

Abstract
Background and aim:
Congenital heart disease (CHD) affects close to 1% of all live births and is considered as the main reason for morbidity and mortality in early childhood. In this study, we aimed to investigate molecular genetic factors associated with Tetralogy of Fallot (TOF), using high throughput technologies in the consanguineous families with at least two affected individuals.
Method:
This family study started from March 2017 to May 2019, in the pediatric cardiovascular research center, Cardiovascular Research Institute, Isfahan, Iran. Families with consanguineous marriage who had at least two patients in pedigree were invited to attend. Genomic DNA was extracted from peripheral blood lymphocytes of the patient and samples were investigated for variations such as deletion or duplication in the genome, using single nucleotide polymorphism array (SNP array). In the next step, if the SNP array was negative, the next generation sequencing (NGS) was performed in the probands. The raw data was analyzed and filtered to identify the genetic cause of the disease.
Results:
In this study, total five families were evaluated. All affected and unaffected individuals of each family included in the pedigree. Fourteen subjects, 9 males and 5 females, 8.92±6.21 years old were included. The prevalence of consanguineous marriage was 92.2% among parents, 71.4% among maternal grandparents and 28.6% among paternal grandparents. Almost, 64.3% of our participants had a sibling with a similar disease. The prevalence of Atrial Septal Defect (ASD), Ventricular Septal Defect (VSD), and arrhythmia and TOF was 7.1%.
Conclusion:
We found some families with two or more CHD cases and with high rate of consanguineous marriage, having a genetic predisposition. In the next step, high throughput techniques such as NGS and SNP-array was performed in order to find any genetic variation. Functional study will be done, to confirm and determine the role of the identified variants in the function of genes involved in disease phenotype. (www.actabiomedica.it)
Keywords: gene mutations, children, adolescents, tetralogy of Fallot, family history
Introduction
Congenital heart disease (CHD) affects almost 1% of all live births and is considered as the main cause of morbidity and mortality in early childhood (1). CHD explains a number of unusual problems affecting the heart, including defects in the structure of the heart, arrhythmia, and cardiomyopathy. CHD is divided into two groups of cyanotic and non-cyanotic (2). Tetralogy of Fallot (TOF) is the most common form of cyanotic CHD, affecting ~3 per 10.000 newborns, and accounts for about 7 to 10% of CHD (3). Population studies recommend a substantial familial recurrence risk in sporadic and non-syndromic TOF (4). Family studies showed that 80% of ‘sporadic’ CHD cases might have a significant, complex genetic condition or a single nucleotide polymorphism (SNP) (5). Therefore, 20% of remaining CHD occur by chromosomal abnormalities or multisystem malformation syndromes (6). Many published papers insist on the critical role of some genetic and environmental factors, such as maternal diabetes, gestational Rubella (or other viral diseases), dietary nutritional deficiencies during pregnancy, use of alcohol during the pregnancy, pregnancy after 40 years, and mothers with phenylalanine deficiencies. However, it seems that genetic factors have the most important effects (7). In the past, only genetic screening has been conducted for common genes in heart disease, but nowadays, with development of the next generation sequencing (NGS), these limitations have been fainted. Previous genome-wide association studies (GWAS) assessed the relationship between common SNP and CHD risk (8-14). In this study, our goal was to investigate molecular genetic factors associated with TOF, using high throughput technologies in the consanguineous families with at least two affected individuals with CHD.
Materials and methods
This family study started from March 2017 to May 2018 in Pediatric Cardiovascular Research Center, Cardiovascular Research Institute, Isfahan University of Medical Sciences, Isfahan, Iran. The study protocol was approved by the Ethical Committee of Cardiovascular Research Institute. After describing the study protocol for subjects, the written consent was signed.
Inclusion criteria
Families who had one patient with non-syndromic TOF, and at least one more affected with CHD in pedigree was included. CHD refers to a range of possible heart defects such as Atrial Septal Defect (ASD), Ventricular Septal Defect (VSD), Patent Ductus Arteriosus (PDA), Pulmonary Stenosis (PS). All subjects with non-syndromic TOF have the same phenotype of the disease.
Exclusion criteria
Single-case CHD in the family, Patients with a known syndrome or metabolic disease leading to CHD and Heart valve regurgitation stated as exclusion criteria.
Considering the inclusion criteria, patients were referred from the clinics of the pediatric cardiologists to the genetics department of Isfahan Cardiovascular Research Center for genetic counseling and blood collection. For subjects who suffer from TOF, echocardiography, ECG and cardiac catheterization were performed or their previous data were reviewed. Eligible patients underwent genetic counseling and advised by their respective physician. personal and familial history of each individual was fully investigated. The pedigree was drawn and the number of afflicted and healthy individuals were located in this.
Sample collection: Up until now, we investigated five families. First, we look through each patient’s documents and separated the families, according to our inclusion criteria. In the second step, the families were called out, asked for genetic counseling and advised to do genetic tests without paying the cost. Some cooperated in a very good manner; however, some families rejected our suggestion. In the next step, pedigree was designed for the families with the affected members. Questions were asked about the medical history of the patients and the mother during the pregnancy, such as diabetes and other environmental factors. Ten ml of venous blood were obtained in EDTA-containing tube for DNA testing.
DNA Extraction
Genomic DNA was extracted from peripheral blood lymphocytes of the patient by salting out method (15). The quality and quantity of the DNA sample were measured, using a NanoDrop spectrophotometer (Thermo Fisher Scientific Inc, Waltham, MA). For genetic testing 3 μg DNA of each sample was used. Selected high throughput technologies depending on the study was performed by The Omid Generation Center in Tehran (Iran).
Data Analysis
Data were analyzed for investigating disease-causing variants using SNP array. In the next step, if SNP array was negative, whole exome sequencing (WES) was performed in probands. After analyzing and filtering the data for investigating rare pathogenic variants, co-segregation analyses were performed, if disease-causing variant was found in pedigree.
Results
In this study, total five families were evaluated. All affected and unaffected individuals of each family included in the pedigree. This study comprised 5 relatively large families who had totally14 patients with TOF (9 males and 5 females; 8.92±6.21 years old).
Baseline characteristics and clinical data of the study subjects are presented in Table 1. The prevalence of consanguineous marriage was 92.2% among parents, 71.4% among maternal grandparents and 28.6% among paternal grandparents. In total, 64.3 % of our participants had a sibling with a similar disease.
Table 1.
Demographic and clinical characteristics of participants
| Characteristics | |
| Gender (%) | |
| 35.7 | Female |
| 64.3 | Male |
| 8.92±6.21 | Mean age (Mean (SD)) |
| Age at the first referral (%) | |
| 78.6 | ≤ 2 years, |
| 14.3 | 2-7 |
| 7.1 | 7-12 |
| (%)Consanguineous marriage | |
| 71.4 | Grandparents (maternal) |
| 28.6 | Grandparents (paternal) |
| 64.3 | Having sibling |
| 7.1 | ASD |
| (%) The cause of the first visit | |
| 50 | History of sudden death in low age in the family |
| (%)History of disease in pedigree | |
| 7.1 | Coronary heart disease |
| 7.1 | Favism |
| 7.1 | Arrhythmia |
| 14.3 | TOF is going to be operated |
| 50.0 | TOF after surgery |
| 7.1 | TOF without surgery |
| Other disease in Patents | |
| (%)Lifestyle habits in mother (during pregnancy) | |
| 0 | (%)Alcohol drinking |
| 7.1 | (%)smoking |
| (%)Disease during Pregnancy | |
| 7.1 | Gestational diabetes |
| 0 | Viral infection |
| 0 | Pharmacological treatment during pregnancy |
ASD: Atrial septal defect, VSD: ventricular septal defect, TOF: tetralogy of Fallot
Family 1: The phenotypes of two probands (V:5 and V:8) are somewhat different, so the question is if we will find the same genetic defect in both patients. So, SNP array is the best approach in all available individuals (8 in total) and WES in the 2 affected patients (V:5 and V:8).
Family 2: For the second family, SNP array was performed in all available individuals (4 in total).
Family 3: In the third family, targeted NGS for cardiomyopathy was performed to exclude known causes of DCM or 2) WES was performed for the affected boy (proband).
Family 4: SNP array was performed for all available individuals (5 in total). Cardiomyopathy NGS panel was run for the probands.
Family 5: SNP array was performed for all available individuals (3 in total), and cardiomyopathy panel NGS or WES were run for the affected girl.
Figure 1.
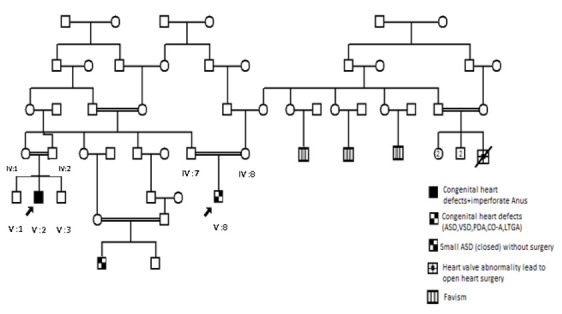
Family 1
Figure 2.
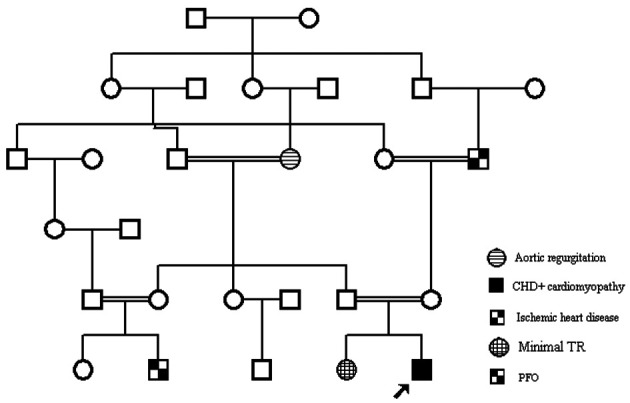
Family 2
Figure 3.
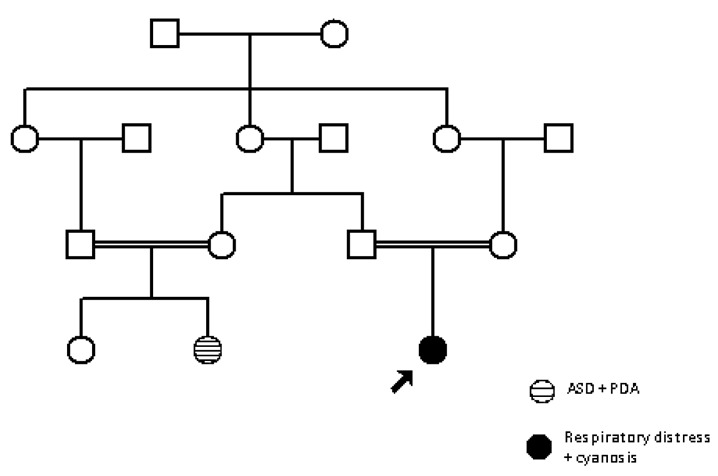
Family 3
Figure 4.
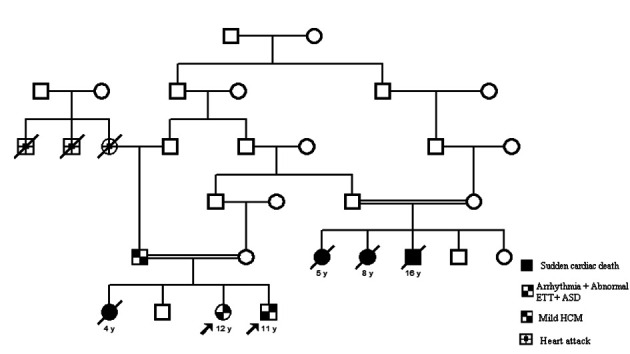
Family 4
Figure 5.
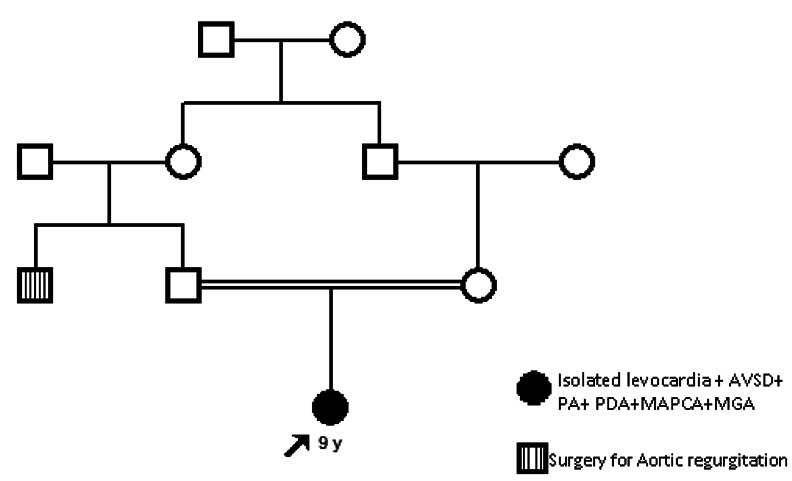
Family 5
Discussion
With the rapid growth of technology, many approaches had been used to find the disease-causing genes, such as SNP array, targeted NGS, and WES. Targeted NGS suggests opportunities for genetic testing and is able to examine large amounts of genetic data precipitously. Targeted NGS to be additional clinically useful than whole exome sequencing, due to speeder turnaround time (reduced sequencing volume and related data analysis), better and more consistent coverage, in addition to avoiding incidental findings. In recent years, many investigators have published reports on gene mutations, determined by targeted sequencing, for many genetic diseases. If there are other people in the family, their phenotypes should be within the same range of structural abnormalities of CHD. To the best of our knowledge, this study is the first study performed in Iran on CHD, with the priority of TOF patients, using the NGS technique.
Limitations: At the first level, this study had several limitations.
NGS studies still considered as the most expensive studies, so the sample size was limited. Therefore, in this family study, we objectively have focused on one phenotype instead of collecting different phenotypes. The next problem was the accessibility of the biopsies, which was limited for some phenotypes.
Conclusion
This study is the first in our region, to examine the large amounts of genetic data precipitously. high throughput techniques such as NGS and SNP-array helps us to better understand the genetic defect in subjects, suffering from TOF. To determine the effect of the identified variants or probably new genes, we will do functional study such as making a mouse model as long as we find a novel gene in our families in further studies.
Conflict of interest:
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article
References
- 1.Yun SW. Congenital heart disease in the newborn requiring early intervention. Korean J Pediatr. 2011;54(5):183–91. doi: 10.3345/kjp.2011.54.5.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A, Bhargava K. Spectrum of cyanotic congenital heart disease diagnosed by echocardiographic evaluation in patients attending a tertiary cardiac care center of South Rajasthan. Ann Pediatr Cardiol. 2017;10(1):97–98. doi: 10.4103/0974-2069.197050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgenthau A, Frishman WH. Genetic Origins of Tetralogy of Fallot. Cardiol Rev. 2018 Mar/Apr;26(2):86–92. doi: 10.1097/CRD.0000000000000170. doi: 10.1097/CRD.0000000000000170. [DOI] [PubMed] [Google Scholar]
- 4.Akhirome E, Walton NA, Nogee JM, Jay PY. The Complex Genetic Basis of Congenital Heart Defects. Circ J. 2017;81(5):629–634. doi: 10.1253/circj.CJ-16-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards AA, Garg V. Genetics of congenital heart disease. Curr Cardiol Rev. 2010;6(2):91–7. doi: 10.2174/157340310791162703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rman MJ, Priori SG, Willems S, et al. HRS_EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 7.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Europace. 2011;13(8):1077–1109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Muzny DM, Reid JG, et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N Engl J Med. 2013;369(16):1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10(10):571–583. doi: 10.1038/nrcardio.2013.108. [DOI] [PubMed] [Google Scholar]
- 10.Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, leftventricular noncompaction, and sudden death. BMC Med Genet. 2014;15:99. doi: 10.1186/s12881-014-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karam S, Raboisson MJ, Ducreux C, et al. A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit Heart Dis. 2008;3(2):138–143. doi: 10.1111/j.1747-0803.2008.00165.x. [DOI] [PubMed] [Google Scholar]
- 12.Norton N, Li D, Rieder MJ, Siegfried JD, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as acause of dilated cardiomyopathy. Am J Hum Genet. 2011 Mar 11;88(3):20–21. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell N, Sinagra G, Jones KL, et al. Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One. 2013 Oct 29;8(10):e78104. doi: 10.1371/journal.pone.0078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsman RF, Barc J, Beekman L, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood andadolescence. J Am Coll Cardiol. 2014 Jan 28;63(3):20–21. doi: 10.1016/j.jacc.2013.07.091. [DOI] [PubMed] [Google Scholar]
- 15.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]