

Abstract
So far, most techniques for modifying perovskite solar cells (PSCs) focus on either the perovskite or electron transport layer (ETL). For the sake of comprehensively improving device performance, a dual‐functional method of simultaneously passivating trap defects in both the perovskite and ETL films is proposed that utilizes guidable transfer of Eu3+ in SnO2 to perovskite. Europium ions are distributed throughout the SnO2 film during the formation process of SnO2, and they can diffuse directionally through the SnO2/perovskite interface into the perovskite, while most of the europium ions remain at the interface. Under the synergistic effect of distributed Eu3+ in the SnO2 and aggregated Eu3+ at the interface, the electron mobilities of ETLs are evidently improved. Meanwhile, diffused Eu3+ ions passivate the perovskite to reduce trap densities at the grain boundaries, which can dramatically elevate the open‐circuit voltage (V oc) of PSCs. Finally, the mainly PSCs coated on SnO2:Eu3+ ETL achieve a power conversion efficiency of 20.14%. Moreover, an unsealed device degrades by only 13% after exposure to ambient atmosphere for 84 days.
Keywords: europium, perovskite, photovoltaics, solar cells
A dual‐functional method of simultaneously passivating trap defects in both perovskite and electron transport layer (ETL) films is proposed. Europium ions distribute throughout SnO2 film and diffuse into perovskite, while most of Eu3+ remain at the interface. Under the synergistic effect of distributed Eu3+, the electron mobility of ETL is improved and the trap density of perovskite is also reduced.
1. Introduction
The power conversion efficiency and device lifetime are both key factors for the assessing efficient perovskite solar cells. Recently, the certified power conversion efficiency (PCE) of perovskite solar cells (PSCs) has risen steeply to 23.7%,[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 ] however, in comparison with commercial solar cells, such as crystalline silicon, polycrystalline silicon and Cu(In, Ga)Se2 solar cells, the poor device stability of PSCs is still the obstacle to obtaining a market share.[ 11 , 12 , 13 ]
Most of the reported methods aiming to improve either or both the PCE and stability of PSCs focus on optimizing the perovskite or electron transport layer (ETL). Regarding perovskite photon absorber layers, their soft crystal lattices tend to easily deform, particularly under various stresses, such as moisture, oxygen, and ultraviolet light exposure, and even under the electric field and thermal stress during device operation.[ 14 , 15 , 16 , 17 ] Various techniques, such as encapsulation, ultraviolet filtration, and modification have been used to delay the degradation of perovskite materials under the stress of environmental and device operational factors for maintaining long‐term stability of PSCs. Additionally, methods that introduce additives into perovskite films to promote PCE are widely used.[ 18 , 19 ] For instance, goethite quantum dots interact with iodine, lead and methylamine, resulting in the retardation of crystallization kinetics to achieve perovskite films with high crystallinity and large grain size;[ 20 ] Imidazole sulfonate zwitterions are introduced to regulate the crystal orientation of MAPbI3 film so it is highly ordered to passivate trap states;[ 21 ] and the conjugated polymer poly(bithiophene imide) is incorporated within grain boundaries to improve the crystallinity of perovskite film for reducing its defects.[ 22 ]
Besides optimizing the photon absorber, as an important part of a PSC, the ETL must possess high electron mobility to extract photo‐induced carriers because effectively transferring carriers to the external circuitry can promote the PCE of devices. Meanwhile, a suitable ETL should present decent optical transmittance for ensuring enough light reaches the perovskite absorber. Various strategies for optimizing ETLs are reported, such as ethylene diamine tetraacetic acid complexing SnO2,[ 23 ] using [6,6]‐Phenyl C61 butyric acid to modify ZnO,[ 24 ] or doping TiO2 with Sm3+ and Eu3+ ions.[ 25 ] Fabricating an ETL with organic chemicals or rare‐earth ions can not only tune the Fermi level of the ETL to better match the conduction band of the perovskite for facilitating charge carrier transfer but also modify the interface between the perovskite and the ETL to induce the perovskite to crystalize with better quality and larger grain size. However, more effective and convenient techniques need to be developed to improve both the perovskite and ETL by simultaneously repairing the different trap defects in the photon absorber and ETL, finally achieving effective photon‐induced charge carrier separation and transfer for higher PCE. This one‐step technique can reduce the cost of PSC engineering by a significant margin.
Europium ions can perform as a redox shuttle to selectively oxidize Pb0 and reduce I0 defects simultaneously in MAPbI3 thin films, and the elimination of both Pb0 and I0 defects promotes the photovoltaic properties of MAPbI3 PSCs with PCE up to 19.67%.[ 10 ] This MAPbI3 film is deposited by a traditional two‐step method. In this process, Eu(acac)3 additive is added to the PbI2/DMF (dimethylformamide) precursor solution. In addition to modifying the perovskite materials, Eu3+ and Sm3+ co‐doped TiO2 are prepared by the pulsed laser deposition method. The incorporated Eu3+ ions in cooperation with Sm3+ optimize the TiO2 ETL, achieving higher electron extraction and lower interfacial recombination; therefore, power conversion efficiency as high as 19.01% can be obtained for a MAPbI3 solar cell.[ 25 ] Evidently, for the purpose of simultaneously reducing the trap defects in perovskite and ETL films, europium additive is a good choice; however, the above‐discussed preparation methods present restrictive europium oxidation effects or high‐temperature/cost synthesis processes.
We demonstrate a guidable transfer method to achieve Eu3+ incorporation in both the ETL and perovskite in one step. Europium and tin ions are simultaneously deposited to form SnO2:Eu3+ film on FTO (F‐doped SnO2) glass. We observed directional diffusion of Eu3+ from the SnO2 ETL to the MAPbI3 perovskite film, which leads to accumulation of large amount of Eu3+ at the perovskite/ETL interface. Eu3+ ions synergistically eliminate the trap defects in both the ETL and perovskite films, resulting in an improved electron mobility of the SnO2 and grain boundary passivation within MAPbI3 films. The champion fabricated PSC attains a PCE as high as 20.14%, and, when exposed to the ambient atmosphere, the unsealed PSC presents a slow degradation by only 13% after 84 days. All these results indicate that our dual‐functional technique of europium passivation is extremely effective and convenient.
2. Results and Discussion
Since the refractive indices of FTO substrates and SnO2 films are different, the reflectance can be influenced by the modification of SnO2 films. Doping with Y3+ ions improves the antireflection ability of the SnO2 films and results in an increase in the optical transmittance in the region of 350 to 625 nm for the substrates.[ 26 ] The optical transmittance spectra of SnO2 and SnO2:Eu3+ films on FTO substrates are shown in Figure 1a. It is interesting to note that doping Eu3+ ions can improve the optical transmission properties of FTO/SnO2 substrates. In both the 350–410 nm and 440–600 nm regions, the optical transmittances of FTO/SnO2 substrates are enhanced with increasing concentration of Eu3+ dopant.
Figure 1.
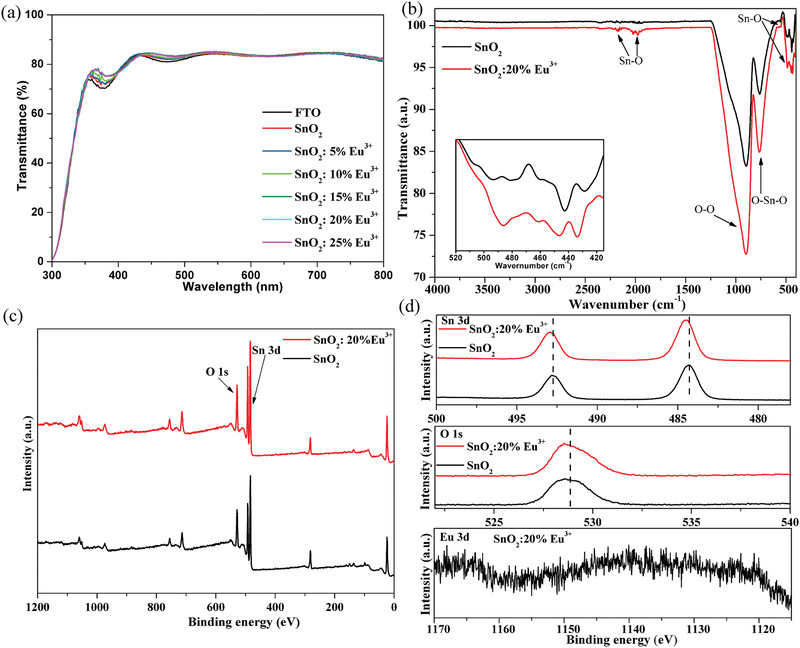
a) Optical transmission spectra of FTO substrates, SnO2 and SnO2:Eu3+ films on FTO substrates. b) Fourier‐transform infrared spectroscopy (FTIR) spectra of SnO2 and SnO2:20% Eu3+ films. c) Typical X‐ray photoelectron spectroscopy (XPS) spectra of SnO2 and SnO2:20% Eu3+ films. d) High‐resolution XPS spectra of Sn 3d, O 1s, and Eu 3d of SnO2 and SnO2:Eu3+ films.
The variation of optical transmittance is caused by the reduction of light scattering on the surface of SnO2:Eu3+ films, which should be related to the variable morphology of the homogeneously distributed SnO2:Eu3+ grains. Thus, the top‐view scanning electron microscopy (SEM) images of SnO2 and SnO2:Eu3+ films were measured and are shown in Figure S1, Supporting Information. The images show that the SnO2 film appears to be flat, uniform and pinhole‐free. After the introduction of Eu3+ ions, many nanoparticles appear on the grain surface. Figure S2, Supporting Information shows atomic force microscopy images of SnO2 and SnO2:Eu3+ films deposited on FTO substrates. The calculated data reveal that the root‐mean‐square roughness decreases from 29.9 to 22.1 nm with Eu3+ doping. Note that the smoother surface is beneficial to film‐forming of the perovskite layer.[ 23 ]
FTIR spectra is used to study the interaction between the dopant Eu3+and matrix SnO2. As shown in Figure 1b, the peaks around 760 cm−1 belong to the O—Sn—O symmetric stretch, and the peaks at 895 cm−1 are attributed to the O—O stretching about vibration of the oxygen adsorbed on the surface of SnO2 films. All the weaker peaks at ≈2180, ≈2027, and ≈1977 cm−1 are due to Sn—O stretching vibrations.[ 23 , 27 ] All of these absorption peaks are unaffected by Eu3+ doping; however, for the SnO2:20% Eu3+ sample, the characteristic asymmetric stretching peaks on the SnO2 surface shift to 486, 446, and 434 cm−1,[ 27 ] which demonstrates that Eu3+ ions might enter into the SnO2 crystal lattice and affect the SnO2 surface.
To further clarify the interaction between Eu3+ and SnO2, the XPS spectra for SnO2 and SnO2:20% Eu3+ films are measured and shown in Figure 1c. Clearly, the two Sn peaks and O peak are centered at ≈484, ≈493, and ≈529 eV, respectively. Meanwhile, high‐resolution Sn 3d, O 1s, and Eu 3d spectra are displayed in Figure 1d. In comparison with pristine SnO2 films, the shifts of ≈ 0.2 and 0.05 eV of the Sn 3d and O 1s peaks, respectively, can be observed in the SnO2:20% Eu3+ films, indicating that the Eu3+ dopant affects the SnO2 surface. Furthermore, the presence of trivalent Eu3+ can be confirmed by the observed binding energies at ≈1167.2 and 1139.8 eV, which are attributed to the 3d3/2 and 3d5/2 orbitals of Eu3+.
The electrical properties of semi‐conductive films can be characterized by the Hall Effect measurements, such as conductivity type, resistivity, mobility and carrier concentration. The average Hall coefficient, resistivity, mobility, and carrier concentration of SnO2 and SnO2:20% Eu3+ were measured and shown in Table S1, Supporting Information, respectively. The average Hall coefficients indicate that SnO2 and SnO2:20% Eu3+ are both well ETL films. Meanwhile the recorded resistivities, mobilities, and carrier concentrations prove Eu3+ doping can reduce resistivity and promote mobility in SnO2 films. It is known that the electron mobility of ETLs is the key factor for the performance improvement of PSCs. The various ETLs are also measured by the space charge‐limited current method,[ 23 ] and the results shown in Figure 2a. It is found that the electron mobility of SnO2:20% Eu3+ is 1.48 × 10−5 cm2 V−1 s−1, which is about eight larger than that of SnO2 (1.97 × 10−6 cm2 V−1 s−1). High electron mobility can effectively promote electron transfer in an ETL, thus the charge consumption at the interface between the ETL and perovskite is reduced, finally resulting in improved efficiency of PSCs.
Figure 2.
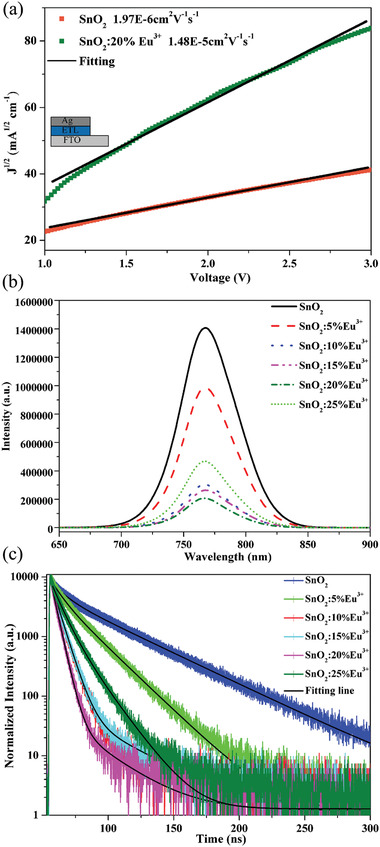
a) Electron mobility for SnO2 and SnO2:20% Eu3+ films; the inset shows the device structure of FTO/ETL/Ag. b) Steady‐state photoluminescence (PL) and c) time‐resolved photoluminescence (TRPL) spectra of perovskite films deposited on different ETL substrates.
The PL spectra of the perovskite deposited on different ETL substrates are presented in Figure 2b. Compared with the FTO/SnO2/perovskite sample, significant PL quenching is observed with increasing Eu3+ concentration in the samples on SnO2:Eu3+ ETL substrates. The data demonstrate optimized SnO2:20% Eu3+ presents the most appealing merits with the highest electron mobility. Figure 2c displays the normalized TRPL for perovskites on various ETLs. The lifetimes and corresponding proportions are listed in Table S2, Supporting Information. The lifetime decay curves have two parts: a slow decay component τ 1 and a fast decay component τ 2. Generally, τ 1 is attributed to the radiative recombination of free carriers captured by the traps in bulk materials and τ 2 originates from the quenching of charge carriers in the transportation process through the interfaces.[ 23 , 28 ] The FTO/SnO2/perovskite sample presents a long τ 2 lifetime of 37.55 ns, but it quickly decreases with increasing Eu3+ in SnO2 films. Smaller τ 2 dominating PL decay indicates that electrons can be effectively extracted from the perovskite layer to the ETL with minimal recombination loss at the interface. In the sample of perovskite deposited on SnO2:20% Eu3+, τ 1 was increased slightly to 7.65 ns, but τ 2 was considerably shortened to 3.53 ns; moreover, the lifetime contributions of τ 1 and τ 2 are 13.05% and 86.95%, respectively. These results indicate that the Eu3+ dopant in SnO2:20% Eu3+ film leads to suppressed charge carrier recombination at the interfaces, which remarkably dominates the overall charge carrier transport process. Moreover, sharply decreased τ 2 indicates that the carrier transfer efficiency at the interfaces can be significantly promoted by doping Eu3+ into SnO2. To summarize, the reduced loss of charge carriers at the interfaces drastically improves the carrier transfer efficiency in SnO2:20% Eu3+, which can be considered as a potential electron extraction layer for planar‐type PSCs.
The doped europium ions in MAPbI3 perovskite films can simultaneously reduce Pb0 and I0 defects for achieving high PCE, and they more easily concentrate at surfaces and grain boundaries or intercalate into two adjacent lattices.[ 10 ] In our research, X‐ray diffraction (XRD) patterns of MAPbI3 on various ETL substrates (SnO2 and SnO2:Eu3+) in Figure 3a,b indicate that europium ions can spread from SnO2:Eu3+ films into MAPbI3 perovskite films. When the europium ion concentration in SnO2 films is adjusted from 5 to 20 mol%, an obvious shift of the MAPbI3 (110) diffraction peak is observed. Because the radii of Eu3+ and Pb2+ are 94.7 and 119 pm, respectively, the shifts of the diffraction peak indicate that Eu3+ may prefer to intercalate into two adjacent lattices than to enter into the perovskite crystal lattice, in accordance with the reported density functional theory result.[ 10 ] When the doping concentration of Eu3+ exceeds 20%, excess Eu3+ might enter Pb2+ sites of the perovskite crystal lattice. Therefore, the shift of diffraction peak would reverse, and the corresponding photovoltaic properties of PSCs would be affected.
Figure 3.
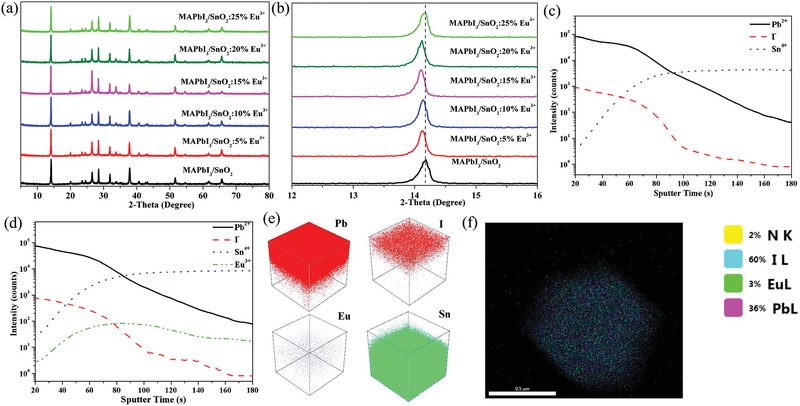
a) XRD patterns and b) enlarged XRD patterns of MAPbI3 on SnO2 and SnO2:Eu3+ ETL substrates. Secondary ion mass spectroscopy (SIMS) of c) perovskite/SnO2 and d) perovskite/SnO2:20% Eu3+ films. e) The spatial distribution of Pb, I, Eu, and Sn in perovskite/SnO2:Eu3+ films. f) Energy dispersivespectroscopy (EDS) mapping image of perovskite/SnO2:Eu3+ film.
The SIMS curves shown in Figure 3c,d present the distribution of Pb, I, Sn, and Eu along the depth direction of perovskite/SnO2 and perovskite/SnO2:20% Eu3+ films, respectively, and their spatial distribution images are presented in Figure 3e. In the crossing regions of the SIMS profiles, sharply varying Pb, I, and Sn contents means those regions are the interfaces between the perovskite and SnO2 (or SnO2:Eu3+) films. It is observed that Eu3+ ions are evenly distributed in the SnO2 film, but they tend to aggregate at the interface between the perovskite and SnO2:20% Eu3+ films, and subsequently, through the process of diffusion, a few Eu3+ ions enter the perovskite film. Therefore, the shifts of the asymmetric stretching Sn—O peaks in the FTIR spectra (Figure 1b) and the dramatically increased proportions of τ 2 lifetime in the TRPL spectra (Figure 2c) are observed, which are both related to the aggregation of Eu3+ at the interface between the perovskite and SnO2:Eu3+ films. An EDS mapping image of perovskite/SnO2:Eu3+ film is presented in Figure 3f. Although the amount of Eu3+ ions pervading the perovskite film is very low, their distribution is uniform throughout the perovskite film.
The surface coverage of perovskite films is also very important for high‐performance PSCs.[ 27 , 28 , 29 ] The aggregation of Eu3+ on the top interface of SnO2:Eu3+ films may affect the nucleation and growth of perovskite films. A smaller contact angle can result in a reduced Gibbs free energy facilitating heterogeneous nucleation; meanwhile, the formation of more crystal nuclei will accelerate the process of thin film growth from nuclei to island structures, then to networked, and finally into a continuous film.[ 23 , 30 , 31 ] The contact angles of SnO2 and SnO2:Eu3+ films are measured and presented in Figure S3, Supporting Information. All calculated contact angles indicate that increased concentration of dopant Eu3+ in SnO2:Eu3+ films reduces the contact angle on the surface, and moreover, the contact angle is a minimum of 10.2° on the surface of SnO2:20% Eu3+ films, which can result in lower surface energy and accelerated perovskite crystallization during the growth of the networked structure.[ 30 , 32 ] However, the grain sizes of perovskite films are not obviously affected by the different substrates, as shown in the SEMs of Figure S4, Supporting Information.
Grain boundary plays a critical role in determining the charge collection efficiency and stability of PSCs. The passivation of grain boundary can reduce the trap densities of perovskite films to improve the performance of PSCs. Different grain boundary passivation methods are being studied, such as introducing the PbI2‐rich phase at grain boundaries in MAPbI3 PSCs,[ 33 ] or using carbon quantum dots additive to passivate the uncoordinated lead ions on grain boundaries of MAPbI3 PSCs.[ 34 ] In our work, the aggregated Eu3+ at the grain boundaries and the interfaces can reduce Pb0 and I0 defects further to passivate grain boundaries. Thus the trap densities of perovskites on different ETLs are evaluated by the space charge‐limited current measurements of electron‐only devices fabricated with the structure ITO (indium tin oxide)/ETL/perovskite/PCBM (phenyl‐C61‐butyric acid methyl ester)/Ag, and the corresponding dark current–voltage (I–V) curves are shown in Figure S5, Supporting Information. At low bias voltage, the linear correlation shown as red lines indicates an ohmic response. When the bias voltage increases above the kink point, the current suddenly increases with a nonlinear correlation (cyan line), which reveals that the traps in the perovskite film are totally filled. The bias voltage corresponding to the kink point between the linear and nonlinear correlation is defined as the trap‐filled limit voltage (V TFL). The trap densities (N t) of the perovskites on different ETLs are calculated by the equation N t = (2V TFL εε 0)/(eL 2),[ 35 , 36 ] where ε and ε 0 are the relative dielectric constant for MAPbI3 perovskite and vacuum permittivity, respectively, e is the electron charge and L is the thickness of the MAPbI3 perovskite film. It is obvious that the trap densities of the perovskites were reduced from 1.69 × 1016 to 1.31 × 1016 cm−3 by increasing the amount of Eu3+ dopant to 20% in the SnO2 substrate, and this reduction is attributed to the passivation of grain boundaries caused by increased Eu3+ at grain boundaries and interfaces.
According to the above discussion, SnO2:Eu3+ is expected to be a better ETL for PSCs than the pristine SnO2 film. Therefore, planar‐type PSCs with different SnO2 films as ETL substrates are designed with the structure shown in Figure 4a. MAPbI3 and Spiro‐OMeTAD are used as the photon absorber layer and hole‐transport layer, respectively. The thicknesses of the perovskites are nearly unchanged by doping 20% Eu3+ into SnO2 film, as shown in the cross‐sectional SEM images of PSCs in Figure S6, Supporting Information.
Figure 4.
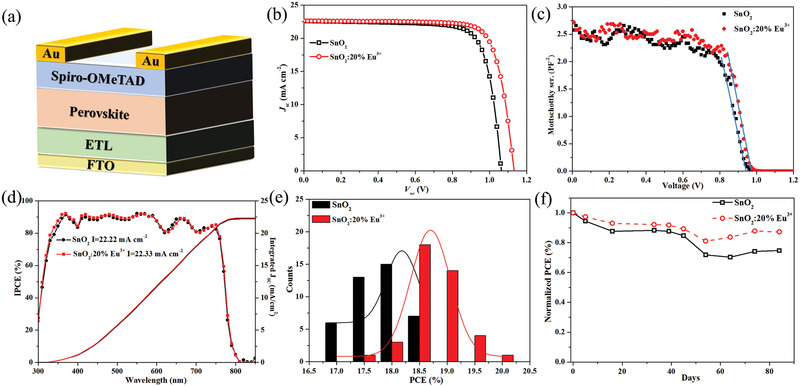
a) The designed device configuration. b) Current density−voltage (J–V) curves, c) Mott–Schottky plots and d) The incident‐photon‐to‐charge conversion efficiency (IPCE) of the PSCs with different ETL substrates. The integrated current densities from the IPCE curves were recorded under the AM1.5G spectrum. e) The PCE distribution of the PSCs based on the different ETLs. f) Long‐term stability test for planar‐type PSC devices with different ETLs without any encapsulation under ambient conditions (35% humidity in the dark).
Figure 4b and Figure S7, Supporting Information present the J–V curves of PSCs on various ETL substrates, and the corresponding key parameters, such as the open‐circuit voltage (V oc), the short‐circuit current (J sc), fill factor (FF), and PCE are all summarized in Table S3, Supporting Information. The maximum PCE of the devices based on the SnO2 ETL substrate is 18.66%, with the corresponding V oc = 1.06 V, J sc = 22.57 mA cm−2, and FF = 77.77%. It is exciting that the optimal PCE can be increased to 20.14% by changing the SnO2 ETL to SnO2:20% Eu3+ ETL, with corresponding V oc and FF dramatically increased to 1.13 V and 78.76%, respectively. The increased FF are due to the reduced trap density in perovskite film; however, the improved V oc should be attributed to the reduced energy loss in MAPbI3 PSCs.[ 37 ]
Mott–Schottky analysis based on capacitance–voltage (C–V) measurement can be used to investigate the change of built‐in electric field and is carried out to further understand the charge carrier trapping and accumulating behaviors with the incorporation of Eu3+ dopant.[ 38 , 39 , 40 , 41 ] Figure 4c presents the C −2–V plots for MAPbI3 perovskite PSCs, and the corresponding values of the built‐in electric fields (V bi) in the PSCs based on different ETLs are obtained from the Mott–Schottky equation C −2 = (2(V bi − V))/(A 2 eɛɛ 0 N), where C is the capacitance under applied voltage, V bi is the built‐in potential, V is the applied bias, A is the device area, ɛ is the relative permittivity, ɛ 0 is the vacuum permittivity, and N is the free carrier concentration at the edge of the depletion layer.[ 42 , 43 ] The V bi for the PSC coated on SnO2:20% Eu3+ ETL is 0.972 V, which is larger than 0.942 V of the control device without europium dopant. The enhanced built‐in potential provides more driving force to separate the photogenerated charge carriers, resulting in an extended depletion region, which suppresses electron‐hole recombination, finally contributing to the increase of V oc.
On the other hand, the V oc is also determined by the quasi‐Fermi level separation of electrons and holes in the light‐dependent dynamic equilibrium condition, and the reduced charge carrier recombination and fewer traps within the bandgap can narrow the distribution of defect states and elevate the quasi‐Fermi levels of electrons.[ 44 ] As shown in Figure S5, Supporting Information, the reduced trap density from 1.69 × 1016 to 1.31 × 1016 cm−3 can be obtained by coating MAPbI3 perovskite film on SnO2:20% Eu3+ substrate. The significantly reduced trap‐assisted recombination elevates the quasi‐Fermi level of electrons, and thus greater quasi‐Fermi level separation can be obtained. As a result, the PSCs fabricated on the SnO2:20%Eu3+ substrate exhibit a higher V oc compared to the control PSCs on the SnO2 substrate.
Electrical impedance spectroscopy is carried out to monitor the transfer resistance in PSCs. The Nyquist plots of the PSCs fabricated on different SnO2 or SnO2:Eu3+ films recorded at V oc under dark conditions are shown in Figure S8, Supporting Information, and the corresponding equivalent circuit is shown in Figure S9, Supporting Information. It is known that the recombination resistance (R rec) is in the low‐frequency range,[ 42 ] and, in our fabricated PSCs, it increases with increased Eu3+ doping in the SnO2 films. Compared to the control PSCs, the device fabricated on SnO2:20% Eu3+ substrate shows the largest R rec of 289 Ω, which can effectively inhibit charge recombination at grain boundaries and the interface, because the aggregated Eu3+ at grain boundaries and the interface decrease the amount of negative defects Pb0 and I0.
Figure 4d shows the IPCE spectra and the integrated J sc values versus wavelength for the PSCs based on different ETLs. The effect on IPCE of Eu3+ doping in the ETL is divided into two parts: 1) In the UV region of 300–370 nm, the IPCE intensity is enhanced by Eu3+ doping due to the f–f transitions absorption of Eu3+.[ 45 , 46 , 47 , 48 , 49 ] The absorption of Eu3+ reduces the damage to the perovskite film from UV light, resulting in the obvious increase of the IPCE; 2) The enhancement of IPCE in the region of 370–550 nm is attributed to the increased transmittance of ITO/SnO2:20% Eu3+ substrate. The highest observed IPCE value reaches 92%. Meanwhile, the integrated J sc increases from 22.22 to 22.33 mA cm−2 by using SnO2:20% Eu3+ film as the ETL substrate, which indicates SnO2:20% Eu3+ is an excellent ETL for application in PSCs. The stabilized power output of the device with SnO2:20% Eu3+ ETL is shown in Figure S10, Supporting Information. While maintaining an external bias near the maximum power output point (0.88 V), the stabilized photocurrent for the PSC with SnO2:20% Eu3+ ETL is 21.37 mA cm−2. The results indicate that SnO2:20% Eu3+ ETL is beneficial to the illumination stability of the MAPbI3 perovskite device.
Stability and repeatability are also very important characteristics for PSCs. The PCE distribution histograms for devices with different ETLs are presented in Figure 4e. The device based on SnO2:20% Eu3+ substrate exhibits excellent repeatability in contrast to that based on the pristine SnO2 substrate. Figure 4f shows normalized PCE of the different device exposed to an ambient atmosphere (≈35% humidity) during 84 days in the dark. It is clear that the device based on SnO2:20% Eu3+ substrate maintains 87% of its initial PCE on the 84th day, but the device coated on SnO2 substrate has decreased to 75% of its initial PCE under the same storage conditions. Their stabilities at higher temperature in N2 atmosphere, even under continuous illumination and higher humidity are both investigated and shown in Figure S11a,b, Supporting Information, respectively. After storing devices in dark with N2 atmosphere at 80 °C for 500 min, the PCE of control device has dropped to 30% of its initial value. But the decrease can be retarded to be very slow by 20% Eu3+ doped in SnO2, thus the device coated on SnO2:20% Eu3+ substrate just decreases to 65% of its initial PCE under the same storage conditions. Even under continuous 100 mW cm−2 illumination and 40–50% humidity at 60 °C for 300 min, the SnO2:20% Eu/Perovskite device can still keep 50% of its initial PCE, but the PCE of SnO2/Perovskite device has drop to 40% of its initial value. The comparison demonstrates that the PSCs coated on SnO2:20% Eu3+ show more excellent stability, which is due to the europium ions can effectively passivate the defects at the grain boundaries and interfaces preventing moisture permeation, further resulting in the improved environmental stability.[ 50 ]
3. Conclusion
This work describes a novel dual‐functional method to simultaneously optimize charge transport characteristics of the perovskite and ETL layers which account for the enhancement of performance of the corresponding PSCs. An effective SnO2:Eu3+ ETL is developed, and the champion device incorporating it achieves a PCE of 20.14%, showing excellent stability by maintaining 87% of its initial efficiency after storage in ambient atmosphere for 84 days. The excellent performance of the PSCs is attributed to the dual‐passivation effect of europium ions in SnO2. The uniformly distributed europium dopants reduce the trap defects in the SnO2 film, resulting in increased electron mobility of the ETL. The aggregation of europium ions at the interface between the perovskite and SnO2 films is beneficial for improving electron transport through the interface by reducing the charge accumulation at the interface. Moreover, the aggregated europium ions passivate the perovskite by reducing the trap density in the grain boundaries, which is favorable to the V oc and FF of PSCs. Meanwhile, the aggregated europium ions both on the grain boundaries and interface suppress perovskite degradation by preventing moisture permeation. Our dual‐functional method provides a promising direction toward simultaneously optimizing the ETL and perovskite films, and we believe that the present work will facilitate the development of perovskite photovoltaics.
4. Experimental Section
Materials
SnCl2·2H2O (98%) was purchased from Macklin. Thioglycolic acid was purchased from Sigma‐Aldrich. EuCl3·6H2O (99.99%) was purchased from CIVI‐CHEM. PbI2 (99.9985%) was purchased from Alfa Aesar. MAI (99.5%) was purchased from Xi'an Polymer Light Technology Corp. Urea, 4‐tert‐butylpyridine (TBP) and bis(trifluoromethane) sulfonamide lithium salt (Li‐TFSI) were purchased from Aladdin. HCl (37 wt%) and chlorobenzene (≥99.0%) were purchased from Sinopharm Chemical Reagent Corporation Co., Ltd. Spiro‐OMeTAD (≥99.0%) was ordered from Youxuan Tech. 4‐Hydroxybutanoic acid lactone (GBL) and dimethyl sulfoxide (DMSO) were ordered from Alfa‐Aesar.
Device Fabrication
The FTO‐coated glass (2.5 × 2.5 cm) was cleaned by sequential sonication in acetone, isopropanol, and ethanol for 30 min each time and then dried under air flow and treated by ozone plasma for 6 min. The undoped and Eu‐doped SnO2 layers were prepared by the chemical bath deposition method. Briefly, for the preparation of undoped SnO2 ETL, 0.5 g of urea was dissolved in 40 mL deionized water, and then 10 mL thioglycolic acid and 500 mL HCl were added to the aqueous solution. Finally, SnCl2·2H2O was dissolved in the solution at 0.002 m concentration followed by stirring for 2 min. The clean FTO substrates were immersed in the aqueous solution at 70 °C for 3 h, followed by rinsing in a deionized water sonication bath for 2 min. Then they were dried with flowing air and heat‐treated for 1 h at 180 °C in air. For the preparation of (5, 10, 15, 20, and 25 mol%) Eu‐doped SnO2 ETLs, EuCl3⋅6H2O was added directly to the prepared aqueous solution before immersing the cleaned FTO substrates.
The MAPbI3 perovskite solution (1.4 m) was comprised of MAI and PbI2 in 1 mL of GBL/DMSO = 7:3 (v/v). The solution was stirred at room temperature for 12 h. Then the FTO/ETL substrates were treated by ozone plasma for 6 min. The solution was spin‐coated onto the FTO/ETL substrate by a consecutive two‐step process at 1000 rpm for 10 s and followed by 3000 rpm for 40 s. During the second step, 200 mL of chlorobenzene was dropped onto the substrate. The films were then annealed at 100 °C for 10 min in a nitrogen‐filled glovebox. 90 mg mL−1 spiro‐OMeTAD in 1 mL chlorobenzene with the addition of 36 mL TBP and 22 mL Li‐TFSI solution (520 mg in 1 mL acetonitrile) was spin‐coated onto the perovskite films at 5000 rpm for 40 s. The samples were kept in a desiccator overnight. Finally, 80 nm gold electrodes were deposited on the top of each cell by a thermal evaporator.
Device Characterization
XRD spectra were obtained using a D/MAX 2400 diffractometer with Cu Ka radiation (Rigaku). Transmittance spectra were acquired on a PerkinElmer UV‐Lambda 950 instrument. PL (excitation at 510 nm, front‐side excitation) and TRPL spectra (excitation at 510 nm and emission at 768 nm, front‐side excitation) were measured with a PicoQuant FT‐300. Water contact angles were measured using a DataPhysics OCA 20. The surface morphologies of the perovskite films and the SnO2 films were characterized by SEM (FE‐SEM; SU‐8020, Hitachi) at an acceleration voltage of 5 kV. XPS measurements were carried out by using a photoelectron spectrometer (ESCALAB 250Xi, Thermo Fisher Scientific). SIMS curves were recorded by the time of flight secondary ion mass spectrometry (TOF SIMS IV, ION TOF GmbH). The J−V performance of the perovskite solar cells was analyzed using a Keithley 2400 Source Meter under ambient conditions at room temperature, and the illumination intensity was 100 mW cm−2 (AM 1.5G Oriel solar simulator) with scan rate 0.2 V s−1. The device area of 0.09 cm2 was defined by a metal aperture to avoid light scattering from the metal electrode into the device during the measurement. TheIPCE was characterized on a QTest Station 2000ADI system (Crowntech Inc., USA), and the light source was a 300 W xenon lamp. The monochromatic light intensity for the IPCE measurement was calibrated with a reference silicon photodiode. The Hall Effect measurements were recorded by the Hall Effect Measurement System (HMS‐3000).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This research was supported by the National Key Research Program of China (2016YFA0202403), the National Natural Science Foundation of China (21603140) and the 111 Project (B14041).
Chen Y., Zuo X., He Y., Qian F., Zuo S., Zhang Y., Liang L., Chen Z., Zhao K., Liu Z., Gou J., S. (Frank) Liu, Dual Passivation of Perovskite and SnO2 for High‐Efficiency MAPbI3 Perovskite Solar Cells. Adv. Sci. 2021, 8, 2001466. 10.1002/advs.202001466
Contributor Information
Jing Gou, Email: goujing@snnu.edu.cn.
Shengzhong (Frank) Liu, Email: liusz@snnu.edu.cn.
References
- 1. Kojima A., Teshima K., Shirai Y., Miyasaka T., J. Am. Chem. Soc. 2009, 131, 6050. [DOI] [PubMed] [Google Scholar]
- 2. Lee M. M., Teuscher J., Miyasaka T., Murakami T. N., Snaith H. J., Science 2012, 338, 643. [DOI] [PubMed] [Google Scholar]
- 3. Zhou H., Chen Q., Li G., Luo S., Song T., Duan H. S., Hong Z., You J., Liu Y., Yang Y., Science 2014, 345, 542. [DOI] [PubMed] [Google Scholar]
- 4. Im J. H., Jang I. H., Pellet N., Grätzel M., Park N. G., Nat. Nanotechnol. 2014, 9, 927. [DOI] [PubMed] [Google Scholar]
- 5. Yang M., Zhou Y., Zeng Y., Jiang C. S., Padture N. P., Zhu K., Adv. Mater. 2015, 27, 6363. [DOI] [PubMed] [Google Scholar]
- 6. Malinkiewicz O., Yella A., Lee Y. H., Espallargas G. M., Graetzel M., Nazeeruddin M. K., Bolink H. J., Nat. Photonics 2014, 8, 128. [Google Scholar]
- 7. Yang W. S., Noh J. H., Jeon N. J., Kim Y. C., Ryu S., Seo J., Il Seok S., Science 2015, 348, 1234. [DOI] [PubMed] [Google Scholar]
- 8. Yang W. S., Park B. W., Jung E. H., Jeon N. J., Kim Y. C., Lee D. U., Shin S. S., Seo J., Kim E. K., Noh J. H., Il Seok S., Science 2017, 356, 1376. [DOI] [PubMed] [Google Scholar]
- 9. National Renewable Energy Laboratory (NREL) Efficiency chart 2018; www.nrel.gov/pv/assets/pdfs/pv-efficiency-chart.20181214.pdf. Accessed January 2021.
- 10. Wang L., Zhou H., Hu J., Huang B., Sun M., Dong B., Zheng G., Huang Y., Chen Y., Li L., Xu Z., Li N., Liu Z., Chen Q., Sun L., Yan C., Science 2019, 363, 265. [DOI] [PubMed] [Google Scholar]
- 11. Tiep N. H., Ku Z., Fan H. J., Adv. Energy Mater. 2016, 6, 1501420. [Google Scholar]
- 12. Rong Y., Liu L., Mei A., Li X., Han H., Adv. Energy Mater. 2015, 5, 1501066. [Google Scholar]
- 13. Berhe T. A., Su W. N., Chen C. H., Pan C. J., Cheng J. H., Chen H. M., Tsai M. C., Chen L. Y., Dubaleb A. A., Hwang B. J., Energy Environ. Sci. 2016, 9, 323. [Google Scholar]
- 14. Frost J. M., Butler K. T., Brivio F., Hendon C. H., Schilfgaarde M., Walsh A., Nano Lett. 2014, 14, 2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu Z., Sun L., Adv. Energy Mater. 2015, 5, 1500213. [Google Scholar]
- 16. Li W., Li J., Niu G., Wang L., J. Mater. Chem. A 2016, 4, 11688. [Google Scholar]
- 17. Li W., Zhang W., Reenen S. V., Sutton R. J., Fan J., Haghighirad A. A., Johnston M. B., Wang L., Snaith H. J., Energy Environ. Sci. 2016, 9, 490. [Google Scholar]
- 18. Niu T., Lu J., Munir R., Li J., Barrit D., Zhang X., Hu H., Yang Z., Amassian A., Zhao K., Liu S. F., Adv. Mater. 2018, 30, 1706576. [DOI] [PubMed] [Google Scholar]
- 19. Niu T., Lu J., Tang M. C., Barrit D., Smilgies D. M., Yang Z., Li J., Fan Y., Luo T., McCulloch I., Amassian A., Liu S. F., Zhao K., Energy Environ. Sci. 2018, 11, 3358. [Google Scholar]
- 20. Chen H., Luo Q., Liu T., Ren J., Li S., Tai M., Lin H., He H., Wang J., Wang N., Small 2019, 15, 1904372. [DOI] [PubMed] [Google Scholar]
- 21. Zhou W., Li D., Xiao Z., Wen Z., Zhang M., Hu W., Wu X., Wang M., Zhang W. H., Lu Y., Yang S., Yang S., Adv. Funct. Mater. 2019, 29, 1901026. [Google Scholar]
- 22. Chen W., Wang Y., Pang G., Koh C. W., Djurišic´ A. B., Wu Y., Tu B., Liu F., Chen R., Woo H. Y., Guo X., He Z., Adv. Funct. Mater. 2019, 29, 1808855. [Google Scholar]
- 23. Yang D., Yang R., Wang K., Wu C., Zhu X., Feng J., Ren X., Fang G., Priya S., Liu S. F., Nat. Commun. 2018, 9, 3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. An Q., Fassl P., Hofstetter Y. J., Koch D. B., Bausch A., Hopkinson P. E., Vaynzof Y., Nano Energy 2017, 39, 400. [Google Scholar]
- 25. Zhang B., Song Z., Jin J., Bi W., Li H., Chen C., Dai Q., Xu L., Song H., J. Colloid Interface Sci. 2019, 553, 14. [DOI] [PubMed] [Google Scholar]
- 26. Yang G., Lei H., Tao H., Zheng X., Ma J., Liu Q., Ke W., Chen Z., Xiong L., Qin P., Chen Z., Qin M., Lu X., Yan Y., Fang G., Small 2017, 13, 1601769. [DOI] [PubMed] [Google Scholar]
- 27. Zhuang S., Xu X., Feng B., Hu J., Pang Y., Zhou G., Tong L., Zhou Y., ACS Appl. Mater. Interfaces 2014, 6, 613. [DOI] [PubMed] [Google Scholar]
- 28. Li Y., Meng L., Yang Y. M., Xu G., Hong Z., Chen Q., You J., Li G., Yang Y., Li Y., Nat. Commun. 2016, 7, 10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhumekenov A. A., Burlakov V. M., Saidaminov M. I., Alofi A., Haque M. A., Turedi B., Davaasuren B., Dursun I., Cho N., El‐Zohry A. M., Bastiani M. D., Giugni A., Torre B., Fabrizio E. D., Mohammed O. F., Rothenberger A., Wu T., Goriely A., Bakr O. M., ACS Energy Lett. 2017, 2, 1782. [Google Scholar]
- 30. Zhao H., Wang S., Sun M., Zhang F., Li X., Xiao Y., J. Mater. Chem. A 2018, 6, 10825. [Google Scholar]
- 31. Salim T., Sun S., Abe Y., Krishna A., Grimsdale A. C., Lam Y. M., J. Mater. Chem. A 2015, 3, 8943. [Google Scholar]
- 32. Fu P., Huang L., Yu W., Yang D., Liu G., Zhou L., Zhang J., Li C., Nano Energy 2015, 13, 275. [Google Scholar]
- 33. Hoque M. N. F., He R., Warzywoda J., Fan Z., ACS Appl. Mater. Interfaces 2018, 10, 30322. [DOI] [PubMed] [Google Scholar]
- 34. Ma Y., Zhang H., Zhang Y., Hu R., Jiang M., Zhang R., Lv H., Tian J., Chu L., Zhang J., Xue Q., Yip H. L., Xia R., Li X., Huang W., ACS Appl. Mater. Interfaces 2019, 11, 3044. [DOI] [PubMed] [Google Scholar]
- 35. Yang D., Yang R., Ren X., Zhu X., Yang Z., Li C., Liu S. F., Adv. Mater. 2016, 28, 5206. [DOI] [PubMed] [Google Scholar]
- 36. Jiang H., Yan Z., Zhao H., Yuan S., Yang Z., Li J., Liu B., Niu T., Feng J., Wang Q., Wang D., Yang H., Liu Z., Liu S. F., ACS Appl. Energy Mater. 2018, 1, 900. [Google Scholar]
- 37. Yang S., Zhao H., Han Y., Duan C., Liu Z., Liu S. F., Small 2019, 15, 1904387. [DOI] [PubMed] [Google Scholar]
- 38. Murgatroyd P. N., J. Phys. D: Appl. Phys. 1970, 3, 151. [Google Scholar]
- 39. Bube R. H., J. Appl. Phys. 1962, 33, 1733. [Google Scholar]
- 40. Luther J. M., Law M., Beard M. C., Song Q., Reese M. O., Ellingson R. J., Nozik A. J., Nano Lett. 2008, 8, 3488. [DOI] [PubMed] [Google Scholar]
- 41. Jin Z., Wang A., Zhou Q., Wang Y., Wang J., Sci. Rep. 2016, 6, 37106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bai D., Zhang J., Jin Z., Bian H., Wang K., Wang H., Liang L., Wang Q., Liu S. F., ACS Energy Lett. 2018, 3, 970. [Google Scholar]
- 43. Yang D., Yang R., Zhang J., Yang Z., Liu S. F., Li C., Energy Environ. Sci. 2015, 8, 3208. [Google Scholar]
- 44. Wang K., Zheng L., Zhua T., Yao X., Yi C., Zhang X., Cao Y., Liu L., Hu W., Gong X., Nano Energy 2019, 61, 352. [Google Scholar]
- 45. Gou J., Fan J., Zuo S., Lou M., Chen Y., Zhou X., Yang Y., Yu B., Liu S. F., J. Am. Chem. Soc. 2017, 100, 4011. [Google Scholar]
- 46. Moon T., Hwang S. T., Jung D. R., Son D., Kim C., Kim J., Kang M., Park B., J. Phys. Chem. C 2007, 111, 4164. [Google Scholar]
- 47. Kong J., Zheng W., Liu Y., Li R., Ma E., Zhu H., Chen X., Nanoscale 2015, 7, 11048. [DOI] [PubMed] [Google Scholar]
- 48. Meetei S. D., Singh S. D., J. Alloys Compd. 2014, 587, 143. [Google Scholar]
- 49. Yu X., Zhang L., Xu X., Wang T., Yu H., Jiang T., Jiao Q., Yang Z., Zhou D., Qiu J., J. Lumin. 2014, 145, 114. [Google Scholar]
- 50. Chu Z., Yang M., Schulz P., Wu D., Ma X., Seifert E., Sun L., Li X., Zhu K., Lai K., Nat. Commun. 2017, 8, 2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information