

Inhaled bacteriophage (phage) therapy is a potential alternative to conventional antibiotic therapy to combat multidrug-resistant (MDR) Pseudomonas aeruginosa infections. However, pharmacokinetics (PK) and pharmacodynamics (PD) of phages are fundamentally different from antibiotics and the lack of understanding potentially limits optimal dosing. The aim of this study was to investigate the in vivo PK and PD profiles of antipseudomonal phage PEV31 delivered by pulmonary route in immune-suppressed mice.
KEYWORDS: bacteriophages, phage, Pseudomonas, pulmonary delivery, pulmonary infection, pharmacokinetics, pharmacodynamics, multidrug-resistant infection, MDR infection, Pseudomonas aeruginosa
ABSTRACT
Inhaled bacteriophage (phage) therapy is a potential alternative to conventional antibiotic therapy to combat multidrug-resistant (MDR) Pseudomonas aeruginosa infections. However, pharmacokinetics (PK) and pharmacodynamics (PD) of phages are fundamentally different from antibiotics and the lack of understanding potentially limits optimal dosing. The aim of this study was to investigate the in vivo PK and PD profiles of antipseudomonal phage PEV31 delivered by pulmonary route in immune-suppressed mice. BALB/c mice were administered phage PEV31 at doses of 107 and 109 PFU by the intratracheal route. Mice (n = 4) were sacrificed at 0, 1, 2, 4, 8, and 24 h posttreatment and various tissues (lungs, kidney, spleen, and liver), bronchoalveolar lavage fluid, and blood were collected for phage quantification. In a separate study combining phage with bacteria, mice (n = 4) were treated with PEV31 (109 PFU) or phosphate-buffered saline (PBS) at 2 h postinoculation with MDR P. aeruginosa. Infective PEV31 and bacteria were enumerated from the lungs. In the phage-only study, the PEV31 titer gradually decreased in the lungs over 24 h, with a half-life of approximately 8 h for both doses. In the presence of bacteria, in contrast, the PEV31 titer increased by almost 2-log10 in the lungs at 16 h. Furthermore, bacterial growth was suppressed in the PEV31-treated group, while the PBS-treated group showed exponential growth. Of the 10 colonies tested, four phage-resistant isolates were observed from the lung homogenates sampled at 24 h after phage treatment. These colonies had a different antibiogram to the parent bacteria. This study provides evidence that pulmonary delivery of phage PEV31 in mice can reduce the MDR bacterial burden.
INTRODUCTION
Since the discovery of penicillin, much effort has been targeted toward understanding the pharmacokinetics (PK) and pharmacodynamics (PD) of antibiotics to guide safe and effective treatment regimens. While bacteria can be intrinsically resistant to antibiotics, the use of antibiotics has subjected bacteria to high selective pressure, leading to the advent of resistant strains at an alarming rate that now poses a serious global threat to human health. The severe threat of antimicrobial resistance remains imminent (1) and the World Health Organization has called for global action to tackle this crisis (2). Unfortunately, the antibiotic discovery pipeline is drying, with a lack of novel antimicrobial agents against Gram-negative bacteria (3). In particular, the emergence of multidrug-resistant (MDR) Pseudomonas aeruginosa strains presents a major public health risk due to their prevalent intrinsic and acquired resistance to most antibiotics (4). MDR P. aeruginosa causes complications of respiratory infections associated with high morbidity and mortality rates in many diseases, including bronchiectasis, cystic fibrosis, chronic obstructive pulmonary disease, and pneumonia (4).
Bacteriophages (phages) are naturally occurring bactericidal viruses that infect targeted host bacteria. They have been recently rediscovered and reintroduced as potential antimicrobial treatment and are considered an attractive solution to the increasing failure of antibiotics (5). Phage therapy predominantly relies on the lytic life cycle of phages. Virulent (lytic) phages recognize and attach to surface receptors of host bacterium, inject their genetic material, and then utilize the metabolic machineries of the host for self-replication (5). Up to hundreds of progenies can be produced and then released into the surroundings during bacteriolysis. Phage therapy has distinct advantages over conventional antibiotic treatment in that phages are (i) a naturally occurring antibacterial, (ii) self-replicating, (iii) self-limiting upon resolution of infection, (iv) effective against both MDR or antibiotic-sensitive bacteria, (v) highly specific with low inherent toxicity, (vi) able to coevolve with bacteria, and (vii) able to penetrate biofilms (5). The potential use of phages as antibacterial agents has been demonstrated in in vitro (6, 7) and preclinical studies (8–11), and in compassionate single-case studies (12–14).
Despite these advantages and potential, development and application of phage therapy has been relatively slow. A possible reason is that the current understanding and paradigm associated with antibiotic treatment cannot be transferred directly to phages (15). The PK and PD of phages are fundamentally different from those of conventional antibiotics. While many antibiotics are small molecules, phages are nano-sized viruses composed of proteins, nucleic acids (DNA or RNA), and sometimes lipids. In addition, phages have a unique dynamic with their bacterial host as self-replicating biopharmaceuticals (15). The PK/PD of phages are determined by their antibacterial activities that feature self-replication and phage/bacteria coevolution, as well as the human immune system in response to the two concurrent events (16).
Inhaled phage therapy holds remarkable potential to treat respiratory infections caused by bacteria, including MDR isolates (5). With an oral inhalation route for delivery to the lung, high concentration of phages can be delivered to the site of infection in the respiratory tract, achieving high pulmonary bioavailability. Although inhaled phage therapy has been practiced in Eastern Europe for many decades, the phage viability in nebulized aerosol droplets has only recently been investigated. Our group and others have demonstrated that inhalable aerosolized Pseudomonas phages remain biologically active when a suitable nebulizer system is used (17–20). Although the feasibility of producing infective phage aerosols have been well established, there is a lack of understanding of in vivo PK and PD profiles of phage and bacteria in the lungs. The aim of this study is to investigate the PK and time-kill profiles of Pseudomonas phage PEV31 administered by pulmonary delivery in a neutropenic murine model of respiratory infection.
RESULTS
Phage preparation and in vitro phage susceptibility.
The titer of purified PEV31 enumerated against the reference strain (PAV237) used for phage amplification was 4 × 1010 PFU/ml. Phage PEV31 formed a clear zone of lysis on top of an overlay plate containing the MDR P. aeruginosa isolate FADDI-001. PEV31 was highly efficacious in vitro against FADDI-001 with an efficiency of plating of 1. The endotoxin level in the purified phage lysate was 3.8 EU/ml (i.e., 0.095 EU in 25 μl). The spike recovery was 130% and the coefficient of variation of the assay was 4%, which were all considered acceptable as per the manufacturer’s recommendations.
Pharmacokinetics of intratracheally administered PEV31.
Infectious PEV31 gradually decreased in the lungs (lung homogenate and bronchoalveolar lavage fluid [BALF] combined) over time, regardless of the administered dose (Fig. 1, Fig. 2, and Fig. 3). At 24 h after IT administration, the phage titer dropped to 12.3% and 15.2% of the administered dose for the low (107 PFU) and high (109 PFU) doses, respectively. The elimination of active phage could be adequately described using a simple exponential decay model, with the adjusted weighted being 0.931 and 0.907 for the low and high dose, respectively. The rate constant was estimated to be 0.0876 h−1 (95% confidence interval [CI] 0.0719 to 0.106) for the low dose and 0.0878 h−1 (95% CI 0.0667 to 0.113) for the high dose, which are equivalent to half-lives of 7.92 (95% CI 6.54 to 9.64) and 7.90 (95% CI 6.12 to 10.4) hours, respectively. Infective PEV31 titers in other organs, including kidney, liver, blood, and spleen, were extremely low and accounted for less than 0.01% of the administered doses (Fig. 4). The phage titer gradually increased over 24 h in the liver of mice that received 109 PFU of PEV31. PEV31 suspension was well tolerated at a low dose without changes in inflammatory cytokine levels (Fig. 5). On the contrary, a transient upregulation of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) activity was observed at 4 and 8 h postadministration, respectively, when the mice were given a high dose of PEV31. Both cytokines returned to baseline at 24 h after the single IT dose.
FIG 1.
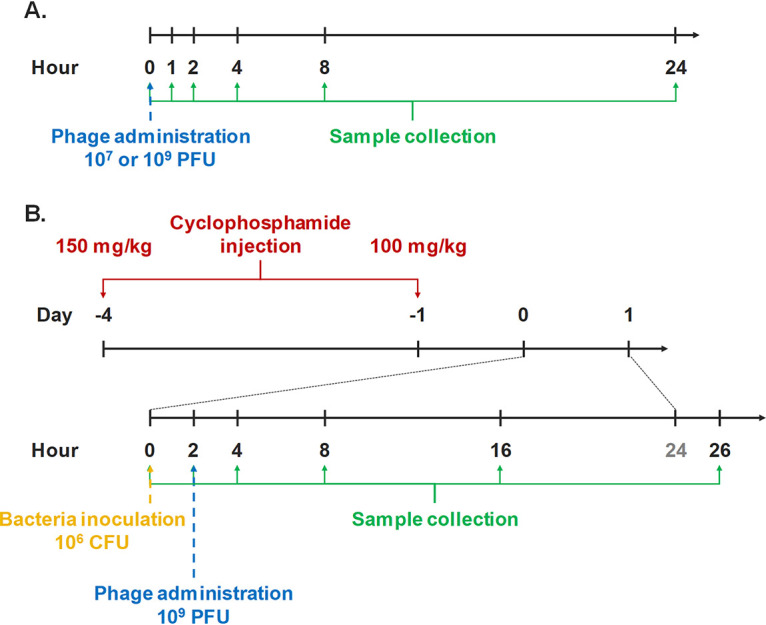
Timeline of experimental procedures to investigate the pharmacokinetics (A) and pharmacodynamics (B) of intratracheally administered PEV31 in mice.
FIG 2.

The effect of viricides on PEV31 phage titer (A) and P. aeruginosa FADDI-PA001 (B). Phages (n = 3) and bacteria (n = 2) were treated with PBS (control) or viricides (15 mg/liter of tannic acid and 2.5 mM ferrous sulfate).
FIG 3.

Phage titer in the lungs (lung tissues and BALF combined) of healthy mice after intratracheal administration of phage PEV31 at doses of 107 and 109 PFU. Phage titer is expressed as PFU per lung homogenate and BALF combined (left) and as PFU relative to the administered dose (right). Lines of regression (superimposed) are shown in the right panel. Error bars denote standard deviation (n ≥ 4 except for t = 2 h of the 107 PFU group and t = 1 h and 4 h of the 109 PFU group, for which n = 3).
FIG 4.

Infective phage titers in kidney, liver, blood, and spleen after intratracheal administration of phage PEV31 at doses of 107 and 109 PFU. Phage titers are expressed as PFU per organ. Error bars denote standard deviation (n ≥ 4).
FIG 5.

Levels of TNF-α and IL-6 in BALF in healthy mice after phage administration at doses of 107 and 109 PFU. Error bars denote standard deviation (n ≥ 4 except for t = 2 h of the 107 PFU group, for which n = 3).
Pharmacodynamics of intratracheally administered PEV31.
In the infected-only group, the bacteria continued to replicate without any significant stationary period (Fig. 6). Initially, the bacteria grew exponentially for up to about 8 h postinfection (hpi), after which the growth rate decreased. In the infected mice treated with 109 PFU of PEV31 at 2 hpi, the bacterial load in lung remained mostly unchanged except for the initial drop at 4 hpi (2 h post-phage-IT administration). Treatment with phage resulted in stasis, with greater than 4-log difference in bacterial load at 26 hpi compared with PBS-treated control. Some of the surviving bacterial colonies at 26 hpi in the phage-treated group showed a different antibiogram profile in comparison with the parent bacteria used to inoculate each mouse (Table 1). A reduction in MIC values of amikacin (from >64 to 16 μg/ml), ciprofloxacin (from 32 to 0.125 μg/ml), and tobramycin (from 32 to 8 μg/ml) was observed, while the MIC of colistin increased from 8 to 64 μg/ml. There were no apparent sensitivity changes to other antibiotics tested and all the tested bacterial colonies from PBS-treated group had the same MIC as the parent stock.
FIG 6.
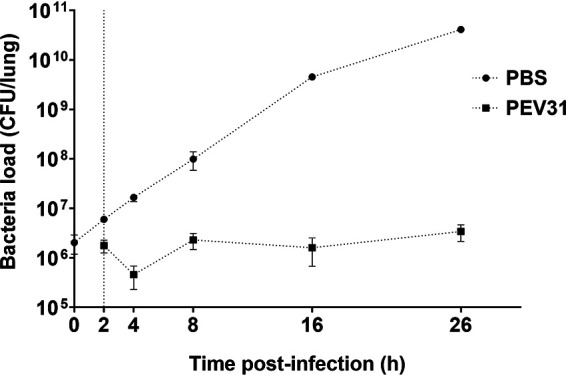
Bacterial load in the lungs of mice treated with PBS and phage over 26 h. Dotted vertical line represents time of phage administration (t = 2 h). Error bars denote standard deviation (n = 2 to 4).
TABLE 1.
Antibiotic and phage susceptibility of bacterial colonies isolated from the lung homogenate before and after treatment with phage PEV31 or PBS
| Time postinfection (h) | Treatmentb | No. of colonies observed (tested) | MIC (μg/ml)a |
PEV31 susceptibilityb | ||||
|---|---|---|---|---|---|---|---|---|
| Amikacin (R > 16) | Ciprofloxacin (R > 0.5) | Tobramycin (R > 2) | Aztreonam (R > 16) | Colistin (R > 2) | ||||
| 0 | NA | 4 (4) | >64 | 32 | 32 | >64 | 8 | S |
| 26 | PBS | 4 (4) | >64 | 32 | 32 | >64 | 8 | S |
| 26 | Phage | 4 (10) | 16 | 0.125 | 8 | >64 | 64 | R |
| 26 | Phage | 6 (10) | >64 | 32 | 32 | >64 | 8 | S |
MIC breakpoints (R) from EUCAST (v. 10.0) for Pseudomonas spp. are given under each antibiotic.
NA, not applicable; PBS, phosphate-buffered saline; S, susceptible; R, resistant.
The phage-mediated bacterial killing was evident by increase of infectious phage particles over time (around 2-log10) at 24 h after phage administration (Fig. 7). Inflammatory cytokine activity (TNF-α, IL-1β, and IL-6) in BALF were also measured as an evaluation of lung inflammation. In the bacteria-infected only group, a substantial upregulation of all three cytokines was observed. TNF-α peaked at 4 hpi, while the other two cytokines displayed peak activity at later time points. The upregulation of IL-1β activity considerably diminished at 26 hpi and to a lesser extent the same was seen for TNF-α, but not for IL-6. Interestingly, the upregulation in cytokines was only partially suppressed by the phage treatment for IL-1-β (21), but not for TNF-α and IL-6. In the phage-treated group, the peak of TNF-α appeared delayed to 8 hpi.
FIG 7.
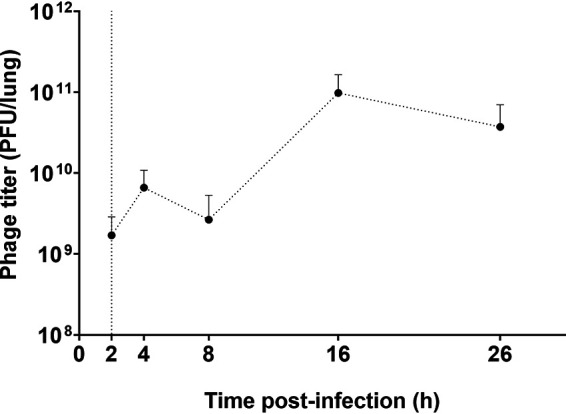
Phage titer in the lungs of mice infected with P. aeruginosa. Dotted vertical line represents the time (t = 2 h) of phage administration (109 PFU). Error bars denote standard deviation (n = 3 to 4).
DISCUSSION
Inhaled phage therapy has been used in Eastern European countries to treat bacterial respiratory infections that were otherwise untreatable with antibiotics. A 7-year-old cystic fibrosis patient received inhaled phage therapy in 2011 that dramatically reduced the MDR P. aeruginosa and Staphylococcus aureus numbers in the lungs (12). Despite promising clinical case studies of inhaled phage therapy, there is a lack of understanding of the behavior of phages and bacteria in the lungs. This is the first study investigating PK and time-kill profiles of intratracheally administered Pseudomonas phages in vivo. In previous studies with mice, the intranasal route has been widely used for initiation of lung infection and then phage treatment (22–24), likely due to the ease of administration. These studies provide strong support for inhaled phage therapy with reduction in bacterial load and inflammation in the mouse lung infection model. Compared with the intranasal route, intratracheal administration enables direct application of bacteria and phages to the mouse lungs with minimal loss in other parts of the respiratory route, including nose, throat, and upper airways (8, 25). Hence, the exact phage doses of interest were given in the PK study, and in the PD study. Despite these advantages, studies on intratracheal administration of phages for lung infections have been scarce (8, 26). In this study, intratracheal instillation was used to administer and assess the PK of phage PEV31 at two different doses.
The infectious phage PEV31, as quantified using plaque assays, exhibited an exponential decay in the lung at both low and high doses with equal half-life (rate constant, Fig. 3). After oral inhalation of phages to the lung, the phage titer dropped by 1-log10 over 24 h at both doses. Liu et al. studied the PK profile of Siphoviridae lytic mycobacteriophage D29 after administering 5 × 108 PFU via the intratracheal route (26). The titer of D29 dropped to 1.2-log10 by 24 h postadministration. Using the same regression methodology on the titers reported by Liu et al., we determined the half-life of D29 to be 5.8 h, which is lower than our values of 7.9 h for PEV31. Phage D29 belongs to the Siphoviridae family and has a longer phage tail compared with PEV31 (Podoviridae). Whether there is a correlation between the family and/or the geometry of the phage particle and the rate of elimination warrants further investigations. Compared with phage delivered via intravenous injection (10), intratracheal route administration resulted in reduced systemic exposure (Fig. 4).
Our current work has shown that the total titer of administered PEV31 phages in various organs do not add up to 100% of the delivered dose. No phage titer reduction was observed during the sample processing, including homogenization, filtration (0.22-μm PES membrane and BagPage filter), and sample dilution. This implies that phage inactivation or degradation in the lungs and/or other organs are likely. Hence, both biodistribution of phages as well as degradation/inactivation may contribute to the titer reduction observed in the lungs over time. Plaque assay is the method of choice for quantifying infectious phages (27, 28). In a plaque assay, a zone of clearance (plaques) are formed on top of a bacterial lawn as a result of cycles of infection of the bacterial cells with phage progeny radiating from the original source of infection (29). It follows that only infectious phages can be enumerated. To evaluate the total number of viral particles, i.e., infectious, noninfectious, and defective, genome quantification using quantitative PCR (qPCR) can potentially be utilized (30, 31). The combination of qPCR and plaque assay could potentially help understand the biodistribution of infective phages, as well as those that have been broken down or inactivated in different organs. This information may be particularly useful for estimating the total phage burden over time and correlating any long-term side effects associated with accumulation of nano-sized virus particles in the various cavities during a prophylactic treatment. Nano-sized particles (<10 nm) can easily enter human tissues and disrupt the biochemical environment of normal cells (32–36). Nanoparticles mostly accumulate in the liver tissues and adverse unpredictable health outcomes have recently surfaced (37, 38). Our current study has shown that infective phage titer gradually increases in the liver over time, and there may be inactive or degraded phage particles further accumulated in the organ. The most significant phage phagocytosis function is thought to be played by the liver. Phages mostly accumulate in the liver (99%) after intravenous administration and the rate of phagocytosis by Kupffer cells are four times faster than splenic macrophages (39). The current study has shown that by delivering phages directly to the lungs, systemic exposure and liver-induced phagocytosis is substantially minimized.
In the current study, phage PEV31 at a high dose (0.095 EU; 4.75 EU/kg) resulted in an upregulation of the inflammatory cytokine TNF-α at 2 h postadministration, which then substantially increased at 4 h (Fig. 5). The upregulation of IL-6 followed and then peaked at a later time point of 8 h. Upon phage administration, TNF-α and other inflammatory cytokines (e.g., IL-1) secreted from resident macrophages stimulated the release of other chemoattractant factors such as MIP-2, MCP-1, and IL-6, promoting the adherence of circulating inflammatory cells to the endothelium (26). The upregulation of both cytokines subsided at 24 h. Overexpression of these cytokines was absent when the mice were administered a lower dose. It has been reported that phages could trigger both inflammatory and anti-inflammatory responses (40), and endotoxin alone cannot explain all the observed upregulation of cytokines in the current study. Liu et al. reported no significant differences in leukocyte, neutrophils, lymphocytes, and TNF-α levels in the BALF at 24 h postadministration of D29 in healthy mice (26). However, the levels of these cytokines between 0 and 24 h postadministration is unknown and, unfortunately, the endotoxin levels in the phage preparations were not reported. In both earlier studies and the current study, the kinetics of phage in lungs has been the primary endpoint reported, typically presented as PFU per lung over time (8, 22, 24, 26). To improve and enhance comparison on BALF between studies, standardization by means of urea dilution correction can be performed to better account for the variations in the lavage procedure, particularly for the quantification of proteins such as inflammatory mediators, where the concentration is more relevant. In another study, intranasal administration of phages with an endotoxin level of 0.0063 EU/mice (approximately 0.3 EU/kg) did not result in appreciable levels of TNF-α at 48 h posttreatment (9). Extremely low TNF-α and IL-6 levels were similarly observed in the lungs of mice that received Pseudomonas phages via the intranasal route at 24 h postadministration, although the endotoxin level of the phage lysate was unreported (22). These findings aligned with our observation, where the upregulation of inflammatory cytokines subsided at 24 h postadministration. Hence, phage preparations with endotoxin levels even lower than that for parenteral and free of bacterial impurities should be considered for respiratory delivery (40) to ensure minimum toxicity (41), particularly in the case of prophylactic use. The current consensus is that phage therapy is safe (and has been so for decades) provided the phage preparation is sufficiently purified to ensure low endotoxin levels and remove other bacterial impurities (5, 42, 43). However, phage lysates originating from Gram-negative pathogens can be contaminated with endotoxins (lipopolysaccharides) and other proteins that are toxic to humans. Endotoxins are highly immunogenic and may cause septic shock by triggering cytokine signaling (44–46). The highest permitted endotoxin concentration for injection is 5 EU/kg/h. Even purified phage preparations with extremely low endotoxin levels (<0.1 EU) may induce some proinflammatory responses, which are caused by other bacterial proteins and nucleic acids present in the phage lysate (40). Despite low endotoxin level in our PEV31 preparation, bacterial proteins and other contaminants may have caused the proinflammatory response in the murine lungs. Hence, not only should the phage preparation have low endotoxin levels, but also be free of bacterial genes, proteins, and other debris that may trigger host immune response. On the flip side, host immune system is considered essential for efficacious phage therapy as it can control phage-sensitive and emergent phage-resistant bacteria (16). Lung infection with P. aeruginosa caused significant increase in inflammatory cytokines (TNF-α, IL-1β, and IL-6), with levels of IL-1β partially suppressed by phage treatment between 4 to 16 h (P < 0.05) (Fig. 8). Immune responses to phages are phage specific and some phages can even be anti-inflammatory, such that bacterial clearance is reduced to promote phage propagation, as well as to minimize the clearance of phages from the site of infection (40). Anti-inflammatory cytokines (e.g., IL-10) can be investigated in future studies to assess the potential role of phages as an anti-inflammatory agent.
FIG 8.

Levels of IL-6, TNF-α, and IL-1β in BALF of P. aeruginosa-infected mice treated with PBS or phage PEV31 (109 PFU) over time. Error bars denote standard deviation (n = 2 to 4). Statistically significant differences between the two groups are indicated with asterisks (P < 0.05).
The MDR P. aeruginosa isolate FADDI-001 used in this study is a clinical strain isolated from an intensive care unit patient and is resistant to multiple antibiotics, including rifampin, doxycycline, ciprofloxacin, amikacin, aztreonam, and tobramycin. When PEV31 was administered 2 h postinfection, the growth of FADDI-001 was suppressed, suggesting the rate of bacteria replication was similar to that of bacteria removal through phage-killing or clearance by host immune response, with the former evident by the increase in phage titer (Fig. 7). FADDI-001 was highly susceptible to PEV31 in vitro but developed phage resistance over time (upon overnight incubation), as reported for many naturally occurring phages under in vitro conditions. Bacteria can resist phage infection through different mechanisms, including (i) spontaneous mutations to prevent phage adsorption or phage DNA entry, (ii) restriction modification systems to cut phage nucleic acids, and (iii) CRISPR-Cas system-mediated adaptive immunity (47–51). When bacteria are pressured with a high number of infective phages, resistance can develop rapidly (52) by changing the bacterial surface components that act as phage-binding receptors. These receptors can be blocked by producing extracellular matrix or competitive inhibitors, or even be removed to prevent phage adsorption (47). Contrary to in vitro results (data not shown), some but not all bacteria at 24 hpi remained susceptible to the phage despite high initial MOI. This may be due to fundamental differences between in vitro and in vivo systems, such as the involvement of mammalian immune responses and heterogeneous mixing. For the latter, it is possible that bacteria and phages were not evenly mixed within the mouse lungs during administration (i.e., spatial constraint). Hence, not all the bacteria may have been exposed to the same stress and selective pressure despite high phage titer used in this study. Those colonies that became resistant to phage PEV31 showed a different antibiogram to phage-susceptible bacteria. In the fight to become phage-resistant, FADDI-001 developed increased sensitivity to ciprofloxacin (quinolone), amikacin, and tobramycin (both aminoglycosides), but also developed increased resistance to colistin (polymyxin) (Table 1). In fact, these phage-resistant isolates were highly susceptible to ciprofloxacin with MIC values of only a quarter of the breakpoint for resistance (53), suggesting the possibility of clinically relevant reversal of antibiotic resistance through inhaled phage therapy. The changes to the antibiogram suggests possible modifications in the bacterial cell envelope as a result of acquiring phage resistance. Phages can use a number of cell surface moieties as a binding receptor, such as outer membrane proteins (e.g., efflux pumps and porins) and glycolipids (e.g., lipopolysaccharide) (54). Reduced expression of multidrug efflux pumps (e.g., Mex systems) in response to phage-mediated stress may reverse the resistance mechanism of aminoglycosides and fluoroquinolones. Alterations of lipopolysaccharides can increase colistin resistance (55), which may have resulted from acquiring phage resistance (i.e., modification of the phage-binding receptor). Reversal of antibiotic resistance of P. aeruginosa under selective pressure of phage has been reported (56). Chan et al. isolated a lytic Pseudomonas phage OMKO1 that binds to outer membrane porin M of the multidrug efflux systems. MDR P. aeruginosa developed resistance to OMKO1 within 24 h of incubation in vitro, while these phage-resistant bacteria regained sensitivity to ciprofloxacin, erythromycin, ceftazidime, and tetracycline. Understanding the phage-mediated mechanisms to antibiotic susceptibility is outside the scope of this work. However, the data suggest the need to assess the impact of phage treatment on antibiotic susceptibilities for each phage-bacteria system, particularly if combined phage-antibiotic treatment is being considered in a clinical setting. Phage therapy could potentially be used early on as a single dose to substantially decrease the bacterial burden, followed by further treatment with other antimicrobials and/or by host immune response.
The current study used an acute lung infection mouse model and does not necessarily inform about chronic infections, such as cystic fibrosis. One of the major challenges in conducting simultaneous PK/PD studies of phage therapy in bacterial infection lies in the continued interactions between bacterial host and phage even after sample collection. Procedures have been taken to minimize lysis of bacteria and phage propagation once the lung tissues have been harvested through physical separation by filtration and chemical inactivation by viricides. However, complete elimination of phage from bacteria in the tissue homogenates and removing phages that have already infected the bacteria could be difficult. Any remaining phages that have not been removed or inactivated could reduce the bacteria count and thus overestimate phage-killing efficacy.
Conclusions. This is the first study investigating the PK and time-kill profiles of antipseudomonal phage in the lungs of healthy and P. aeruginosa-infected mice, respectively. The safety and biodistribution of phage PEV31 over time were assessed in the lungs of healthy mice. Importantly, inhaled phages not only reduced the lung bacterial load, but also suppressed proinflammatory cytokines in the lungs. The bacterial antibiogram was altered upon phage treatment, where bacteria became susceptible to some antibiotics while becoming more resistant to others. Nonetheless, more work is required to examine the influence of phage exposure on antibiotic susceptibility of bacteria. Further in vivo toxicity and PK/PD studies evaluating various dose regimes in both acute and chronic models are urgently needed to better understand the phage and bacteria kinetics in the lungs.
MATERIALS AND METHODS
Bacteriophage.
Anti-Pseudomonas phage PEV31 was isolated from the sewage plant in Olympia (WA, USA) by the Evergreen Phage Lab (Kutter Lab). PEV31 belongs to the Podoviridae family. Stocks of the phage were amplified using the Phage-on-Tap protocol (57) with minor modifications. Briefly, 200 ml of nutrient broth (NB, Amyl Media, Australia) supplemented with 1 mM CaCl2 and MgCl2 was mixed with 0.1 volumes of overnight bacterial host (P. aeruginosa dog-ear strain PAV237). The mixture was incubated for 1 h with continuous shaking (220 rpm) at 37°C. A volume (200 μl) of PEV31 lysate at 109 PFU/ml was added, followed by further incubation for 8 h. The mixture was centrifuged at 4,000 × g for 20 min and the supernatant was filter sterilized using a 0.22-μm polyethersulfone membrane filter. The phage lysate was further purified and concentrated using ultrafiltration (100 kDa Amicon Ultra-15 centrifugal filter, Sigma, Australia), and the medium was replaced with phosphate-buffered saline (PBS) supplemented with 1 mM CaCl2. Bacterial endotoxins were removed by adding 0.4 volumes of 1-octanol, followed by vigorous shaking at room temperature for 1 h. The mixture was centrifuged at 4,000 × g for 10 min and then the aqueous phase was collected. Residual organic solvent was removed by centrifuging the phages at 20,000 × g for 1.5 h and then replacing the buffer with fresh PBS supplemented with 1 mM CaCl2.
Endotoxin level quantification.
The Endosafe Portable Test System (Charles River Laboratories, Boston, USA) was used as per the manufacturer’s instructions to quantify endotoxin level in the resulting phage lysates. The single use LAL assay cartridges contain four channels to which the LAL reagent and a chromogenic substrate have been preapplied. A single cartridge enables duplicate measurements of the sample and positive control with a known endotoxin concentration. The sensitivity of the readouts between 50% and 200% spike recovery are deemed acceptable. The sensitivity of the assay was 1 to 10 EU/ml. Endotoxin-free water, tips, and tubes were used at all times.
Bacterial strain and phage-susceptibility testing.
P. aeruginosa FADDI-PA001 was used to induce bacterial infection in the mice used in this study. The strain is an MDR clinical isolate (isolated from an intensive care unit patient) provided by the Li lab, Monash University, Australia (8). Phage susceptibility of this isolate was assessed using a spot test (6). Briefly, 5 ml of 0.4% nutrient broth top agar was mixed with overnight culture of FADDI-PA001 (approximately 2 × 108 CFU) and then overlaid onto a 1.5% nutrient agar plate. Then, 10 μl of a phage stock solution was spotted on the top agar plate, air-dried, and incubated at 37°C for 24 h. After incubation, the appearance of the lysis zone was assessed for phage susceptibility.
Animals.
Female BALB/c mice of 6 to 8 weeks were obtained from Australian BioResources Ltd. (Moss Vale, New South Wales, Australia). The mice were housed under a 12-h dark-light cycle with ad libitum supply to standard chow diet and water. All animal experiments conducted were approved by the University of Sydney Animal Ethics Committee.
Pharmacokinetics of PEV31 after intratracheal administration.
Healthy (noninfected) mice were anaesthetized by intraperitoneal injection of ketamine/xylazine mixture (80 mg/kg and 10 mg/kg, respectively) in 150 μl PBS. Upon deep anesthesia as confirmed by the absence of pedal reflex, the anaesthetized mouse was suspended on a nylon floss by its incisor teeth and placed on an inclined intubation board. The trachea was then gently intubated with a soft plastic guiding cannula (58). PEV31 at two different doses (107 and 109 PFU) suspended in 25 μl PBS was administered into the trachea through the guiding cannula with a micropipette and a 200-μl gel-loading pipette tip. At 0 (immediately after administration), 1, 2, 4, 8, and 24 h post-phage administration, mice (n ≥ 4) were terminally anaesthetized by intraperitoneal injection of an overdose of ketamine/xylazine mixture (300 mg/kg and 30 kg/mg, respectively). Broncho-alveolar lavage (BAL) fluid followed by blood samples were collected from the anaesthetized animals (Fig. 1A). BAL was performed by instilling 1.5 ml PBS (as three aliquots of 0.5 ml) through the trachea to the lung and collecting the lavage suspension. Approximately 1.2 to 1.3 ml of lavage suspension was recovered. Lung, kidneys, spleen, and liver organs were subsequently collected upon exsanguination of the animal. Harvested tissues were homogenized by TissueRuptor II with plastic probes (Qiagen, Hilden, Germany) in cold PBS under ice-water bath for 30 s. Tissue homogenates were kept at 2 to 8°C until phage quantification by plaque assay (described below). Plaque assay was performed within 3 h of sample collection.
Pseudomonas pulmonary infection.
A neutropenic murine model (8) was used to establish pulmonary P. aeruginosa infection. Two doses of cyclophosphamide were intraperitoneally administered 4 days (150 mg/kg) and 1 day (100 mg/kg) prior to infection. On the day of infection, the FADDI-001 bacterial suspension at its early logarithmic growth phase was inoculated intratracheally at a concentration of 106 CFU in 25 μl, as described above. At 2 h postinfection, a PEV31 suspension (109 PFU in 25 μl PBS) or sterile PBS of equal volume (as untreated control) was intratracheally administered to the infected mice. Following terminal anesthesia as described above, BAL fluid and other tissues were collected at 0 (immediately after bacteria inoculation), 2 (immediately after phage administration), 4, 8, 16, and 26 h postinfection (n = 4) (Fig. 1B). Collected tissues were homogenized in cold PBS. Bacterial load and phage concentrations in the tissue homogenates and BAL fluid were stored on ice at all times and analyzed within 2 h using plaque assay and colony counting, respectively, as described below. Bacteria enumeration was performed as soon as practically possible and was not later than 2 h after sample collections. Plaque assay was performed within 3 h of sample collection.
Plaque assay.
BAL fluid and tissue homogenates were serially diluted in PBS for phage quantification. For samples from infected animals, bacteria were first removed from the homogenates and BAL samples by filtering through a 0.22-μm polyethersulfone membrane filter before dilution. A volume of reference bacterial host (PAV237) containing 2 × 108 CFU at stationary phase was mixed with 5 ml of nutrient broth top agar. The mixture was overlaid on top of a nutrient agar plate and dried for 15 min. Then, 20 μl of serially diluted phage suspension was dropped on top of the top agar plate, left to air dry, and then incubated for 24 h at 37°C. The diluted samples were analyzed in triplicate.
Bacteria enumeration.
Phage inactivation was performed prior to bacteria enumeration to prevent bactericidal activities of phages and impede reduction of CFU counts ex vivo. Furthermore, all samples were kept on ice at all times to minimize the risk of phage-bacteria interactions in vitro. The samples were treated with tannic acid (20 mg/liter) and ferrous sulfate (2.5 mM) to inactivate the phage PEV31 and then treated with 2% Tween 80 in PBS to stop the interaction. Our preliminary study has shown that the two chemicals in combination can inactivate the phage PEV31 without compromising the viability of P. aeruginosa FADDI-PA001 (Fig. 2). The phage-inactivated lung homogenate was filtered through a sterile filter bag (bag stomacher filter with a pore size of 280 μm, Labtek Pty Ltd., Australia). Filtrate samples and BAL fluid were serially diluted in PBS and then spiral plated on nutrient agar plates using an automatic spiral plater (WASP, Don Whitley Scientific, United Kingdom). The plates were air-dried and then incubated for 24 h at 37°C. Colonies were counted using a ProtoCOL automated colony counter (Synbiosis, United Kingdom).
MIC.
Bacterial colonies from the lung homogenate samples (t = 0 and 26 h) were taken and inoculated in cation-adjusted Mueller-Hinton Broth (CAMHB; Becton, Dickinson, and Co., Sparks, MD, USA). The antibiograms of these colonies were assessed by determining the MICs of selected antibiotics, including ciprofloxacin, tobramycin, colistin, and aztreonam, using broth microdilution in CAMHB. The stock solutions of these antibiotics were prepared immediately before the experiment and serially diluted in CAMHB to the desired final concentration. All assays were conducted in 96-well plate microtiter plates in CAMHB with a bacterial inoculum of 5 × 105 CFU/ml. The treated bacterial culture was incubated for 24 h at 37°C with continuous shaking at 220 rpm. Optical density at 600 nm (OD600) was measured using a microplate reader (Victor multilabel Plate Reader, Perkin Elmer, United States).
Cytokine quantification.
The BAL fluid collected was centrifuged at 400 × g for 10 min, and the supernatant was collected as the bronchoalveolar lavage fluid (BALF). The levels of interleukin (IL)-6, tumor necrosis factor alpha (TNF-α), and IL-1β (in infected mice) in BALF were quantified by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s protocol (DY406, DY410, and DY401 from R&D Systems, Minneapolis, MN, USA). The UV absorbances at 450 nm and 570 nm were measured for the primary signal and for plate correction, respectively (Victor multilabel plate reader). The standard curves were constructed by 4-parameter logistic nonlinear regression.
Data analysis.
For the pharmacokinetics study, regression analysis on the phage titer (PFU) in lungs (lung tissues and BALF combined) over time was performed using a simple exponential decay model. The exponential decay takes the form of , where and are the relative phage titer at time and the rate constant (in h−1), respectively ( denoted the phage titer at time = 0). It follows that the half-life of the decay was given by . The regression was performed using Prism software version 8.3 for Windows (GraphPad Software Inc., CA, USA).
ACKNOWLEDGMENT
This study was financially supported by the National Health and Medical Research Council (project grant number APP1140617).
REFERENCES
- 1.Centers for Disease Control and Prevention. 2019. Antibiotic resistance threats in the United States, 2019. Centers for Disease Control and Prevention, Atlanta, GA. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf. [Google Scholar]
- 2.World Health Organization. 2015. Global action plan on antimicrobial resistance. https://www.who.int/antimicrobial-resistance/publications/global-action-plan/en/. [DOI] [PubMed]
- 3.Theuretzbacher U, Outterson K, Engel A, Karlen A. 2020. The global preclinical antibacterial pipeline. Nat Rev Microbiol 18:275–285. doi: 10.1038/s41579-019-0288-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z. 2019. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 37:177–192. doi: 10.1016/j.biotechadv.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 5.Chang RYK, Wallin M, Lin Y, Leung SSY, Wang H, Morales S, Chan HK. 2018. Phage therapy for respiratory infections. Adv Drug Deliv Rev 133:76–86. doi: 10.1016/j.addr.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang RY, Wong J, Mathai A, Morales S, Kutter E, Britton W, Li J, Chan HK. 2017. Production of highly stable spray dried phage formulations for treatment of Pseudomonas aeruginosa lung infection. Eur J Pharm Biopharm 121:1–13. doi: 10.1016/j.ejpb.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallavali RR, Degati VL, Lomada D, Reddy MC, Durbaka VRP. 2017. Isolation and in vitro evaluation of bacteriophages against MDR-bacterial isolates from septic wound infections. PLoS One 12:e0179245. doi: 10.1371/journal.pone.0179245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang RYK, Chen K, Wang J, Wallin M, Britton W, Morales S, Kutter E, Li J, Chan HK. 2017. Proof-of-principle study in a murine lung infection model of Antipseudomonal activity of phage PEV20 in a dry-powder formulation. Antimicrob Agents Chemother 62:e01714-17. doi: 10.1128/AAC.01714-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmody LA, Gill JJ, Summer EJ, Sajjan US, Gonzalez CF, Young RF, LiPuma JJ. 2010. Efficacy of bacteriophage therapy in a model of Burkholderia cenocepacia pulmonary infection. J Infect Dis 201:264–271. doi: 10.1086/649227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin YW, Chang RY, Rao GG, Jermain B, Han ML, Zhao JX, Chen K, Wang JP, Barr JJ, Schooley RT, Kutter E, Chan HK, Li J. 2020. Pharmacokinetics/pharmacodynamics of antipseudomonal bacteriophage therapy in rats: a proof-of-concept study. Clin Microbiol Infect doi: 10.1016/j.cmi.2020.04.039. [DOI] [PubMed] [Google Scholar]
- 11.Wang JL, Kuo CF, Yeh CM, Chen JR, Cheng MF, Hung CH. 2018. Efficacy of φkm18p phage therapy in a murine model of extensively drug-resistant Acinetobacter baumannii infection. Infect Drug Resist 11:2301–2310. doi: 10.2147/IDR.S179701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvachadze L, Balarjishvili N, Meskhi T, Tevdoradze E, Skhirtladze N, Pataridze T, Adamia R, Topuria T, Kutter E, Rohde C, Kutateladze M. 2011. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb Biotechnol 4:643–650. doi: 10.1111/j.1751-7915.2011.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhukov-Verezhnikov NN, Peremitina LD, Berillo EA, Komissarov VP, Bardymov VM. 1978. Therapeutic effect of bacteriophage preparations in the complex treatment of suppurative surgical diseases (in Russian). Sov Med :64–66. [PubMed] [Google Scholar]
- 14.Ujmajuridze A, Chanishvili N, Goderdzishvili M, Leitner L, Mehnert U, Chkhotua A, Kessler TM, Sybesma W. 2018. Adapted bacteriophages for treating urinary tract infections. Front Microbiol 9:1832. doi: 10.3389/fmicb.2018.01832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Principi N, Silvestri E, Esposito S. 2019. Advantages and limitations of bacteriophages for the treatment of bacterial infections. Front Pharmacol 10:513. doi: 10.3389/fphar.2019.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roach DR, Leung CY, Henry M, Morello E, Singh D, Di Santo JP, Weitz JS, Debarbieux L. 2017. Synergy between the host immune system and bacteriophage is essential for successful phage Ttherapy against an acute respiratory pathogen. Cell Host Microbe 22:38–47 e4. doi: 10.1016/j.chom.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 17.Leung SSY, Carrigy NB, Vehring R, Finlay WH, Morales S, Carter EA, Britton WJ, Kutter E, Chan HK. 2019. Jet nebulization of bacteriophages with different tail morphologies—structural effects. Int J Pharm 554:322–326. doi: 10.1016/j.ijpharm.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 18.Carrigy NB, Chang RY, Leung SSY, Harrison M, Petrova Z, Pope WH, Hatfull GF, Britton WJ, Chan HK, Sauvageau D, Finlay WH, Vehring R. 2017. Anti-tuberculosis bacteriophage D29 delivery with a vibrating mesh nebulizer, jet nebulizer, and soft mist inhaler. Pharm Res 34:2084–2096. doi: 10.1007/s11095-017-2213-4. [DOI] [PubMed] [Google Scholar]
- 19.Astudillo A, Leung SSY, Kutter E, Morales S, Chan HK. 2018. Nebulization effects on structural stability of bacteriophage PEV 44. Eur J Pharm Biopharm 125:124–130. doi: 10.1016/j.ejpb.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Marqus S, Lee L, Istivan T, Kyung Chang RY, Dekiwadia C, Chan HK, Yeo LY. 2020. High frequency acoustic nebulization for pulmonary delivery of antibiotic alternatives against Staphylococcus aureus. Eur J Pharm Biopharm 151:181–188. doi: 10.1016/j.ejpb.2020.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Dinarello CA. 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281:8–27. doi: 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debarbieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, Balloy V, Touqui L. 2010. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis 201:1096–1104. doi: 10.1086/651135. [DOI] [PubMed] [Google Scholar]
- 23.Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L. 2011. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One 6:e16963. doi: 10.1371/journal.pone.0016963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alemayehu D, Casey PG, McAuliffe O, Guinane CM, Martin JG, Shanahan F, Coffey A, Ross RP, Hill C. 2012. Bacteriophages phiMR299-2 and phiNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio 3:e00029-12–e00012. doi: 10.1128/mBio.00029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bivas-Benita M, Zwier R, Junginger HE, Borchard G. 2005. Non-invasive pulmonary aerosol delivery in mice by the endotracheal route. Eur J Pharm Biopharm 61:214–218. doi: 10.1016/j.ejpb.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Liu KY, Yang WH, Dong XK, Cong LM, Li N, Li Y, Wen ZB, Yin Z, Lan ZJ, Li WP, Li JS. 2016. Inhalation study of mycobacteriophage D29 aerosol for mice by endotracheal route and nose-only exposure. J Aerosol Med Pulm Drug Deliv 29:393–405. doi: 10.1089/jamp.2015.1233. [DOI] [PubMed] [Google Scholar]
- 27.Imamovic L, Serra-Moreno R, Jofre J, Muniesa M. 2010. Quantification of Shiga toxin 2-encoding bacteriophages, by real-time PCR and correlation with phage infectivity. J Appl Microbiol 108:1105–1114. doi: 10.1111/j.1365-2672.2010.04664.x. [DOI] [PubMed] [Google Scholar]
- 28.Anderson B, Rashid MH, Carter C, Pasternack G, Rajanna C, Revazishvili T, Dean T, Senecal A, Sulakvelidze A. 2011. Enumeration of bacteriophage particles: comparative analysis of the traditional plaque assay and real-time qPCR- and nanosight-based assays. Bacteriophage 1:86–93. doi: 10.4161/bact.1.2.15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You L, Yin J. 1999. Amplification and spread of viruses in a growing plaque. J Theor Biol 200:365–373. doi: 10.1006/jtbi.1999.1001. [DOI] [PubMed] [Google Scholar]
- 30.Refardt D. 2012. Real-time quantitative PCR to discriminate and quantify lambdoid bacteriophages of Escherichia coli K-12. Bacteriophage 2:98–104. doi: 10.4161/bact.20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duyvejonck H, Merabishvili M, Pirnay JP, De Vos D, Verbeken G, Van Belleghem J, Gryp T, De Leenheer J, Van der Borght K, Van Simaey L, Vermeulen S, Van Mechelen E, Vaneechoutte M. 2019. Development of a qPCR platform for quantification of the five bacteriophages within bacteriophage cocktail 2 (BFC2). Sci Rep 9:13893. doi: 10.1038/s41598-019-50461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla S, Wen AM, Ayat NR, Commandeur U, Gopalkrishnan R, Broome AM, Lozada KW, Keri RA, Steinmetz NF. 2014. Biodistribution and clearance of a filamentous plant virus in healthy and tumor-bearing mice. Nanomedicine (Lond) (Lond) 9:221–235. doi: 10.2217/nnm.13.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruckman MA, Randolph LN, VanMeter A, Hern S, Shoffstall AJ, Taurog RE, Steinmetz NF. 2014. Biodistribution, pharmacokinetics, and blood compatibility of native and PEGylated tobacco mosaic virus nano-rods and -spheres in mice. Virology 449:163–173. doi: 10.1016/j.virol.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang YY, Hu KQ. 2015. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J Med Virol 87:1989–1999. doi: 10.1002/jmv.24270. [DOI] [PubMed] [Google Scholar]
- 35.Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. 2016. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release 240:332–348. doi: 10.1016/j.jconrel.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 36.Dai J, Chen EQ, Bai L, Gong DY, Zhou QL, Cheng X, Huang FJ, Tang H. 2012. Biological characteristics of the rtA181T/sW172* mutant strain of Hepatitis B virus in animal model. Virol J 9:280. doi: 10.1186/1743-422X-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mostafalou S, Mohammadi H, Ramazani A, Abdollahi M. 2013. Different biokinetics of nanomedicines linking to their toxicity; an overview. Daru 21:14. doi: 10.1186/2008-2231-21-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B. 2002. Passage of inhaled particles into the blood circulation in humans. Circulation 105:411–414. doi: 10.1161/hc0402.104118. [DOI] [PubMed] [Google Scholar]
- 39.Inchley CJ. 1969. The activity of mouse Kupffer cells following intravenous injection of T4 bacteriophage. Clin Exp Immunol 5:173–187. [PMC free article] [PubMed] [Google Scholar]
- 40.Van Belleghem JD, Clement F, Merabishvili M, Lavigne R, Vaneechoutte M. 2017. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci Rep 7:8004. doi: 10.1038/s41598-017-08336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Belleghem JD, Merabishvili M, Vergauwen B, Lavigne R, Vaneechoutte M. 2017. A comparative study of different strategies for removal of endotoxins from bacteriophage preparations. J Microbiol Methods 132:153–159. doi: 10.1016/j.mimet.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 42.Speck P, Smithyman A. 2016. Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol Lett 363:fnv242. doi: 10.1093/femsle/fnv242. [DOI] [PubMed] [Google Scholar]
- 43.Wienhold SM, Lienau J, Witzenrath M. 2019. Towards inhaled phage therapy in Western Europe. Viruses 11:295. doi: 10.3390/v11030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosadini CV, Kagan JC. 2017. Early innate immune responses to bacterial LPS. Curr Opin Immunol 44:14–19. doi: 10.1016/j.coi.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skirecki T, Cavaillon JM. 2019. Inner sensors of endotoxin—implications for sepsis research and therapy. FEMS Microbiol Rev 43:239–256. doi: 10.1093/femsre/fuz004. [DOI] [PubMed] [Google Scholar]
- 47.Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 48.Le S, Yao X, Lu S, Tan Y, Rao X, Li M, Jin X, Wang J, Zhao Y, Wu NC, Lux R, He X, Shi W, Hu F. 2014. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci Rep 4:4738. doi: 10.1038/srep04738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Azam AH, Tanji Y. 2019. Bacteriophage-host arm race: an update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl Microbiol Biotechnol 103:2121–2131. doi: 10.1007/s00253-019-09629-x. [DOI] [PubMed] [Google Scholar]
- 50.Latino L, Midoux C, Vergnaud G, Pourcel C. 2019. Investigation of Pseudomonas aeruginosa strain PcyII-10 variants resisting infection by N4-like phage Ab09 in search for genes involved in phage adsorption. PLoS One 14:e0215456. doi: 10.1371/journal.pone.0215456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim WS, Ho PL, Li SF-Y, Ow DS-W. 2019. Clinical implications of Pseudomonas aeruginosa: antibiotic resistance, phage & antimicrobial peptide therapy. Proceedings of the Singapore National Academy of Science 13:65–86. doi: 10.1142/S2591722619400076. [DOI] [Google Scholar]
- 52.Lin Y, Chang RYK, Britton WJ, Morales S, Kutter E, Chan HK. 2018. Synergy of nebulized phage PEV20 and ciprofloxacin combination against Pseudomonas aeruginosa. Int J Pharm 551:158–165. doi: 10.1016/j.ijpharm.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.European Committee on Antimicrobial Susceptibility Testing. 2020. Breakpoint tables for interpretation of MICs and zone diameters, version 10.0. EUCAST, Växjö Sweden. https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_10.0_Breakpoint_Tables.pdf. [Google Scholar]
- 54.Chaturongakul S, Ounjai P. 2014. Phage-host interplay: examples from tailed phages and Gram-negative bacterial pathogens. Front Microbiol 5:442. doi: 10.3389/fmicb.2014.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo Sciuto A, Cervoni M, Stefanelli R, Mancone C, Imperi F. 2020. Effect of lipid A aminoarabinosylation on Pseudomonas aeruginosa colistin resistance and fitness. Int J Antimicrob Agents 55:105957. doi: 10.1016/j.ijantimicag.2020.105957. [DOI] [PubMed] [Google Scholar]
- 56.Chan BK, Sistrom M, Wertz JE, Kortright KE, Narayan D, Turner PE. 2016. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci Rep 6:26717. doi: 10.1038/srep26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bonilla N, Rojas MI, Netto Flores Cruz G, Hung SH, Rohwer F, Barr JJ. 2016. Phage on tap—a quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ 4:e2261. doi: 10.7717/peerj.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu Y, Liao Q, Chow MYT, Lam JKW. 2020. Intratracheal administration of dry powder formulation in mice. J Vis Exp 161:e61469. doi: 10.3791/61469. [DOI] [PubMed] [Google Scholar]