

Abstract
Von Willebrand factor (VWF) plays an important role in ischemic stroke. However, the exact mechanism by which VWF mediates progression of ischemic stroke brain damage is not completely understood. Using flow cytometric analysis of single cell suspensions prepared from brain tissue and immunohistochemistry, we investigated the potential inflammatory mechanisms by which VWF contributes to ischemic stroke brain damage in a mouse model of cerebral ischemia/reperfusion injury. Twenty-four hours after stroke, flow cytometric analysis of brain tissue revealed that overall white blood cell recruitment in the ipsilesional brain hemisphere of VWF knockout mice was two times lower than that in wildtype mice. More detailed analysis showed a specific reduction of proinflammatory monocytes, neutrophils and T cells in the ischemic brain of VWF knockout mice compared to wild-type mice. Interestingly, histological analysis revealed a substantial number of neutrophils and T cells still within the microcirculation of the stroke brain, potentially contributing to the noreflow phenomenon. Specific therapeutic targeting of the VWF A1 domain in the wild-type mice resulted in reduced numbers of immune cells in the affected brain and protected mice from ischemic stroke brain damage. More specifically, recruitment of proinflammatory monocytes was reduced two-fold, neutrophil recruitment was reduced five-fold and T-cell recruitment was reduced two-fold in mice treated with a VWF A1-targeting nanobody compared to the recruitment in mice receiving a control nanobody. In conclusion, our data identify a potential role for VWF in the recruitment of proinflammatory monocytes, neutrophils and T cells to the ischemic brain through a mechanism that is mediated by its A1 domain.
Introduction
Ischemic stroke is caused by a blood clot occluding one or multiple cerebral arteries, often leading to irreversible brain damage. Unfortunately, treatment options are limited and not always successful. In 2015, stroke deaths accounted for 11.8% of total deaths worldwide, making stroke the second leading cause of death.1 The pathogenesis of ischemic stroke brain damage remains largely unclear and better understanding of the underlying mechanisms is crucial to meet the critical demand for improved stroke therapy.
Remarkably, while successful recanalization of the occluded blood vessel is necessary to alleviate ischemic stroke brain injury, it is often not sufficient. Indeed, there is not a strict correlation between vessel recanalization and overall neurological outcome 3 months after a stroke.2 The reasons why efficient recanalization is not always associated with good clinical outcomes are unclear and most likely multifactorial. One key contributor is cerebral ischemia/reperfusion injury. The complex pathophysiology of cerebral ischemia/reperfusion injury includes both thrombotic and inflammatory pathways causing microvascular obstructions, increased blood-brain barrier permeability, and overall neurological deterioration.3
In recent years, the pathophysiological role of von Willebrand factor (VWF) in ischemic stroke has become apparent from both clinical and experimental studies.4,5 VWF is a large multimeric glycoprotein that recruits platelets at sites of vascular injury. Several case-control studies demonstrated increased VWF levels in ischemic stroke patients6-11 and high plasma levels of VWF were found to be an independent risk factor for ischemic stroke.12 Preclinical experiments have shown that mice lacking VWF show significantly reduced brain injury and better functional outcome in experimental models of cerebral ischemia/reperfusion injury.13,14 Remarkably, initial VWF-mediated platelet adhesion rather than subsequent platelet aggregation contributes to ischemic brain injury with a prominent role for the interaction between the VWF A1 domain and platelet glycoprotein (GP)Ibα.15-19 It is currently not known exactly how platelets and VWF contribute to stroke progression in a way that is not strictly related to platelet-thrombus formation. Most likely, VWFmediated acute inflammation also aggravates acute ischemic stroke brain injury,20 but the exact mechanisms of VWF-mediated inflammatory responses in stroke remain poorly understood.
In this study, we used flow cytometry and a unique nanobody that blocks the VWF A1 domain to investigate the precise role of VWF in the acute cerebral inflammatory response during stroke. We specifically found that neutrophils, monocytes and T cells were recruited to the brain after stroke through a mechanism that involves the VWF A1 domain.
Methods
A detailed description of the methods can be found in the Online Supplement.
Animals and nanobodies
For this study, 10-week old VWF knockout (KO)21 and littermate wild-type (WT) C57BL/6 mice were used. All animal experiments were approved by the local ethical committee (P050/2017 KU Leuven, Leuven, Belgium) and were performed following the ARRIVE guidelines (www.nc3rs.org.uk), including randomization of treatment and analysis blind to the treatment. Mice were treated with a well-characterized nanobody targeting the VWF A1 domain (KB-VWF-006 bv; 10 mg/kg) or a control nanobody (KB-VWF-004 bv; 10 mg/kg).22
Cerebral ischemia and reperfusion injury model
Transient occlusion of the middle cerebral artery was performed as described previously.17 Briefly, a standardized silicon rubbercoated 6.0 nylon monofilament (6021; Doccol Corp, Redlands, CA, USA) was inserted via the right internal carotid artery to occlude the origin of the right middle cerebral artery. The suture was left in situ for 60 min. Immediately after the start of reperfusion, nanobodies were administered intravenously.
Neurological tests
Twenty-four hours after induction of transient middle cerebral artery occlusion, mice were subjected to the modified Bederson test23 and the grip test24 to assess global neurological and motor function, respectively, as described previously.25
Cerebral lesion quantification and assessment of bleeding
To measure cerebral infarct volumes, mice were euthanized 24 h after induction of transient middle cerebral artery occlusion. Coronal brain sections (2 mm thick) were stained with 2% 2,3,5- triphenyl-tetrazolium chloride (TTC, Sigma-Aldrich, St Louis, MO, USA) to visualize cerebral infarcts. The presence of cerebral hemorrhages was assessed macroscopically.
Table 1.
Antibody cocktails used to discriminate between different white blood cell subtypes.
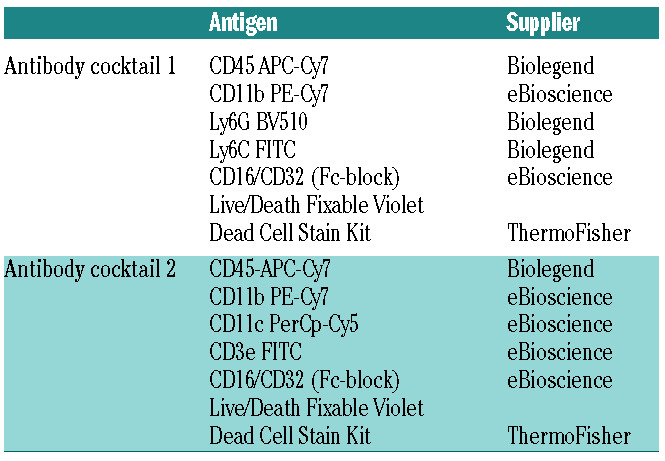
Flow cytometry
Twenty-four hours after stroke, mice were euthanized and perfused with 20 mL of phosphate-buffered saline. Single-cell suspensions were made of the ischemic (ipsilateral) and non-ischemic (contralateral) hemispheres as previously described.26 Next, cells were incubated with appropriate antibody cocktails (Table 1) containing Fc-blocker. Live cells were stained with Live/Death violet cell viability staining (L34963; ThermoFisher; Waltham, MA, USA) after which they were fixed and analyzed with a FACSVerse flow cytometer (BD, Franklin Lakes, NJ, USA) and BD Facs Suite software.
Immunofluorescence
Twenty-four hours after stroke, mice were euthanized, and the brains were dissected. Nine micron thick sections were stained for the presence of neutrophils (rat anti-mouse Ly6G, 1/500, eBioscience, San Diego, CA, USA), T cells (Armenian hamster antimouse CD3e, 1/500, Biolegend, San Diego, CA, USA), VWF (rabbit anti-human VWF, 1/1500, Dako, Santa Clara, CA, USA) or platelets (rat anti-mouse GPIX, 1/100, emfret, Würzburg, Germany). A lectin dye (FITC conjugated lectin from Lycopersicon esculentum, 1/500, Sigma-Aldrich) was used to stain the microvasculature.
Statistical analysis
Statistical analysis was performed with Graph Pad Prism Version 8.1.2. Prior to statistical analysis, a D’Agostino and Pearson normality test was used to check data distribution. One-way analysis of variance with a Dunnett post-hoc test or a Mann- Whitney test was used for statistical comparison of infarct size and immune cell infiltration when applicable. In the case of non-parametric data (Bederson and grip-test scores) a Kruskal-Wallis test with post-hoc Dunn correction was performed. Infarct size is represented as mean ± standard deviation. Bederson and grip-test scores are shown as scatter plots with the median. Immune cell recruitment is shown as a minimum-maximum box plot, with the median.
Results
von Willebrand factor deficiency reduces immune cell recruitment to the brain after ischemic stroke
To examine the cerebral immune cell response mediated by VWF, we performed flow cytometric analysis of single-cell suspensions prepared from brain tissue isolated from WT and VWF KO mice subjected to stroke. After stroke, mice were perfused, their brains harvested and subsequently divided into ipsilateral (affected by stroke) and contralateral (unaffected) hemispheres.
In agreement with previous reports,13,14 we observed significantly reduced cerebral infarcts in VWF KO mice, compared to WT mice (P<0.01) (Figure 1A). Twenty-four hours after stroke, the ipsilateral hemisphere showed increased numbers of infiltrated white blood cells (WBC) compared to the contralateral hemisphere in both the WT (P<0.005) and VWF KO (P<0.05) mice. However, average recruitment of WBC in the ipsilateral hemisphere of VWF KO mice was two-fold lower than in WT mice (5596 ± 1644 vs. 12435 ± 2083, respectively; P<0.05) (Figure 1B). The numbers of both myeloid and lymphoid WBC were significantly reduced in the brains of VWF KO mice compared to those in WT mice (P<0.05) (Figure 1C and D).
von Willebrand factor deficiency leads to reduced recruitment of inflammatory monocytes, neutrophils and T cells
To better determine which inflammatory cells were potentially recruited by VWF to the affected brain tissue during stroke, we used two antibody cocktails that allowed discrimination and quantification of recruited inflammatory monocytes, neutrophils, T cells and CD3neg lymphocytes (B cells and natural killer cells) (Table 1).
As shown in Figure 1E-H, all four subsets of immune cells were significantly increased in the ipsilateral brain of WT mice 24 h after stroke, compared to their numbers in the unaffected contralateral hemisphere. However, despite the presence of an infarct core in the affected ipsilateral hemisphere of VWF KO mice, the numbers of neutrophils and T cells did not increase and remained similar to the baseline values of the contralateral hemisphere (Figure 1E and F). Hence, the absolute numbers of recruited neutrophils and T cells was significantly higher in the ischemic brain of WT mice than in VWF KO mice (2017 ± 733 vs. 512 ± 203, P<0.05 and 974 ± 184 vs. 244 ± 44, P<0.01, respectively). These results suggest an important role for VWF in recruiting both neutrophils and T cells to the affected brain tissue during ischemic stroke.
A similar, but less marked, trend was observed for inflammatory monocytes (Figure 1G). In VWF KO mice, the ischemic hemisphere contained significantly more inflammatory monocytes than did the unaffected hemisphere, but still significantly fewer than the number of inflammatory monocytes that were recruited to the ischemic brain of WT mice (2466 ± 955 vs. 6760 ± 1414; P< 0.05).
No differences regarding CD3neg lymphocytes were observed between WT and VWF KO mice as similar numbers were recruited in the ischemic hemispheres of both groups (1162 ± 245 vs. 585 ± 188 respectively, P>0.05) (Figure 1H). Leukocyte blood counts and circulating platelet-leukocyte complexes were similar between VWF KO and VWF WT mice 24 h after stroke (data not shown).
Visualization of von Willebrand factor-mediated thromboinflammation in the ischemic stroke brain
For visualization of VWF-mediated thromboinflammation, immunofluorescent staining of platelets and leukocytes was performed on brains obtained from VWF WT and KO mice 24 h after stroke (Figures 2 and 3). In VWF KO mice, very few platelet accumulations were found within the ipsilateral, stroke-affected brain (Figure 2A). In contrast, platelet/VWF-rich microthrombi were found frequently throughout the ipsilateral brain of VWF WT mice, underscoring the importance of VWF-mediated platelet adhesion in the ischemic stroke brain (Figure 2BD). Platelet/VWF-rich microthrombi were absent in the contralateral hemisphere of both WT and VWF KO mice (data not shown).
Next, we visualized immune cell recruitment in VWF WT and KO mice. Since previous studies found no major role for monocytes,27 but an important detrimental role for both T cells28 and neutrophils29 in the acute phase of ischemic stroke, we focused on visualizing neutrophils and T cells in the stroke brain. To stain the vasculature in both VWF WT and KO mice, a sensitive lectin staining of the endothelium was performed (Figure 3). Using a specific histological marker for neutrophils (Ly6G), we observed that neutrophils were more frequently present within the ipsilateral side of the brain of VWF WT mice than in that of VWF KO mice (Figure 3A and B). Since the smaller infarct sizes observed in VWF KO mice might bias neutrophil quantification by flow cytometry, we also quantified neutrophil recruitment in the ischemic infarct core in both VWF KO and WT mice by histology. Importantly, analysis of fixed areas of 1 mm2 in the infarct core corroborated our flow cytometric data, arguing against a nonspecific effect related to smaller infarct sizes. Quantification of neutrophil recruitment to the infarct core revealed a two-fold reduction of neutrophil density in VWF KO brains compared to that in WT mice (Figure 3C). Intriguingly, neutrophils were frequently observed within the microcirculation (Figure 3A and B). To investigate this further, both intravascular and extravascular neutrophils were counted in brain sections of VWF WT mice. On average, 66 ± 4% of neutrophils were found within the vasculature of the ischemic hemisphere, while the remaining neutrophils were already extravasated (Figure 3D). A comparable observation was made when we stained for T cells, which were also occasionally found within the microvasculature of VWF WT mice (Figure 3E). Due to the low number of T cells within the ischemic brain, quantification of T cells was not feasible. Lastly, T cells were virtually absent in the ipsilateral brain hemisphere of VWF KO mice (Figure 3F), confirming our flow cytometric data.
Inhibition of the von Willebrand factor A1 domain reduces the recruitment of neutrophils, monocytes and T cells and limits ischemic stroke brain injury
Given the central role of the VWF A1 domain in cerebral ischemia/reperfusion injury,30 we next wanted to unravel its potential inflammatory contribution in ischemic stroke. To this end, we used a nanobody (KBVWF- 006bv) that specifically binds the VWF A1 domain, inhibiting its interaction with the platelet receptor GPIbα.22 Intravenous treatment with 10 mg/kg of the nanobody was started immediately after establishment of reperfusion. The mean residence time of the nanobody is 3.5 h which allows blocking of the VWF-A1 domain during the acute reperfusion phase. As a control, a nonspecific nanobody (KB-VWF-004bv) was administered. Twenty-four hours after stroke, mice treated with the anti-VWF A1 nanobody KB-VWF-006bv had significantly less ischemic stroke brain damage than had control-treated mice (Figure 4A and B). This translated into an improved motor score (Figure 4C) and neurological behavior (Figure 4D), although the difference was only statistically significant for the latter. Of note, no intracranial bleeding was observed in any of the mice treated with the VWF A1 nanobody. These data further corroborate the crucial involvement of the VWF-GPIbαaxis in cerebral ischemia/reperfusion injury.15-17,19
Targeting the VWF A1 domain also significantly reduced inflammatory cell recruitment to the ischemic brain (Figure 5). Indeed, similar to our results in VWF KO mice, flow cytometric analysis revealed that inhibition of VWF A1 also led to a two-fold reduction of immune cell recruitment in the brains of treated mice compared to that in control mice (7095 ± 2550 vs. 16310 ± 3980, respectively; P<0.005) (Figure 5A). Both myeloid (5195 ± 2259 vs. 11978 ± 3322; P<0.05) (Figure 5B) and lymphoid WBC counts (1529 ± 269 vs. 3017 ± 514; P<0.05) (Figure 5C) were reduced in the ipsilateral hemispheres of VWF A1 nanobody-treated mice. Specifically, inhibition of the VWF A1 domain reduced the number of infiltrated inflammatory monocytes (3726 ± 1824 vs. 8266 ± 2651; P<0.05) (Figure 5D), neutrophils (304 ± 94 vs. 1557 ± 317; P<0.0005) (Figure 5E) and T cells (487 ± 93 vs. 1111 ± 248; P<0.05) (Figure 5F) in the ipsilateral hemisphere of the brain, 24 h after stroke. These data suggest that the inflammatory effect of VWF in ischemic stroke is mediated through the VWF A1 domain.
Figure 1.

von Willebrand factor deficiency leads to a reduction of the number of monocytes, neutrophils and T cells to the brain after acute ischemic stroke. (A-H) Transient focal cerebral ischemia was induced by occlusion of the right middle cerebral artery for 60 min in wild-type (WT) and von Willebrand factor (VWF) knockout (KO) mice. This was followed by 23 h of reperfusion, after which edema-corrected brain infarct volumes were quantified by planimetric analysis (A) and white blood cell recruitment to each hemisphere was determined by flow cytometry (B-H). The total number of white blood cells (CD45high) was analyzed (B) as well as the myeloid (CD45high; CD11b+) (C) and lymphoid white blood cells (CD45high; CD11b-; CD11c-) (D). More specifically, neutrophils (CD45high; CD11b+; Ly6G+) (E), T cells (CD45high; CD11b-; CD11c-; CD3e+) (F); inflammatory mo - nocytes (CD45high; CD11b+; Ly6C+; Ly6G-) (G) and CD3neg lymphocytes (CD45high; CD11b-; CD11c-; CD3e-) (H) were quantified. Data are represented as box plots showing all data points and the median value, except for infarct size which is shown as mean ± standard deviation. *P<0.05; **P<0.01; ***P<0.005; ****P<0.001; ns: not statistically significant. (n=8-9). WBC: white blood cells; Ipsi: ipsilateral cerebral hemisphere; contra: contralateral cerebral hemisphere.
Discussion
As the main finding from this study, we report that VWF mediates an inflammatory response during cerebral ischemia/reperfusion via the recruitment of inflammatory monocytes, neutrophils and T cells, through a mechanism that is dependent on the VWF A1 domain. Blocking the A1 domain reduced the inflammatory response and improved stroke outcome.
Current ischemic stroke treatment is aimed at achieving reperfusion of the ischemic brain as soon as possible. When thrombolysis or thrombectomy is initiated, an unsalvageable infarct core often already exists. Reperfusion therapy is therefore aimed at salvaging the penumbra to limit further ischemic brain damage. Unfortunately, rescue of the perfused salvageable penumbra is not always achieved, which has led to the concept of cerebral ischemia/reperfusion injury.31,32 In recent years, VWF has emerged as an important mediator of cerebral ischemia/reperfusion injury.30 We and others reported that absence of VWF protected mice from ischemic stroke brain injury, without increasing the risk of cerebral bleeding. 13,14 Intriguingly, the detrimental role of VWF in the ischemic brain appears to be distinct from its role in hemostasis. Whereas hemostatic thrombus formation requires both platelet adhesion and platelet aggregation, the latter does not seem to play a major role in reperfusion injury after ischemic stroke.15,16 Besides thrombotic events, ischemic stroke is also characterized by a strong inflammatory response that occurs in the brain after reperfusion.33 Since VWF is a known modulator of inflammation, 20 a thromboinflammatory role for VWF in the ischemic stroke brain was proposed. Yet, the inflammatory mechanisms by which VWF mediates reperfusion injury in the ischemic stroke brain remain poorly understood. Using flow cytometric analysis of single-cell suspensions prepared from brain tissue from VWF KO and WT mice and histology experiments, we found that the acute immune response in the brain after stroke was greatly reduced in the absence of VWF. These results further corroborate the notion that VWF contributes to cerebral inflammation in stroke.34-36 In agreement with Khan et al., we found that neutrophil recruitment to the ischemic stroke brain was significantly reduced in VWF-deficient mice compared to that in WT mice.35 Interestingly, we additionally identified a two-fold decrease in the recruitment of inflammatory monocytes and a four-fold decrease in the recruitment of T cells to the ischemic stroke brain of VWF-deficient mice. Importantly, not only the overall number but also the density of neutrophils in the affected brain tissue was reduced. Furthermore, there was no direct link between the reduction in infarct size (2-fold) and the reduction of all subsets of leukocytes (2- to 5-fold). Together, these observations go against a potential nonspecific effect due to smaller infarct volumes.
Figure 2.

Immunofluorescent visualization of thromboinflammation in the ipsilateral hemisphere of mice 24 hours after ischemic stroke brain injury. Transient focal cerebral ischemia was induced in von Willebrand factor (VWF) wild-type (WT) or knockout (KO) mice by occluding the right middle cerebral artery for 60 min. This was followed by 23 hours of reperfusion, after which brain sections were stained for VWF (red), platelets (green) and nuclei (blue). (A) Only a few platelets were found within the ischemic brain of VWF KO mice. (B-D) Clumps of VWF together with platelets were frequently found attached to the vessel wall within the ipsilateral, stroke-affected hemisphere. Panel (D) is a magnification of the white box in panel C. Scale bars are 50 m except for that in panel (D) in which the scale bar is 25 m. Images are representative of three animals per genotype.
Figure 3.

Immunofluorescent visualization of neutrophils and T cells in the ipsilateral hemisphere of mice 24 hours after ischemic stroke brain injury. Transient focal cerebral ischemia was induced in von Willebrand factor (VWF) wild-type (WT) or knockout (KO) mice by occluding the right middle cerebral artery for 60 min. This was followed by 23 h of reperfusion, after which, brain sections were stained for blood vessels and neutrophils or T cells. (A, B) Neutrophils were stained with a marker for Ly6G (red) and blood vessels with a lectin stain (green). Neutrophils are marked with an *. (C) Quantification of the number of neutrophils/mm2 in the infarct core of WT and VWF KO mice (n=3). Data are the mean ± standard deviation. (D) The number of neutrophils within and outside the vasculature (n=3). Data are the mean ± standard deviation. (E, F) T cells were stained with a marker for CD3 (red) and blood vessels with a lectin stain (green). T cells are marked with an *. Scale bars are 20 m. Images are representative of three mice per genotype.
Ischemic stroke induces an extensive inflammatory response, with various types of immune cells transmigrating over the activated endothelium to invade the damaged brain.33 The key steps mediating the initial phase of leukocyte-mediated stroke brain damage are not yet fully resolved. During cerebral ischemia, the brain endothelium becomes rapidly activated, resulting in the upregulation of cell adhesion molecules.37 Together with transient disruption of the blood-brain barrier, this allows entry of leukocytes into the brain. Our results now suggest that VWF is an important molecule involved in the initial recruitment of inflammatory monocytes, T cells and neutrophils leading to reperfusion injury. The potential role of monocytes in the acute phase of ischemic stroke is not clear27 but interactions between monocytes and VWF-platelet complexes have been shown to contribute to monocyte diapedesis in vitro.38 Both neutrophils and T cells have been found to cause cerebral ischemia/reperfusion injury by obstructing the microcirculation, contributing to the no-reflow phenomenon.37,39,40 Interestingly, we observed a significant number of neutrophils and T cells within the microvasculature of WT mice during the early phase after stroke. Whether these cells are in the process of extravasating or really plugging the microcirculation is hard to ascertain from our current study. Together with VWF/platelet aggregates, these intravascular immune cells can occlude brain capillaries and impair microcirculatory reperfusion. Our observations are in line with those of other studies on neutrophil recruitment to the brain after stroke. Indeed, neutrophils have been found trapped in the cerebral microcirculation in both murine41,42 and baboon39 stroke models. Importantly, also in human stroke patients, intravascular neutrophil accumulation has been observed in postmortem brain tissue.41,43 Similarly, regulatory T cells were found to increase cerebral thrombus formation, impair cerebral reperfusion and cause overall microvascular dysfunction in the acute reperfusion phase in a murine stroke model.44 Accordingly, depletion of neutrophils or T cells in the acute phase of ischemic stroke has a protective effect.28,35,44-46 Our results now identify VWF as a key adhesion molecule that could be implicated in the recruitment of these immune cells, most likely via a platelet-dependent mechanism, potentially triggering microvascular obstructions in the reperfused ischemic stroke brain.
A key finding of this study is that we could pinpoint the pathogenic contribution of VWF to its A1 domain using a nanobody that specifically targets the platelet GPIbαbinding site in the VWF A1 domain.22 Indeed, anti- VWF A1 nanobody treatment resulted in a similar reduction of leukocyte recruitment as that present in VWF-deficient animals (Online Supplementary Table S1). This nanobody was previously shown to reduce overall leukocyte recruitment in a model of immune complex-mediated vasculitis and a model of irritant contact dermatitis, suggesting that the pro-inflammatory properties of the VWF A1 domain can play an important role in several settings. 22 Pendu et al. previously demonstrated that VWF, potentially via its A1 domain, was able to directly interact with PSGL-1 and several 2 integrins on leukocytes.47 PSGL-1 and 2 integrins are present on monocytes, neutrophils and T cells and their interaction with VWF is one possible mechanism by which VWF could recruit leukocytes in the ischemic brain. Another mechanism by which VWF most likely recruits leukocytes involves platelets that are bound to the VWF A1 domain via their GPIbαreceptor. Platelets can subsequently bind leukocytes through well-established interactions.48 This VWF-platelet mediated leukocyte recruitment was previously presented in an elegant study by Petri and colleagues.49 They observed that peritoneal inflammation triggered the release and cell surface deposition of endothelial VWF which allowed platelets to adhere to the endothelium via GPIbαand subsequently recruit leukocytes. Blocking VWF or depleting platelets had an equally strong anti-inflammatory effect in their model, highlighting the inflammatory role of VWFassociated platelets. A similar course of events is most likely also occurring in the ischemic brain. The detrimental interaction between VWF and platelets has already been extensively studied in the setting of stroke.15,17,19 Our results again highlight the importance of the inflammatory component of the VWF-GPIbαinteraction in the ischemic stroke brain. Furthermore, our findings corroborate the observations of Schuhmann et al., who, after inhibition of platelet GPIbα, also observed an attenuated inflammatory response, which protected mice from cerebral ischemiareperfusion injury.50
Figure 4.

Inhibition of the von Willebrand factor A1 domain protects mice from acute ischemic stroke. Transient focal cerebral ischemia was induced by occluding the right middle cerebral artery for 60 min, followed by 23 h of reperfusion. Immediately at the start of reperfusion, mice were intravenously treated with 10 mg/kg of either control (KB-VWF-004 bv) or inhibitory anti-VWF A1 nanobody (KB-VWF-006 bv). (A) Edema-corrected brain infarct volumes were quantified by planimetric analysis 24 h after stroke. (B) Representative TTC staining of three consecutive brain sections. (C) Motor function was examined using the grip test. (D) Neurological outcome 24 h after stroke was assessed using the Bederson test. Data are represented as scatter plots showing all data points and the median value, except for infarct size which is shown as mean ± standard deviation. *P<0.05; **P<0.01 (n=10-11).
Figure 5.

The von Willebrand factor A1 domain recruits monocytes, neutrophils and Tcells to the brain after acute ischemic stroke. Transient focal cerebral ischemia was induced by occluding the right middle cerebral artery for 60 min, followed by 23 h of reperfusion. Immediately at the start of reperfusion, mice were intravenously treated with 10 mg/kg of either control (KBVWF- 004 bv) or inhibitory anti- VWF A1 nanobody (KB-VWF- 006 bv). Twenty-four hours after the transient arterial occlusion, recruitment of specific subsets of white blood cells (WBC) to each hemisphere was determined by flow cytometry. (A) WBC (CD45high). (B) Myeloid WBC (CD45high; CD11b+). (C) Lymphoid WBC (CD45high; CD11b-; CD11c-). (D) Inflammatory monocytes (CD45high; CD11b+; Ly6C+; Ly6G- ). (E) Neutrophils (CD45high; CD11b+; Ly6G+). (F) T cells (CD45high; CD11b-; CD11c-; CD3e+). (G) CD3neg lymphocytes (CD45high; CD11b-; CD11c-; CD3e-). *P<0.05; **P<0.01; ***P<0.005; ****P<0.001 (n=10-11). Ipsi: ipsilateral cerebral hemisphere; contra: contralateral cerebral hemisphere.
Our study has some limitations that need to be addressed. First, all outcomes were measured at 24 h after ischemic stroke. This time-point was chosen to specifically investigate the early, acute inflammatory response after stroke. Nevertheless, it was previously shown that leukocyte infiltration peaks at 72 h after ischemic stroke in mice.51 It would be interesting to investigate VWF-mediated inflammatory responses at later time-points or even during initial ischemia. Second, the set-up of our study did not allow identification of the specific interactions that mediate leukocyte-platelet or leukocyte-VWF binding in the setting of ischemic stroke. Specific inhibitors of platelet-leukocyte or VWF-leukocyte interactions would be needed to further elucidate the specific leukocyte interactions. Lastly, histology or flow cytometric analysis of brain tissue will not perfectly reflect the dynamic inflammatory processes going on in the ischemic stroke brain. Future studies are needed to visualize VWFplatelet- leukocyte interactions in vivo by, for example, intravital microscopy.
In conclusion, we found that VWF is involved in the recruitment of inflammatory monocytes, neutrophils and T cells to the ischemic brain via its A1 domain. Inhibition of VWF-mediated thromboinflammation, for example by blocking the VWF A1-GPIbαinteraction, or by cleaving VWF with ADAMTS13,4 could become a promising treatment strategy for the prevention of cerebral ischemia/reperfusion injury.
Supplementary Material
Funding Statement
Funding: This work was supported by Fonds voor Wetenschappelijk Onderzoek - Vlaanderen (research grants G.0A86.13, G.0785.17 and 1509216N to SFDM), by research grants from KU Leuven (OT/14/099 and ISP/14/02L2 to SFDM) and by a research grant from the Queen Elisabeth Medical Foundation (to SFDM). FD is a postdoctoral fellow of Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO, 12U7818N). KM was a H2020 Marie Skłodowska-Curie Actions fellow (under agreement number 747993, “VWF and NETs”).
References
- 1.Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics - 2018 update: a report from the American Heart Association. Circulation. 2018;137(12):1-426. [DOI] [PubMed] [Google Scholar]
- 2.Goyal M, Menon BK, van Zwam WH, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387(10029):1723-1731. [DOI] [PubMed] [Google Scholar]
- 3.De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. 2016;47(4):1165-1172. [DOI] [PubMed] [Google Scholar]
- 4.De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: an emerging target in stroke therapy. Stroke. 2012;43(2):599-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonneveld MAH, de Maat MPM, Leebeek FWG. Von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta-analysis. Blood Rev. 2014;28(4):167-178. [DOI] [PubMed] [Google Scholar]
- 6.Bongers TN, de Maat MPM, van Goor MLPJ, et al. High von Willebrand factor levels increase the risk of first ischemic stroke: influence of ADAMTS13, inflammation, and genetic variability. Stroke. 2006;37(11): 2672-2677. [DOI] [PubMed] [Google Scholar]
- 7.Andersson HM, Siegerink B, Luken BM, et al. High VWF, low ADAMTS13, and oral contraceptives increase the risk of ischemic stroke and myocardial infarction in young women. Blood. 2012;119(6):1555-1160. [DOI] [PubMed] [Google Scholar]
- 8.Tóth NK, Székely EG, Czuriga-Kovács KR, et al. Elevated factor VIII and von Willebrand factor levels predict unfavorable outcome in stroke patients treated with intravenous thrombolysis. Front Neurol. 2018;8:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catto AJ, Carter AM, Barrett JH, Bamford J, Rice PJ, Grant PJ. von Willebrand factor and factor VIII: C in acute cerebrovascular disease. Relationship to stroke subtype and mortality. Thromb Haemost. 1997;77(6): 1104-1108. [PubMed] [Google Scholar]
- 10.Kraft P, Drechsler C, Gunreben I, et al. Von Willebrand factor regulation in patients with acute and chronic cerebrovascular disease: a pilot, case-control study. PLoS One. 2014;9(6):e99851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanson E, Jood K, Karlsson S, Nilsson S, Blomstrand C, Jern C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J Thromb Haemost. 2011;9(2):275-281. [DOI] [PubMed] [Google Scholar]
- 12.Wieberdink RG, van Schie MC, Koudstaal PJ, et al. High von Willebrand factor levels increase the risk of stroke: the Rotterdam study. Stroke. 2010;41(10):2151-2156. [DOI] [PubMed] [Google Scholar]
- 13.Kleinschnitz C, De Meyer SF, Schwarz T, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. 2009;113(15):3600-3603. [DOI] [PubMed] [Google Scholar]
- 14.Zhao BQ, Chauhan AK, Canault M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114(15): 3329-3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Meyer SF, Schwarz T, Deckmyn H, et al. Binding of von Willebrand factor to collagen and glycoprotein Ibalpha, but not to glycoprotein IIb/IIIa, contributes to ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2010;30(10):1949-1951. [DOI] [PubMed] [Google Scholar]
- 16.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115(17):2323-2330. [DOI] [PubMed] [Google Scholar]
- 17.De Meyer SF, Schwarz T, Schatzberg D, Wagner DD. Platelet glycoprotein Ibα is an important mediator of ischemic stroke in mice. Exp Transl Stroke Med. 2011;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraft P, Schuhmann MK, Fluri F, et al. Efficacy and safety of platelet glycoprotein receptor blockade in aged and comorbid mice with acute experimental stroke. Stroke. 2015;46(12):3502-3506. [DOI] [PubMed] [Google Scholar]
- 19.Li T-T, Fan M-L, Hou S-X, et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br J Pharmacol. 2015;172(15):3904-3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15(7):1285-1294. [DOI] [PubMed] [Google Scholar]
- 21.Denis C, Methia N, Frenette PS, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95(16):9524-9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aymé G, Adam F, Legendre P, et al. A novel single-domain antibody against von Willebrand factor A1 domain resolves leukocyte recruitment and vascular leakage during Inflammation. Arterioscler Thromb Vasc Biol. 2017;37(9):1736-1740. [DOI] [PubMed] [Google Scholar]
- 23.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472-476. [DOI] [PubMed] [Google Scholar]
- 24.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 1995;92(12):5341-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhenne S, Denorme F, Libbrecht S, et al. Platelet-derived VWF is not essential for normal thrombosis and hemostasis but fosters ischemic stroke injury in mice. Blood. 2015;126(14):1715-1722. [DOI] [PubMed] [Google Scholar]
- 26.Denorme F, Manne BK, Portier I, et al. Platelet necrosis mediates ischemic stroke outcome in mice. Blood. 2020;135(6):429-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt A, Strecker J-K, Hucke S, et al. Targeting different monocyte/macrophage subsets has no impact on outcome in experimental stroke. Stroke. 2017;48(4):1061-1069. [DOI] [PubMed] [Google Scholar]
- 28.Kleinschnitz C, Schwab N, Kraft P, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115(18):3835-3842. [DOI] [PubMed] [Google Scholar]
- 29.Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denorme F, De Meyer SF. The VWF-GPIb axis in ischaemic stroke: lessons from animal models. Thromb Haemost. 2016;116(4): 597-604. [DOI] [PubMed] [Google Scholar]
- 31.Kidwell CS, Saver JL, Starkman S, et al. Late secondary ischemic injury in patients receiving intraarterial thrombolysis. Ann Neurol. 2002;52(6):698-703. [DOI] [PubMed] [Google Scholar]
- 32.Mizuma A, You JS, Yenari MA. Targeting reperfusion injury in the age of mechanical thrombectomy. Stroke. 2018;49(7):1796-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res. 2016;119(1):142-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujioka M, Nakano T, Hayakawa K, et al. ADAMTS13 gene deletion enhances plasma high-mobility group box1 elevation and neuroinflammation in brain ischemia-reperfusion injury. Neurol Sci. 2012;33(5):1107-1115. [DOI] [PubMed] [Google Scholar]
- 35.Khan MM, Motto DG, Lentz SR, Chauhan AK. ADAMTS13 reduces VWF-mediated acute inflammation following focal cerebral ischemia in mice. J Thromb Haemost. 2012;10(8):1665-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhanesha N, Prakash P, Doddapattar P, et al. Endothelial cell-derived von Willebrand factor is the major determinant that mediates von Willebrand factor-dependent acute ischemic stroke by promoting postischemic thrombo-inflammation. Arterioscler Thromb Vasc Biol. 2016;36(9):1829-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23(8):879-894. [DOI] [PubMed] [Google Scholar]
- 38.Popa M, Tahir S, Elrod J, et al. Role of CD40 and ADAMTS13 in von Willebrand factormediated endothelial cell–platelet–monocyte interaction. Proc Natl Acad Sci U S A. 2018;115(24):E5556-E5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Poly - morphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22(10):1276-1283. [DOI] [PubMed] [Google Scholar]
- 40.Okada Y, Copeland BR, Fitridge R, Koziol JA, del Zoppo GJ. Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke. 1994;25(9):1847-1853. [DOI] [PubMed] [Google Scholar]
- 41.Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129(2):239-257. [DOI] [PubMed] [Google Scholar]
- 42.Otxoa-de-Amezaga A, Gallizioli M, Pedragosa J, et al. Location of neutrophils in different compartments of the damaged mouse brain after severe ischemia/reperfusion. Stroke. 2019;50(6):1548-1557. [DOI] [PubMed] [Google Scholar]
- 43.Ritzel RM, Lai Y-J, Crapser JD, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 2018;136(1):89-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinschnitz C, Kraft P, Dreykluft A, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121(4):679-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-in ischemic stroke. Circulation. 2006;113(17):2105-2112. [DOI] [PubMed] [Google Scholar]
- 46.Herz J, Sabellek P, Lane TE, Gunzer M, Hermann DM, Doeppner TR. Role of neutrophils in exacerbation of brain injury after focal cerebral ischemia in hyperlipidemic mice. Stroke. 2015;46(10):2916-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pendu R, Terraube V, Christophe OD, et al. P-selectin glycoprotein ligand 1 and 2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108(12):3746-3752. [DOI] [PubMed] [Google Scholar]
- 48.Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol. 2018;9:2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petri B, Broermann A, Li H, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116(22):4712-4719. [DOI] [PubMed] [Google Scholar]
- 50.Schuhmann MK, Guthmann J, Stoll G, Nieswandt B, Kraft P, Kleinschnitz C. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J Neuroinflammation. 2017;14(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849-1857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.