

Abstract
Diabetic wounds are characterized by persistent accumulation of pro-inflammatory monocytes/macrophages (Mo/MΦ) and impaired healing. However, the mechanisms underlying the persistent accumulation of Mo/MΦ remain poorly understood. Here, we report that Ly6C+F4/80lo/- Mo/MΦ proliferate at higher rates in wounds of diabetic mice compared to non-diabetic mice, leading to greater accumulation of these cells. Unbiased scRNAseq analysis of combined non-diabetic and diabetic wound Mo/MΦ revealed a cluster, populated primarily by cells from diabetic wounds, for which genes associated with the cell cycle were enriched. In a screen of potential regulators, CCL2 levels were increased in wounds of diabetic mice, and subsequent experiments showed that local CCL2 treatment increased Ly6C+F4/80lo/- Mo/MΦ proliferation. Importantly, adoptive transfer of mixtures of CCR2−/− and CCR2+/+ Ly6Chi Mo indicated that CCL2/CCR2 signaling is required for their proliferation in the wound environment. Together, these data demonstrate a novel role for the CCL2/CCR2 signaling pathway in promoting skin Mo/MΦ proliferation, contributing to persistent accumulation of Mo/MΦ and impaired healing in diabetic mice.
Introduction
Diabetes and related complications represent a growing public health crisis throughout the world (1). Among diabetes-related complications, non-healing ulcers are a major concern, often leading to amputation and even death. The socioeconomic burden of chronic wounds is escalating, with annual Medicare costs as high as $13 billion (1, 2). Despite the heavy toll of chronic wounds, the causes of impaired healing in diabetes are not well understood (3).
Monocytes (Mo) and macrophages (MΦ) play critical roles in wound healing, by regulating inflammation, removing debris and dead cells and producing cytokines and growth factors that promote healing (3). However, a common characteristic of poorly healing diabetic wounds is chronic inflammation, associated with persistent accumulation of inflammatory Mo/MΦ (3). Our lab and others have shown that diabetes leads to dysregulation of myelopoiesis in bone marrow which may contribute to persistent accumulation of inflammatory Mo/MΦ in skin wounds (4, 5). Moreover, our recent findings show that proliferation of skin wound Ly6C+F4/80lo/- Mo/MΦ contributes to their accumulation during normal healing in non-diabetic mice (6). However, whether diabetes influences proliferation of inflammatory Mo/MΦ remains to be established.
Many factors, both cell intrinsic and environmental, have been reported to induce proliferation of Mo/MΦ, but these appear to be context-dependent, varying between tissues and inflammatory conditions (6–9). In our recent study, the data indicated that the wound environment likely regulates Mo/MΦ proliferation (6). We screened a number of potential regulators that have been reported to influence Mo/MΦ proliferation, including IL-1ß, IL-6, IL-4, and M-CSF (6–10). However, our data did not support a role for any of these candidates in regulating Mo/MΦ proliferation in skin wounds (6). Other cytokines including CCL2, CCL3, TNF-α, IL-10 and IL-33 have been reported to influence Mo/MΦ activity, although their role in regulating wound Mo/MΦ proliferation has not been explored (11–13). Thus, the factor(s) that regulate Mo/MΦ proliferation during wound healing remain to be elucidated.
The purposes of this study were to determine whether diabetes influences Mo/MΦ proliferation in skin wounds, and to identify potential factor(s) that regulate Mo/MΦ proliferation in wounds. Indeed, we found that inflammatory Ly6C+F4/80lo/- Mo/MΦ exhibit significantly higher rates of proliferation in wounds of diabetic compared to non-diabetic mice. Moreover, our data demonstrate a novel role for CCL2/CCR2 signaling in promoting proliferation of inflammatory Mo/MΦ in wounds.
Materials and Methods
Animals
Male C57Bl/6 (Non-diabetic controls, CD45.2), BKS.Cg-Dock7m +/+ Leprdb/J (db/db, diabetic), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1), and B6(C)-Ccr2tm1.1Cln/J (CCR2−/−EGFP) mice (age 9 – 12 weeks) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in environmentally controlled conditions with a 12-hr light/dark cycle. Water and food were available ad libitum. With the exception of the single cell RNA sequencing, each experiment was performed at least twice with N= 3–6 mice per group/condition. To minimize bias, mice were randomly assigned to experimental groups and resulting samples were coded and analyzed in a blinded fashion. All animal studies were approved by the Animal care and se Committee of the University of Illinois at Chicago.
Wound model
Age matched non-diabetic and diabetic mice were subjected to excisional wounding (two wounds per mouse) of the dorsal skin with an 8mm biopsy punch as previously described (6). Tegaderm (3M,1626W) was used to cover the wounds until tissue harvest.
Wound closure was evaluated by digital pictures using Fiji Image J. Skin wounds were collected using a 12mm biopsy punch on day 3, 6, and 10 post-injury, followed by enzymatic digestion to obtain a single cell suspension for flow cytometry analysis, or snap frozen in liquid nitrogen for protein measurements as previously described (6).
Flow cytometry
Skin wound, femoral bone marrow cells and peripheral blood cells were used for flow cytometry analysis. For skin wound cells, Zombie Violet or Aqua (Biolegend, San Diego, CA, USA) were used to assess cell viability. Cells from bone marrow and blood were not stained with Zombie dyes due to low percentage of dead cells (<3%). After Fc receptor blocking with anti-CD16/32 antibody (Biolegend, clone S17011E), skin cells were labeled with anti-Ly6G–BV605 (Biolegend, clone 1A8), CD11b–APC/Fire750 (Biolegend, clone CBRM1/5), F4/80–PE (Biolegend, clone BM8), and Ly6C–Percp/cy5.5 (BD Biosciences, San Jose, CA, USA, clone AL–21) antibodies; cells from bone marrow and blood were stained with anti-Ly6G–BV605, CD11b–APC/Fire750, CD115–PE (clone AFS98), Ly6C–BV421 (BD Biosciences, clone AL–21), and/or cKit-Percp/cy5.5 (Biolegend, clone 2B8) antibodies.
For proliferation/cell cycle analysis, all cells were fixed and permeabilized, then intracellularly labeled with anti-Ki67 antibody (Abcam, Cambridge, MA, USA; ab15580) or its corresponding isotype control (Abcam, ab171870) followed by Alexa Fluor 488 anti-rabbit secondary antibody (Abcam, ab150077) incubation using BD Cytofix/Cytoperm™ kit. Finally, cells were incubated with FxCycle™ Far Red (ThermoFisher Scientific, Grand Island, NY, USA) 30 minutes before acquisition following the manufacturer’s instructions.
For single cell RNA sequencing, single cells were collected and pooled from skin wounds of two non-diabetic or two diabetic mice on day 6 post-injury as described above. Target cell population was sorted out as Zombie-CD45+CD11b+Ly6G- cells on BD FACSAria™ III sorter (BD Biosciences). Cells were first stained with Zombie Violet then blocked Fc receptors by anti-CD16/32 antibody followed by incubation of anti-Ly6G-BV605, CD11b–APC/Fire750, and anti-CD45-FITC (Biolegend, clone 30-F11).
For adoptive transfer experiments, cells were collected from 6 long bones of each donor mouse (2 femurs + 2 tibiae + 2 humeri). After Zombie Aqua staining and Fc receptor blocking, cells were labeled with anti-Ly6G-BV510 (Biolegend, clone 1A8), CD11b–APC/Fire750, CD115-PE, Ly6C-BV421, and cKit-Percp/cy5.5. Next, bone marrow immature monocytes were sorted out as Zombie-Ly6G-CD11b+cKit-CD115+Ly6Chi from CD45.1 mice or Zombie-Ly6G-CD11b+cKit-CD115+Ly6Chi EGFP+ from CCR2 KO EGFP mice on MoFlo Astrios EQ sorter (Beckman Coulter, Brea, California, USA).
All samples were analyzed on either LSR Fortessa with HTS (BD Biosciences) or CytoFLEX S (Beckman Coulter) cytometers. Data were analyzed using FlowJo (FlowJo LLC, Ashland, OR, USA).
For single cell imaging, skin Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ were pre-labeled and FAC sorted. 2000 cells from each population were collected on Amnis Flowsight (Luminex, Chicago, IL, USA) with further confirmed surface labeling of either anti-Ly6C-FITC (BD Biosciences, clone AL–21) or anti-F4/80-PE (Biolegend, clone 1A8) together with DNA dye DRAQ5 (Biolegend). Cell images were captured using CCD camera with 20x magnification on Flowsight and analyzed using IDEAS Software ((Luminex, Chicago, IL, USA). Five representative pictures of either Ly6C+F4/80lo/- Mo/MΦ or Ly6C-F4/80+ MΦ were presented.
Single cell RNA sequencing
Cells from skin wounds of either non-diabetic or diabetic mice were collected and sorted as described above. Fresh cells with viability over 85% were resuspended in cold DPBS with 10% FBS at concentration approximately 200 cells/μL then immediately processed using the Chromium platform (10X Genomics, San Francisco, CA, USA) for HMW gDNA extraction, GEM generation and barcoding. Library construction was prepared according to the manufacturer’s instructions and sequenced on HiSeq™ Sequencing Systems (Illumina, San Diego, CA, USA) with paired-end reads. Fastq files were generated and demultiplexed into single cells using Cell Ranger software (10X Genomics).
Single cell RNA analysis
Cell barcodes with < 10% mitochondrial expression were removed from the data set, and genes with fewer than 50 counts across all cells or expressed in fewer than 5% of the cells were removed. Gene expression was normalized to counts per million using TMM normalization with edgeR (14) and log-scaled. Principle component analysis of cells was computed, and the top 10 PCs, which captured 47% of total variance, were used downstream for clustering analysis.
Cells were clustered using k-means clustering with ten random initializations on a range of cluster numbers k (2 to 20). For each k, we evaluated the reproducibility of the repeated clustering runs by comparing the pair-wise distance between clustering results, computed as the fraction of co-clustered feature pairs between two clustering results relative to the number of co-clustered feature pairs within each result individually. This difference was averaged across all result pairs for each value of k to evaluate cluster robustness. k=4 was determined to be an optimal cluster count and used for downstream analysis.
Differentially expressed genes per cluster were detected using three statistical tests: Kruskal-Wallis, on log-scaled normalized gene expression levels; Fisher’s Exact Test, between the count of expressed vs unexpressed cells for each gene; and the area under the receiver operating characteristic curve (AUROC), treating each gene as a classifier for each cluster.
CCL2 protein measurement in skin wounds
Whole skin wounds from non-diabetic and diabetic mice were homogenized as previously described (6). Total protein levels were measured using Pierce™ Rapid Gold BCA Protein Assay Kit (ThermoFisher Scientific). Protein levels of CCL2, CCL3, TNF-α, IL-4, IL-10, M-CSF and IL-33 were determined using BioLegend LEGENDplex™ kit. All samples were analyzed on CytoFLEX S (Beckman Coulter) cytometer.
Local CCL2 protein treatment
On day 2 post-injury, recombinant mouse CCL2 protein (carrier-free, Biolegend) was administrated at a single dose of 60ng per wound by intradermal injection at four equally spaced sites around the periphery of each wound in non-diabetic mice (15 ng/injection, n=12 per group) (15). Control mice received equal volume of DPBS injection (n=12 per group). On day 3 post-injury, wounds treated with either CCL2 or DPBS were collected and processed as described above for flow cytometry analysis.
Adoptive transfer
Bone marrow Ly6Chi monocytes from donor mice were collected as described above by FAC sorting. Approximately 1.5–2 million cells from each donor strain were resuspended in cold DPBS and injected intradermally into skin wounds of the recipient mice at four equally spaced sites by the edge on day 2 (cells combined from CCR2 KO mice + CD45.1 mice to non-diabetic mice, n=5 per group) or day 2 and day 5 (cells from CD45.1 mice to non-diabetic or diabetic mice, n=3 per group). Approximately 18 hours post-adoptive transfer, skin wounds from recipient mice were collected on day 3 or day 6 post-injury and processed as described above for flow cytometry analysis to evaluate cell proliferation of the donor cells.
Statistics
Data are expressed as mean ± SEM. Statistical significance of differences was evaluated by Mann–Whitney test or ANOVA. A value of P < 0.05 was considered statistically significant.
Data Availability
Single-cell RNA sequencing data in this study are available in Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) with the accession code GSE154400. All other relevant data are available from the corresponding authors.
Results
Enhanced proliferation of Ly6C+ Mo/MΦ in skin wounds of diabetic versus non-diabetic mice
Consistent with previous work by us and other groups, diabetic mice showed significantly delayed wound closure compared to non-diabetic controls (3, 6, 16). By day 10 post-injury, wounds in diabetic mice achieved an average of only ~40% closure while wounds in non-diabetic mice reached an average of ~95% closure (Fig 1A).
Figure 1. Enhanced proliferation of Ly6C+ Mo/MΦ in skin wounds of diabetic versus non-diabetic mice.

(A) Wound closure was calculated as 100% minus the percentage of open area relative to the initial wound size on day 0 (n=4/group). (B) Gating strategy for identifying proliferation of Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ in skin wounds. (C) Numbers of Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ in non-injured skin (NS, n=4) and wounds on days 3 (n=10), 6 (n=12), and 10 (n=7) post-injury in non-diabetic (black bar) and diabetic (open bar) mice. (D) Numbers of Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ in S/G2/M phases of cell cycle (Ki67+FxCycle+) in non-injured skin and wounds. Data are mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001 diabetic vs non-diabetic groups by ANOVA.
Next, we measured the accumulation and proliferation of two major Mo/MΦ populations in skin wounds by flow cytometry, namely live Ly6G-CD11b+Ly6C+F4/80lo/- (inflammatory Mo/MΦ) and live Ly6G-CD11b+Ly6C-F4/80+ (mature MΦ) cells (Fig 1B). Both cell populations accumulated in skin wounds by day 3 post-injury with significantly higher cell numbers in diabetic mice than in non-diabetic mice on day 6 and day 10 post-injury (Fig 1C). Moreover, percentages of Ly6C+F4/80lo/- Mo/MΦ were also significantly higher in diabetic wounds than in non-diabetic wounds on day 6 and day 10 post-injury while percentages of Ly6C-F4/80+ MΦ were comparable between strains at all time point (Supplementary Fig 1A).
Importantly, and consistent with our previous findings, there were significantly higher numbers and percentages of Ly6C+F4/80lo/- Mo/MΦ in the S/G2/M phases of the cell cycle (Ki67+FxCycle+) compared to Ly6C-F4/80+ MΦ over the course of healing, in both diabetic and non-diabetic mice (Fig 1D and Supplementary Fig 1B). Although both diabetic and non-diabetic mice showed a peak of Ly6C+F4/80lo/- Mo/MΦ in S/G2/M phases on day 6 post-injury, diabetic mice had a significantly higher peak of proliferating Ly6C+F4/80lo/- Mo/MΦ which lasted through day10 post-injury (Fig 1D, Supplementary Fig 1B and 1C). In addition to extremely low numbers and percentages of Ly6C-F4/80+ MΦ in S/G2/M phases, there were no differences in proliferation of these latter cells at any time point between diabetic and non-diabetic mice (Fig 1D, Supplementary Fig 1B and 1D). Analysis of the forward and side scatter (FSC-A and SSC-A) characteristics and morphology of these cells on day 3 and day 6 post-injury indicated that Ly6C+F4/80lo/- Mo/MΦ were slightly smaller and clearly less granular than Ly6C-F4/80+ MΦ, indicating that the former exhibit a more monocytic morphology (Supplementary Fig 1E and 1F). Note that the full cell cycle analysis of Ly6C+F4/80lo/- MΦ /Mo and Ly6C-F4/80+ MΦ in both mouse strains can be found in the Supplementary Fig 1C and 1D.
Taken together, these data indicate that wounds in diabetic mice contain higher levels of proliferating Ly6C+ Mo/MΦ compared to their non-diabetic counterparts.
Single cell RNA sequencing analysis indicates elevated expression of cell cycle genes in a specific cluster of Mo/MΦ populated primarily by cells from diabetic wounds
To determine whether enhanced Mo/MΦ proliferation in skin wounds of diabetic mice is associated with altered expression of cell cycle genes in these cells, we performed single cell RNA sequencing (scRNAseq) analysis on Mo/MΦ isolated from wounds on day 6 post-injury, which we identified as the peak of proliferation within our time course.
Mo/MΦ from skin wounds of diabetic and non-diabetic mice were pre-enriched as live Ly6G-CD45+CD11b+ by FACS sorting before capturing. Data were pre-filtered which resulted in a total pool of 2904 cells (Diabetic: 1767 vs Non-diabetic: 1137). Using k-means clustering analysis on the combined pool of cells, we identified 4 clusters of Mo/MΦ. As shown in Fig 2A -2C, cluster 1 was enriched in cells from non-diabetic wounds with relatively high expression levels of several NLRP3-inflammasome related genes, such as Cxcl2, Nlrp3 and Il1b (17, 18), along with other genes associated with inflammation such as Clec4d, Ets2, Vegfa, and Thbs1 (19–23). Cluster 2 contained similar levels of cells from both strains, but most genes were expressed only at low levels, suggesting an inactive cell population. In contrast, cluster3, which was enriched in cells from diabetic mice, expressed genes associated with anti-inflammatory functions including Coro1a, Cst3, Ly86 and Pld4 (24–28), as well as markers for other myeloid cells, such as Cd52 and H2-DMa (29–31). Importantly, cluster 4, which was populated primarily by cells from diabetic mice, contained cells that expressed high levels of several genes related to cell proliferation, apoptosis, oxidative stress, energy homeostasis and acute inflammation, such as Ppia, mt-Nd1, Pfn1, Calm2, Pltp and Prdx1 (32–39). Thus cluster 4 may represent a subset of pro-inflammatory Mo/MΦ undergoing proliferation and/or apoptosis that warranted further investigation. Interestingly, differential gene expression analysis indicated that common M1 and M2 markers were not specifically expressed in any one cluster and most clusters co-expressed M1 and M2 markers (Supplementary Fig 3), indicating that these clusters do not fit nicely into the M1/M2 classification scheme.
Figure 2. Single cell RNA sequencing analysis indicates elevated expression of cell cycle genes in a specific cluster of Mo/MΦ populated primarily by cells from diabetic wounds.
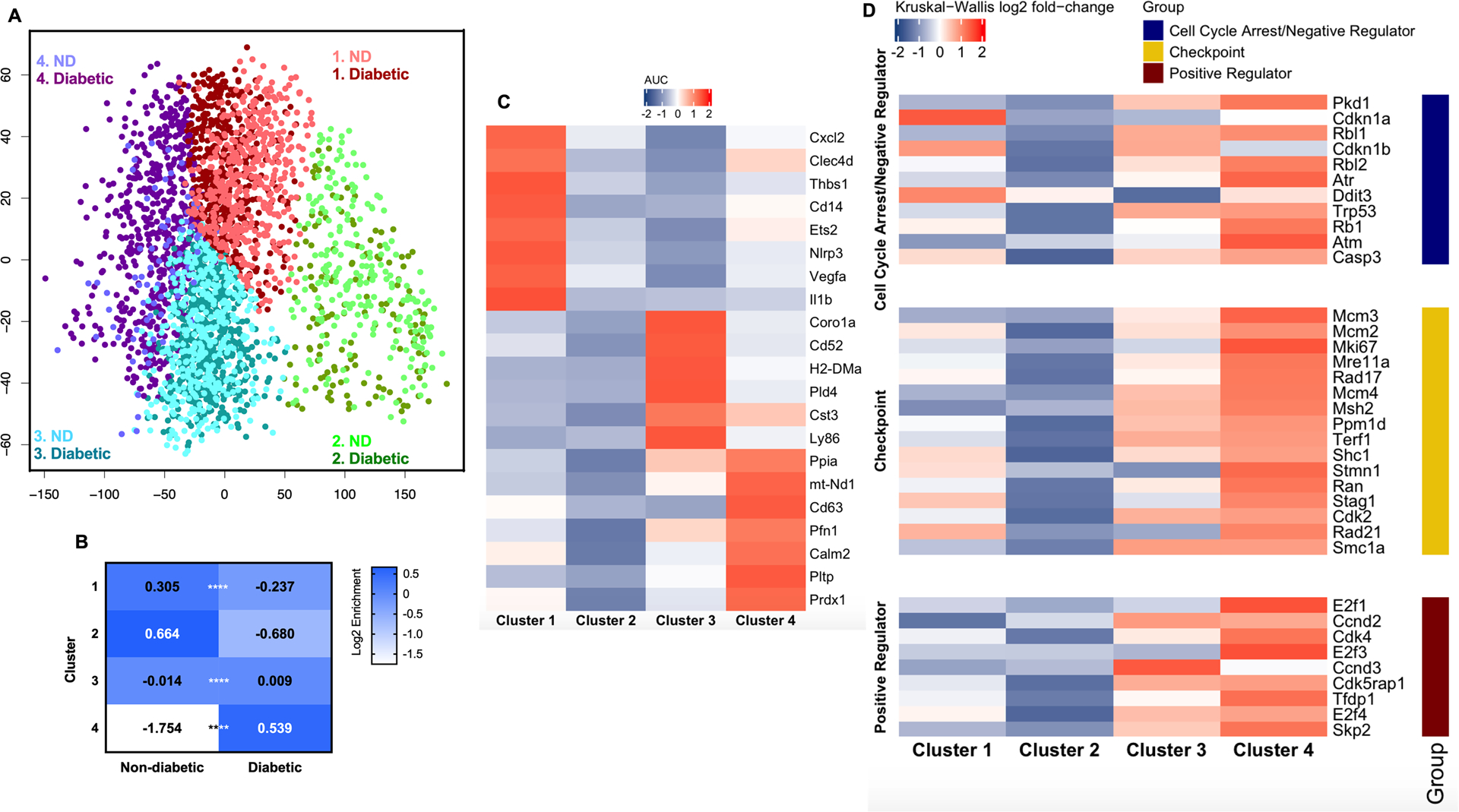
(A) Principal component analysis of single cell RNA sequencing data using Mo/MΦ isolated from day 6 skin wounds from non-diabetic (ND) and diabetic mice reveals four distinct clusters, each with differing composition of cells from each strain. (B) Contributions of cells from each sample to each cluster. In particular, note enrichment of cells from diabetic wounds in cluster 4. (C) Expression of signature genes for each cluster determined as AUC using the ROC curve. (D) Expression of genes associated with progression and regulation of cell cycle within each cluster. Data are presented as log2 fold-changes by Kruskal-Wallis test. ****P < 0.0001 vs corresponding control group by Fisher’s Exact Test.
Next, we compiled a list of genes associated with progression and regulation of the cell cycle from previous publications and analyzed them in our scRNAseq dataset (40). As shown in Fig 2D, the majority of proliferation-related genes were highly expressed in cluster 4, which was significantly enriched in cells from diabetic mice (diabetic 88% vs non-diabetic 12%). There was significant upregulation of checkpoint genes of S phase (Mcm2, Mcm3, Mcm4, Mik67, Mre11a, Msh2 and Rad17), G2 phase or G2/M Transition (Ppm1d) and M phase (Cdk2, Rad21, Ran, Shc1, Smc1a, Stag1, Stmn1 and Terf1). Moreover, expression of most cell cycle-promoting genes was also enhanced in cluster 4, including Skp2, Cdk4, E2f3 and Tfdp1. Interestingly, along with the upregulation of these positive regulator genes of the cell cycle, genes associated with cell cycle arrest/negative regulation were also upregulated, suggesting that feedback inhibition may be taking place simultaneously with the promotion of proliferation in this population.
Taken together, the scRNAseq data revealed four Mo/MΦ populations with different phenotypes on day 6 post-wounding, of which cluster 4 was populated primarily by cells from diabetic mice and showed high expression of genes related to cell cycle, consistent with enhanced proliferation of wound Mo/MΦ in diabetic mice.
Mo proliferate in bone marrow in both diabetic and non-diabetic mice
Wound Mo/MΦ are derived primarily from infiltrated blood monocytes, which in turn, are generated in the bone marrow (3). Thus, we explored Mo proliferation in bone marrow and peripheral blood during wound healing. We first assessed two Mo populations in bone marrow -- Ly6G-CD11b+cKit-CD115+Ly6Chi and Ly6G-CD11b+cKit-CD115+Ly6Clo cells (Fig 3A). Consistent with previous reports (5, 6), Ly6Chi cells were the dominant Mo population in bone marrow in both mouse strains (Fig 3B). At steady state, diabetic and non-diabetic mice had comparable levels of Ly6Chi Mo at baseline, while injury increased these cells in diabetic mice on day 3 post-wounding, resulting in a significantly higher percentage compared to the non-diabetic controls. Meanwhile, levels of mature Ly6Clo Mo remained stable and did not differ between mouse strains throughout the healing process (Fig 3B).
Figure 3. Mo proliferate in bone marrow but not in peripheral blood in diabetic and non-diabetic mice.

(A) Gating strategy for assessing proliferation of Ly6Chi Mo and Ly6Clo Mo in bone marrow. (B) Percentages of total Ly6Chi vs Ly6Clo Mo in bone marrow in non-diabetic (black bar) and diabetic (open bar) mice. (C) Percentages of proliferating Ki67+FxCycle+ cells (S/G2/M phases) in Ly6Chi and Ly6Clo Mo in bone marrow. (D) Gating strategy for assessing proliferation of Ly6Chi Mo and Ly6Clo Mo in peripheral blood. (E) Percentages of total Ly6Chi vs Ly6Clo Mo in peripheral blood in non-diabetic (black bar) and diabetic (open mice). (F) Percentages of Ki67-FxCycle- cells (G0 phase, black), Ki67+FxCycle- cells (G1 phase, grey), and Ki67+FxCycle+ cells (S/G2/M phases, empty) in Ly6Chi Mo and Ly6Clo Mo in peripheral blood in non-diabetic (left) and diabetic (right) mice. Data are mean ± SEM; n= 4–8 mice/group; *P < 0.05 or **P < 0.01 diabetic vs non-diabetic groups by ANOVA.
Next, at baseline, both Ly6Chi and Ly6Clo Mo proliferate in bone marrow with larger percentages of Ly6Chi cells in the S/G2/M phases (Ki67+FxCycle+) compared to Ly6Clo cells. In response to skin injury, both diabetic and non-diabetic mice showed a trend towards lower percentages of Mo in S/G2/M phases in bone marrow on day 3 post-wounding, then slowly recovered toward baseline levels on day 10. However, there were no significant differences between diabetic and non-diabetic mice in the percentage of Ly6Chi Mo in S/G2/M phases at any time point. Diabetic mice tended to have lower percentages of Ly6Clo Mo in S/G2/M phases compared to non-diabetic mice, and this difference reached statistical significance on day 6 post-wounding (Fig 3C, Supplementary Fig 2A and 2B).
Mo do not proliferate in peripheral blood of either diabetic or non-diabetic mice
At baseline, percentages of peripheral blood Ly6Chi and Ly6Clo Mo did not differ between diabetic and non-diabetic mice. However, after injury, diabetic mice maintained relatively high percentages of Ly6Chi Mo throughout the healing process, while non-diabetic mice showed a decrease especially on day 3 post-injury, which resulted in significantly lower percentages of Ly6Chi Mo in non-diabetic compared to diabetic mice on day 3 and day 6 (Fig 3E). Different from skin wounds and bone marrow, virtually no blood Mo were found in S/G2/M phases of the cell cycle in either diabetic or non-diabetic mice; these cells were found primarily in the G1 phase (Ki67+FxCcyle-) (Fig 3F and Supplementary Fig 2C).
Thus, whereas bone marrow Mo actively proliferate before and after skin wounding, mobilization into peripheral blood results in transition to the G1 phase. Subsequent infiltration into the wound then triggers reentry into the S/G2/M phases. Combined, these data suggest that environmental factors drive the proliferation of Mo/MΦ in diabetic skin wounds.
Wound environment promotes Mo/MΦ proliferation
To establish the importance of the wound environment in driving Mo/MΦ proliferation, we used a local Mo adoptive transfer approach that involved isolating Ly6Chi Mo from bone marrow of CD45.1 congenic mice and injecting into wounds of diabetic and non-diabetic mice that express CD45.2 on day 2 or day 5 post-injury. The proliferative status of the transferred cells was then assessed by flow cytometry 18 hours later on day 3 or day 6 post-injury.
Importantly, Ly6Chi Mo transferred to day 6 diabetic wounds showed a significantly higher percentage of cells in the S/G2/M as compared to those transferred to non-diabetic wounds on day 6 post-injury while having comparable percentages on day 3 post-injury (Fig 4B). Interestingly, some Ly6Chi Mo differentiated to Ly6C- Mo/MΦ in the wound environment (Supplementary Fig 4A) and those Ly6Chi Mo derived Ly6C-F4/80+ MΦ in skin wounds maintained some proliferative capability as percentages of these latter cells in S/G2/M phases were higher than what we previously found in skin wounds (Fig 1D and 4B). In short, these experiments confirm that wound environmental factors likely contribute to the increased Mo/MΦ proliferation in diabetic mice.
Figure 4. Wound environment promotes Mo/MΦ proliferation.
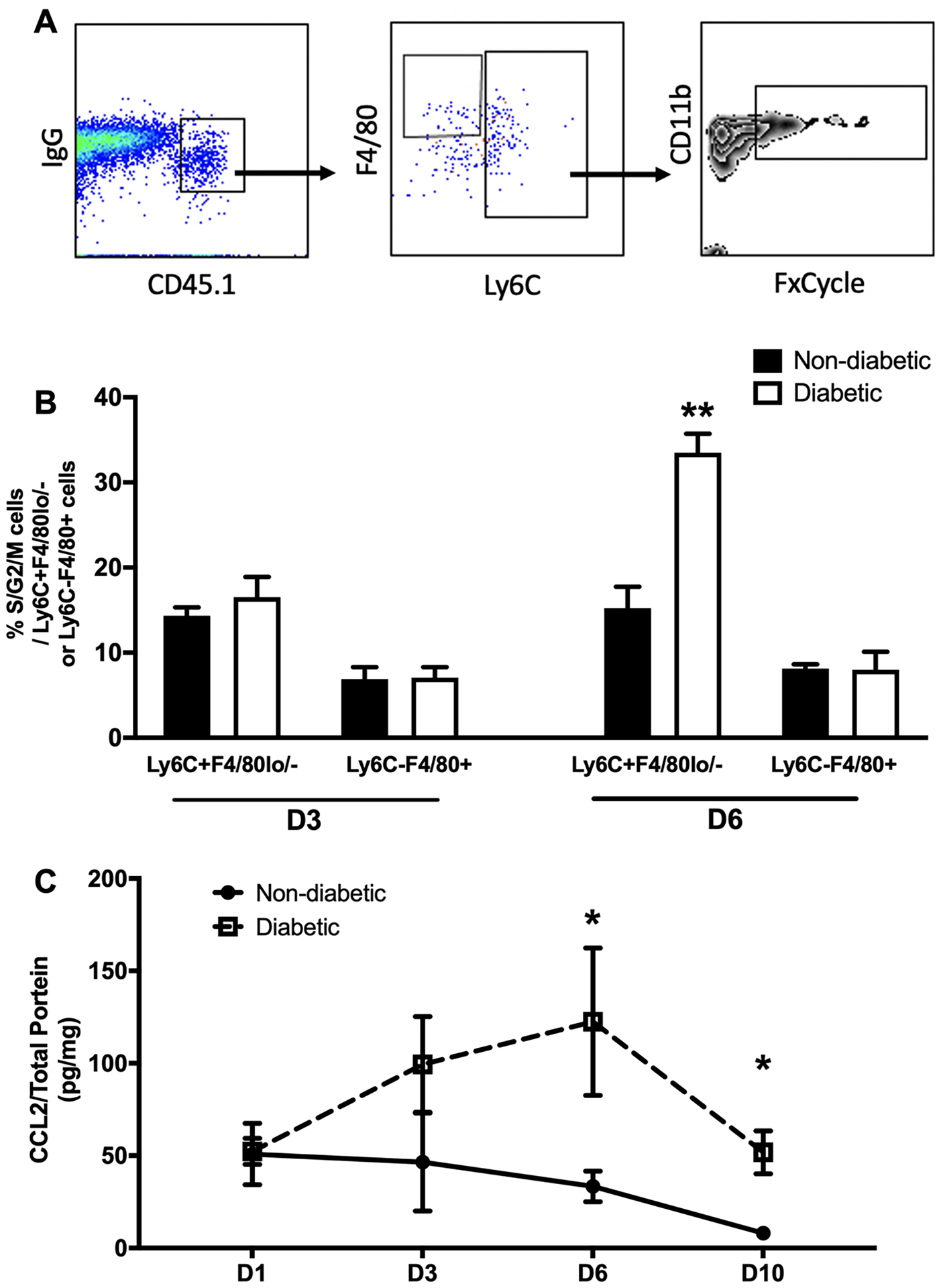
(A) Adoptive transfer of equal numbers of Ly6Chi Mo obtained from non-diabetic mice into wounds of non-diabetic and diabetic mice shows that diabetic wound environment mediates proliferation of these cells in wounds on day 6 post-injury (n = 3–4/group). Gating strategy for assessing proliferation of donor Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ in skin wounds on day 3 and 6 post-injury. (B) Higher percentages of proliferating cells in S/G2/M phases of cell cycle on day 6 post-injury when cells were transferred into wounds of diabetic mice compared to those of non-diabetic mice. (C) Protein levels of CCL2 measured via custom multiplex kit via flow cytometry using homogenates of wounds harvested on day 1 (n=3), 3 (n=4), 6 (n=4), and 10 (n=4). Data are mean ± SEM; n= 4–8 mice/group; *P < 0.05 or **P < 0.01 diabetic vs non-diabetic groups by ANOVA.
Search for potential regulators of proliferation in skin wound environment
We previously screened a number of cytokines which have been reported to play a role in regulation of Mo/MΦ proliferation in other tissues and pathological conditions, such as IL-6, IL-4, M-CSF, and IL-1ß (6). However, our data did not support a role for any of these cytokines in regulation of Mo/MΦ proliferation in skin wounds. Here, we expanded our screen to other potential regulators of proliferation including TNF-α, IL-10, CCL2, CCL3, and IL-33 which have been reported regulating proliferation or other functions of Mo/MΦ (6–9, 41).
Among the proteins screened, we found that levels of CCL2, which is known to recruit Mo/MΦ to damaged tissue, was significantly upregulated post-wounding and higher in wound homogenates from diabetic mice on day 6 and day 10 compared to non-diabetic controls (Fig 4C). In contrast, although skin injury altered levels of TNF-α , IL-10, CCL3, IL-33, M-CSF, and IL-4 in wounds, there were no significant differences between non-diabetic and diabetic mice (Supplementary Fig 4B–4G).
CCL2 promotes Mo/MΦ proliferation in skin wounds
To explore whether CCL2 promotes Mo/MΦ proliferation during wound healing, we treated wounds in non-diabetic mice with a single dose of 60ng CCL2 or equal volume of PBS on day 2 post-injury and assessed Mo/MΦ proliferation 24 hours later. As expected, CCL2 treatment induced a 1.6-fold higher accumulation of Mo/MΦ (CD11b+Ly6G-) in the wounds compared to PBS treatment (not shown). Importantly, in response to CCL2 stimulation, Ly6C+F4/80lo/- Mo/MΦ showed a significant increase in the percentage of cells in S/G2/M phases compared to PBS treatment (Fig 5B).
Figure 5. CCL2 promotes Mo/MΦ proliferation in skin wounds.

(A) and (B) Local treatment of 60ng CCL2 recombinant protein on day 2 post-injury upregulated percentage of proliferating Ly6C+F4/80lo/- Mo/MΦ in S/G/M phase of cell cycle compared to those treated with PBS; cells collected and analyzed 18 hours after treatment (n = 12 / group). (C) Adoptive transfer of equal numbers of Ly6Chi Mo obtained from CCR2−/−EGFP and CCR2+/+CD45.1 mice into wounds of wild-type C57Bl/6 mice shows that CCR2 mediates proliferation of these cells in wounds. Gating strategy for assessing proliferation of donor (CCR2−/−EGFP and CCR2+/+CD45.1) Ly6C+F4/80lo/- Mo/MΦ and Ly6C-F4/80+ MΦ in skin wounds on day 3 post-injury. (D) Baseline levels of proliferating Ly6Chi Mo in S/G2/M phases in bone marrow (left) were comparable between CCR2−/− and CCR2+/+ mice, while 18h after transfer into skin wounds, CCR2+/+ cells showed increased proliferation whereas CCR2−/− cells did not and had a significantly lower percentage of cells in S/G2/M phases compared to CCR2+/+ controls (n = 5 /group). Data are mean ± SEM; **P < 0.01 or ****P < 0.0001 vs corresponding control group by Mann–Whitney test.
Next, since CCR2 is the major receptor for CCL2 signaling, we performed adoptive transfer experiments with Ly6Chi Mo from CCR2−/− EGFP mice, in which an EGFP sequence was inserted into the translation initiation site of the Ccr2 gene thereby abolishing Ccr2 gene expression, to investigate the effect of loss of CCL2/CCR2 signaling on wound Mo/MΦ proliferation. Ly6Chi Mo were collected from bone marrow from CCR2−/− EGFP and CD45.1 (control CCR2+/+) mice, and equal numbers from each donor strain were transferred to wounds of C57BL6 mice on day 2 post-injury. Proliferation of both CCR2−/− and CCR2+/+ Ly6Chi Mo were then analyzed at 18 hours post-transfer. As shown in Fig 5D, live CCR2−/− or CD45.1+ cells were both observed in the wounds of recipient mice, indicating successful tracking of these cells. At baseline prior to transfer, percentages of bone marrow Ly6Chi Mo in S/G2/M phases were comparable between CCR2−/− and CCR2+/+ mice. However, after adoptive transfer to wounds, CCR2+/+ cells showed a significant increase in the percentage in S/G2/M phases, whereas CCR2−/− cells did not, indicating that proliferation was blocked by the loss of CCR2 signaling (Fig 5D).
In summary, our data demonstrate that the wound environment contributes to increased proliferation of Ly6C+ Mo/MΦ post-injury and that CCL2/CCR2 signaling is a key factor in promoting Mo/MΦ proliferation during skin wound healing in mice.
Discussion
We investigated whether factors in the diabetic wound environment enhance Mo/MΦ proliferation thereby contributing to persistent accumulation of these cells and impaired healing. Our data indicate that diabetic mice indeed exhibit higher rates of proliferation in skin wound Ly6C+F4/80lo/- Mo/MΦ compared to non-diabetic controls. In addition, unbiased analysis of scRNAseq data revealed the presence of a Mo/MΦ cluster that exhibited upregulation of genes associated with progression and regulation of cell cycle. Notably, this Mo/MΦ cluster is populated primarily by cells from diabetic mice. Moreover, consistent with our previous study (6), Ly6Chi Mo proliferate in bone marrow of both diabetic and non-diabetic mice, while in peripheral blood, Ly6Chi Mo are maintained in the G1 phase. Regarding the mechanism underlying increased proliferation in wounds of diabetic mice, diabetic mice showed higher CCL2 protein levels in skin wounds compared to non-diabetic controls and local wound treatment in non-diabetic mice with CCL2 protein increased proliferation of Ly6C+F4/80lo/- Mo/MΦ. Importantly, adoptive transfer experiments using Ly6Chi Mo lacking CCR2 showed that CCL2/CCR2 signaling is required for proliferation of these cells in the wound environment. Together, these data demonstrate that the diabetic wound environment enhances Mo/MΦ proliferation which likely contributes to persistent accumulation of these cells and impaired healing in diabetic mice. These data also demonstrate a critical role of the CCL2 signaling pathway in driving Mo/MΦ proliferation during skin wound healing, in addition to its known function as a chemokine.
Diabetes leads to impaired wound healing in both humans and in animal models (3, 42). Associated with impaired healing, we found persistently high levels of Ly6C+F4/80lo/- Mo/MΦ in wounds of diabetic mice which are thought to be pro-inflammatory cells (3, 42), while mature Ly6C-F4/80+ MΦ levels were comparable between diabetic vs non-diabetic mice. Several previous studies demonstrated that mature, resident MΦ can proliferate, contributing to their accumulation in in lung, aorta, peritoneal cavity and adipose tissue (7–9, 43). In contrast, we previously reported that inflammatory Ly6C+F4/80lo/- Mo/MΦ but not mature Ly6C-F4/80+ MΦ proliferate in skin wounds and in the current study, we demonstrate that wounds in diabetic mice contained persistently higher levels of proliferating Ly6C+F4/80lo/- Mo/MΦ compared to non-diabetic controls. We also provide evidence that skin Ly6C+F4/80lo/- Mo/MΦ exhibit a more monocyte-like morphology than Ly6C-F4/80+ MΦ cells. Dixit et al. and Rolot et al. have also reported that Ly6C+ Mo/MΦ proliferate in a unitary tract infection model and hepatic S. mansoni infection model in mice, respectively (6, 10, 44). Thus, the proliferation of different Mo/MΦ subsets may depend on tissue and/or pathological context.
We and others have reported that transition from a pro-inflammatory Mo/MΦ phenotype to a pro-healing phenotype is critical for normal wound healing, while diabetes leads to dysregulation of this process that leads to persistent accumulation of pro-inflammatory Mo/MΦ and impaired healing (3, 45). Single cell RNA sequencing allows for a more comprehensive and unbiased description of cell heterogeneity than methods used previously by us and other to characterize Mo/MΦ phenotypes. Therefore, we conducted scRNAseq analysis in live Ly6G-CD45+CD11b+ cells isolated from wounds of diabetic and non-diabetic mice at the time point of peak wound Mo/MΦ proliferation. Using k-means clustering cells were separated into four clusters, but no cluster exhibited clear M1 or M2 phenotypes, indicating that this classification system is not sufficient to characterize skin wound Mo/MΦ. Nonetheless, cells in cluster 4 exhibited high expression of genes related to checkpoints and regulators of cell cycle, consistent with an actively proliferating Mo/MΦ population. Notably, this cluster was characterized by higher abundance of cells from diabetic wounds compared to non-diabetic controls. Together, data from flow cytometry and scRNAseq demonstrated that a subset of Mo/MΦ proliferate in skin wounds and that this subset in increased in diabetic mice.
Importantly, we identified a novel regulator of Ly6C+ Mo/MΦ proliferation in skin wounds. First, we confirmed our previous suggestion that the wound environment regulates Mo/MΦ proliferation (6) using adoptive transfer of Ly6Chi Mo from non-diabetic mice; non-diabetic Mo proliferated at a higher rate when transferred into wounds of diabetic mice than when transferred into wounds of non-diabetic mice. As our previous data did not support a role for IL-1ß, IL-6, IL-4, nor M-CSF in regulating proliferation of Mo/MΦ in skin wounds (6), we expanded our search to other cytokines/chemokines that play important roles in regulating Mo/MΦ during wound healing including TNF-α, IL-10, CCL2, CCL3 and IL-33.
Among the cytokines we screened, CCL2 is a well-characterized chemokine that attracts Mo/MΦ to inflammatory sites including skin wounds (15, 46). In our study, CCL2 protein levels were elevated in skin wounds following injury and diabetic mice had significantly higher CCL2 protein levels in wounds compared to non-diabetic controls. In addition, administration of a single dose of exogenous CCL2 directly to wounds enhanced proliferation of Ly6C+F4/80lo/- Mo/MΦ compared to controls treated with PBS. Interestingly, CCL2 has previously been reported to promote proliferation of adipose tissue F4/80+ MΦ and several other types of cells in mice (9, 47–50). In addition to chemotaxis and proliferation of Mo/MΦ, CCL2 can affect the function of many other major cell types in wounds, including fibroblasts and endothelial cells (51–54). Therefore, to avoid off-target effects of CCL2/CCR2 signaling in our studies, we used adoptive transfer of Ly6Chi Mo from CCR2−/− EGFP mice to skin wounds to determine whether CCL2/CCR2 signaling promotes Mo/MΦ proliferation. Importantly, transferred Ly6Chi Mo lacking CCR2 showed lower proliferation compared to CCR2 competent cells. Together, these data show that CCL2 is a key regulator of Mo/MΦ proliferation during skin wound healing and that persistently high levels of CCL2 and the resulting sustained proliferation of inflammatory Mo/MΦ contribute to chronic inflammation and impaired wound healing in diabetic mice.
Finally, evidence in the literature suggests that wound Mo/MΦ are derived primarily from Ly6Chi Mo produced in the bone marrow, which are mobilized into the blood and then infiltrate into damaged tissue (42). Diabetes leads to alteration of several critical steps in this process including myeloid lineage progenitor bias in bone marrow, which in turn, promote excess accumulation of Mo/MΦ and prolonged inflammation in wounds (5, 42). In the present study, in agreement with previous studies, we detected significantly higher levels of Ly6Chi Mo in bone marrow and peripheral blood of diabetic mice following skin injury (5, 42). Moreover, similar to what we reported previously for non-diabetic mice, Ly6Chi Mo proliferated in bone marrow but not in peripheral blood of diabetic mice (6). However, proliferation of bone marrow Ly6Chi Mo did not differ between diabetic and non-diabetic mice. Combined with our previous studies, our data suggests that diabetes affects myelopoiesis but not Mo proliferation in bone marrow.
One limitation of this study is that we used a single time point for the scRNAseq analysis. Therefore, we are planning future studies to generate a time course of scRNAseq data for Mo/MΦ to produce a more comprehensive picture of Mo/MΦ characteristics over the course skin wound healing. Once we have an unbiased atlas of Mo/MΦ phenotypes over the course of healing, we can start to better understand the regulation of cell cycle in these cells as well as other cellular functions. Another limitation is that other molecules in addition to CCL2 could promote monocyte/macrophage proliferation in wounds. Although we did not detect significant differences of either IL-4 nor M-CSF levels in skin wound homogenates between non-diabetic and diabetic mice and these cytokines did not increase after injury, previous studies have implicated these cytokines in MΦ proliferation (7) and lack of differences in measured protein levels does not necessarily mean lack of a role in MΦ proliferation. Finally, our study did not distinguish between resident vs infiltrated Mo/MΦ in the skin wound, though current understanding states that Mo/MΦ accumulate in skin wounds mainly by the recruitment of inflammatory Ly6C+ Mo from the circulation (55).
In conclusion, we have demonstrated that Ly6C+ Mo/MΦ proliferate at a higher rate in wounds of diabetic compared to their non-diabetic counterparts. Moreover, we demonstrate that CCL2, known primarily for its chemoattractant function, promotes Ly6C+ MΦ /Mo proliferation and sustained high levels of CCL2 in diabetic wounds likely drives proliferation of these inflammatory cells, contributing to chronic inflammation and impaired healing. Therefore, future studies should focus on potential for inhibiting the enhanced Ly6C+ Mo/MΦ proliferation in diabetic wounds as a potential therapeutic approach to dampen chronic inflammation and improve healing of diabetic wounds.
Supplementary Material
Key points.
Mo/MΦ proliferate at higher rates in wounds of diabetic compared to non-diabetic mice
CCL2/CCR2 signaling promotes wound Mo/MΦ proliferation in mice
Acknowledgements
All authors thank Dr. Luisa A. DiPietro and Dr. Giamila Fantuzzi for their input on aspects of presentation of this manuscript.
Funding statement
This study was supported by NIGMS grants R01GM092850 and R35GM136228 to TJK. MMC supported in part by NCATS through Grant UL1TR002003.
Footnotes
Competing interests
All authors have no financial conflicts of interest to report.
References
- 1.Magliano DJ, Islam RM, Barr ELM, Gregg EW, Pavkov ME, Harding JL, Tabesh M, Koye DN, and Shaw JE. 2019. Trends in incidence of total or type 2 diabetes: systematic review. BMJ 366: l5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geraghty T, and LaPorta G. 2019. Current health and economic burden of chronic diabetic osteomyelitis. Expert Rev Pharmacoecon Outcomes Res 19: 279–286. [DOI] [PubMed] [Google Scholar]
- 3.Koh TJ, and DiPietro LA. 2011. Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med 13: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, Schmidt AM, Orchard TJ, Fisher EA, Tall AR, and Goldberg IJ. 2013. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 17: 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barman PK, Urao N, and Koh TJ. 2019. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J Pathol 249: 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang J, Urao N, and Koh TJ. 2019. Proliferation of Ly6C+ monocytes/macrophages contributes to their accumulation in mouse skin wounds. J Leukoc Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, Maizels RM, Hume DA, and Allen JE. 2013. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med 210: 2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, and Merad M. 2013. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, and Aouadi M. 2014. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixit A, Bottek J, Beerlage AL, Schuettpelz J, Thiebes S, Brenzel A, Garbers C, Rose-John S, Mittrucker HW, Squire A, and Engel DR. 2018. Frontline Science: Proliferation of Ly6C(+) monocytes during urinary tract infections is regulated by IL-6 trans-signaling. J Leukoc Biol 103: 13–22. [DOI] [PubMed] [Google Scholar]
- 11.Ridiandries A, Tan JTM, and Bursill CA. 2018. The Role of Chemokines in Wound Healing. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin H, Li X, Hu S, Liu T, Yuan B, Gu H, Ni Q, Zhang X, and Zheng F. 2013. IL-33 accelerates cutaneous wound healing involved in upregulation of alternatively activated macrophages. Mol Immunol 56: 347–353. [DOI] [PubMed] [Google Scholar]
- 13.Ashcroft GS, Jeong MJ, Ashworth JJ, Hardman M, Jin W, Moutsopoulos N, Wild T, McCartney-Francis N, Sim D, McGrady G, Song XY, and Wahl SM. 2012. Tumor necrosis factor-alpha (TNF-alpha) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen 20: 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MD, McCarthy DJ, and Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood S, Jayaraman V, Huelsmann EJ, Bonish B, Burgad D, Sivaramakrishnan G, Qin S, DiPietro LA, Zloza A, Zhang C, and Shafikhani SH. 2014. Pro-inflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response. PLoS One 9: e91574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, Behl Y, Alikhani M, Yang J, Braasch C, and Graves DT. 2010. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 53: 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boro M, and Balaji KN. 2017. CXCL1 and CXCL2 Regulate NLRP3 Inflammasome Activation via G-Protein-Coupled Receptor CXCR2. J Immunol 199: 1660–1671. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Hara H, and Nunez G. 2016. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M, Dennehy KM, Hornsell WG, Guler R, Campanero-Rhodes MA, Palma AS, Feizi T, Kim SK, Sobieszczuk P, Willment JA, and Brown GD. 2012. The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem 287: 25964–25974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao M, Zhang J, Chen W, and Chen W. 2018. M1-like tumor-associated macrophages activated by exosome-transferred THBS1 promote malignant migration in oral squamous cell carcinoma. J Exp Clin Cancer Res 37: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Xiong Z, Lechner EJ, Klenotic PA, Hamburg BJ, Hulver M, Khare A, Oriss T, Mangalmurti N, Chan Y, Zhang Y, Ross MA, Stolz DB, Rosengart MR, Pilewski J, Ray P, Ray A, Silverstein RL, and Lee JS. 2014. Thrombospondin-1 triggers macrophage IL-10 production and promotes resolution of experimental lung injury. Mucosal Immunol 7: 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou D, Huang X, Xie Y, Deng Z, Guo J, and Huang H. 2019. Astrocytes-derived VEGF exacerbates the microvascular damage of late delayed RBI. Neuroscience 408: 14–21. [DOI] [PubMed] [Google Scholar]
- 23.Boulukos KE, Pognonec P, Sariban E, Bailly M, Lagrou C, and Ghysdael J. 1990. Rapid and transient expression of Ets2 in mature macrophages following stimulation with cMGF, LPS, and PKC activators. Genes Dev 4: 401–409. [DOI] [PubMed] [Google Scholar]
- 24.Kim YI, Shin HW, Chun YS, Cho CH, Koh J, Chung DH, and Park JW. 2018. Epithelial cell-derived cytokines CST3 and GDF15 as potential therapeutics for pulmonary fibrosis. Cell Death Dis 9: 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Schnorrer P, Proietto A, Kowalski G, Febbraio MA, Acha-Orbea H, Dickins RA, and Villadangos JA. 2011. IL-10 controls cystatin C synthesis and blood concentration in response to inflammation through regulation of IFN regulatory factor 8 expression. J Immunol 186: 3666–3673. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki K, Takeshita F, Nakata N, Ishii N, and Makino M. 2006. Localization of CORO1A in the macrophages containing Mycobacterium leprae. Acta Histochem Cytochem 39: 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X, Kong B, Shuai W, Shen C, Yang F, Fu H, and Huang H. 2019. Loss of MD1 exacerbates myocardial ischemia/reperfusion injury and susceptibility to ventricular arrhythmia. Eur J Pharmacol 844: 79–86. [DOI] [PubMed] [Google Scholar]
- 28.Gavin AL, Huang D, Huber C, Martensson A, Tardif V, Skog PD, Blane TR, Thinnes TC, Osborn K, Chong HS, Kargaran F, Kimm P, Zeitjian A, Sielski RL, Briggs M, Schulz SR, Zarpellon A, Cravatt B, Pang ES, Teijaro J, de la Torre JC, O’Keeffe M, Hochrein H, Damme M, Teyton L, Lawson BR, and Nemazee D. 2018. PLD3 and PLD4 are single-stranded acid exonucleases that regulate endosomal nucleic-acid sensing. Nat Immunol 19: 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rashidi M, Bandala-Sanchez E, Lawlor KE, Zhang Y, Neale AM, Vijayaraj SL, O’Donoghue R, Wentworth JM, Adams TE, Vince JE, and Harrison LC. 2018. CD52 inhibits Toll-like receptor activation of NF-kappaB and triggers apoptosis to suppress inflammation. Cell Death Differ 25: 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Su H, Shen X, Du J, Zhang X, and Zhao Y. 2017. The immunological function of CD52 and its targeting in organ transplantation. Inflamm Res 66: 571–578. [DOI] [PubMed] [Google Scholar]
- 31.Menges PR, Jenks SA, Bikoff EK, Friedmann DR, Knowlden ZA, and Sant AJ. 2008. An MHC class II restriction bias in CD4 T cell responses toward I-A is altered to I-E in DM-deficient mice. J Immunol 180: 1619–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue C, Sowden M, and Berk BC. 2017. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler Thromb Vasc Biol 37: 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun S, Guo M, Zhang JB, Ha A, Yokoyama KK, and Chiu RH. 2014. Cyclophilin A (CypA) interacts with NF-kappaB subunit, p65/RelA, and contributes to NF-kappaB activation signaling. PLoS One 9: e96211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Zhai Q, Bharadwaj U, Wang H, Li F, Fisher WE, Chen C, and Yao Q. 2006. Cyclophilin A is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer 106: 2284–2294. [DOI] [PubMed] [Google Scholar]
- 35.Wallace L, Mehrabi S, Bacanamwo M, Yao X, and Aikhionbare FO. 2016. Expression of mitochondrial genes MT-ND1, MT-ND6, MT-CYB, MT-COI, MT-ATP6, and 12S/MT-RNR1 in colorectal adenopolyps. Tumour Biol 37: 12465–12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pae M, and Romeo GR. 2014. The multifaceted role of profilin-1 in adipose tissue inflammation and glucose homeostasis. Adipocyte 3: 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu J, Shi D, Ding M, Huang T, Gu R, Xiao J, Xian CJ, Dong J, Wang L, and Liao H. 2019. Calmodulin-dependent signalling pathways are activated and mediate the acute inflammatory response of injured skeletal muscle. J Physiol 597: 5161–5177. [DOI] [PubMed] [Google Scholar]
- 38.Desrumaux C, Lemaire-Ewing S, Ogier N, Yessoufou A, Hammann A, Sequeira-Le Grand A, Deckert V, Pais de Barros JP, Le Guern N, Guy J, Khan NA, and Lagrost L. 2016. Plasma phospholipid transfer protein (PLTP) modulates adaptive immune functions through alternation of T helper cell polarization. Cell Mol Immunol 13: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong SJ, Kim S, Park JG, Jung IH, Lee MN, Jeon S, Kweon HY, Yu DY, Lee SH, Jang Y, Kang SW, Han KH, Miller YI, Park YM, Cheong C, Choi JH, and Oh GT. 2018. Prdx1 (peroxiredoxin 1) deficiency reduces cholesterol efflux via impaired macrophage lipophagic flux. Autophagy 14: 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiagen. 2011. Qiagen RT2 Profile PCR Array Mouse Cell Cycle. Qiagen, Germantown, MD. Available at: https://geneglobe.qiagen.com/biologygenesapi/genelist/file?catalogNumber=PAMM-020Z. Accessed: September 18, 2019. [Google Scholar]
- 41.Jackson-Jones LH, Ruckerl D, Svedberg F, Duncan S, Maizels RM, Sutherland TE, Jenkins SJ, McSorley HJ, Benezech C, MacDonald AS, and Allen JE. 2016. IL-33 delivery induces serous cavity macrophage proliferation independent of interleukin-4 receptor alpha. Eur J Immunol 46: 2311–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Zhang L, Yu C, Yang XF, and Wang H. 2014. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng C, Yang Q, Cao J, Xie N, Liu K, Shou P, Qian F, Wang Y, and Shi Y. 2016. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis 7: e2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rolot M, M. D. A, Javaux J, Lallemand F, Machiels B, Martinive P, Gillet L, and Dewals BG. 2019. Recruitment of hepatic macrophages from monocytes is independent of IL-4Ralpha but is associated with ablation of resident macrophages in schistosomiasis. Eur J Immunol 49: 1067–1081. [DOI] [PubMed] [Google Scholar]
- 45.Krzyszczyk P, Schloss R, Palmer A, and Berthiaume F. 2018. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front Physiol 9: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isbel NM, Nikolic-Paterson DJ, Hill PA, Dowling J, and Atkins RC. 2001. Local macrophage proliferation correlates with increased renal M-CSF expression in human glomerulonephritis. Nephrol Dial Transplant 16: 1638–1647. [DOI] [PubMed] [Google Scholar]
- 47.Ishida Y, Kuninaka Y, Nosaka M, Furuta M, Kimura A, Taruya A, Yamamoto H, Shimada E, Akiyama M, Mukaida N, and Kondo T. 2019. CCL2-Mediated Reversal of Impaired Skin Wound Healing in Diabetic Mice by Normalization of Neovascularization and Collagen Accumulation. J Invest Dermatol 139: 2517–2527 e2515 . [DOI] [PubMed] [Google Scholar]
- 48.Boniakowski AE, Kimball AS, Joshi A, Schaller M, Davis FM, denDekker A, Obi AT, Moore BB, Kunkel SL, and Gallagher KA. 2018. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur J Immunol 48: 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinojosa AE, Garcia-Bueno B, Leza JC, and Madrigal JL. 2011. CCL2/MCP-1 modulation of microglial activation and proliferation. J Neuroinflammation 8: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macanas-Pirard P, Quezada T, Navarrete L, Broekhuizen R, Leisewitz A, Nervi B, and Ramirez PA. 2017. The CCL2/CCR2 Axis Affects Transmigration and Proliferation but Not Resistance to Chemotherapy of Acute Myeloid Leukemia Cells. PLoS One 12: e0168888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gharaee-Kermani M, Denholm EM, and Phan SH. 1996. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem 271: 17779–17784. [DOI] [PubMed] [Google Scholar]
- 52.Weber KS, Nelson PJ, Grone HJ, and Weber C. 1999. Expression of CCR2 by endothelial cells : implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol 19: 2085–2093. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto T, Eckes B, Mauch C, Hartmann K, and Krieg T. 2000. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol 164: 6174–6179. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto T, and Nishioka K. 2003. Role of monocyte chemoattractant protein-1 and its receptor,CCR-2, in the pathogenesis of bleomycin-induced scleroderma. J Invest Dermatol 121: 510–516. [DOI] [PubMed] [Google Scholar]
- 55.Burgess M, Wicks K, Gardasevic M, and Mace KA. 2019. Cx3CR1 Expression Identifies Distinct Macrophage Populations That Contribute Differentially to Inflammation and Repair. Immunohorizons 3: 262–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA sequencing data in this study are available in Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) with the accession code GSE154400. All other relevant data are available from the corresponding authors.