

Abstract
Objectives
Although axitinib has achieved a preferable response rate for advanced renal cell carcinoma (RCC), patient survival remains unsatisfactory. In this study, we evaluated the efficacy and safety of a combination treatment of axitinib and a low dose of pembrolizumab‐activated autologous dendritic cells–co‐cultured cytokine‐induced killer cells in patients with advanced RCC.
Methods
All adult patients, including treatment‐naive or pretreated with VEGF‐targeted agents, were enrolled from May 2016 to March 2019. Patients received axitinib 5 mg twice daily and pembrolizumab‐activated dendritic cells–co‐cultured cytokine‐induced killer cells intravenously weekly for the first four cycles, every 2 weeks for the next four cycles, and every month thereafter.
Results
The 43 patients (22 untreated and 21 previously treated) showed a median progression‐free survival (mPFS) of 14.7 months (95% CI, 11.16–18.30). mPFS in treatment‐naive patients was 18.2 months, as compared with 14.4 months in pretreated patients (log‐rank P‐value = 0.07). Overall response rates were 25.6% (95% CI, 13.5–41.2%). Grade 3 or higher adverse events occurred in 5% of patients included hypertension (11.6%) and palmar‐plantar erythrodysesthesia (7.0%). Peripheral blood lymphocyte immunophenotype and serum cytokine profile analyses demonstrated increased antitumor immunity after combination treatment particularly in patients with a long‐term survival benefit, while those with a minimal survival benefit demonstrated an elevated proportion of peripheral CD8+TIM3+ T cells and lower serum‐level immunostimulatory cytokine profile.
Conclusions
The combination therapy was active and well tolerated for treatment of advanced RCC, either as first‐ or second‐line treatment following other targeted agents. Changes in immunophenotype and serum cytokine profile may be used as prognostic biomarkers.
Keywords: advanced renal cell carcinoma, axitinib, dendritic cells and cytokine‐induced killer cells immunotherapy, pembrolizumab
This study demonstrated that the combination of axitinib plus pembrolizumab‐activated autologous DC‐cytokine‐induced killer cells contributed to encouraging clinical outcomes in patients with advanced renal cell carcinoma who were treatment‐naive or targeted agents‐pretreated. Such combination therapy led to superior antitumor immunity, including increased lymphocyte infiltration and cellular responses of CD8+ T cells, especially in patients who acquired long‐term survival benefit. An increased percentage of peripheral CD8+TIM3+ T cells and a lower serum‐level immunostimulatory cytokine profile were identified as a potential resistance mechanism to this combination therapy in patients with minimal survival benefit.

Introduction
Each year, there are an estimated 403 300 new cases of renal cell carcinoma (RCC) worldwide, resulting in 175 100 deaths. 1 Approximately one‐third of RCC patients present with metastatic disease at their initial diagnosis and 30–40% of patients with localised tumors will develop metastases after surgery. 2 RCC is often resistant to conventional therapy regimens such as chemotherapy and radiotherapy. 3 Immunotherapy using high‐dose interleukin (IL)‐2 is the most successful single‐agent therapy for metastatic RCC, although clinical benefits are limited to patients with excellent performance status and normal organ function. 4 Recently, the introduction of agents targeting the vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) pathway has supplanted IL‐2 as the mainstay therapy and has improved the survival of advanced RCC. 5 Several active inhibitors targeting the VEGF/VEGF‐receptor and the mTOR in metastatic RCC (mRCC) have also been approved. 5 However, the survival rate among patients with mRCC has plateaued. 6
Axitinib, a selective, second‐generation inhibitor of VEGFR‐1, VEGFR‐2 and VEGFR‐3, 7 is a standard first‐ or second‐line therapy for mRCC. Previous clinical studies demonstrated that the median progression‐free survival (PFS) seen in patients with mRCC treated with axitinib was 10.1 and 6.7 months when used as first‐ and second‐line therapies, respectively. 8 , 9 Although antiangiogenic drugs such as axitinib have changed the therapeutic landscape for this disease, they are associated with a limited ability of durable tumor control once resistance to therapy is developed. 8 , 9 , 10 , 11 , 12 Thus, the identification of more effective treatment strategies to improve patient care and increase survival for patients with mRCC is urgently needed.
Immunotherapy is the fourth pillar of treatment for malignant tumors due to our improved understanding of the underlying principles of tumor biology and immunology. 13 As a typical representative of immune checkpoint therapy, pembrolizumab is an anti‐programmed death (PD)‐1 antibody that selectively blocks the interaction between PD‐1 and PD‐1 ligands 1 and 2 (PD‐L1/L2), thus resulting in activation of the cellular immune response. 14 , 15 Previous reports have demonstrated that pembrolizumab shows extensive antitumor activity in patients with various cancers 16 and elicits long‐term tumor regression. 17 Rini et al. 18 also reported that patients with mRCC treated with first‐line pembrolizumab plus axitinib showed a significantly increased median PFS compared to those treated with sunitinib alone (15.1 versus 11.1 months). Moreover, axitinib was also reported to play an immunomodulatory role within the tumor microenvironment beyond its antiangiogenic activity. For example, axitinib could increase the infiltration of immune cells and reduces the suppressive capacity of monocytic myeloid‐derived suppressor cells in mouse model 19 and reverse tumor‐induced immunosuppression via modulation of myeloid and mast cells to induce anticancer immunity. 20 These results provide a theoretical rationale for immunotherapy combined with axitinib.
Besides immune checkpoint inhibitors, several adoptive cell immunotherapies using various killer cells have been investigated as new immunotherapeutic options, including tumor‐infiltrating lymphocytes (TIL), antigen‐specific T lymphocytes, cytokine‐induced killer (CIK) cells/dendritic cells–co‐cultured CIK (DC‐CIK) cells and chimeric antigen receptor T cells. 21 , 22 , 23 , 24 , 25 Among them, CIK/DC‐CIK cells exhibited potent cytotoxic activity against a broad spectrum cancer cells in vitro and in vivo. 26 , 27 A series of clinical studies have demonstrated the safety and therapeutic efficacy of CIK/DC‐CIK cell treatment for several types of cancer, 28 , 29 including RCC. 23 , 25 However, our and others previous studies showed that a fraction of CIK/DC‐CIK cells are PD‐1 positive and that antitumor activity of CIK/DC‐CIK cells is restricted by PD‐1/PD‐L1 pathways in the tumor microenvironment. 30 , 31 , 32 In addition, anti–PD‐1 antibody systemic therapy is associated with immune‐related adverse effects and may induce an immunosuppressive program by interacting with various immunosuppressive cells (e.g. myeloid cells, Treg cells and macrophage) in the tumor bed. 33 , 34 , 35 It is therefore logical to assume that using a low dose of pembrolizumab in vitro to directly block the PD‐1 epitope in CIK/DC‐CIK cells may reduce the negative effects of pembrolizumab systemic therapy and enhance the cytotoxicity of CIK/DC‐CIK cells against tumor cells. 30 , 31 , 36 , 37 Our previous phase I clinical study showed the feasibility and low toxicity of a low dose of PD‐1 blockade‐activated DC‐CIK cell immunotherapy in patients with advanced cancers, including mRCC. 30 Based on these considerations, we conducted an investigator‐initiated, single‐arm, phase 2 study of axitinib plus a low dose of pembrolizumab‐activated autologous DC‐CIK cells in patients with advanced RCC.
Results
Patient characteristics
From May 2016 to March 2019, 53 patients were assessed for eligibility, of whom 10 were deemed ineligible [previous treatment with axitinib (three patients) or anti‐PD‐1 antibody (one patient), non‐renal cell carcinoma (two patients), uncontrolled hypertension (two patients), failure to obtain informed consent (one patient), and no measurable disease at screening (one patient)] and 43 patients were enrolled for safety and activity analysis (Figure 1). Baseline and disease characteristics are shown in Table 1. Overall, 36 patients had histology of clear cell RCC (ccRCC), and seven patients had papillary RCC (pRCC). Thirty patients (69.8%) were categorised with favorable or intermediate‐risk disease according to the International Metastatic Renal Cell Carcinoma Database Consortium model (determined at the time of study entry). Twenty‐two patients had not received previous treatment, while the other 21 had received previous VEGF‐targeted treatment, including 10 (23.2%) with sunitinib, 7 (16.3%) with sorafenib, and 4 (9.3%) with pazopanib. All the enrolled patients received at least four cycles of pembrolizumab‐activated DC‐CIK cell treatment; the median cycle of cell treatment was 18 cycles (range, 4–33 cycles). At the cut‐off date (29 April 2020), 74.4% of patients had discontinued study treatment. Disease progression was the most common reason for discontinuation (Figure 1).
Figure 1.
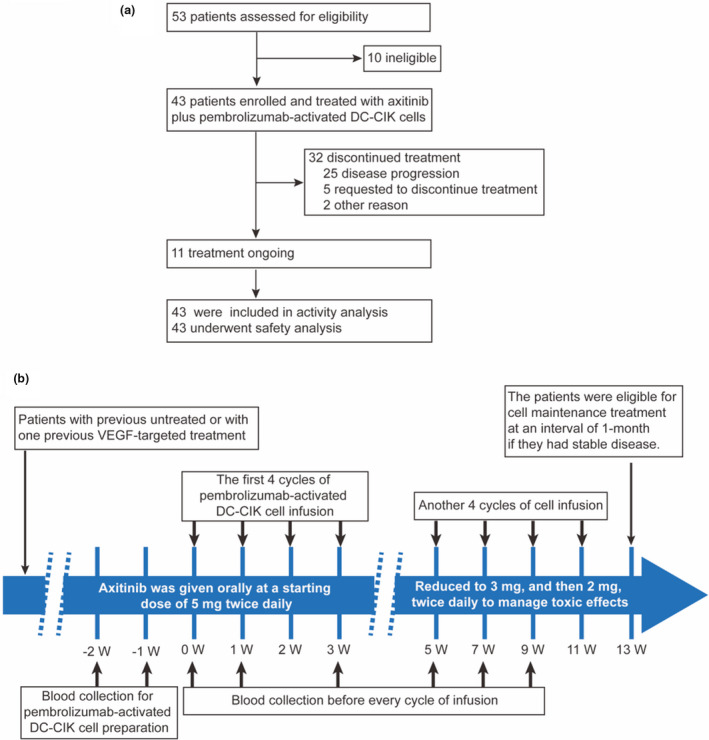
Clinical trial diagram and protocol. (a) Flow diagram illustrating the enrolment and assignment protocols and the analyses performed in the patients in this trial. (b) Overview of the treatment protocol. DC‐CIK, dendritic cells–co‐cultured with cytokine‐induced killer cells; VEGF, vascular endothelial growth factor; W, week.
Table 1.
Baseline demographic and clinical characteristics
| Variable | No. of patients | % |
|---|---|---|
| Sex | ||
| Male | 31 | 72.1 |
| Female | 12 | 27.9 |
| Age (years) | ||
| ≥ 60 | 14 | 32.6 |
| < 60 | 29 | 67.4 |
| Histology | ||
| ccRCC | 36 | 83.7 |
| pRCC | 7 | 16.3 |
| ECOG performance status | ||
| 0 | 17 | 39.5 |
| 1 | 22 | 51.2 |
| 2 | 4 | 9.3 |
| Common sites of metastases | ||
| Lung | 34 | 79.1 |
| Live | 10 | 23.3 |
| Bone | 21 | 48.8 |
| Lymph nodes | 30 | 69.8 |
| No. of evaluable disease sites | ||
| 1 | 16 | 37.2 |
| ≥ 2 | 27 | 62.8 |
| IMDC prognostic risk | ||
| Favorable | 6 | 14.0 |
| Intermediate | 24 | 55.8 |
| Poor | 13 | 30.2 |
| Previous nephrectomy | ||
| Yes | 38 | 88.4 |
| No | 5 | 11.6 |
| Previous systemic therapy | ||
| Treatment naive | 22 | 51.2 |
| Pazopanib | 4 | 9.3 |
| Sorafenib | 7 | 16.3 |
| Sunitinib | 10 | 23.2 |
ccRCC, clear cell renal cell carcinoma; ECOG, Eastern Cooperative Oncology Group; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; pRCC, papillary renal cell carcinoma.
IMDC risk groups were defined as favorable (0 factors), intermediate (1 or 2 factor) or poor (3–6 factors).
Characteristics of final pembrolizumab‐activated autologous DC‐CIK cells
Pembrolizumab‐activated autologous DC‐CIK cells were successfully generated in all cycles of treatment. The median count of DC‐CIK cells after 14 days of expansion reached 1.2 × 1010 (range, 0.8 × 1010 to 1.5 × 1010) for all cycles. The median percentage of CD3+, CD3+CD4+, CD3+CD8+, CD3−CD56+ and CD3+CD56+ population in the infused DC‐CIK cells was 98.3% (range, 88.6–99.2%), 28.5% (range, 5.3–80.4%), 71.2% (range, 21.9–85.5%), 1.5% (range, 0.4–11.2%), and 16.5% (range, 5.4–50.8%), respectively (Supplementary figure 1). Based on trypan blue staining, the cellular vitality of infused DC‐CIK cells was 95% or higher. The cells were without any bacterial, fungal, or mycoplasma contamination. Following quality testing and cultured with pembrolizumab for 30–40 min, all numbers of harvested autologous DC‐CIK cells (range, 0.8 × 1010 to 1.5 × 1010) per cycle were infused back to patients at one time.
Efficacy
At the date of the analysis (the cut‐off date), the median follow‐up for the 43 patients in this study was 23.6 months. Twenty‐six (60.5%) patients had disease progression, and 2 (4.7%) patients had died from disease progression. In the treatment‐naive and VEGFR‐targeting TKI‐pretreated subpopulations, 11 and 15 PFS events were recorded, respectively. Median PFS was 14.7 months (95% CI 11.16–18.30, Figure 2a) in the overall study population. The percentages of patients without disease progression at 6 and 12 months were 72.1% and 62.1%, respectively (Table 2). Median PFS was 18.2 and 14.4 months for the patients untreated versus previously treated with targeted therapy, respectively, and no statistically significant difference was found between the two groups (Figure 2a; Log‐rank P‐value = 0.07). PFS rate at 12 months for patients untreated versus previously treated with targeted therapy were 71.6% and 52.4%, respectively (Figure 2a). Median PFS in ccRCC and pRCC were 17.3 and 14.4 months, respectively (Supplementary figure 2; Log‐rank P‐value = 0.1069). The median overall survival (OS) was not reached in the overall study population. Final OS results will be reported in a subsequent paper.
Figure 2.

Clinical outcomes in 43 patients who were treated with axitinib plus pembrolizumab‐activated DC‐CIK cells. (a) Kaplan–Meier survival curve of progression‐free survival (PFS) in (left) overall study population, (middle) treatment‐naive population and (right) targeted agents‐pretreated population. (b) Waterfall plot of the best response in all eligible patients. Twenty‐seven patients (62.8%) had measurable tumor reduction.
Table 2.
Efficacy outcomes
| Parameter | |
|---|---|
| Objective response, n (%) | |
| Best overall response | |
| Complete response | 1 (2.3) |
| Partial response | 10 (23.2) |
| Stable disease | 21 (48.8) |
| Progressive disease | 11 (25.6) |
| Response rate (CR + PR) | 11 (25.6) |
| 95% CI | 13.5–41.2 |
| Treatment subgroup | |
| Treatment‐naive | 22 |
| Response rate (CR + PR), n (%) | 7 ( 31.8) |
| 95% CI | 13.9–54.9 |
| One previous treatment | 21 |
| Response rate (CR + PR), n (%) | 4 (19) |
| 95% CI | 5.6–41.9 |
| Histology subgroup | |
| ccRCC | 36 |
| Response rate (CR + PR), n (%) | 10 (27.8) |
| 95% CI | 14.2–45.2 |
| pRCC | 7 |
| Response rate (CR + PR), n (%) | 1 (14.3) |
| 95% CI | 0.4–57.9 |
| Progression‐free survival | |
| Median (95% CI) progression‐free survival (months) | 14.7 (11.16–18.30) |
| Progression‐free survival rate (95% CI) | |
| At 6 months | 72.1% (59.9–86.8) |
| At 12 months | 62.1% (49.1–78.7) |
ccRCC, clear cell renal cell carcinoma; pRCC, papillary renal cell carcinoma.
Treatment efficacy for the overall study population, as well as for the subgroups determined by previously treatment and histology classification, is detailed in Table 2. The observed objective response rate (ORR) in the entire population was 25.6% (95% CI, 13.5–41.2%, including 1 complete response (CR) and 10 partial responses ([PR], Figure 2b), with a median duration of response of 18.2 months. Twenty‐one of 43 patients (48.8%) had stable disease (SD). Patients in the treatment‐naive subpopulations had an ORR of 31.8% (95% CI, 13.9–54.9%), which was not significantly improved than that in the VEGFR‐targeting TKI‐pretreated subpopulation (19%). Similarly, there was no statistically significant difference in ORR between patients with ccRCC (ORR, 27.8%, 95% CI, 14.2–45.2%) and pRCC (ORR, 14.3%, 95% CI, 0.4–57.9%) (Table 2).
Safety
Treatment‐related toxicities for all 43 patients are listed in Table 3. Most patients only had mild (grade 1–2) adverse events similar to those previously reported with single‐agent axitinib. Diarrhoea (48.8%), palmar‐plantar erythrodysesthesia (37.2%), hypertension (30.2%), and proteinuria (25.6%) were the most common adverse events (AEs). The most common grade 3–4 AEs were hypertension (11.6%), palmar‐plantar erythrodysesthesia (7.0%), diarrhoea (4.7%), and proteinuria (4.7%). Immune‐related AEs as potential toxicities of interest were also recorded during the administration of autologous pembrolizumab‐activated DC‐CIK cells. In our study, only five (11.6%) patients developed grade 1 chill and fever, which were related to the infusion of pembrolizumab‐activated DC‐CIK cells and recovered spontaneously without any medical treatment. No immune‐related serious adverse events such as pneumonitis, colitis, hepatitis, nephritis, and myocarditis appeared in any of the patients. No patients died for AEs, and no treatment was discontinued in any patients because of treatment‐related AEs.
Table 3.
Treatment‐related toxicities for the entire cohort
| Adverse events | All grades (%) | ≥ Grade 3 (%) |
|---|---|---|
| Diarrhoea | 21 (48.8) | 2 (4.7) |
| Palmar‐plantar erythrodysesthesia | 16 (37.2) | 3 (7.0) |
| Hypertension | 13 (30.2) | 5(11.6) |
| Proteinuria | 11 (25.6) | 2 (4.7) |
| Nausea | 6 (14.0) | 0 |
| Decreased appetite | 6 (14.0) | 0 |
| Fatigue | 6 (14.0) | 0 |
| Increased aspartate aminotransferase | 6 (14.0) | 1 (2.3) |
| Increased alanine aminotransferase | 6 (14.0) | 1 (2.3) |
| Pruritus | 6 (14.0) | 0 |
| Chills | 5 (11.6) | 0 |
| Fever | 5 (11.6) | 0 |
| Hypothyroidism | 5 (11.6) | 0 |
| Mucosal inflammation | 4 (9.3) | 1 (2.3) |
| Rash | 4 (9.3) | 1 (2.3) |
| Anaemia | 3 (7.0) | 0 |
| Thrombocytopenia | 3 (7.0) | 0 |
| Vomiting | 3 (7.0) | 0 |
| Constipation | 2 (4.7) | 0 |
| Arthralgia | 2 (4.7) | 0 |
| Abdominal pain | 2 (4.7) | 0 |
Peripheral blood lymphocyte immunophenotype analysis
Next, we investigated whether the combination of axitinib and pembrolizumab‐activated autologous DC‐CIK cells contributed to the changes in peripheral blood lymphocyte immunophenotype. Compared with the baseline before treatment, the percentage of CD3+CD8+ cell subsets, the main population of cytotoxic lymphocyte (CTL) in antitumor immunity, and their central memory (CD8+CD62L+CD45RA–), effector memory (CD8+CD62L–CD45RA–) and proliferation (CD8+Ki67+) phenotypes, and expression of PD‐1 and co‐stimulation molecule CD40 were significantly increased after 2 and 4 cycles of treatment, while the frequency of naive CD8+ T cells (CD8+CD62L+CD45RA+) was decreased (Figure 3a). For the CD3+CD4+ subset, increased PD‐1 expression was observed, with no significant change in their other immunophenotypes (Figure 3a).
Figure 3.

Peripheral blood lymphocyte immunophenotype assessment via flow cytometry. (a) Peripheral blood lymphocyte immunophenotype before treatment and after 2 and 4 cycles of pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib treatment (n = 28). (b, c) Peripheral blood lymphocyte immunophenotype after 2 (b) and 4 (c) cycles of pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib treatment in patients with long‐term (n = 18) and minimal (n = 10) survival benefits. DC‐CIK, dendritic cells–co‐cultured with cytokine‐induced killer cells. ns, not significant. *P < 0.05. **P < 0.01. ***P < 0.001. Data are representative of three independent experiments.
Using 12 months of PFS as a cut‐off value, patients were divided into a long‐term survival benefit (PFS ≥ 12 months) and minimal survival benefit (PFS < 12 months) cohorts. Intriguingly, all lymphocyte immunophenotypes between the two cohorts showed no significant differences before treatment according to the baseline measurement (Supplementary figure 3). The activation profile of the adoptive cells between the two cohorts had no significant difference in good survivors versus poor survivors at baseline prior to transfer, with an increased trend in good survivors (Supplementary figure 4). However, the long‐term survival benefit cohort exhibited higher frequencies of peripheral CD3+CD8+ CTL cell subsets and their effector memory (CD8+CD62L−CD45RA−) and proliferation (CD8+Ki67+) phenotypes than the minimal survival benefit cohort after either 2 (Figure 3b) or 4 (Figure 3c) cycles of treatment. In the minimal survival benefit cohort, peripheral CD8+ cells were enriched for TIM3 expression and trended to express more PD‐1 after 2 and 4 cycles of treatment (Figure 3b and c).
Serum cytokine profile analysis
Serum samples were obtained from patients before and after 2 and 4 cycles of pembrolizumab‐activated DC‐CIK cell infusion in combination with axitinib treatment. High‐throughput antibody microarray analysis was performed to assess human inflammatory factors, growth factors, chemokines, and receptors. Compared to the baseline measurement before infusion, we found that the majority of these factors were increased after 2 and 4 cycles of treatment, particular at the timepoint of 4 cycles, according to the fold‐change (Figure 4a). The top 30 up‐regulated factors included 11 pro‐inflammatory/immunostimulatory cytokines or their receptors (IL‐12p70, IL‐28A, MICB, ICAM‐3, MICA, IFN‐γ, IL‐29, IL‐1RA, IL‐2, OPG, TNF‐α, and IL‐5), 3 anti‐inflammatory/immunosuppressive cytokines (IL‐13, IL‐4, and VEGF‐D), 3 chemokines (I‐309 (CCL1), I‐TAC (CXCL11) and MCP‐1 (CCL2)), and some regulatory growth factors such as BMP‐4, HB‐EGF, MCSF and LIF. Based on these treatment‐induced differential factors, Gene Ontology (GO) analysis (before treatment versus after 2 cycles of treatment versus after 4 cycles of treatment) demonstrated that they are mainly enriched in leucocyte migration, cellular response to interferon‐gamma, response to interleukin‐1, lymphocyte migration, monocyte chemotaxis, cellular response to chemokines, neutrophil chemotaxis, granulocyte chemotaxis, and so on. (Figure 4b).
Figure 4.

Serum cytokine profiles analysis. Serum levels of various cytokines were determined using antibody microarrays, and the fold‐change of each cytokine after pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib treatment versus before treatment was calculated. (a) Heatmap showed differential cytokines of after versus before treatment at the timepoints after 2 and 4 cycles of combination treatment according to the median fold‐change (n = 11). (b) GO analysis for the significant terms enriched by the differential cytokines of before versus after 2 cycles' versus 4 cycles' combination treatment (n = 11). (c) Heatmap showed differential cytokines in the patients with long‐term survival benefit (n = 8) versus minimal survival benefit (n = 3) at timepoints before and after 2 and 4 cycles of combination treatment according to the median fold‐change. (d, e) GO analysis for the significantly top terms enriched by differentially expressed serum cytokines in long‐term survival benefit patients (n = 8) in comparison with minimal survival benefit patients (n = 3) at timepoints of after 2 (d) and 4 (e) cycles of combination treatment. FC, fold‐change. Data are representative of three independent experiments.
Furthermore, we also evaluated the differences of all these serum factors between the two outcome cohorts of long‐term and minimal survival benefit. For the baseline measurement before treatment, slight differences were found between the two cohorts (Figure 4c) and these minor differences are unavailable for GO analysis (data not shown). However, some significant differences appeared after 2 cycles of treatment; more differences were observed after 4 cycles of treatment (Figure 4c). At the timepoint of after 2 cycles of treatment, the highly up‐regulated 30 factors in long‐term survival cohort were mainly pro‐inflammatory/immunostimulatory cytokines or their receptors (IL‐1α, IL‐18 BPa, IL‐2 Rg, TNF‐α, IL‐17F, IL‐1β, IL‐17R, IL‐1R4, LIGHT, IL‐1 RI, 4‐1BB, uPAR, LIMPII, TIM‐1 and IFN‐γ) and chemokines (MIP‐3a (CCL20), Eotaxin‐3(CCL26), MIG (CXCL9) and 6Ckine (CCL21)) compared with the minimal survival cohort, with a few up‐regulation of anti‐inflammatory/immunosuppressive cytokines (IL‐10RB, Dtk, IL‐8 and TGF‐β3) and regulatory growth factors (bFGF, PIGF, IGFBP‐6, SCF, G‐CSF and NRG1‐β1). Moreover, the measurement after 4 cycles of treatment showed more significant increase of multiple factors in the long‐term survival cohort. The majority of them were pro‐inflammatory/immunostimulatory cytokines or their receptors (IL‐2, IL‐12p70, TNF‐α, MICB, IL‐28A, IL‐1RA, IL‐15, IFN‐γ, ICAM‐3, IL‐1β, IL‐29, MICA, IL‐5, TNF‐β, IL‐2 Rg, IL‐1α, IL‐17F, and so on) and chemokines (I‐309 (CCL1), I‐TAC (CXCL11), MCP‐1 (CCL2), MIG (CXCL9), and so on), together with some anti‐inflammatory/immunosuppressive cytokines (IL‐13, VEGF‐D, IL‐4, VEGF, and so on) and regulatory growth factors (MCSF, G‐CSF, LIF, NGFR, NT‐3, GDNF, NT‐4, and so on. GO analysis for the differential factors between the two cohorts of long‐term and minimal survival benefit demonstrated similar results at the timepoints of after 2 (Figure 4d) and 4 (Figure 4e) cycles of treatment, and response to tumor necrosis factor, lymphocyte chemotaxis/migration, response to interleukin 1, neutrophil chemotaxis/migration, leucocyte chemotaxis/migration, granulocyte chemotaxis/migration, and monocyte/mononuclear cell chemotaxis/migration were commonly enriched. Positive regulation of response to external stimulus and chemokine‐mediated signalling pathway were additionally enriched by the differential factors at the timepoints of after 4 cycles of treatment (Figure 4e).
Discussion
During the past several years, multiple VEGFR‐targeted inhibitors have become available for the treatment of advanced RCC. 5 However,these antiangiogenic targeted agents are palliative treatments and rarely produce durable disease control. 38 Nowadays, the combination of targeted agents (axitinib) and anti‐PD1 antibody (pembrolizumab) have shown significantly longer OS and PFS in patients with mRCC as compared with a single targeted agent (sunitinib). 18 This might indicate that the combination of targeted agents and immunotherapy can be a novel therapeutic regimen for mRCC. Besides, both of our and other previous clinical studies demonstrated that adoptive cell immunotherapy produced clinical activity for patients with mRCC. 30 , 39 Therefore, in this phase II clinical trial, we sought to investigate the efficacy and safety of the combination of axitinib and a low dose of pembrolizumab‐activated autologous DC‐CIK cell immunotherapy for patients with mRCC who had not received any previous systemic anticancer therapy or who had only received one prior treatment with targeted agents.
As anticipated, the combination of axitinib and pembrolizumab‐activated autologous DC‐CIK cells was an active regimen. In our previous study, we found that a fraction of DC‐CIK cells were PD‐1 positive and that antitumor activity of DC‐CIK cells was significantly increased after pre‐activating with pembrolizumab at a dose of 1 μg per million cells. 30 This dose (8–15 mg per infusion) of pembrolizumab is far less than what was used in clinical setting, and has little clinical activity. 40 , 41 Thus, we consider the clinical activity of pembrolizumab‐activated DC‐CIK cells were mainly attributed to in vitro pre‐activated DC‐CIK cells, but not this low dose of pembrolizumab. In the overall population of 43 patients treated, 25.6% patients had ORR (CR, 2.3%, PR, 23.5%), and 62.8% experienced a measurable reduction in tumor size. The median PFS was 14.7 months (95% CI, 11.16–18.30 months), and 62.1% of patients were progression‐free at 12 months. This met the predefined threshold for a successful result. The efficacy of this regimen in untreated patients, or those previously treated with one targeted inhibitor, was similar. In the 22 previously untreated patients, the ORR was 31.8%, with an 18.2‐month median PFS. In patients previously treated with targeted therapy, the ORR was 19%, with a 14.4‐month median PFS. Similar result was also observed between patients with pRCC and ccRCC. Although the response rate of this combination therapy was not higher than that of single axitinib, the median PFS was longer than that of single axitinib for either first‐ or second‐line therapy, 8 , 9 which is a very suggestive of immunotherapeutic response with lasting survival benefit. Moreover, the median PFS for the patients in the second‐line therapy in our study was still similar to that observed in previously published data on the combination of axitinib and pembrolizumab which was administered in the first‐line therapeutic setting. 18 Altogether, these results collectively indicate that the combination of axitinib and pembrolizumab‐activated DC‐CIK cells may be an alternative therapeutic option for patients with mRCC.
Either axitinib or pembrolizumab‐activated DC‐CIK cells might reprogram host immune status towards a more immune supportive environment in cancer patients. 19 , 20 , 31 Axitinib was also contradictorily reported to decrease in T‐cell proliferation 42 and impair DC activation, differentiation, and function. 43 In the current study, combination of axitinib and pembrolizumab‐activated DC‐CIK cells generally enhanced the number of peripheral CD3+CD8+ CTL cells, promoted their proliferation and differentiation from naive to effector memory and central memory phenotypes, and up‐regulated their expression of CD40, a receptor providing strong co‐stimulatory signal for augmenting T‐cell responses. 44 Meanwhile, elevated frequencies of CD4+PD‐1+ and CD8+PD‐1+ T cells together with other changed immunophenotypes indicated that the combination therapy might more or less activate peripheral T cells. In‐depth work will be needed to clarify how CD8+ T cells accomplish such kind of changes. Interestingly, although there were no significant differences between the two outcome cohorts at the baseline before treatment, higher frequencies of peripheral CD3+CD8+ cells and their effector memory and proliferation phenotypes were observed in patients with long‐term survival benefits after combination treatment, suggesting that the peripheral T cells in this cohort might be phenotypically and functionally activated and thus contributed to therapeutic benefit. However, stronger expression of TIM3, a ‘brake’ molecule up‐regulated on exhausted T cells, 45 , 46 was observed on peripheral CD8+ T cells in minimal survival cohort than that in long‐term survival cohort. It might be a potential resistance mechanism by synergistically inducing CD8+ T‐cell exhaustion with PD‐1 that also trended to be higher expressed in minimal survival benefit cohort. Dual targeting of PD‐1 and TIM3 could be applied with DC‐CIK infusion in the future. As a result, we inferred that the combination treatment appropriately activated the peripheral T cells in long‐term survival cohort, and such activation did not reach the threshold of over‐activation or exhaustion, therefore resulting in an encouraging outcome. But those T cells in minimal survival cohort probably were over‐activated and became exhausted, therefore leading to an unsatisfactory outcome. Our work will be dedicated towards addressing these issues and finding a way how most patients can benefit from this combination therapy.
Adoptive cell transfer provided the active in vivo development of sufficient numbers of antitumor lymphocytes, and trafficking and subsequent infiltration to the tumor microenvironment were also crucial for the persistence of durable antitumor responses. 47 Serum cytokine profile analysis in the present study demonstrated that at the timepoint of 2 or 4 cycles of treatment, about half of significantly up‐regulated factors caused by this combination therapy were pro‐inflammatory/immunostimulatory cytokines and chemokines (such as immunostimulatory IL‐12p70, IL‐28A, IL‐29, and so on, classical cytotoxic IFN‐γ, IL‐2, and TNF‐α, NKG2D ligands MICA and MICB, and chemotactic CCL1, CXCL11, and CCL2), together with some anti‐inflammatory/immunosuppressive cytokines, such as IL‐13 and IL‐4, and immunomodulatory growth factors, such as BMP‐4, HB‐EGF, and MCSF. These differential cytokines were tightly associated with leucocyte (such as lymphocyte, monocyte, neutrophil, and granulocyte) chemotaxis and response to the cytotoxic interferon‐gamma and pro‐inflammatory interleukin‐1, indicating a potentially general immune activation at serum level after receiving the combination treatment. More importantly, we also identified highly significant differences in serum cytokine profile between the two outcome cohorts of long‐term and minimal survival benefit at the timepoints after 2 and 4 cycles' treatment, but not the baseline before treatment. The up‐regulation in long‐term survival cohort at the two timepoints particularly after 4 cycles of treatment, was mainly pro‐inflammatory/immunostimulatory cytokines, chemokines, and their receptors (such as the most up‐regulated cytotoxic IL‐2, TNF‐α, and IFN‐γ, immunostimulatory IL‐12p70, IL‐1RA, IL‐15, ICAM‐3, IL‐1β, IL‐29, IL‐5, and so on, NKG2D ligands MICA and MICB, and chemotactic CCL1, CXCL11, CCL2, and CXCL9), and a few anti‐inflammatory/immunosuppressive cytokines (such as IL‐13, IL‐4, VEGF, and so on) as well as some immunomodulatory growth factors (MCSF, G‐CSF, LIF, and so on). They were extensively implicated with response to external stimulus, pro‐inflammatory tumor necrosis factor and interleukin 1, immune cell chemotaxis, and so on. These underlying changes in immunobiology trended to be a signature of increased or stimulatory host immunity and might be potentially linked to treatment benefit of the long‐term survival cohort in this study.
The observed safety profiles of axitinib plus pembrolizumab‐activated autologous DC‐CIK cells were as expected based on the known toxicity profiles of these two agents. In general, combination therapy was well tolerated by most patients. The most frequently observed drug‐related AEs were diarrhoea, palmar‐plantar erythrodysesthesia, hypertension, and proteinuria. The incidence of grade ≥ 3 AEs was generally less than 5%, with the exceptions of grade 3 hypertension and grade 3 palmar‐plantar erythrodysesthesia, which were observed in 11.6% and 7.0% of patients, respectively. Other than these targeted therapy‐related AEs, chills and fever were observed in this trial, which was related to the infusion of the pembrolizumab‐activated DC‐CIK cells. The overall incidence of grade 3 or higher‐grade AEs was lower in our study (27.9%) than that in axitinib plus pembrolizumab (62.9%) treatment. 18 In addition, in patients treated with axitinib plus pembrolizumab, AEs of any cause led to the discontinuation of drugs in 41.2% of patients. 18 In contrast, no patient discontinued treatment because of AEs in our study. This low frequency of higher‐grade AEs and immune‐related serious AEs in our treatment may result from the administration of autologous DC‐CIK cells and the low dose of pembrolizumab we used. These findings demonstrate that the combination of axitinib and pembrolizumab‐activated DC‐CIK cells was safe and might be a potential treatment option for patients with advanced RCC.
Our study had several limitations. First, this was a single‐arm study, and no control group was designed to compare the efficacy of the combination therapy. Thus, the results should interpret carefully. Second, longer‐term follow‐up evaluations should be used to assess overall survival and response durability in remission. Third, the specific cellular and molecular mechanisms that mediate the effects of combination therapy with axitinib plus pembrolizumab‐activated DC‐CIK cells are not comprehensively understood, and more clinical validation is required. As we could not obtain the tumor biopsies of these patients, an additional limitation is a lack tumor biopsy material to further corroborate local infiltration events and immune cell composition associated with treatment benefit and with the identified biomarkers. However, we will continue to follow our patients to assess efficacy and safety.
Conclusions
This study demonstrated that the combination of axitinib plus pembrolizumab‐activated autologous DC‐CIK cells contributed to encouraging clinical outcomes in patients with advanced RCC who were treatment‐naive or targeted agents‐pretreated. Although overall survival data are not mature at this time, survival analyses will help to elucidate the utility of this combination therapy as a first‐ or second‐line therapy for patient with advanced RCC. This combination therapy was well tolerated and manageable. Additionally, such combination therapy led to superior antitumor immunity, including increased lymphocyte infiltration and cellular responses of CD8+ T cells, especially in patients who acquired long‐term survival benefit. An increased percentage of peripheral CD8+TIM3+ T cells and a lower serum‐level immunostimulatory cytokine profile were identified as a potential resistance mechanism to this combination therapy in patients with minimal survival benefit. A prospective, randomised trial will be carried out to validate our study findings.
Methods
Study design and participants
This study was a single‐arm, phase 2 study conducted at Sun Yat‐sen University Cancer Center, Guangzhou, China. Subjects were patients with irresectable or metastatic RCC. Eligible patients were 18 years of age or older with histologically or cytologically confirmed metastatic RCC and at least one measurable disease [according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1)]. Patients included those who had not have received any previous systemic anticancer therapy, or those who had received one treatment with sorafenib, pazopanib, sunitinib, or other VEGFR‐targeting tyrosine kinase inhibitor (other than axitinib) for advanced disease and had radiographic progression. Other eligibility criteria included an Eastern Cooperative Oncology Group performance status of 0–2; a life expectancy of greater than 12 weeks; adequate renal, hepatic and haematological organ function; and no HIV infection or chronic active hepatitis. We excluded patients with malignancies other than RCC; brain metastasis; concomitant system corticosteroid therapy; uncontrolled hypertension; myocardial infarction, uncontrolled angina, congestive heart failure, or those who had sustained a cerebrovascular accident within the previous 12 months. We also excluded patients previously treated with an immune checkpoint inhibitor.
The trial was approved by the institutional review board or ethics committee of Sun Yat‐sen University Cancer Center (Approval No. B2017‐027‐01) and was registered at www.clinicaltrials.gov. (NCT 02886897). All methods and procedures associated with this study complied with Good Clinical Practice guidelines, the Declaration of Helsinki, and local laws. All patients provided written informed consent before any trial procedure or receiving study treatment.
Treatments
The enrolled patients received axitinib and pembrolizumab‐activated autologous DC‐CIK cell immunotherapy until disease progression, development of intolerable toxicity, or physician or patient decision to discontinue. Axitinib was given orally at a starting dose of 5 mg twice daily. The dose could be increased to 7 mg, then 10 mg, twice daily if there were no adverse reactions above grade 2 of Common Terminology Criteria for Adverse Events. The axitinib dose could be reduced to 3 mg, then 2 mg, twice daily to manage toxic side‐effects. The pembrolizumab‐activated autologous DC‐CIK cells were infused intravenously at the Biotherapy Center of Sun Yat‐sen University Cancer Center. In general, patients received at least 4 cycles of cell infusion with a 1‐week interval between each cycle. Then, another 4 cycles of treatment would be given at 2‐week intervals. Patients who achieved good disease control were eligible for cell maintenance treatment once every month. Patients received a median of 1.2 × 1010 (range, 0.8 × 1010 to 1.5 × 1010) pembrolizumab‐activated autologous DC‐CIK cells per cycle. No patient who received pembrolizumab‐activated DC‐CIK cell immunotherapy required dose modification. If one drug was discontinued because of toxic effects, the other treatment could be continued. The design and procedures used in the clinical trial are shown in Figure 1.
Efficacy and safety assessments
Patients were evaluated for tumor response and progression according to RECIST v1.1 48 using computed tomography or magnetic resonance imaging every 8 weeks during the first 6 months, and every 12 weeks after that. The baseline tumor burden was assessed within 28 days before the first treatment. Routine safety evaluations primarily consisted of clinical and laboratory abnormalities. AE severity was graded by the investigator using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
Generation of pembrolizumab‐activated autologous DC‐CIK cells
There are two steps necessary to generate pembrolizumab‐activated autologous DC‐CIK cells, as described in our previous studies. 30 Firstly, autologous DC‐CIK cells were generated according to our previous report. 49 Briefly, peripheral blood mononuclear cells were separated using Ficoll‐Hypaque density centrifugation, and cultured in X‐VIVO 15 medium (Lonza, Visp, Switzerland) for 1 h. The suspended cells were then collected to induce CIK cells using 1000 U mL−1 recombinant human IFN‐γ (ShangClone, Shanghai, China) for the first 24 h followed by stimulation with 100 ng mL−1 mouse anti‐human CD3 monoclonal antibody (R&D Systems, MN, USA), 1000 U mL−1 IL‐2 (Beijing Sihuan Pharm, Beijing, China), and 100 U mL−1 IL‐1α (Life Technologies, CA, USA). The adherent cells were cultured using DC medium (X‐VIVO 15 medium, supplemented with 1000 U mL−1 GM‐CSF and 30 ng mL−1 IL‐4) to induce DCs. After matured with 10 ng mL−1 of TNF‐α on the sixth day, DCs were mixed with the CIK cells at a ratio of 1:20 and cultured in fresh medium containing 1000 U mL−1 IL‐2 for another 7 days to induce DC‐CIK cells. Secondly, on day 14, the DC‐CIK cells were harvested and analysed for number, viability, and phenotype. Before the DC‐CIK cells were transferred to patients, they were incubated with pembrolizumab (1 μg 10−6 cell) for 30–40 min at 37°C to induce pembrolizumab‐activated DC‐CIK cells. All products were free of bacterial, mycoplasma, or fungal contamination. The endotoxin was less than 5 EU.
Flow cytometry and protein microarray analysis
Additional informed consent was obtained from patients to collect peripheral blood samples for flow cytometry and protein microarray analysis. For flow cytometry analysis, blood samples were collected from 28 patients before treatment and after 2 and 4 cycles of pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib. Red Blood Cell Lysis Buffer (Sigma, Santa Clara, CA) was used to remove red blood cells from fresh peripheral blood. Cells were washed twice using PBS and stained with anti‐human CD3, CD4, CD8, CD56, CD62L, CD45RA, PD‐1, TIM3, LAG3, CD40, OX40, CD25 or CD127 antibodies (BD, San Diego, CA, USA). According to the manufacturer's instructions, the anti‐human Ki67 antibody (BD) was used for intracellular staining with a Fixation/Permeabilization Solution Kit (BD). Next, cells were incubated in the dark for 15 min at 4°C, washed with PBS, and measured by flow cytometry.
For protein microarray analysis, serum samples were obtained from 11 patients before treatment and after 2 and 4 cycles of pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib. The total protein concentration of the serum samples was determined using a bicinchoninic acid assay kit (Biyuntian, Jiangsu, China). According to the manufacturer's instructions, the concentrations of multiple proteins, including human inflammatory factors, growth factors, and chemokines in the serum were measured using antibody microarrays (RayBiotech, GA, USA). The detailed protocol used for the antibody microarrays has been described elsewhere. 50 Then, we calculated the fold‐change of each cytokine after pembrolizumab‐activated DC‐CIK cell infusion combination with axitinib treatment, compared to before treatment.
Statistical analyses
The primary endpoint was PFS, defined as the time from initiation of treatment to either first documentation disease progression or death due to any cause. Secondary endpoints were OS and ORR. Previous experience with single‐agent axitinib as a first‐ or second‐line treatment produced PFS rates at 12 months of 40% and 25%, respectively. 8 , 9 The sample size calculation assumed a 20% improvement in PFS at 12 months with a one‐sided α‐risk of 5% and a β‐risk of 20%. PFS rates at 12 months were estimated to be 60 % under a null hypothesis that PFS rates at 12 months ≤ 40% would have no interest. Under these assumptions, 35 patients were required to detect this significant difference.
Distributions of survival time and rate were estimated using the Kaplan–Meier method; median PFS and 1‐year PFS rates along with 95% confidence intervals (CI) were reported. Descriptive statistics were used to summarise the patient baseline characteristics, treatment‐related AEs, overall responses and pembrolizumab‐activated DC‐CIK cell phenotypes. The differences of peripheral blood lymphocyte immunophenotype before and after pembrolizumab‐activated DC‐CIK cell infusion combined with axitinib treatment were analysed by the Student's t‐test as a post hoc analysis. Based on the 12 months of PFS, we divided patients into subgroups consisting of those who demonstrated a long‐term survival benefit (PFS ≥ 12 months) and those with a minimal survival benefit (PFS < 12 months). The association between survival benefit and immunophenotyping, or the cytokine level, was compared using the Student's t‐test. In all cases, a difference of P‐value less than 0.05 using two‐sided tests indicates statistical significance. All calculations were conducted using SPSS version 23 or GraphPad Prism 5 software.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Jian‐Chuan Xia: Conceptualization; Funding acquisition; Resources; Supervision. Meng‐Jia Song: Data curation; Formal analysis; Investigation; Project administration; Software; Visualization; Writing‐original draft. Qiu‐Zhong Pan: Conceptualization; Data curation; Investigation; Resources; Validation; Visualization; Writing‐review & editing. Ya Ding: Formal analysis; Methodology; Project administration; Resources; Software. Jianxiong Zeng: Data curation; Investigation; Project administration; Validation. Pei Dong: Data curation; Investigation; Resources. Jing‐Jing Zhao: Methodology; Resources; Validation. Yan Tang: Conceptualization; Methodology; Project administration; Software. Jingjing Li: Data curation; Formal analysis; Investigation. Zhiling Zhang: Data curation; Resources; Software; Validation. Junyi He: Investigation; Methodology; Project administration. Jieying Yang: Formal analysis; Software; Validation; Visualization. Yue Huang: Formal analysis; Software. Ruiqing Peng: Formal analysis; Methodology. Qi‐Jing Wang: Data curation; Project administration; Resources. Jia‐Mei Gu: Resources; Validation. Jia He: Formal analysis; Software. Yong‐Qiang Li: Data curation; Visualization. Shi‐Ping Chen: Methodology; Project administration. Rongxing Huang: Investigation; Methodology. Zi‐Qi Zhou: Formal analysis; Project administration. Chaopin Yang: Software; Validation. Yulong Han: Data curation; Visualization. Hao Chen: Formal analysis; Investigation. Heping Liu: Data curation; Methodology. Shangzhou Xia: Resources; Validation. Yang Wan: Data curation; Formal analysis. De‐Sheng Weng: Investigation; Methodology. Liming Xia: Resources; Software; Supervision. Fang‐Jian Zhou: Data curation; Supervision.
Supporting information
Acknowledgments
This study was funded by the National Key R&D Program of China for J‐CX (grant number 2018YFC1313400), the National Natural Science Foundation of China for J‐CX (grant number 81773110) and Q‐ZP (grant number 81803079), Science and Technology Planning Project of Guangdong Province, China for J‐CX (grant number 2017B020227003), Science and Technology Planning Project of Guangzhou, Guangdong Province, China, for J‐CX (grant number 201704020215), the Guangdong Natural Science Foundation, Guangdong Province, China, for Q‐ZP (grant number 2018A030310237), and District High Tech R&D Funds of Guangzhou, Guangdong Province, China, for J‐CX (grant number 2017‐L179). We are grateful to our patients who participated and contributed to this study for the benefit of all patients with advanced RCC. We also thank the investigators, nurses and research staff for their dedication to this project.
Trial registration: trial register, Jian‐Chuan Xia; ClinicalTrials.gov identifier: NCT02886897.
Contributor Information
Liming Xia, Email: 527109307@qq.com.
Fang‐Jian Zhou, Email: zhoufj@sysucc.org.cn.
Jian‐Chuan Xia, Email: xiajch@mail.sysu.edu.cn.
References
- 1. Ferlay J, Colombet M, Soerjomataram I et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019; 144: 1941–1953. [DOI] [PubMed] [Google Scholar]
- 2. Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008; 34: 193–205. [DOI] [PubMed] [Google Scholar]
- 3. Rini BI. Metastatic renal cell carcinoma: many treatment options, one patient. J Clin Oncol 2009; 27: 3225–3234. [DOI] [PubMed] [Google Scholar]
- 4. McDermott DF, Regan MM, Clark JI et al. Randomized phase III trial of high‐dose interleukin‐2 versus subcutaneous interleukin‐2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 2005; 23: 133–141. [DOI] [PubMed] [Google Scholar]
- 5. Bedke J, Gauler T, Grunwald V et al. Systemic therapy in metastatic renal cell carcinoma. World J Urol 2017; 35: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsimafeyeu I, Zolotareva T, Varlamov S et al. Five‐year survival of patients with metastatic renal cell carcinoma in the Russian Federation: results from the RENSUR5 registry. Clin Genitourin Cancer 2017; 15: e1069–e1072. [DOI] [PubMed] [Google Scholar]
- 7. Kelly RJ, Rixe O. Axitinib (AG‐013736). Recent Results Cancer Res 2010; 184: 33–44. [DOI] [PubMed] [Google Scholar]
- 8. Rini BI, Escudier B, Tomczak P et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011; 378: 1931–1939. [DOI] [PubMed] [Google Scholar]
- 9. Hutson TE, Lesovoy V, Al‐Shukri S et al. Axitinib versus sorafenib as first‐line therapy in patients with metastatic renal‐cell carcinoma: a randomised open‐label phase 3 trial. Lancet Oncol 2013; 14: 1287–1294. [DOI] [PubMed] [Google Scholar]
- 10. Motzer RJ, Michaelson MD, Redman BG et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet‐derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 2006; 24: 16–24. [DOI] [PubMed] [Google Scholar]
- 11. Meng M. Tyrosine kinase inhibitors in renal cell carcinoma. Clin Cancer Res 2004; 10 (18 Pt 2): 6371S‐6376S. Urol Oncol 2017; 35: 313–314. 10.1016/j.urolonc.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 12. Atkins MB, Hidalgo M, Stadler WM et al. Randomized phase II study of multiple dose levels of CCI‐779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 2004; 22: 909–918. [DOI] [PubMed] [Google Scholar]
- 13. Kalkman HO, Neumann V, Brauner V. Supersensitivity of atherosclerotic rabbit aorta to ergometrine is mediated by 5‐HT2 receptors. J Pharm Pharmacol 1989; 41: 876–878. [DOI] [PubMed] [Google Scholar]
- 14. Hamanishi J, Mandai M, Iwasaki M et al. Programmed cell death 1 ligand 1 and tumor‐infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA 2007; 104: 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamid O, Carvajal RD. Anti‐programmed death‐1 and anti‐programmed death‐ligand 1 antibodies in cancer therapy. Expert Opin Biol Ther 2013; 13: 847–861. [DOI] [PubMed] [Google Scholar]
- 16. Kwok G, Yau TC, Chiu JW, Tse E, Kwong YL. Pembrolizumab (Keytruda). Hum Vaccin Immunother 2016; 12: 2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamid O, Robert C, Daud A et al. Five‐year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE‐001. Ann Oncol 2019; 30: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rini BI, Plimack ER, Stus V et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019; 380: 1116–1127. [DOI] [PubMed] [Google Scholar]
- 19. Du Four S, Maenhout SK, De Pierre K et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015; 4: e998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laubli H, Muller P, D'Amico L, Buchi M, Kashyap AS, Zippelius A. The multi‐receptor inhibitor axitinib reverses tumor‐induced immunosuppression and potentiates treatment with immune‐modulatory antibodies in preclinical murine models. Cancer Immunol Immunother 2018; 67: 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brouwenstijn N, Gaugler B, Kruse KM et al. Renal‐cell carcinoma‐specific lysis by cytotoxic T‐lymphocyte clones isolated from peripheral blood lymphocytes and tumor‐infiltrating lymphocytes. Int J Cancer 1996; 68: 177–182. [DOI] [PubMed] [Google Scholar]
- 22. Lamers CH, Sleijfer S, van Steenbergen S et al. Treatment of metastatic renal cell carcinoma with CAIX CAR‐engineered T cells: clinical evaluation and management of on‐target toxicity. Mol Ther 2013; 21: 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jakel CE, Hauser S, Rogenhofer S, Muller SC, Brossart P, Schmidt‐Wolf IG. Clinical studies applying cytokine‐induced killer cells for the treatment of renal cell carcinoma. Clin Dev Immunol 2012; 2012: 473245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou J, Weng D, Zhou F et al. Patient‐derived renal cell carcinoma cells fused with allogeneic dendritic cells elicit anti‐tumor activity: in vitro results and clinical responses. Cancer Immunol Immunother 2009; 58: 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao X, Zhang Z, Li H et al. Cytokine induced killer cell‐based immunotherapies in patients with different stages of renal cell carcinoma. Cancer Lett 2015; 362: 192–198. [DOI] [PubMed] [Google Scholar]
- 26. Schmidt‐Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine‐induced killer cells with potent antitumor cell activity. J Exp Med 1991; 174: 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Schmidt‐Wolf IGH. Ten‐year update of the international registry on cytokine‐induced killer cells in cancer immunotherapy. J Cell Physiol 2020; 235: 9291–9303. [DOI] [PubMed] [Google Scholar]
- 28. Mata‐Molanes JJ, Sureda Gonzalez M, Valenzuela Jimenez B, Martinez Navarro EM, Brugarolas MA. Cancer immunotherapy with cytokine‐induced killer cells. Target Oncol 2017; 12: 289–299. [DOI] [PubMed] [Google Scholar]
- 29. Schmidt‐Wolf IG, Lefterova P, Mehta BA et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine‐induced killer cells. Exp Hematol 1993; 21: 1673–1679. [PubMed] [Google Scholar]
- 30. Chen CL, Pan QZ, Weng DS et al. Safety and activity of PD‐1 blockade‐activated DC‐CIK cells in patients with advanced solid tumors. Oncoimmunology 2018; 7: e1417721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poh SL, Linn YC. Immune checkpoint inhibitors enhance cytotoxicity of cytokine‐induced killer cells against human myeloid leukaemic blasts. Cancer Immunol Immunother 2016; 65: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai C, Lin F, Geng R et al. Implication of combined PD‐L1/PD‐1 blockade with cytokine‐induced killer cells as a synergistic immunotherapy for gastrointestinal cancer. Oncotarget 2016; 7: 10332–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dodagatta‐Marri E, Meyer DS, Reeves MQ et al. α‐PD‐1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α‐TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer 2019; 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Antonios JP, Soto H, Everson RG et al. Immunosuppressive tumor‐infiltrating myeloid cells mediate adaptive immune resistance via a PD‐1/PD‐L1 mechanism in glioblastoma. Neuro Oncol 2017; 19: 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arlauckas SP, Garris CS, Kohler RH et al. In vivo imaging reveals a tumor‐associated macrophage‐mediated resistance pathway in anti‐PD‐1 therapy. Sci Transl Med 2017; 9: eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang W, Song Z, Xiao J et al. Blocking the PD‐1/PD‐L1 axis in dendritic cell‐stimulated cytokine‐induced killer cells with pembrolizumab enhances their therapeutic effects against hepatocellular carcinoma. J Cancer 2019; 10: 2578–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nie W, Wei W, Zuo L et al. Magnetic nanoclusters armed with responsive PD‐1 antibody synergistically improved adoptive T‐cell therapy for solid tumors. ACS Nano 2019; 13: 1469–1478. [DOI] [PubMed] [Google Scholar]
- 38. Albiges L, Choueiri T, Escudier B et al. A systematic review of sequencing and combinations of systemic therapy in metastatic renal cancer. Eur Urol 2015; 67: 100–110. [DOI] [PubMed] [Google Scholar]
- 39. Liu L, Zhang W, Qi X et al. Randomized study of autologous cytokine‐induced killer cell immunotherapy in metastatic renal carcinoma. Clin Cancer Res 2012; 18: 1751–1759. [DOI] [PubMed] [Google Scholar]
- 40. Patnaik A, Kang SP, Rasco D et al. Phase I study of pembrolizumab (MK‐3475; anti‐PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015; 21: 4286–4293. [DOI] [PubMed] [Google Scholar]
- 41. Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stehle F, Schulz K, Fahldieck C et al. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J Biol Chem 2013; 288: 16334–16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heine A, Held SA, Daecke SN et al. The VEGF‐receptor inhibitor axitinib impairs dendritic cell phenotype and function. PLoS One 2015; 10: e0128897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Croft M. The role of TNF superfamily members in T‐cell function and diseases. Nat Rev Immunol 2009; 9: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J, Zhang S, Hu Y et al. Targeting PD‐1 and Tim‐3 pathways to reverse CD8 T‐cell exhaustion and enhance ex vivo T‐cell responses to autologous dendritic/tumor vaccines. J Immunother 2016; 39: 171–180. [DOI] [PubMed] [Google Scholar]
- 46. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med 2010; 207: 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348: 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–247. [DOI] [PubMed] [Google Scholar]
- 49. Wang QJ, Wang H, Pan K et al. Comparative study on anti‐tumor immune response of autologous cytokine‐induced killer (CIK) cells, dendritic cells‐CIK (DC‐CIK), and semi‐allogeneic DC‐CIK. Chinese J Cancer 2010; 29: 641–648. [DOI] [PubMed] [Google Scholar]
- 50. Xu MD, Liu L, Wu MY et al. The combination of cantharidin and antiangiogenic therapeutics presents additive antitumor effects against pancreatic cancer. Oncogenesis 2018; 7: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials