (h), and the malformation of the sixth ventricles (g) were noted with the axial T2-weighted imaging. Arrows indicated the bi-lateral ventricles and the sixth ventricles in (g) and the third ventricle in (h)
Beginning at the age of 4 months, the patient received two courses of physical therapy every three months including exercise training, low-frequency electrical stimulation, and water therapy. Beginning at the age of 24 months, the patient received individualized cognitive/linguistic training including eye contact, imitation training, linguistic expression, verbal comprehension, finger fine motor, and so on. In the meantime, she also received applied behavior analysis therapy with behavior analyst in the one-on-one manner. Neuropsychological evaluation was performed to patient at different ages. The evaluation was based on the Chinese version of child neuropsychological development checklist for ages 0–6 years [15]. Specifically, the checklist has 211 items including five categories (gross motor, fine motor, adaptive strengths, language, and social behaviors). Developmental quotient was used to indicate the results of neuropsychological evaluation. As shown in Table 1, no evident improvement was observed in the developmental delays after therapy, except for gross motor function.
Genetic analysis
Methods
Peripheral blood samples were collected from the patient and her parents. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The DNA concentration was determined by measuring the absorbance at 260 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher, Waltham, MA, USA).
The DNA sample of the patient was analyzed by commercial whole-exome sequencing (WES, Fulgent Genetics, Fuzhou, Fujian, China). In short, the exome sequences from 1.0 μg of genomic DNA were enriched using a liquid capture system (Agilent SureSelect Human All Exon V5; Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol, and sequenced on the Hiseq4000 platform (Illumina, San Diego, CA, USA) to generate 150-bp paired-end reads. Detail information of WES results is listed in Additional file 1: Table S1.
The candidate variant was examined with Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk), ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), and gnomAD Browser (https://gnomad.broadinstitute.org/). The functional importance of the mutation was predicted using SIFT (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster databases (http://www.mutationtaster.org/).
Results
Among three WES-identified candidate variants (Additional file 1: Table S1), a novel heterozygous c.1366_1367dup (p.K457Rfs*54) variant in exon 9 of ANKRD11 (NM_013275.6) was suggested as the target variant, and this variant was confirmed by subsequent Sanger sequencing. Moreover, Sanger sequencing revealed that the variant should be de novo since it was absent in the genomes of patient’s parents (Fig. 4). The variant was speculated to result in a truncated protein with a frameshift starting from the Lysine 457 (Fig. 4).
Fig. 4.
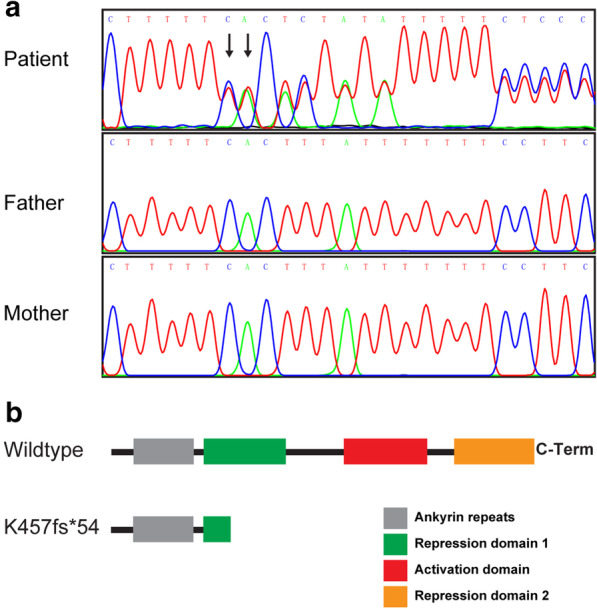
Nucleic acid and protein changes for c.1366_1367dup variant. a Sanger sequencing results of patient and her parents. Arrows indicates the duplicated nucleotides. b Predicted protein change for c.1366_1367dup variant
The ANKRD11 c.1366_1367dup variant has not been previously reported and was absent in the HGMD, ClinVar, dbSNP, and gnomAD databases. Based on the SIFT (score: 0.007) and Polyphen-2 (score: 0.982 for HumDiv model and 0.952 for HumVar model, respectively) results, the p.K457Rfs*54 variant was predicted to be probably damaging to the structure/function of the protein. Similarly, based on the Mutation Taster results (probability: 1.000), this variant was predicted to be disease causing.
Eventually, the variant was interpreted as pathogenic according to the ACMG Standards and Guidelines (PVS1 + PS2 + PM2 + PP3 + PP4) [16].
Discussion and conclusions
ANKRD11 encodes ankyrin repeat domain-containing protein 11, which contains two transcriptional repression domains, a transcriptional activation domain, and an ankyrin repeat domain (Fig. 4) [13, 17]. ANKRD11 functions in transcriptional regulation by binding to chromatin-modifying enzymes, e.g., histone deacetylases. The protein is essential for the development and function of the nervous system and plays crucial roles in embryogenesis, skeletogenesis, and bone turnover [14, 18]. Heterozygous pathogenic variants in ANKRD11 or deletions involving ANKRD11 are the major causes of KBG syndrome [8]. To date, more than 300 variants in ANKRD11 have been identified and deposited in the ClinVar database, including 104 frameshift, 193 missense, 63 nonsense, and 6 splicing variants (accessed on June 20, 2020). To our knowledge, the c.1366_1367dup variant in our case has not been reported previously. Similar to other frameshift variants identified in repression domain 1 (ClinVar database), in silico analysis predicted that the c.1366_1367dup variant is likely pathogenic. However, the explicit mechanism underlying the pathogenicity of ANKRD11 variants in KBG syndrome remains unclear. Previous studies have suggested that variants leading to premature stop codons, especially in the N-terminal region, could trigger nonsense-mediated decay, resulting in haploinsufficiency [18, 19]. The findings for some variants that affect the N-terminal region of ANKRD11 suggest a dominant-negative effect could also be involved [19]. We speculated that haploinsufficiency was most likely responsible for the pathogenicity observed in our case due to nonsense-mediated decay or a non-functional truncated protein. However, further studies are needed to fully elucidate the mechanism underlying the pathogenicity of these ANKRD11 variants in KBG syndrome.
Although accurate diagnosis of KBG syndrome relies on the identification of pathogenic changes in ANKRD11, early identification of suspect phenotypes is important, since it could direct the appropriate genetic evaluation and facilitate more effective prospective care of medical problems and genetic counseling. The phenotypic spectrum of KBG syndrome is wide, and there are no consensus clinical diagnostic criteria for KBG syndrome [20, 21]. Based on previous cases, the common phenotypes of KBG syndrome at an early age include craniofacial, skeletal, and neurologic abnormalities [22]. In terms of the associated craniofacial abnormalities, wide eyebrows, hypertelorism, a long philtrum, and macrodontia of the upper central incisors are frequently observed [8], and these were also present in our case (Fig. 2). However, compared to previous cases [8], the craniofacial abnormalities were mild in our case and was not a first clue to the diagnosis. In terms of skeletal anomalies, short stature is characteristic. In addition, delayed bone age, spinal anomalies, brachydactyly, and a large anterior fontanelle with delayed closure have been noted; these skeletal anomalies were also present in our case (Fig. 3). Specifically, brachydactyly and fifth finger clinodactyly were characteristic in our case and previous cases [8]. Moreover, caudal appendage, which was rare but specific in previous cases [8], was also identified in our case. These skeletal anomalies could be important clues leading to the further diagnosis. In terms of neurologic anomalies, developmental delays are characteristic. Seizures, various brain abnormalities, and behavioral issues have also been noted [8]. In our case, the dilation or malformation of several ventricles was observed (Fig. 3), suggesting the abnormal brain developments, which could result in the prominent developmental delay (Table 1) and isolated EEG-indicated abnormal electroencephalic activities. The ANKRD11 might participate in the regulation of cellular mechanisms that underlie activity-dependent plasticity [13]. However, the underlying mechanism of various brain abnormalities induced with ANKRD11 variants needs further investigation. Although seizures had not been noted, the EEG indicated abnormal electroencephalic activities, suggesting a close attention should be taken for potential seizures in the family care. Therefore, our case provides supportive evidence that combined presentations of short stature, developmental delay, and characteristic craniofacial and skeletal anomalies might be effective indicators for the genetic evaluation of KBG syndrome at an early age, and WES is one of the most valuable tools for genetic evaluation of clinically rare Mendelian diseases, owing to its low cost and high yield of clinical variants.
Current treatment of KBG syndrome depends on targeted management for its specific manifestations [20]. The patient in our case received physical therapy, individualized cognitive/linguistic training, and applied behavior analysis therapy at early age, and there was evident improvement in gross motor dysfunction (Table 1). Moreover, she would benefit from early physical therapy to maximize mobility and reduce the risk of later-onset orthopedic complications, such as scoliosis and hip dislocation. Growth hormone therapy and surgical correction will be performed to address the short stature and skeletal issues, respectively. Routine monitoring of hearing, vision, growth, and cognitive development are important for timely management of disease-associated conditions.
In conclusion, we reported a novel heterozygous frameshift ANKRD11 c.1366_1367dup variant in a Chinese girl with KBG syndrome and the corresponding phenotypes. Our study expands the ANKRD11 variant spectrum in KBG patients and provides clinical phenotypic data regarding KBG syndrome at an early age. Our study further demonstrates that WES is an effective method for the diagnosis of rare genetic disorders.
Supplementary Information
Additional file 1: Candidates variants identified by whole exome sequencing.
Acknowledgements
We thank the patient and their families for their contributions in this study.
Abbreviations
- ANKRD11
Ankyrin repeat domain-containing protein 11
- WES
Whole-exome sequencing
Authors’ contributions
All authors contributed significantly to this study. JC collected clinical samples, performed clinical examinations, and collected patient’s blood sample and clinical data. ZMX retrieved the literature, and prepared the figures and tables. ZMX and YLZ performed some of the experiments and analyzed the data. XMM performed the radiographic examinations, collected and analyzed the imaging data. XDW and QWG designed the experiments, drafted the manuscript, and revised it. All authors have read and approved the final manuscript.
Funding
This work was supported by Guiding project of the Natural Science Foundation of Fujian Province (Grant No. 2019D010), and Young and Middle-aged Talent Cultivation Projects of Fujian Province (Grant Nos. 2019-ZQNB-31 and 2019-ZQNB-32). The funders had no role in the study design, data collection, analysis, decision to publish, or manuscript preparation.
Availability of data and materials
The raw data of whole-exome sequencing of the patient in this study are not publicly available in order to protect participant confidentiality but are available from the corresponding author on reasonable request. Reference sequence for ANKRD11 (NC_000016.9) is available in the Genbank repository (https://www.ncbi.nlm.nih.gov/nuccore/NC_000016.9?report=genbank&from=89334038&to=89556969&strand=true). Databases used in this study were Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk), ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), gnomAD Browser (https://gnomad.broadinstitute.org/), SIFT (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/).
Declarations
Ethics approval and consent to participate
This study was approved by the Research Ethics Committees of the Women and Children’s Hospital at the School of Medicine of Xiamen University (No. KY-2018-013). Written informed consent was obtained from the parents.
Consent for publication
Written informed consent for publication were obtained from the patient’s parents.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jing Chen, Zhongmin Xia and Yulin Zhou have contributed equally to this work
Contributor Information
Xudong Wang, Email: wangxudong0524@163.com.
Qiwei Guo, Email: guoqiwei@xmu.edu.cn.
References
- 1.Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975;11(5):7–18. [PubMed] [Google Scholar]
- 2.Swols DM, Foster J, Tekin M. KBG syndrome. Orphanet J Rare Dis. 2017;12(183):1–7. doi: 10.1186/s13023-017-0736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi PM, Bianchi A, Digilio MC, Tucci FM, Sitzia E, De Vincentiis GC. Audiological findings in a de novo mutation of ANKRD11 gene in KBG syndrome: report of a case and review of the literature. Int J Pediatr Otorhi. 2017;103:109–112. doi: 10.1016/j.ijporl.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Kang YB, He DY, Li YY, Zhang YH, Shao Q, Zhang M, et al. A heterozygous point mutation of the ANKRD11 (c.2579C>T) in a Chinese patient with idiopathic short stature. Mol Genet Genom Med. 2019;7(12):1–8. doi: 10.1002/mgg3.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smithson SF, Thompson EM, McKinnon AG, Smith IS, Winter RM. The KBG syndrome. Clin Dysmorphol. 2000;9(2):87–91. doi: 10.1097/00019605-200009020-00002. [DOI] [PubMed] [Google Scholar]
- 6.van Dongen LCM, Wingbermuhle E, van der Veld WM, Vermeulen K, Bos-Roubos AG, Ockeloen CW, et al. Exploring the behavioral and cognitive phenotype of KBG syndrome. Genes Brain Behav. 2019;18(4):e12553. doi: 10.1111/gbb.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SJ, Yang A, Park JS, Kwon DG, Lee JS, Kwon YS, et al. Two novel mutations of ANKRD11 gene and wide clinical spectrum in KBG syndrome: case reports and literature review. Front Genet. 2020;11:579805. doi: 10.3389/fgene.2020.579805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldenberg A, Riccardi F, Tessier A, Pfundt R, Busa T, Cacciagli P, et al. Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. 2016;170(11):2847–2859. doi: 10.1002/ajmg.a.37878. [DOI] [PubMed] [Google Scholar]
- 9.Low K, Ashraf T, Canham N, Clayton-Smith J, Deshpande C, Donaldson A, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016;170(11):2835–2846. doi: 10.1002/ajmg.a.37842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayed ISM, Abdel-Hamid MS, Abdel-Salam GMH. KBG syndrome in two patients from Egypt. Am J Med Genet A. 2020;182(6):1309–1312. doi: 10.1002/ajmg.a.61552. [DOI] [PubMed] [Google Scholar]
- 11.Kleyner R, Malcolmson J, Tegay D, Ward K, Maughan A, Maughan G, et al. KBG syndrome involving a single-nucleotide duplication in ANKRD11. CSH Mol Case Stud. 2016;2(6):a001131. doi: 10.1101/mcs.a001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim JH, Seo EJ, Kim YM, Cho HJ, Lee JO, Cheon CK, et al. A de novo microdeletion of ANKRD11 gene in a Korean patient with KBG syndrome. Ann Lab Med. 2014;34(5):390–394. doi: 10.3343/alm.2014.34.5.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89(2):289–294. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbaric I, Perry MJ, Dear TN, Salopek D, Marusic A, et al. An ENU-induced mutation in the Ankrd11 gene results in an osteopenia-like phenotype in the mouse mutant Yoda. Physiol Genom. 2008;32(3):311–321. doi: 10.1152/physiolgenomics.00116.2007. [DOI] [PubMed] [Google Scholar]
- 15.Bao XL. Best beginning of 0–3 years old children's life: guidelines for early childhood education and potential development in China (high-risk children volume) Beijing: China Women Publishing Press; 2013. [Google Scholar]
- 16.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang A, Li CW, Chen JD. Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem Biophys Res Commun. 2007;358(4):1034–1040. doi: 10.1016/j.bbrc.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell. 2015;32(1):31–42. doi: 10.1016/j.devcel.2014.11.031. [DOI] [PubMed] [Google Scholar]
- 19.Walz K, Cohen D, Neilsen PM, Foster J, Brancati F, Demir K, et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum Genet. 2015;134(2):181–190. doi: 10.1007/s00439-014-1509-2. [DOI] [PubMed] [Google Scholar]
- 20.Morel Swols D, Tekin M, et al. KBG Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle: University of Washington; 2018. [Google Scholar]
- 21.Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M, et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet. 2017;62(8):741–746. doi: 10.1038/jhg.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gnazzo M, Lepri FR, Dentici ML, Capolino R, Pisaneschi E, Agolini E, et al. KBG syndrome: common and uncommon clinical features based on 31 new patients. Am J Med Genet A. 2020;182(5):1073–1083. doi: 10.1002/ajmg.a.61524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Candidates variants identified by whole exome sequencing.
Data Availability Statement
The raw data of whole-exome sequencing of the patient in this study are not publicly available in order to protect participant confidentiality but are available from the corresponding author on reasonable request. Reference sequence for ANKRD11 (NC_000016.9) is available in the Genbank repository (https://www.ncbi.nlm.nih.gov/nuccore/NC_000016.9?report=genbank&from=89334038&to=89556969&strand=true). Databases used in this study were Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk), ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), gnomAD Browser (https://gnomad.broadinstitute.org/), SIFT (http://provean.jcvi.org/index.php), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/).