

Abstract
Identifying genetic biomarkers for brain connectivity helps us understand genetic effects on brain function. The unique and important challenge in detecting associations between brain connectivity and genetic variants is that the phenotype is a matrix rather than a scalar. We study a new concept of super‐variant for genetic association detection. Similar to but different from the classic concept of gene, a super‐variant is a combination of alleles in multiple loci but contributing loci can be anywhere in the genome. We hypothesize that the super‐variants are easier to detect and more reliable to reproduce in their associations with brain connectivity. By applying a novel ranking and aggregation method to the UK Biobank databases, we discovered and verified several replicable super‐variants. Specifically, we investigate a discovery set with 16,421 subjects and a verification set with 2,882 subjects, where they are formed according to release date, and the verification set is used to validate the genetic associations from the discovery phase. We identified 12 replicable super‐variants on Chromosomes 1, 3, 7, 8, 9, 10, 12, 15, 16, 18, and 19. These verified super‐variants contain single nucleotide polymorphisms that locate in 14 genes which have been reported to have association with brain structure and function, and/or neurodevelopmental and neurodegenerative disorders in the literature. We also identified novel loci in genes RSPO2 and TMEM74 which may be upregulated in brain issues. These findings demonstrate the validity of the super‐variants and its capability of unifying existing results as well as discovering novel and replicable results.
Keywords: brian connectivity, GWAS, UK Biobank
The unique and important challenge in detecting associations between brain connectivity and genetic variants is that the phenotype is a matrix rather than a scalar. We study a new concept of super‐variant for genetic association detection. By applying a novel ranking and aggregation method to the UK Biobank databases, we discovered and verified several replicable super‐variants.
1. INTRODUCTION
There have been a number of genome‐wide association studies (GWAS) conducted to identify genetic risk variants for brain functional disorders. However, those studies provide only limited understanding of brain structure and function, and pathways to neurological disorders (Jahanshad et al., 2013). With the advances of functional magnetic resonance imaging (fMRI), the human brain's intrinsic functional connectivity network architecture has been delineated (Seeley, Crawford, Zhou, Miller, & Greicius, 2009). Brain connectivity is known to be associated with a wide range of neurological disorders, such as Alzheimer's disease (Greicius, Srivastava, Reiss, & Menon, 2004; Supekar, Menon, Rubin, Musen, & Greicius, 2008), autism (Belmonte et al., 2004) and working memory performance (Hampson, Driesen, Skudlarski, Gore, & Constable, 2006). GWAS of the brain connectivity can guide the development and evaluation of treatments by identifying potential mechanisms and circuits promoting disease risk (Medland, Jahanshad, Neale, & Thompson, 2014). Using the method in Elliott et al., 2018, we find that the estimated heritability of the nuclear norm of resting‐state functional partial correlation matrix from the UK Biobank (UKB) databases (Data field: 25753) is 0.21 (p < .001), indicating that genetic variation may indeed have impacts on the brain connectivity matrix. Despite the progress that has been made in this regard (Jahanshad et al., 2012; Kong, An, Zhang, & Zhu, 2019), the existing studies generally are based on datasets of limited sizes (less than 1,000) and without convincing replications.
The unique and important challenge in detecting associations between brain connectivity and genetic variants is that the phenotype is a matrix rather than a scalar. To address this challenge, we introduce a novel method, matrix regression with local ranking and aggregation (MLRA). The MLRA adopts the concept of super‐variant to aggregate weak signals in individual single nucleotide polymorphisms (SNPs) and to include potential interactions between SNPs. Similar to the classic concept of gene, a super‐variant is a combination of alleles in multiple loci, but the loci contributing to a super‐variant can be anywhere in the genome. A gene is a biologically defined concept, but a super‐variant is identified from data through GWAS. To search for super‐variants, SNPs are divided into generally local, but not necessarily, blocks and ranked within each block by their importance based on a matrix regression analysis of brain connectivity matrix against the SNPs. To accommodate the fact that the response variable is matrix, we adopt a proper matrix norm when measuring importance of individual SNP.
Super‐variants constructed from local blocks could capture group effects of SNPs within blocks. While there have been several existing methods based on group‐wise penalization in genetics and imaging‐genetics literature (see Sliver et al., 2012; Lu et al., 2017; Ramanan et al., 2012; and see Shen & Thompson, 2019 for a comprehensive review), it is worthy pointing out that some existing methods use imaging‐derived phenotypes (IDPs) as the traits, and in contrast, the proposed method can directly deals image (or network) data as matrix responses. The formation of IDPs requires prior knowledge. In addition, because these derived phenotypes are summary data, they may lose information from the original data. Other existing methods perform regression analysis on each voxel, which completely ignores the spatial structure. To the best of our knowledge, this is the first work to consider the group‐wise structure of SNPs and brain connectivity network in matrix form at the same time.
In this study, we analyze genetic and brain connectivity network data from the UKB database. Recently, fMRI data for more than 19,000 participants were collected and released by the UKB team. This database has a large number of participants with multimodal imaging data acquired using homogeneous hardware and software (Elliott et al., 2018). With the large number of participants in the UKB database, we have the opportunity to use the same database to discover and validate the findings independently, instead of adopting the ensemble methods to stabilize and validate the findings using the discovery set alone (Asahchop et al., 2018; Tu et al., 2020). Several GWAS of brain imaging based phenotypes have been reported (Elliott et al., 2018; Zhao et al., 2019) to take advantage of this large and rich database, but, a study of brain connectivity has not been conducted to the best of our knowledge.
2. SUBJECTS AND METHODS
2.1. Matrix regression with local ranking and aggregation
MLRA is designed to find associations between matrix response (brain functional connectivity matrix in this study) and ultra‐high dimensional variable (SNPs in this study). Let Yi, i ∈ {1, …, n}, denote the matrix response and xi, i ∈ {1, …, n}, be a p dimensional variable. The original data are standardized so that for each dimension of variable the mean equals zero and the variance equals one. Each element of Yi has also been standardized; that is, the mean of Yi, jk equals 0 and variance of Yi, jk equals 1 for j = 1, …, d1 and k = 1, …, d2, where Yi, jk is the element on jth row and kth column of matrix Yi.
Segmentation: We divide all SNPs to form local blocks. We currently consider a partition based on the physical location with fixed length (1 × 106 bp in this study). There could be other ways to incorporate biological information to form information‐rich blocks. While it is convenient to consider physically neighboring regions in identifying the super variants, super variants can be identified in any regions. An advantage of super variants is to be able to assess potential SNP by SNP interactions that are in linkage disequilibrium or not. Moreover, although it is straightforward to define an initial set of variants for consideration, how to identify a group, or super variant, is challenging. Our approach, as one of the solutions that complement the existing methods, is convenient and provides intuitive results.
Marginal ranking: In line with the work of Song & Zhang, 2014, we order the SNPs according to their marginal effect sizes. Because the true effect sizes are unknown, we estimate the marginal effect of each SNP using a linear model:
where for the ith participant, xi0u is the value of the uthenvironmental covariate (u = 1, …, s), and xigv is the number of minor alleles of SNP v in block g. In this article, we work with bi‐allelic SNPs. We take nuclear norm of the coefficient matrix, as the marginal effect size for the vth SNP in block g with Jg variants, and 1 ≤ v ≤ Jg, and the estimated marginal effects are ordered in a descending order. Let dgv be the indices of the SNPs with the vth largest marginal effect size. Define
where 1 ≤ j ≤ Jg. Recall that xigj > 0 indicates the minor allele of SNP j in block g is present in participant i. When there exist missing data in some of the SNPs, we can calculate the marginal effects by excluding the individuals with missing value when estimating the coefficient matrix of the SNP, and then order the SNPs accordingly.
Note that the number of SNPs falling within each block may be different. However, the coefficient matrices Bgv do not depend on the number of SNPs within each block. They depend on the dimension of the matrix phenotypes only. Therefore, we can use ranking in terms of the associated norm.
The main advantage of adopting nuclear norm is that nuclear norm explicitly accounts for the structure of coefficient matrices. It is sensitive to various signal patterns in coefficient matrices and robust against random noise. Moreover, nuclear norm is computationally efficient. Other summary statistics of Bgv such as operator norm and Frobenius norm can also be considered.
Find the best cutting point: To obtain the super‐variants, we inspect all possible cut‐off values for variable Zg with observations {z1g, …, zng}. For each cut‐off value c, the variable is turned into binary; Sg = I ( Zg < c), where I is the indicator function, and c ∈{z1g, …, zng}. A marginal matrix regression is carried out to investigate the marginal effect of the resulting binary variable, and the final cut‐off value is the one that gives the largest nuclear norm of the coefficient matrix among all possible cut‐offs in the block.
Transformation to super‐variant: Finally, with the best cutting point for each block, we transform the derived variables into super‐variant indicators. Suppose the best cutting point for block g is t, for i ∈ {1, 2, …, n},
2.2. The MLRA for pairwise functional connectivity trait
The MLRA can be adopted to investigate a single pairwise functional connectivity trait as follows. We estimate the marginal effect of each SNP using a linear model:
where for the ith participant, xi0u is the value of the uthenvironmental covariate (u = 1, …, s), and xigv is the number of minor alleles of SNP v in block g. We will rank the SNP effect according to the p‐value of its corresponding coefficient βgv when identifying super‐variants.
2.3. Synthetic data
We generate 32 × 32 matrix responses according to:
here BT is the coefficient matrix of signals, S1 = {1,11,21,31,41,51}, and S2 = {(61, 62), (71, 72), (81, 82)}, which are the sets of signals with group‐wise structure of 2 SNPs and S3 = {(111,112,113), (121,122,123)}, which is the set of signals with group‐wise structure of three SNPs. We generate noise matrix Ei from N(0, σ2I), here I is the identity matrix. We generate genetic covariates using haplotype data from the 1,000 Genomes Project on chromosome 22 (Song & Zhang, 2014). Specifically, we delete haplotypes with standard deviation less than 0.5% and randomly select 3,000 haplotypes as genetic covariates. The number of subjects is 1,258. In Simulation 1, we generate the signal coefficient matrix BT with a block structure and set the noise level σ = 1. In Simulation 2, BT is generated such that the structure is closer to be uniform and the noise level is set with σ = 0.2. The true images of BT are presented in Figure 1.
FIGURE 1.
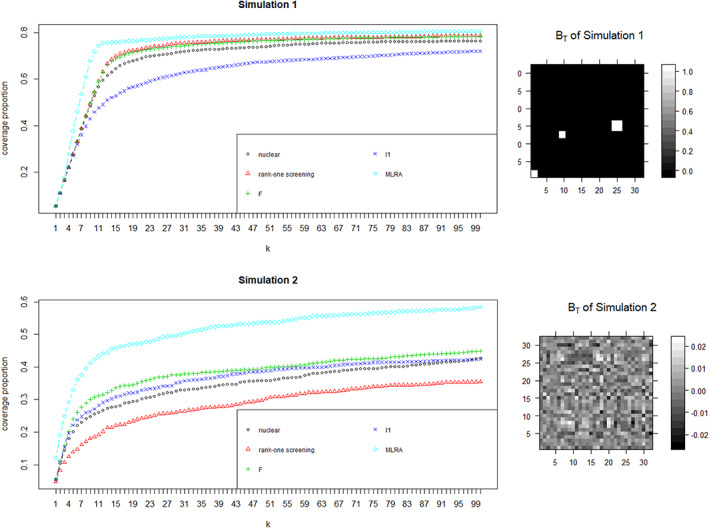
Results on synthetic data. We present the result on synthetic data and the true images of BT used in simulations. We report the average true nonzero coverage proportion as k increases from 1:100. The result is averaged by repeating 100 times. It reveals that MLRA (light blue diamond) outperforms all other four methods. Moreover, the performance of MLRA is stable under different settings of coefficient matrix, while rank‐one screening (red triangle) performs poorly in Simulation 2
Besides the MLRA, we also consider the rank‐one screening method (Kong et al., 2019), the L1 entry‐wise norm screening, the nuclear norm screening, and the Frobenius norm screening, where the last three are compared as the state‐of‐arts in Kong et al., 2019. We divide 3,000 haplotypes into 300 blocks, by every 10 indexes, for MLRA.
We apply screening procedure to each simulated dataset and select the first k largest of all covariates. We report the average true nonzero coverage proportion as k increases from 1:100. The result is averaged by repeating 100 times and is presented in Figure 1.
Figure 1 reveals that MLRA outperforms all other four methods. As expected, MLRA can detect more interactive signals than the other methods. Moreover, the performance of MLRA is stable under different settings of coefficient matrix, while rank‐one screening performs poorly in Simulation 2.
2.4. rfMRI partial correlation matrix
We download the resting‐state rfMRI partial correlation matrix (dimension 100) from the UKB (Data field: 25753). In the dataset, network matrices which represent the functional connectivity (measured by partial correlation) of each pair of nodes have been estimated for all subjects. Nodes that are not neuronally driven are discarded during network connectivity modeling by the UKB. As a result, 55 nodes remain and result in a 55 × 55 connectivity matrix. The list of remaining nodes, the complete original sets of 3D spatial maps and more resources about the dataset can be found at https://www.fmrib.ox.ac.uk/datasets/ukbiobank/index.html.
It is noteworthy that our method is applicable when different maps are available, but it is a different and major undertaking, beyond the scope of this manuscript, to build and assess different connectivity maps.
2.5. Data processing
We apply the MLRA at 41,502,298 SNPs after imputation and genotyping quality controls. Specifically, we removed SNPs with low call rate (missing probability ≥0.1) or that disrupted Hardy–Weinberg equilibrium (p‐value <1 x 10−7). The whole data set is divided into 2,689 local genetic blocks, each with physical length 1 × 106 bp. We use the difference between the date of attendance and the date of birth as individuals' age. Individuals without age information or genetic information are excluded. We also exclude subjects whose genetic gender is inconsistent with self‐reported gender. We primarily use individuals who made an imaging visit prior to first January 2018 as discovery set and the other individuals as the verification set. We create nominally unrelated subsets (without relatives closer than third cousins) of both sets accordingly, following procedures described in Bycroft et al., 2018. In order to reduce the confounding effect of population structure, we also include the first five principal component scores based on the SNP data in the quality control information provided by the UKB as control variables. In the end, we have a discovery set with 16,421 subjects, and a verification set with 2,882 individuals.
We calculate the ordinary least squares estimates of coefficient matrices and compute the corresponding residual matrices for the functional connectivity matrix after adjusting the effects of age, gender and the first five SNP principal component scores on both datasets. Then, we apply the MLRA on each dataset and selected the common blocks where the top 5% important super‐variants are located.
3. RESULTS
We analyze 19,831 individuals with both genetic data and resting‐state networks available from the UKB. Based on the data release dates, we divide the whole dataset into a discovery set with 16,421 subjects, and a verification set with 2,882 subjects as similarly done by Elliott et al., 2018. We use the verification set to evaluate the validity of genetic associations from the discovery phase.
The 41,502,298 SNPs analyzed are grouped into 2,689 sets of one Mbp in length. The matrix response is the partial correlation matrix derived from resting‐state fMRI. We measure the importance of SNPs and candidate super‐variants by the nuclear norm of the regression coefficient matrix. In both discovery and verification stages, we focus on the super‐variants among the top 5% important super‐variants (134/2689). In the end, we find 12 common top super‐variants on both discovery and verification datasets. The probability of getting 12 or more common super‐variants in a pool of 2,689 candidates that are ranked among the top 5% in both datasets by chance is less than 3.32%. The corresponding SNPs of each super‐variant with top 3 largest nuclear norm according to the discovery data are listed in Table 1. The genes, where the SNPs are located, are also reported, if known. The full list of corresponding SNPs of each super‐variant is reported in Supplemental Data.
TABLE 1.
Top SNPs corresponding to 12 verified super‐variants for connectivity matrix
| Super‐variant | SNP name | Position | Major Allele | Minor Allele | Gene |
|---|---|---|---|---|---|
| chr1_119 | rs150587011 | 118,394,978 | CT | C | |
| rs140523673 | 118,104,740 | A | G | ||
| rs187120237 | 118,521,524 | T | C | SPAG17 | |
| chr3_151 | rs754473336 | 150,225,522 | G | T | |
| 3:150010532 | 150,010,532 | G | GTTAC | ||
| rs56911072 | 150,800,599 | G | C | ||
| chr7_139 | rs74888723 | 138,056,520 | A | G | |
| rs76356478 | 138,682,454 | T | C | ||
| rs28644472 | 138,367,105 | C | T | SVOPL | |
| chr8_110 | rs537656432 | 109,182,059 | C | T | |
| rs73316135 | 109,590,330 | T | C | ||
| rs577665281 | 109,352,658 | C | A | ||
| chr9_26 | rs1332432 | 25,214,299 | G | A | |
| rs73471738 | 25,204,838 | G | T | ||
| rs7043237 | 25,201,349 | G | A | ||
| chr9_120 | 9:119683843 | 119,683,843 | G | GGCGACCGAGC | ASTN2 |
| rs564940053 | 119,683,855 | T | A | ASTN2 | |
| rs12002288 | 119,687,073 | T | C | ASTN2 | |
| chr10_30 | rs112305584 | 29,281,784 | A | T | |
| rs58515486 | 29,828,831 | T | C | SVIL | |
| rs73611821 | 29,284,416 | C | T | ||
| chr12_34 | rs7296825 | 33,059,771 | G | C | |
| rs73303683 | 33,050,638 | A | G | PKP2 | |
| rs60059851 | 33,045,241 | G | A | PKP2 | |
| chr15_65 | rs1037847 | 64,279,555 | T | C | DAPK2 |
| rs7168753 | 64,283,625 | T | C | DAPK2 | |
| rs8041460 | 64,279,864 | T | C | DAPK2 | |
| chr16_61 | 16:60145501 | 60,145,501 | T | TG | |
| rs531574432 | 60,145,505 | C | G | ||
| rs144650764 | 60,133,310 | TTA | T | ||
| chr18_71 | rs79191515 | 70,351,615 | C | T | |
| rs17086080 | 70,133,421 | G | A | ||
| rs10514046 | 70,133,865 | C | G | ||
| chr19_55 | rs11881664 | 54,892,237 | G | A | |
| rs113393416 | 54,516,210 | G | A | CACNG6 | |
| rs113772732 | 54,895,558 | A | G |
Our findings gain supports from literature. Among 12 verified super‐variants, 7 of them contain SNPs that reside in 14 genes which have previously been reported to have association with brain structure and function. The consistency with existing results of these 14 genes is presented in detail in Table 2.
TABLE 2.
The concordance with previous results
| Super‐variant | Gene | Papers | Results |
|---|---|---|---|
| Chr3‐151 | SELENOT | Boukhzar et al., 2016 | Gene SELENOT encodes a selenoprotein. This gene has been reported to play a crucial role in the protection of dopaminergic neurons against oxidative stress in mouse model of Parkinson's disease. |
| MED12L |
Risheg et al., 2007 Isidor et al., 2014 Nizon et al., 2019 |
Gene MED12L encodes a subunit of Mediator complex. Mutations in this gene have been identified in several genetic disorders associated with intellectual disabilities. | |
| Chr7‐139 | KIAA1549 |
Jones et al., 2008 Lin et al., 2012 |
KIAA1549:BRAF fusion has been identified in many cases of pilocytic astrocytoma in central neural system. |
| Chr9‐120 | ASTN2 | Fagerberg et al., 2014 |
Gene ASTN2 shows biased expression in brain. |
| Wilson, Fryer, Fang, & Hatten, 2010 | Gene ASTN2 has been reported to regulate glial‐guided neuronal migration. | ||
|
Glessner et al., 2009 Vrijenhoek et al., 2008 Lesch et al., 2008 |
Variations in gene ASTN2 has been identified as a risk factor in neurodevelopmental disorders, including autism spectrum disorder, schizophrenia, attention‐deficit/hyperactivity disorder. | ||
| Chr12‐34 | SYT10 | Moghadam & Jackson, 2013 | Gene SYT10 is a member of synaptotagmin, a family of transmembrane proteins involved in the regulated exocytosis of vesicles. |
|
Mittelsteadt et al., 2009 |
Gene SYT10 is mainly expressed in olfactory bulb neurons. | ||
| Woitecki et al., 2016 | Gene SYT10 has been reported to contribute to activity‐induced neuroprotection against excitotoxic neurodegeneration. | ||
| Chr15‐65 | CSNK1G1 | Martin et al., 2014 | Gene CSNK1G1 encodes a member of the casein kinase I gene family. A mutation in this gene may be associated with non‐syndromic early‐onset epilepsy. |
| Chr18‐71 | CBLN2 | Fagerberg et al., 2014 | Gene CBLN2 shows biased expression in brain. |
| Rong et al., 2012 | Genetic elimination of CBLN2 results in synaptic alterations in cerebellum. | ||
| NETO1 |
Fagerberg et al., 2014 |
Gene NETO1 encodes a transmembrane protein, which shows biased expression in brain. | |
| Ng et al., 2009 | Gene NETO1 has been reported to regulate spatial learning and memory. | ||
| Chr19‐55 | PRKCG | Fagerberg et al., 2014 | Gene PRKCG encodes a member of a family of serine‐ and threonine‐specific protein kinases. This gene shows biased expression in brain. |
|
Chen et al., 2003 Shirafuji et al., 2019 |
Mutations in this gene results in neurodegenerative disorder spinocerebellar ataxia‐14 (SCA14). | ||
|
CACNG6 CACNG7 CACNG8 |
Fagerberg et al., 2014 | These genes encode a type II transmembrane AMPA receptor regulatory protein. Genes CACNG7 and CACNG8 are restrictedly expressed in brain. | |
| Guan et al., 2016 | Genes CACNG6 and CACNG8 have been identified as potential susceptible genes to schizophrenia. | ||
|
CNOT3 |
Martin et al., 2019 | Variations in gene CNOT3 cause a variable neurodevelopmental disorder. | |
| TTYH1 | Fagerberg et al., 2014 | Gene TTYH encodes a member of the tweety family of proteins. This gene shows biased expression in brain. | |
| Halleran et al., 2015 | Gene TTYH is expressed during neuronal development. |
We also conduct the analysis on the pairwise functional connectivity strength between Region of Interest (ROI) 2 and ROI 29 (Net100_0380 in Elliott et al., 2018) based on their partial correlation measure from the connectivity matrix. Elliott et al. (2018) reported that this connectivity had the largest heritability (0.3033) among all the pairwise functional connectivity strengths. From the discovery stage, we select the top 5% important super‐variants (134/2689) for verification, and the smallest p‐value among the 134 selected variants is 0.00595 from the validation dataset, which is greater than 0.05/134.
4. DISCUSSION
Genes that are consistent with existing literature are related to signal transduction and neuronal development, and/or neurodevelopmental and neurodegenerative disorders such as intellectual disabilities (Isidor et al., 2014; Nizon et al., 2019; Risheg et al., 2007), schizophrenia (Guan et al., 2016; Vrijenhoek et al., 2008), and Parkinson's disease (Boukhzar et al., 2016). Our findings indicate that these genes may have impact on these disorders by affecting the brain connectivity. Besides the concordance with previous results, other identified genes, such as RSPO2 and TMEM74, have been reported to have biased expression in the brain (Fagerberg et al., 2014). Therefore, our results may serve as a guide for the further experimental research on association between these genes and brain structure and function.
In addition, the super‐variant on Chromosome 18 involves genes CBLN2 and NETO1, which to the best of our knowledge, have not been reported to be associated with brain function jointly in the literature. Therefore, our method may lead to discoveries of potentially novel mechanisms on how multiple genes affect the brain connectivity collectively.
To visualize the influence of a super‐variant on brain connectivity, we compare the average connectivity matrices by the allele types of the super‐variant. Figure 2 presents the result for the super‐variant on Chromosome 9 block 120 whose top contributing SNPs are from gene ASTN2. From Figure 2, we can see that some regions are affected with clear patterns; for instance, connections including region 6 (region 14) are all weakened if the super‐variant on Chromosome 9 block 120 is present. The details about all of the 55 regions are available on the UKB website listed in Web Source. Although the biological rationale behind such observation is unclear to us, but this finding warrants further investigation, because ASTN2 has been identified as a risk factor in neurodevelopmental disorders. Similar plots for all super‐variants are reported in the Figure S1 in Supplemetal Data.
FIGURE 2.
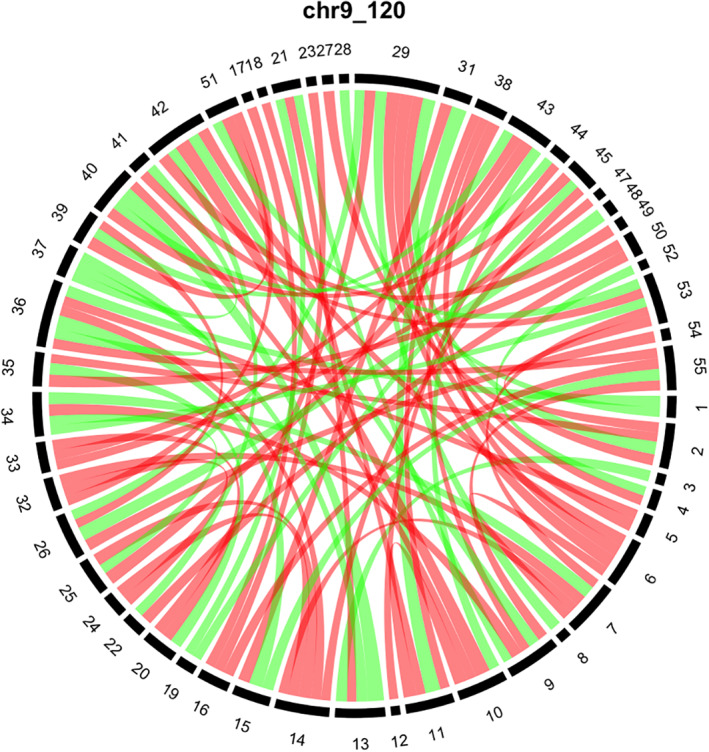
The influence of the super‐variant on Chromosome 9 block 120 on brain connectivity. We standardize the elements of the connectivity matrices to mean 0 and variance 1. Individuals in the discovery set are separated into two groups according to the minor and major variants of the super‐variant on Chromosome 9 block 120. Here, the variant with a lower frequency is referred to as the minor variant. The difference matrix is calculated by subtracting the average connectivity matrix of the group with the major variant from the average connectivity matrix of the group with the minor variant. For visualization, only differences with absolute values in top 5% are plotted in the chord diagram. Red (green) bands indicate the negative (positive) differences and the widths of the bands indicate the magnitudes of the differences. The numbers in the outer circle indicate specific regions in the brain
The MLRA is able to find SNPs that have not been reported in existing studies (Jahanshad et al., 2012; Kong et al., 2019), mainly because that the MLRA orders the SNPs in terms of their importance without screening out any single one (Kong et al., 2019). In this way, it becomes possible to retain and aggregate SNPs with weak and/or interactive effects to discover more associations. Importantly, our results are also verified using a separate dataset, and are more reliable than the existing results (Jahanshad et al., 2012; Kong et al., 2019) which lack of cross verification.
The proposed method still has limitations. When dealing with scalar responses, we can obtain a p‐value for each super‐variate as explained. However, with matrix responses, statistical inference of matrix regression remains a challenging and active research topic. For this reason, we used the cross‐validation method by splitting the samples into two datasets. This method, however, reduces the sample size and hence the power of detecting significant super‐variants. We also analyze the pairwise functional connectivity strength, but do not find any super‐variant that is statistically significant at the 0.05 level after Bonferroni correction.
One may consider adding an explicit sparsity constraint on each Bgv when ranking and finding the best cutting point. In this study, we decided not to add such a constraint based on the following concerns. First, as mentioned in (Kong et al., 2019), one may calculate some other regularized estimates (e.g., Lasso or fused Lasso), but it is computationally infeasible when the number of candidate SNPs is more than 41million. Second, there are hyper‐parameters to be tuned when constraints were introduced. Even if the computation is not a concern, it is difficult to directly tune those parameters in the analysis of real data where the ground truth is unknown. Last, when adding constraints, one should have structural knowledge from the ground truth as rationales for the constraints. However, as far as we know, the specific structure of signals on brain connectivity is not well studied or verified, so it is premature to choose what kind of constraints in brain imaging and genomic analysis.
In summary, the MLRA can capture the structure of response matrices as well as interactions between explanatory variables. We should note that although the proposed method enjoys important advantages, it can be extended and improved. For example, a formal statistical inference on the results needs to be developed. In addition, it may be useful to use biological knowledge to guide the formation of SNP blocks.
Web Resources: 3D‐maps for the brain regions, https://www.fmrib.ox.ac.uk/datasets/ukbiobank/group_means/rfMRI_ICA_d100_good_nodes.html
CONFLICT OF INTEREST
The authors declare no competing interests.
Supporting information
Data S1: Supporting Information
Figure S1 The influences of verified super‐variants on brain connectivity.
Figure S2. The slides from the 3D‐maps for 55 regions.
ACKNOWLEDGMENTS
Zhang's research is supported in part by U.S. National Institutes of Health (R01HG010171 and R01MH116527) and National Science Foundation (DMS1722544). This research has been conducted using the UK Biobank Resource under Application Number 42009. We thank the Yale Center for Research Computing for guidance and use of the research computing infrastructure.
Li T, Hu J, Wang S, Zhang H. Super‐variants identification for brain connectivity. Hum Brain Mapp. 2021;42:1304–1312. 10.1002/hbm.25294
Funding information National Institutes of Health, Grant/Award Numbers: R01HG010171, R01MH116527; National Science Foundation, Grant/Award Number: DMS1722544
DATA AVAILABILITY STATEMENT
The genetic data used in this study are the genotypes with imputation from the UKB (Field ID: 22801‐22822). The imputation was performed by a collaborative group headed by the Wellcome Trust Centre for Human Genetics. More detailed information about the datasets can be found at http://www.ukbiobank.ac.uk/wp‐content/uploads/2014/04/imputation_documentation_May2015‐1.pdf. The resting‐state functional MRI (rfMRI) partial correlation matrix (dimension 100) used in this study are download from the UKB (Field ID: 25753).
REFERENCES
- Asahchop E. L. Branton W. G. Krishnan A. Chen P. A. Yang D., … Kong L. Power C. (2018). HIV‐associated sensory polyneuropathy and neuronal injury are associated with miRNA‐455‐3p induction. JCI Insight, 3(23), e122450. 10.1172/jci.insight.122450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte, M. K. , Allen, G. , Beckel‐Mitchener, A. , Boulanger, L. M. , Carper, R. A. , & Webb, S. J. (2004). Autism and abnormal development of brain connectivity. Journal of Neuroscience, 24(42), 9228–9231. 10.1523/jneurosci.3340-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhzar, L. , Hamieh, A. , Cartier, D. , Tanguy, Y. , Alsharif, I. , Castex, M. , … Falluel‐Morel, A. (2016). Selenoprotein T exerts an essential oxidoreductase activity that protects dopaminergic neurons in mouse models of Parkinson's disease. Antioxidants & Redox Signaling, 24(11), 557–574. 10.1089/ars.2015.6478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft, C. , Freeman, C. , Petkova, D. , Band, G. , Elliott, L. T. , Sharp, K. , et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. 10.3410/f.734198301.793551571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D. H. , Brkanac, Z. , Verlinde, L. C. , Tan, X. J. , Bylenok, L. , Nochlin, D. , … Cimino, P. J. (2003). Missense mutations in the regulatory domain of PKCγ: A new mechanism for dominant nonepisodic cerebellar ataxia. The American Journal of Human Genetics, 72(4), 839–849. 10.1086/373883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, L. T. , Sharp, K. , Alfaro‐Almagro, F. , Shi, S. , Miller, K. L. , Douaud, G. , … Smith, S. M. (2018). Genome‐wide association studies of brain imaging phenotypes in UK biobank. Nature, 562(7726), 210–216. 10.1038/s41586-018-0571-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg, L. , Hallström, B. M. , Oksvold, P. , Kampf, C. , Djureinovic, D. , Odeberg, J. , … Uhlén, M. (2014). Analysis of the human tissue‐specific expression by genome‐wide integration of transcriptomics and antibody‐based proteomics. Molecular & Cellular Proteomics, 13(2), 397–406. 10.1074/mcp.m113.035600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner, J. T. , Wang, K. , Cai, G. , Korvatska, O. , Kim, C. E. , Wood, S. , … Imielinski, M. (2009). Autism genome‐wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459(7246), 569–573. 10.1038/nature07953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius, M. D. , Srivastava, G. , Reiss, A. L. , & Menon, V. (2004). Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proceedings of the National Academy of Sciences, 101(13), 4637–4642. 10.1073/pnas.0308627101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, F. , Zhang, T. , Liu, X. , Han, W. , Lin, H. , Li, L. , … Li, T. (2016). Evaluation of voltage‐dependent calcium channel γ gene families identified several novel potential susceptible genes to schizophrenia. Scientific Reports, 6, 24914. 10.1038/srep24914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halleran, A. D. , Sehdev, M. , Rabe, B. A. , Huyck, R. W. , Williams, C. C. , & Saha, M. S. (2015). Characterization of tweety gene (ttyh1‐3) expression in Xenopus laevis during embryonic development. Gene Expression Patterns, 17(1), 38–44. 10.1016/j.gep.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson, M. , Driesen, N. R. , Skudlarski, P. , Gore, J. C. , & Constable, R. T. (2006). Brain connectivity related to working memory performance. Journal of Neuroscience, 26(51), 13338–13343. 10.1523/jneurosci.3408-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor, B. , Lefebvre, T. , Le Vaillant, C. , Caillaud, G. , Faivre, L. , Jossic, F. , … Pelet, A. (2014). Blepharophimosis, short humeri, developmental delay and hirschsprung disease: expanding the phenotypic spectrum of MED12 mutations. American Journal of Medical Genetics Part A, 164(7), 1821–1825. 10.1002/ajmg.a.36539 [DOI] [PubMed] [Google Scholar]
- Jahanshad, H. , Rajagopalan, P. , Hua, X. , Hibar, D. P. , Nir, T. M. , Toga, A. W. , et al. (2013). Genome‐wide scan of healthy human connectome discovers spon1 gene variant influencing dementia severity. Proceedings of the National Academy of Sciences, 110(12), 4768–4773. 10.1073/pnas.1216206110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanshad, N. , Hibar, D. P. , Ryles, A. , Toga, A. W. , McMahon, K. L. , De Zubicaray, G. I. , … Thompson, P. M. , (2012). Discovery of genes that affect human brain connectivity: a genome‐wide analysis of the connectome. Paper presented at 2012 9th IEEE International Symposium on Biomedical Imaging (ISBI), pp. 542–545. 10.1109/isbi.2012.6235605 [DOI] [PMC free article] [PubMed]
- Jones, D. T. , Kocialkowski, S. , Liu, L. , Pearson, D. M. , Bäcklund, L. M. , Ichimura, K. , & Collins, V. P. (2008). Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Research, 68(21), 8673–8677. 10.1158/0008-5472.can-08-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, D. , An, B. , Zhang, J. , & Zhu, H. (2019). L2rm: Low‐rank linear regression models for high‐dimensional matrix responses. Journal of the American Statistical Association, 115(529), 403–424. 10.1080/01621459.2018.1555092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch, K. P. , Timmesfeld, N. , Renner, T. J. , Halperin, R. , Röser, C. , Nguyen, T. T. , … Freitag, C. (2008). Molecular genetics of adult ADHD: converging evidence from genome‐wide association and extended pedigree linkage studies. Journal of Neural Transmission, 115(11), 1573–1585. 10.1007/s00702-008-0119-3 [DOI] [PubMed] [Google Scholar]
- Lin, A. , Rodriguez, F. J. , Karajannis, M. A. , Williams, S. C. , Legault, G. , Zagzag, D. , … Bar, E. E. (2012). BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549: BRAF fusion variants. Journal of Neuropathology & Experimental Neurology, 71(1), 66–72. 10.1097/nen.0b013e31823f2cb0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z. H. , Khondker, Z. , Ibrahim, J. G. , Wang, Y. , Zhu, H. , & Alzheimer's Disease Neuroimaging Initiative . (2017). Bayesian longitudinal low‐rank regression models for imaging genetic data from longitudinal studies. NeuroImage, 149, 305–322. 10.1016/j.neuroimage.2017.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, H. C. , Kim, G. E. , Pagnamenta, A. T. , Murakami, Y. , Carvill, G. L. , Meyer, E. , … Kronengold, J. (2014). Clinical whole‐genome sequencing in severe early‐onset epilepsy reveals new genes and improves molecular diagnosis. Human Molecular Genetics, 23(12), 3200–3211. 10.1093/hmg/ddu030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, R. , Splitt, M. , Genevieve, D. , Aten, E. , Collins, A. , de Bie, C. I. , … Joss, S. (2019). De novo variants in CNOT3 cause a variable neurodevelopmental disorder. European Journal of Human Genetics, 27(11), 1677–1682. 10.1038/s41431-019-0413-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland, S. E. , Jahanshad, N. , Neale, B. M. , & Thompson, P. M. (2014). Whole‐genome analyses of whole‐brain data: working within an expanded search space. Nature Neuroscience, 17(6), 791–800. 10.1038/nn.3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelsteadt, T. , Seifert, G. , Alvárez‐Barón, E. , Steinhäuser, C. , Becker, A. J. , & Schoch, S. (2009). Differential mRNA expression patterns of the synaptotagmin gene family in the rodent brain. Journal of Comparative Neurology, 512(4), 514–528. 10.1002/cne.21908 [DOI] [PubMed] [Google Scholar]
- Moghadam, P. K. , & Jackson, M. B. (2013). The functional significance of synaptotagmin diversity in neuroendocrine secretion. Frontiers in Endocrinology, 4, 124. 10.3389/fendo.2013.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D. , Pitcher, G. M. , Szilard, R. K. , Sertié, A. , Kanisek, M. , Clapcote, S. J. , … Cortez, M. (2009). Neto1 is a novel CUB‐domain NMDA receptor–interacting protein required for synaptic plasticity and learning. PLoS Biology, 7(2), e1000041. 10.1371/journal.pbio.1000041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizon, M. , Laugel, V. , Flanigan, K. M. , Pastore, M. , Waldrop, M. A. , Rosenfeld, J. A. , … Le Caignec, C. (2019). Variants in MED12L, encoding a subunit of the mediator kinase module, are responsible for intellectual disability associated with transcriptional defect. Genetics in Medicine, 21(12), 2713–2722. 10.1038/s41436-019-0557-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan, V. K. , Kim, S. , Holohan, K. , Shen, L. , Nho, K. , Risacher, S. L. , … Petersen, R. C. , (2012). Genome‐wide pathway analysis of memory impairment in the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort implicates gene candidates, canonical pathways, and networks. Brain Imaging and Behavior, 6(4), 634–648. 10.1007/s11682-012-9196-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risheg, H. , Graham, J. M. , Clark, R. D. , Rogers, R. C. , Opitz, J. M. , Moeschler, J. B. , … Stevenson, R. E. (2007). A recurrent mutation in MED12 leading to R961W causes Opitz‐Kaveggia syndrome. Nature Genetics, 39(4), 451–453. 10.1038/ng1992 [DOI] [PubMed] [Google Scholar]
- Rong, Y. , Wei, P. , Parris, J. , Guo, H. , Pattarini, R. , Correia, K. , … Morgan, J. I. (2012). Comparison of Cbln1 and Cbln2 functions using transgenic and knockout mice. Journal of Neurochemistry, 120(4), 528–540. 10.1111/j.1471-4159.2011.07604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley, W. W. , Crawford, R. K. , Zhou, J. , Miller, B. L. , & Greicius, M. D. (2009). Neurodegenerative diseases target large‐scale human brain networks. Neuron, 62(1), 42–52. 10.3410/f.1165450.628434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, L. , & Thompson, P. M. (2019). Brain Imaging Genomics: Integrated Analysis and Machine Learning. Proceedings of the IEEE, 108(1), 125–162. 10.1109/jproc.2019.2947272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirafuji, T. , Shimazaki, H. , Miyagi, T. , Ueyama, T. , Adachi, N. , Tanaka, S. , … Sakai, N. (2019). Spinocerebellar ataxia type 14 caused by a nonsense mutation in the PRKCG gene. Molecular and Cellular Neuroscience, 98, 46–53. 10.1016/j.mcn.2019.05.005 [DOI] [PubMed] [Google Scholar]
- Silver, M. , Janousova, E. , Hua, X. , Thompson, P. M. , Montana, G. , & Alzheimer's Disease Neuroimaging Initiative . (2012). Identification of gene pathways implicated in Alzheimer's disease using longitudinal imaging phenotypes with sparse regression. NeuroImage, 63(3), 1681–1694. 10.1016/j.neuroimage.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, C. , & Zhang, H. (2014). TARV: Tree‐based analysis of rare variants identifying risk modifying variants in CTNNA2 and CNTNAP2 for alcohol addiction. Genetic Epidemiology, 38, 552–559. 10.1002/gepi.21843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supekar, K. , Menon, V. , Rubin, D. , Musen, M. , & Greicius, M. D. (2008). Network analysis of intrinsic functional brain connectivity in Alzheimer's disease. PLoS Computational Biology, 4(6), e1000100. 10.1371/journal.pcbi.1000100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, W. , Chen, P. A. , Koenig, N. , Gomez, D. , Fujiwara, E. , Gill, M. J. , … Power, C. (2020). Machine learning models reveal neurocognitive impairment type and prevalence are associated with distinct variables in HIV/AIDS. Journal of Neurovirology, 26(1), 41–51. 10.1007/s13365-019-00791-6 [DOI] [PubMed] [Google Scholar]
- Vrijenhoek, T. , Buizer‐Voskamp, J. E. , van der Stelt, I. , Strengman, E. , Sabatti, C. , van Kessel, A. G. , … Veltman, J. A. (2008). Recurrent CNVs disrupt three candidate genes in schizophrenia patients. The American Journal of Human Genetics, 83(4), 504–510. 10.1016/j.ajhg.2008.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, P. M. , Fryer, R. H. , Fang, Y. , & Hatten, M. E. (2010). Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial‐guided neuronal migration. Journal of Neuroscience, 30(25), 8529–8540. 10.1523/jneurosci.0032-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woitecki, A. M. , Müller, J. A. , van Loo, K. M. , Sowade, R. F. , Becker, A. J. , & Schoch, S. (2016). Identification of synaptotagmin 10 as effector of NPAS4‐mediated protection from excitotoxic neurodegeneration. Journal of Neuroscience, 36(9), 2561–2570. 10.1523/jneurosci.2027-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B. , Luo, T. , Li, T. , Li, Y. , Zhang, J. , Shan, Y. , et al. (2019). Genome‐wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co‐architecture with cognitive and mental health traits. Nature Genetics, 51(11), 1637–1644. 10.1038/s41588-019-0516-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Figure S1 The influences of verified super‐variants on brain connectivity.
Figure S2. The slides from the 3D‐maps for 55 regions.
Data Availability Statement
The genetic data used in this study are the genotypes with imputation from the UKB (Field ID: 22801‐22822). The imputation was performed by a collaborative group headed by the Wellcome Trust Centre for Human Genetics. More detailed information about the datasets can be found at http://www.ukbiobank.ac.uk/wp‐content/uploads/2014/04/imputation_documentation_May2015‐1.pdf. The resting‐state functional MRI (rfMRI) partial correlation matrix (dimension 100) used in this study are download from the UKB (Field ID: 25753).